

In this review, Hu and Shilatifard summarize recent advances in our understanding of the role of chromatin modifiers in normal hematopoiesis and their contributions in hematopoietic transformation.
Keywords: chromatin, epigenetics, hematopoiesis, histone
Abstract
Hematological malignancies comprise a diverse set of lymphoid and myeloid neoplasms in which normal hematopoiesis has gone awry and together account for ∼10% of all new cancer cases diagnosed in the United States in 2016. Recent intensive genomic sequencing of hematopoietic malignancies has identified recurrent mutations in genes that encode regulators of chromatin structure and function, highlighting the central role that aberrant epigenetic regulation plays in the pathogenesis of these neoplasms. Deciphering the molecular mechanisms for how alterations in epigenetic modifiers, specifically histone and DNA methylases and demethylases, drive hematopoietic cancer could provide new avenues for developing novel targeted epigenetic therapies for treating hematological malignancies. Just as past studies of blood cancers led to pioneering discoveries relevant to other cancers, determining the contribution of epigenetic modifiers in hematologic cancers could also have a broader impact on our understanding of the pathogenesis of solid tumors in which these factors are mutated.
Hematopoiesis is a highly dynamic developmental process requiring both self-renewal and a well-regulated differentiation process of hematopoietic stem cells (HSCs) to maintain the lifelong regeneration of the mammalian blood cells. The ontogeny of the mouse hematopoietic system involves two waves of hematopoiesis during development, beginning with a transient primitive hematopoiesis, which originates from the embryonic mesoderm and progresses to the extraembryonic yolk sac to produce primitive erythrocytes and some myeloid cells around embryonic day 7.5 (E7.5) of mouse development (Medvinsky et al. 2011). After the first wave of primitive hematopoiesis, HSCs are generated in the aorta–gonad–mesonephros (AGM) around E9.5, resulting in a second wave of definitive hematopoiesis, which contributes to all hematopoietic lineages found in the fetus and adult mice. As embryonic development progresses, HSCs colonize the fetal liver around E12.5 and, shortly before birth, migrate to the bone marrow, where they reside throughout life (Fig. 1A; Moore and Metcalf 1970; Lux et al. 2008; Orkin and Zon 2008; Baron et al. 2012; Cullen et al. 2014). In mammalian adults, HSCs exist in a relatively quiescent state but retain the capabilities of both self-renewal and multipotency, ensuring their lifelong maintenance in the bone marrow while, through a hierarchical cascade of differentiation, giving rise to all types of phenotypically distinct mature blood cells (Fig. 1B). A combination of extrinsic and intrinsic factors—including niche-associated factors, signal transduction pathways, transcription factors, and chromatin modifiers—contributes to the dynamic equilibrium between self-renewal and the multipotent differentiation potential of HSCs. The disruption and misregulation of these processes have the potential to lead to life-threatening hematological disorders (Li and Clevers 2010; Doulatov et al. 2012).
Figure 1.

Hematopoietic systems in mammals. (A) Embryonic tissues of mouse hematopoietic development. Mouse hematopoietic activity arises from the mesoderm and first emerges in the extraembryonic yolk sac around E7.5 followed by the establishment of HSCs in AGM and the placenta around E.9.5. HSCs colonize the fetal liver around E12.5, migrate to the bone marrow before birth, and reside there throughout life. (B) Schematic overview of normal hematopoietic hierarchy in adult mice. HSCs sit at the top of the hierarchy and have both the capacity of self-renewal and the multipotent potential to give rise to all mature hematopoietic cell lineages. After receiving a differentiation signal, HSCs first lose self-renewing capacity and then progressively lose lineage potential, as they are restricted to a certain lineage. (LT-HSC) Long-term HSC; (ST-HSC) short-term repopulating HSC; (MPP) multipotent progenitor; (CMP) common myeloid progenitor; (CLP) common lymphoid progenitor; (LMPP) lymphoid-primed multipotent progenitor; (MEP) megakaryocyte/erythroid progenitor; (GMP) granulocyte-macrophage progenitor.
Hematologic malignancies can arise during any stage of blood cell development and can affect the production and function of blood cells with consequences that include an inability to fight off infections or susceptibility to uncontrolled bleeding. The HSCs in the bone marrow can give rise to immature progenitor cells of either the myeloid or lymphoid lineages. The cells of myeloid lineage include erythrocytes, platelets, and the white blood cells of the innate immune response such as neutrophils, eosinophils, dendritic cells, and macrophages, while the lymphoid lineage produces B and T lymphocytes involved in the adaptive immune response (Fig. 1B). Perturbation of normal hematopoietic differentiation can result in three main types of blood cancers in clinic: leukemia, lymphoma, and myeloma. Leukemia is caused by an excessive production of abnormal white blood cells in the bone marrow, resulting in circulating leukemic cells in the blood. Based on the lineage of the neoplastic cells and the clinical course, leukemia can be categorized into acute lymphocytic leukemia (ALL), acute myeloid leukemia (AML), chronic lymphoid leukemia (CLL), and chronic myeloid leukemia (CML).
Lymphomas are derived from a transformation of lymphocytes residing in lymph nodes that has adverse effects on the lymphatic and immune systems. Lymphomas can result from the transformation of B or T lymphocytes or natural killer (NK) cells and can be divided into two main types: Hodgkin's lymphoma and non-Hodgkin's lymphoma (NHL). The majority of NHLs (∼85%) is B-cell lymphomas, which can be further divided based on their appearance (e.g., follicular vs. diffuse) and how quickly they are likely to grow and spread (indolent vs. aggressive). The indolent NHLs mainly are comprised of follicular lymphoma (FL), mantle cell lymphoma, marginal zone lymphoma, small lymphocytic lymphoma, and cutaneous T-cell lymphoma (CTCL). Any of the indolent lymphomas can demonstrate aggressive behavior or a higher-grade transformation. The aggressive NHLs consist of diffuse large B-cell lymphoma (DLBCL), Burkitt's lymphoma, lymphoblastic lymphoma, and various groups of T-cell and NK-cell lymphomas. The third major type of hematologic malignancies are plasmacytic neoplasms in which abnormal antibodies secreting B lymphocytes, called plasma cells, accumulate in the bone marrow or within other tissues. Based on the site of involvement, the disease burden, and the presence of end organ damage, these neoplasms can be divided into several categories, including plasmacytoma (of bone or extramedullary), monoclonal gammopathy of undetermined significance (MGUS), smoldering (asymptomatic) myeloma, and symptomatic plasma cell myeloma.
Myeloid malignancies also include myeloproliferative neoplasms (MPNs) and myelodysplastic syndrome (MDS). MDS is a group of diverse bone marrow disorders characterized by disorderly and ineffective hematopoiesis, which can lead to cytopenia, low levels of red blood cells (anemia), neutrophils (neutropenia), or platelets (thrombocytopenia). These lower cell counts can result from a failure of progenitor cells to mature, thereby accumulating in the bone marrow, or, alternatively, progenitor cells could mature into blood cells with a shortened life span. Seemingly mature blood cells may not function properly due to an abnormal shape (dysplasia). Around 30% of MDS cases have the possibility of turning into AML (Lindberg 2005). MDS itself can arise as an adverse effect of radiation and chemotherapy. MPNs is a group of diseases characterized by the overproduction of one or more blood cell types in the bone marrow and circulating blood (Saeidi 2016). MPN can also further evolve into AML.
The term “epigenetics” was coined by developmental biologist Conrad Waddington to describe heritable changes in the cellular phenotype and gene expression that are independent of alterations in the DNA sequence during embryonic development (Slack 2002; Berger et al. 2009). Classic examples of the epigenetic regulation of gene expression include genomic imprinting, position effect variegation, and the regulation of homeotic (Hox) gene expression. These processes turn out to be regulated by chromatin modifiers implementing DNA methylation for genomic imprinting, methylation of H3 Lys9 for the establishment of heterochromatin, and methylation of histone H3 at Lys4 or Lys27 for the activation or repression of Hox gene expression, respectively (Piunti and Shilatifard 2016). By controlling chromatin architecture and accessibility, modifications of DNA and histones can convey this epigenetic information and influence gene expression through favoring or antagonizing the recruitment of the activating or repressive complexes.
The primary role of the transcription factors such as RUNX1/AML1, EVI-1, GATA3, IKAROS, and ETS in determining the various stages of normal hematopoiesis is reflected in their misregulation being the most common cause of hematopoietic transformation. The past several years have brought about an increased understanding of the biochemical and cellular functions of chromatin-modifying and remodeling enzymes, specifically as coactivators and corepressors for the regulation of transcription during normal hematopoiesis and in the misregulation of their activities related to hematological malignancies. Indeed, high-throughput, genome-scale sequencing has revealed that chromatin modifiers are among the most frequently mutated in cancer in general, particularly in hematological malignancies, suggesting prominent roles played by the epigenetic alterations in these diseases (Lawrence et al. 2014). In contrast to genetic aberrations, epigenetic alterations are generally reversible and thus may have a more therapeutic value from a clinical standpoint. Indeed, inhibitors targeting chromatin-modifying enzymes are being used in clinical trials (Cai et al. 2015; Brien et al. 2016). In this review, we discuss our current understanding of how epigenetic regulators function in normal hematopoiesis and highlight the consequences of mutations in the DNA and histone lysine methylation machineries in hematological malignancies.
MLL in normal hematopoiesis and in the transcriptional elongation checkpoint defect in leukemia
The mixed-lineage leukemia (MLL or KMT2A) gene was originally identified through cytogenetic studies of infant leukemia patient cells as being involved in chromosomal rearrangements that juxtaposed the N terminus of MLL with a variety of translocation partners. Although it was suspected of being involved as a transcriptional regulator based on its homology with Trithorax, a regulator of homeotic gene expression in Drosophila, insights into its biochemical function came from the purification of the closest yeast homolog Set1 [Su(var)3-9, enhancer of zeste, and trithorax domain 1]. These studies demonstrated that simple model systems such as yeast and Drosophila can provide fundamentally important molecular information about conserved biological processes such as transcription and epigenetics that are relevant to hematopoiesis and hematological malignancies.
Fundamental molecular studies in yeast identified Set1 as biochemically residing in a large macromolecular complex that was named the complex of proteins associated with Set1 (COMPASS). This complex harbors methyltransferase activity specifically toward Lys4 of histone H3 (Miller et al. 2001; Krogan et al. 2002; Shilatifard 2012; Piunti and Shilatifard 2016). The human wild-type MLL gene encodes a protein of 3969 amino acids that is post-translationally cleaved by Taspase I into N-terminal and C-terminal fragments (Hsieh et al. 2003; Shilatifard 2012). The two halves of MLL function together in a COMPASS-like complex with core subunits related to those found in yeast COMPASS as well as additional interactors such as the tumor suppressor menin (Fig. 2A; Hsieh et al. 2003; Yokoyama et al. 2004). As in yeast Set1, the C-terminal SET domain confers histone H3K4 methyltransferase activity to MLL (Milne et al. 2002; Nakamura et al. 2002).
Figure 2.
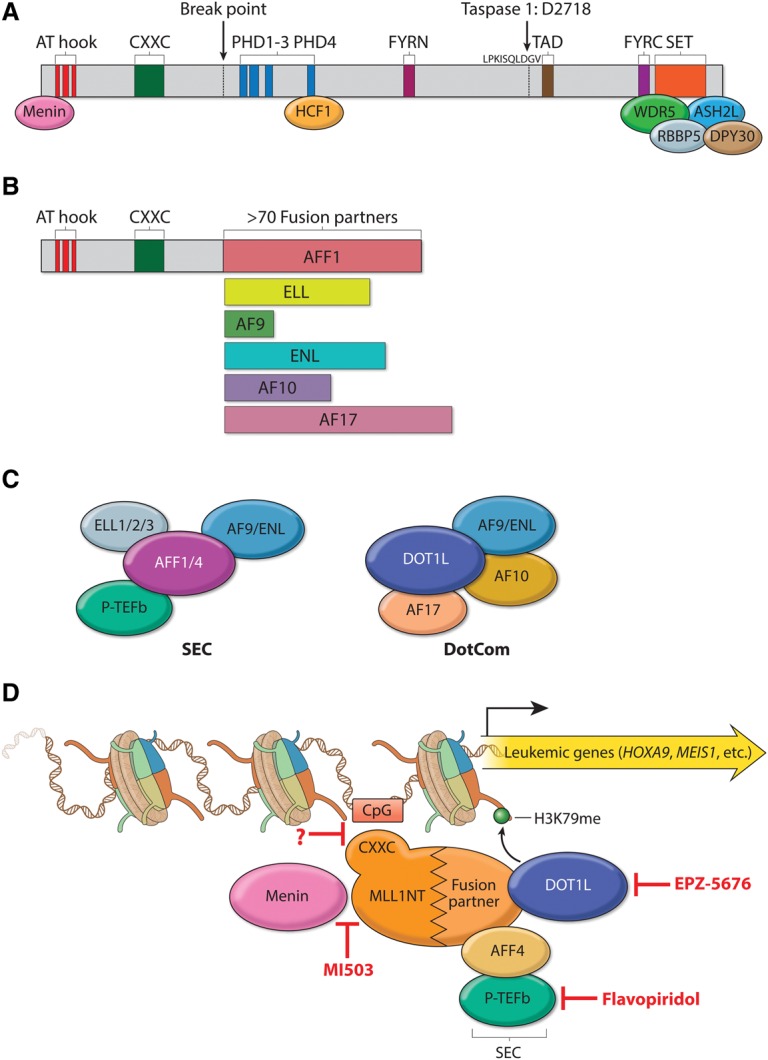
MLL translocation partners function in leukemogenesis. (A) Schematic representation of the domain structure of MLL and its stably associated cofactors in a COMPASS-like complex. MLL contains multiple domains essential for its biochemical and physiological function and is cleaved by Taspase I after aspartic acid 2718 to generate a large N-terminal fragment and a small C-terminal fragment, which subsequently associate noncovalently through FY-rich N-terminal (FYRN) and FY-rich C-terminal (FYRC) domains. (AT hook) Binds to the minor groove of AT-rich DNA sequences; (PHD) Plant homology domain; (SET) Su(var)3-9, enhancer of zeste, and trithorax domain. Core COMPASS subunits WDR5, RBBP5, ASH2L, and DPY30 interact with the SET domain, while Menin and HCF1 associate with more N-terminal regions. (B) Structure of the most frequent MLL chimeras in acute leukemia. A typical MLL fusion protein contains the N terminus of MLL and a C-terminal portion of one of >70 fusion partners. The AT hook and CXXC domains are retained in all MLL chimeras. (C) Two distinct complexes regulating transcription are made up of some of the most frequent MLL fusion partners: SEC (super elongation complex) and DotCom (DOT1L complex). SEC comprises the ELL family members ELL1, ELL2, and ELL3; the MLL translocation partners AF4/FMR2 family (AFF) member 1 (AFF1, also known as AF4) and AFF4; eleven-nineteen leukemia (ENL); the ALL1-fused gene from chromosome 9 (AF9); and the RNA polymerase II (Pol II) elongation factor P-TEFb (containing CDK9 and either cyclin T1 or cyclin T2). DotCom is composed of AF10, AF17, DOT1L, and AF9 or ENL. DOT1L is a methyltransferase specific for H3K79. (D) Therapeutic targeting of MLL translocated leukemia. MLL chimeras recruit SEC and/or DotCom to MLL target genes, which leads to their aberrant activation through misregulation of the transcriptional elongation checkpoint (Smith and Shilatifard 2013). Multiple therapeutic strategies have been developed to target different steps required for the activation of MLL chimera target genes in leukemia: MI503 blocks the association of Menin with MLL, potentially affecting recruitment of the MLL chimera to chromatin (indicated by a question mark); flavopiridol (FP) inhibits the kinase activity of SEC subunit P-TEFb; and EPZ-5676 is a small molecule blocking the enzymatic activity of DOT1L.
Mll (Kmt2a) is required for normal numbers of hematopoietic progenitors. Its deletion in mice causes embryonic lethality at E16.5, with fetal livers having dramatically reduced numbers of HSCs, indicating an essential role for Mll in embryonic hematopoiesis (Hess et al. 1997; Ernst et al. 2004; McMahon et al. 2007). Mll is also essential for sustaining postnatal hematopoiesis, as a conditional deletion in postnatal mice with hematopoietic-specific Vav-Cre leads to a multilineage defect in differentiation and a decrease in adult hematopoietic progenitors, with a fatal bone marrow failure occurring at ∼3 wk of age (Jude et al. 2007; Gan et al. 2010).
Leukemia resulting from MLL translocations follows an aggressive clinical course with a poor response to conventional chemotherapy and often relapses very early. More than 70 translocation partners have been identified, but they share little or no sequence similarities. All of the MLL chimeras retain the N terminus but lose the majority of the C-terminal portion of MLL, which contains the catalytic SET domain for H3K4 methylation (Fig. 2B). However, the molecular mechanisms by which MLL chimeras could contribute to the pathogenesis of leukemia were unknown until the biochemical identification of the translocation partner ELL as a transcription elongation factor for RNA polymerase II (Pol II) (Shilatifard et al. 1996; Shilatifard 1998). ELL was the first MLL translocation partner for which a molecular function was demonstrated. Based on this seminal discovery, it was proposed >20 years ago that the misregulation of transcriptional elongation by RNA Pol II could play a central role in leukemic pathogenesis, a model that is now driving the clinical approaches for the treatment of leukemia associated with MLL translocations (Cai et al. 2015). Purification of some of the most common MLL translocation partners led to the identification of the super elongation complex (SEC) that includes ELL; another previously known transcription elongation factor, P-TEFb; and additional MLL translocation partners AFF1, AFF4, ENL, and AF9 (Fig. 2C; Lin et al. 2010). ELL is also found in the little elongation complex (LEC), which specifically regulates distinct stages of small nuclear RNA transcription, essential housekeeping functions that are likely compensated for by the wild-type allele of ELL in leukemia (Smith et al. 2011; Hu et al. 2013b).
A distinct protein complex that also functions in transcription elongation is DotCom (DOT1L complex), which contains the H3K79 methyltransferase DOT1L and the MLL chromosomal translocation partners AF10, AF17, ENL, and AF9 (Fig. 2C; Okada et al. 2005; Mohan et al. 2010a,b). Dot1l mutant mice are embryonic-lethal between E10.5 and E13.5 and display a severe anemia due to reduced expression of the erythroid master regulator Gata2 (Feng et al. 2010). HoxA9 and Meis1 are the best-characterized targets of the MLL chimeras, and their simultaneous overexpression was shown to be sufficient for leukemic transformation (Kroon et al. 1998; Buske and Humphries 2000; Zeisig et al. 2004). Both SEC and DotCom have been shown to directly associate with HoxA9 and Meis1 loci and are required for their overexpression (Okada et al. 2005; Lin et al. 2010; Neff and Armstrong 2013). Genetic deletion of Dot1l or pharmacological inhibition of its methyltransferase activity renders MLL chimeras unable to activate the malignant transcriptional program in mouse models (Okada et al. 2005; Krivtsov et al. 2008; Chang et al. 2010; Bernt et al. 2011; Nguyen et al. 2011; Deshpande et al. 2013). Consequently, a selective DOT1L small molecule inhibitor compound, EPZ-5676, has been under investigation for the treatment of MLL-rearranged leukemia (Cai et al. 2015). In addition to translocations, partial tandem duplications (PTDs) of MLL have been observed in leukemia with a normal karyotype and linked to an unfavorable prognosis after treatment (Caligiuri et al. 1994, 1998; Schichman et al. 1994). PTD most commonly occurs by insertion of either exons 5–11 or 5–12 into intron 4 of the full-length MLL gene, and this leads to a duplicated N-terminal region that harbors both the AT hooks and the CXXC domains (Whitman et al. 2005). A recent study showed that leukemia cells with MLL-PTD are sensitive to DOT1L small molecule inhibition by EPZ-5676, although the oncogenic mechanisms of MLL-PTD may differ from that of MLL chimeras (Kuhn et al. 2015). Furthermore, since SEC contains the P-TEFb kinase module, specific targeting of this activity or the disruption of its biochemical integrity in leukemic cells may represent additional therapeutic strategies for MLL translocation-based leukemia (Fig. 2D).
Another therapeutic strategy for treating MLL translocation-based leukemia is to prevent the recruitment of MLL chimeras to chromatin. Menin associates directly with an N-terminal region of MLL that is retained in all MLL chimeras. This interaction has been shown to be essential for the leukemic activity of MLL chimeras through facilitating the recruitment to chromatin (Yokoyama et al. 2004, 2005; Caslini et al. 2007). Small molecule disruptors of the MLL–menin interaction were identified through high-throughput screening and structure-based design (Grembecka et al. 2012; Shi et al. 2012). Pharmacological treatment with these inhibitors (MI-2-2 and MI503) in both leukemic cell lines and a mouse model of leukemia led to apoptosis and hematopoietic differentiation, proliferation defects, and reversal of MLL chimera-driven leukemic transcriptional signatures (Fig. 2D; Grembecka et al. 2012; Borkin et al. 2015). However, it is not clear whether or how the inhibition of this mode of recruitment by MI-2-2 and MI503 discriminates between the chimera, the wild-type MLL, or its closest homolog, MLL2 (KMT2B; GeneID 9757). Further molecular studies on the inhibitory role of MI-2-2 and MI503 on endogenous MLL/MLL2/COMPASS and the changes in the pattern of H3K4 methylation should shed further light on this process and determine whether such an approach is feasible or useful for the treatment of MLL translocation-based leukemia.
MLL translocation or PTD in leukemia patients usually occurs in one allele, leaving the second allele unaffected in most cases. Studies have reported that the wild-type MLL is essential for leukemic transformation by MLL chimeras and that its action is dependent on histone methyltransferase activity (Milne et al. 2010; Cao et al. 2014). The methyltransferase activity of MLL requires the formation of a core complex with the WDR5, RBBP5, ASH2L, and DPY30 subunits (abbreviated as WRAD in the literature) (Dou et al. 2006; Patel et al. 2008, 2009). Therefore, small molecules (MM-102 and MM-401) that inhibit the methyltransferase activity of MLL via disruption of its association with WDR5 were developed and have been shown to function by blocking proliferation, induction of apoptosis, and differentiation of the cells expressing MLL-AF9 (Karatas et al. 2013; Cao et al. 2014). However, the basis for the specificity of these inhibitors for MLL versus other members of the COMPASS family is unclear, as WDR5 is a shared component throughout the COMPASS family. Furthermore, mouse genetic studies have demonstrated that the SET domain of the wild-type copy of MLL is dispensable for leukemogenesis (Mishra et al. 2014), raising the question of the efficacy of targeting the wild-type copy of MLL for treating MLL chimera-driven leukemia.
Mutations of MLL3 and MLL4 members of the COMPASS family in enhancer malfunction and hematological malignancies
Transcriptional enhancers were originally defined as noncoding DNA sequences that can increase the transcription of cognate genes in a distance-, position-, and orientation-independent manner (Dorsett 1999; Smith and Shilatifard 2014). A high level of H3K4 monomethylation (H3K4me1) with a relatively low level of H3K4me3 trimethylation (H3K4me3) has been identified as a signature of enhancers (Heintzman et al. 2007, 2009). MLL3 and MLL4 (KMT2C and KMT2D) comprise one of the three major branches of the COMPASS family in mammals, with the other two major branches represented by MLL/MLL2 (KMT2A/KMT2B) and SET1A/SET1B (Mohan et al. 2011; Herz et al. 2012; Shilatifard 2012). MLL3 (KMT2C), MLL4 (KMT2D; GeneID 8085), and their Drosophila ortholog, Trr (Trithorax-related), implement the bulk of H3K4me1 at enhancers and are required for enhancer–promoter communication during development (Fig. 3A; Herz et al. 2012, 2014; Hu et al. 2013a). Further studies by other laboratories have now confirmed these original findings establishing MLL3 and MLL4 COMPASS as major regulators of enhancer H3K4 function (Kanda et al. 2013; Lee et al. 2013; Smith and Shilatifard 2014).
Figure 3.

Misregulation of MLL3/4/COMPASS and polycomb group (PcG) proteins in hematopoietic transformation. (A) In normal HSCs and progenitors, MLL3/4/COMPASS and EZH2/PRC2 control the proper expression of genes involved in self-renewal and differentiation through implementation of H3K4me1 and H3K27me3 at enhancers. UTX, a component of the MLL3/MLL4/COMPASS-like complexes, harbors a demethylase activity specific for H3K27me3 that could antagonize transcriptional repression by the EZH2/PRC2 complex. (B) During lymphoma development, the catalytic subunit of PRC2, EZH2, is frequently mutated at Y641. This mutation results in a switch in substrate preference from nonmethylated to the monomethylated and dimethylated histone H3K27, leading to increased levels of H3K27 dimethylation (H3K27me2) and H3K27me3 at enhancers and promoters. Additionally, inactivating mutations in enhancer monomethyltransferases MLL3 and MLL4/COMPASS can cause loss of H3K4me1 at enhancers. Altogether, these events independently or together could lead to repression of enhancers and promoters of tumor suppressor genes to promote lymphomagenesis. (C) Schematic illustration of mammalian PRC1 and PRC2 complexes. PRC2 is composed of four core components—EED (embryonic ectoderm development), SUZ12 (suppressor of zeste 12 homolog), RBAP46, and RBAP48—and an enzymatic subunit, EZH1 or EZH2. EED and SUZ12 are essential for the stability of EZH1 and EZH2 and therefore for H3K27 methylation (Pasini et al. 2004; Xie et al. 2014). PRC1 complexes include both canonical and noncanonical forms in mammalian cells. Both canonical and noncanonical PRC1 complexes contain the catalytic subunit RING1A or RING1B that implements H2AK119 monoubiquitination. Canonical PRC1 complexes can variably include CBX2, CBX4, CBX7 or CBX8; BMI1 (also known as PCGF4) or MEL18 (also known as PCGF2); and PHC1, PHC2, or PHC3. The chromodomain found in the CBX proteins that participates in canonical PRC1 mediates the interaction with H3K27me3 that is catalyzed by PRC2. Noncanonical PRC1 complexes can contain RYBP or YAF2 and PCGF1, PCGF3, PCGF5, or PCGF6. (D) Inactivating mutations in MLL3, MLL4, UTX, and EZH2 were observed in myeloid leukemias (MDS, chronic myelomonocytic leukemia [CMML], and primary myelofibrosis [PMF]) and T-cell ALL (T-ALL). EZH1 can partially compensate for the loss of EZH2 in depositing H3K27me3 at enhancers and promoters. Loss of function of MLL3 and MLL4 removes H3K4me1 from enhancers, while UTX inactivation may increase H3K27me3 at the enhancers and promoters. All of these epigenetic alterations can collaborate to suppress the enhancers of tumor suppressor genes involved in myeloid leukemia and T-ALL. (E) In myeloid hematopoietic cells, ASXL1 (additional sex combs-like 1) promotes the repression of PRC2 targets via a currently unknown mechanism. (Top panel) Loss-of-function mutations of ASXL1 are frequently detected in CMML, MDS, and MPN, and these mutations could give rise to activation of an oncogene normally repressed by PRC2-mediated H3K27me3. (Bottom panel) BRCA1-associated protein 1 (BAP1) can function to attenuate the activity of PRC2. BAP1 loss results in increased expression of EZH2, which can result in myeloid transformation through the repression of tumor suppressor genes.
Chromatin signatures can be used to further classify enhancers as variably existing in inactive, active, or poised states. Histone H3K4me1 alone marks enhancers before their activation (Heintzman et al. 2007), with the addition of H3K27ac indicating active enhancers. Poised enhancers are distinguished by the presence of H3K27me3 and are found primarily in stem and progenitor cells, transitioning to active enhancers upon differentiation cues (Creyghton et al. 2010; Rada-Iglesias et al. 2011). The MLL3, MLL4 (GeneID 8085), and Trr forms of COMPASS harbor a unique subunit, UTX, which has H3K27 demethylase activity. This raises the possibility that these complexes not only are responsible for H3K4me1 at inactive, active, and poised enhancers but also may facilitate enhancer transitioning from a poised to an active state during hematopoietic differentiation (Fig. 3A; Cho et al. 2007; Herz et al. 2012).
Recent cancer genome sequencing approaches of many types of primary tumors and cancer cell lines have revealed that MLL3 and MLL4/COMPASS are among the most frequently mutated genes in both solid tumors and hematological malignancies (Morgan and Shilatifard 2015). The MLL3 mutation or its deletion is found in hematological malignancies, including 15% of DLBCL, MDS, and AML (Mrozek 2008; Pasqualucci et al. 2011; Zhang et al. 2013). MLL4 (GeneID 8085) mutations are prevalent in NHL (Morin et al. 2011; Pasqualucci et al. 2011; Lohr et al. 2012; Green et al. 2015), ALL (Mar et al. 2012; Lindqvist et al. 2015; Neumann et al. 2015), and AML (Kandoth et al. 2013). Most of the MLL3 and MLL4/COMPASS mutations are heterozygous nonsense, frameshift, and internal insertions/deletions (indels), resulting in protein truncations—which is suggestive of a haploinsufficient tumor suppressor function of MLL3 and MLL4 (GeneID 8085)—in cancer pathogenesis. A tumor suppressor function of MLL3 has been proven in a mouse model of AML, with a 50% reduction in gene dosage cooperating with other genetic alterations to promote leukemogenesis (Chen et al. 2014).
FLs can transform from the indolent to the more aggressive DLBCL, which has allowed the collection and analysis of sequential tumor lymphoid biopsies from the same patient (Okosun et al. 2014). These and related studies suggest that somatic MLL4 (GeneID 8085) mutations are likely to be early events that occur in a common progenitor cell, with additional mutations leading to the more aggressive state (Green et al. 2013, 2015; Okosun et al. 2014; Pasqualucci et al. 2014; Ortega-Molina et al. 2015). Mouse models using Bcl2 overexpression and conditional deletion of Mll4 (Kmt2d; GeneID 381022) demonstrate that Mll4 loss promotes the development of lymphoma through the expansion of germinal centers, likely due to the loss of the expression of known tumor suppressor genes regulating B-cell-activating pathways (Fig. 3B; Ortega-Molina et al. 2015; Zhang et al. 2015).
Although occurring at a lower rate than truncating mutations, missense mutations of MLL4 (GeneID 8085) are found throughout the protein in lymphomas, with many of the missense mutations located in the C-terminal domains impairing the in vitro histone methyltransferase activity of MLL4 (Zhang et al. 2015). However, these mutations have not been characterized in vivo, and their contributions to lymphomagenesis are currently unknown. Recently developed genome-editing tools such as CRISPR will allow the exploration of these missense mutations in mouse models, potentially providing insight into their contribution to hematological malignancies.
In contrast to MLL4-inactivating mutations contributing to lymphomagenesis, recent studies with conditional ablation of Mll4 (Kmt2d; GeneID 381022) in AML cells revealed that Mll4 is essential for MLL-AF9-induced leukemogenesis (Santos et al. 2014). When Mll4 was deleted in HSCs by crossing Mll4f/f mice with transgenic mice that express interferon-inducible MxCre, expansion of HSCs and common myeloid progenitors (CMPs) in the bone marrow were observed. However, these HSCs are impaired in their self-renewing capability due to oxidative stress-induced DNA damage. Therefore, it has been proposed that MLL4/COMPASS helps enforce a differentiation blockade of leukemic stem cells by up-regulating expression of antioxidant genes, with decreased cellular reactive oxygen species (ROS) protecting against oxidative stress-induced DNA damage (Santos et al. 2014).
A potential mechanism for mutations in MLL3 and MLL4/COMPASS family members leading to lymphomagenesis and other hematological cancers is the misregulation of enhancer function during hematopoietic differentiation (Herz et al. 2014). Aberrant transcriptional enhancer activity through either alterations of chromatin modifications, mutations in enhancer-binding factors, or mutations of enhancers themselves could potentially lead to oncogenesis (Herz et al. 2014). A classic example of enhancer-mediated oncogenesis is the chromosomal translocation commonly found in Burkitt's lymphoma that places the MYC oncogene under the control of an immunoglobulin heavy chain (Igh) enhancer, leading to the uncontrolled expression of MYC and the development of lymphoma (Dalla-Favera et al. 1982; Park et al. 2005). More recently, two independent studies of AML with a chromosomal inversion between GATA2 and EVI1 revealed that this inversion allows for promiscuous activation of EVI1 by a GATA2 enhancer (Groschel et al. 2014; Yamazaki et al. 2014). Even a single-nucleotide polymorphism in a PU.1 enhancer leads to reduced PU.1 expression that contributes to the development of AML (Steidl et al. 2007). Furthermore, in a subset of T-cell ALL (T-ALL), heterozygous acquisition of a short DNA sequence at a noncoding intergenic region creates a binding site for the transcription factor MYB, thereby creating a de novo enhancer to activate allele-specific expression of TAL1 (Mansour et al. 2014). A long-range NOTCH1-dependent oncogenic enhancer was found to undergo focal amplifications to aberrantly activate MYC expression to drive T-ALL (Herranz et al. 2014). The pervasive contribution of enhancer malfunction in hematological malignancies and the high rate of the mutation of the MLL3 and MLL4 (GeneID 8085) forms of COMPASS suggest that alterations in enhancer activity by MLL3 and MLL4 mutations help drive an oncogenic program (Fig. 3B). Further studies aimed at identifying MLL4-dependent enhancers and how these factors are specifically recruited to their cognate site using methods such as ChIP-seq (chromatin immunoprecipitation [ChIP] combined with high-throughput sequencing), genome editing, and gene expression profiling will be required for testing this hypothesis.
Polycomb group (PcG) proteins in normal and malignant hematopoiesis
PcG proteins were initially identified in Drosophila by mutations that cause homeotic transformations, such as leg-to-antenna transformations or the appearance of additional sex combs on the second and third pairs of legs (Lewis 1978; Struhl 1981). These transformations result from derepression of the homeotic genes (called Hox genes in vertebrates) due to loss-of-function mutations in genes encoding PcG proteins. In mammals, PcG proteins are also critical regulators of developmental gene expression and tissue homeostasis. Their misregulation has been widely linked to multiple disease states in a variety of contexts, particularly in cancers, including hematological malignancies (Varambally et al. 2002; Bracken et al. 2003; Schlesinger et al. 2007; Kondo et al. 2008; Martinez et al. 2009). PcG proteins operate in at least two distinct multiprotein complexes, PRC1 and PRC2; each harbors an enzymatic activity, with PRC1 implementing H2AK119 monoubiquitination, and PRC2 implementing H3K27 methylation (Margueron and Reinberg 2011).
The mammalian PRC2 complex consists of four core components: EZH1 or its paralog, EZH2; embryonic ectoderm development (EED); suppressor of zeste 12 homolog (SUZ12); and RBAP46/48 (also known as RBBP7/4) (Margueron and Reinberg 2011; Aranda et al. 2015; Piunti and Shilatifard 2016). EZH2 or EZH1 forms the enzymatic core and implements H3K27 methylation. EZH1 is present in both dividing and differentiated cells, whereas EZH2 is found only in actively dividing cells. The PCR2 complex with EZH1 has low methyltransferase activity compared with the PRC2 complex containing EZH2 (Fig. 3C; Margueron et al. 2008). The composition of mammalian PRC1 complexes is more variable and is comprised of the E3 ubiquitin ligases for H2A monoubiquitination (RING1A or RING1B) together with either canonical (CBX2/4/7/8, PHC1/2/3, and BMI1/MEL18) or noncanonical (RYBP/YAF2 and PCGF1/3/5/6) proteins (Fig. 3C; Gao et al. 2012; Tavares et al. 2012; Aranda et al. 2015; Piunti and Shilatifard 2016). PRC1 and PRC2 co-occupy many PcG target loci and play crucial roles in their reciprocal recruitment to chromatin (Boyer et al. 2006; Schwartz et al. 2006; Ku et al. 2008). The chromobox (CBX) proteins have a chromodomain that recognizes the H3K27me3 mark implemented by PRC2 and mediates the chromatin targeting of the canonical PRC1 complex (Cao et al. 2002; Fischle et al. 2003; Min et al. 2003; Agger et al. 2007; Lee et al. 2007). Conversely, noncanonical PRC1-catalyzed H2AK119 monoubiquitination is capable of targeting PRC2 to chromatin (Blackledge et al. 2014; Kalb et al. 2014).
PcG proteins are essential regulators of hematopoiesis and can orchestrate the expression of genes that control the balance between self-renewal and the multipotency of HSCs. Transgenic and conditional knockout studies in mice have provided valuable information on the physiological role of PcG proteins in the hematopoietic system. Ezh1 is required for HSC maintenance, and its conditional loss leads to HSC senescence and impairment of B-cell lymphopoiesis (Hidalgo et al. 2012). Forced expression of Ezh2 was found to prevent the exhaustion of the long-term repopulating capacity of HSCs in a serial transplantation assay and cause development of myeloproliferative disease in recipient mice (Kamminga et al. 2006; Herrera-Merchan et al. 2012). Several studies have shown that PRC2 restricts HSC activity, as a partial loss of the core components such as Ezh2, Eed, or Suz12 in mice enhances HSC function (Majewski et al. 2008, 2010). However, a full loss of PRC2 activity has a distinct effect on HSC maintenance and function. Mice with an Eed deficiency are normal in fetal liver HSC numbers but display defects in the maintenance and differentiation of adult HSCs (Xie et al. 2014). Homozygous deletion of Suz12 also results in failures in both embryonic and adult hematopoiesis (Lee et al. 2015). Collectively, these studies indicate a dosage-dependent impact of PRC2 activity on HSC function. In addition to HSC regulation, PRC2 also has an essential role in lymphoid development. Conditional deletion of EZH2 in the lymphoid lineage revealed its crucial role in early B-cell development, rearrangement of the Igh gene, germinal center formation, and differentiation and survival of CD4+ T help1 (Th1) and Th2 cells (Su et al. 2003; Beguelin et al. 2013; Caganova et al. 2013; Tumes et al. 2013; Zhang et al. 2014). Similarly, Suz12 deficiency causes severe defects in B and T lymphopoiesis (Lee et al. 2015). Future work should clarify the mechanisms underlying the dosage-dependent impact of PRC2 activity on HSC functioning and the role and mechanisms of PRC2 in the specification of other hematopoietic lineages.
PRC1 is also essential for normal hematopoiesis, with its core subunit, BMI1, being the most-studied component of PRC1 in hematopoiesis. Ectopic expression of Bmi1 in mouse embryonic stem cells has been shown to promote primitive hematopoiesis (Ding et al. 2012). Bmi1 is required for HSC self-renewal in both mice and humans. The homozygous deletion of Bmi1 in mice decreases the number of HSCs by inducing symmetrical division, derepression of the Ink4a–Arf locus (encoding p16INK4A and p19ARF proteins), and generation of ROS and DNA damage (Park et al. 2003; Iwama et al. 2004; Oguro et al. 2006; Liu et al. 2009). Bmi1−/− cells exhibit an accelerated differentiation into the B-cell lineage due to premature transcriptional activation of B-cell regulators Ebf1 and Pax5 (Oguro et al. 2010). Although homologous to Bmi1, Mel18 loss has very little impact on the repopulating capacity of fetal HSCs, with the self-renewing capacity of adult HSCs being enhanced in the absence of Mel18, suggesting that Bmi1 and Mel18 have distinct functions in HSCs (Iwama et al. 2004; Kajiume et al. 2004). However, Mel18 was demonstrated to have important roles in the proliferation and maturation of B cells, indicating a function in lymphoid differentiation (Akasaka et al. 1997; Tetsu et al. 1998).
Multiple members of the CBX family have been identified within the canonical PRC1 complex, and several of them have been shown to be vital for hematopoietic homeostasis. Cbx7 is highly expressed in long-term HSCs, and its overexpression enhances HSC self-renewal and induces leukemia, whereas an ectopic expression of Cbx2, Cbx4, or Cbx8 results in differentiation and exhaustion of HSCs, revealed by the competitive transplantation assay (Klauke et al. 2013). Although the repopulating activity of fetal liver cells deficient for Cbx2 is comparable with wild-type cells, homozygous deletion of Cbx2 leads to reduced cellularity in postnatal bone marrow, spleen, and thymus (Core et al. 1997; Iwama et al. 2004). The knockdown of each member of the CBX family in human CD34+ core blood cells leads to a profound reduction in the proliferation of hematopoietic stem and progenitor cells (van den Boom et al. 2013).
Studies on the role of the catalytic components of the PRC1 complex, RING1A and RING1B, are still limited, and H2A monoubiquitination function in fetal and adult hematopoiesis remains unclear. Mice with homozygous deletion of Ring1a are viable but display skeletal abnormalities (del Mar Lorente et al. 2000). Conditional deletion of Ring1b in adult hematopoietic lineages revealed its restrictive role in the proliferation of hematopoietic stem and progenitor cells and its favorable function in the proliferation of differentiated progenies (Cales et al. 2008). In vitro studies revealed that HSCs with a deletion of both Ring1a and Ring1b show a severe defect in their self-renewing capacity (Piunti et al. 2014). Future studies using mice with a conditional deletion of Ring1a and/or Ring1b will shed light on the role of H2A monoubiquitination in normal hematopoiesis.
EZH2 is the most frequently mutated PcG member in the pathogenesis of hematological malignancies (Kroeze et al. 2012; Woods and Levine 2015). EZH2 overexpression is commonly observed in various epithelial malignancies, including breast and prostate cancer, suggesting that EZH2 functions as an oncogene (Varambally et al. 2002; Kleer et al. 2003; Li et al. 2009a). Indeed, recurrent somatic mutations that result in increased EZH2 enzymatic activity have been observed in lymphoma (Morin et al. 2011; Yap et al. 2011; McCabe et al. 2012a). Monoallelic mutations that result in substitution of Y641 within the SET domain of EZH2 lead to increased conversion of H3K27me1 to H3K27me2 (H3K27 dimethylation) and H3K27me3 and have been identified in 22% of patients with germinal center diffuse B-cell lymphoma (Morin et al. 2011). Transgenic mouse models initially demonstrated that a specific overexpression of EZH2Y641F/N in lymphocytes is insufficient for lymphomagenesis on its own but can cooperate with Bcl2 or Myc overexpression (Beguelin et al. 2013; Berg et al. 2014). These studies suggested that the carcinogenic activity of EZH2 Y641 mutations rely on other oncogenic events to drive malignant transformation. However, a more recent study with the Y641F mutation knocked in the endogenous Ezh2 locus in mice showed that Ezh2Y641F itself could cause development of lymphoma and melanoma (Souroullas et al. 2016). In this mouse model, Ezh2Y641F was found to cooperate with the loss of p53 or overexpression of Bcl2, but not Myc, to accelerate lymphoma progression (Souroullas et al. 2016). These discrepancies resulting from the different mouse models used in these studies demonstrate that the oncogenic function of somatic mutations may not be faithfully recapitulated by transgenic mouse models that introduce an extra copy of mutant alleles expressed under the control of exogenous promoters. Despite the overall higher abundance of H3K27me3 observed in lymphoma cells induced in knock-in Ezh2Y641F mice, many loci lose H3K27me3 and exhibit increased transcription (Souroullas et al. 2016). Therefore, the aberrantly high level of H3K27me3 may lead to persistent repression of some tumor suppressor genes (Fig. 3B), while its less focused distribution could allow derepression of other loci contributing to lymphomagenesis.
The importance of the H3K37me3 mark in promoting epigenetic reprogramming in hematological malignancies is further suggested by the high frequency of inactivating mutations of the H3K27me3 demethylase UTX that resides within MLL3/MLL4 COMPASS-like complexes (Agger et al. 2007; Cho et al. 2007; Lan et al. 2007). Exome and whole-genome sequencing has identified homozygous and heterozygous inactivating mutations of UTX in multiple myeloma and T-ALL but not in lymphomas that harbor the EZH2 Y641 activating mutation (van Haaften et al. 2009; Morin et al. 2011). Studies of Utx function in T-ALL found that Utx can act as a tumor suppressor in a Notch1-induced murine model of leukemia (Ntziachristos et al. 2014). More recent studies found that UTX can also act as an oncogene in TAL1-positive T-ALL, while UTX can inhibit cell growth when overexpressed in TAL1-negative T-ALL cells (Benyoucef et al. 2016). Therefore, potential therapeutic options for hematological cancers can be aimed at recovering normal cellular H3K27 methylation levels with small molecule inhibitors of PRC2 or, in a subset of cases, activating molecules of H3K27 demethylases (Fig. 3D; Knutson et al. 2012; McCabe et al. 2012b).
Although an excess of H3K27me3 activity generally has an oncogenic role in cancer, loss-of-function mutations in EZH2 have been detected in a subset of myeloid malignancies, most commonly in MDSs, chronic myelomonocytic leukemia (CMML), primary myelofibrosis (PMF), and T-ALL (Ernst et al. 2010; Nikoloski et al. 2010; Ntziachristos et al. 2012; Score et al. 2012; Simon et al. 2012; Zhang et al. 2012). These inactivating mutations of EZH2 predict a poorer overall outcome in CMML, MDS, and PMF (Grossmann et al. 2011; Guglielmelli et al. 2011). Conditional loss of Ezh2 in a hematopoietic system contributes to the pathogenesis of MDS and accelerates the onset of the early T-cell precursor ALL (ETP-ALL) induced by oncogenic NRASQ61K (Score et al. 2012; Muto et al. 2013; Danis et al. 2016). Besides EZH2 inactivating mutations, loss-of-function mutations of the PRC2 core components SUZ12 and EED have also been detected in T-ALL, all of which lead to lower levels of H3K27me3 (Ntziachristos et al. 2012; Simon et al. 2012). Intriguingly, myeloid leukemia can have inactivating mutations of both PRC2 components and UTX, raising the question of how simultaneous mutations of factors with opposing enzymatic activity can contribute to leukemogenesis (van Haaften et al. 2009; Jankowska et al. 2011). A very recent study demonstrated that Ezh1 function is essential for the pathogenesis of myeloid malignancies induced by Ezh2 loss of function (Mochizuki-Kashio et al. 2015). Therefore, inactivating mutations of UTX could facilitate EZH1-dependent repression of tumor suppressor genes that would otherwise be derepressed by EZH2 loss of function (Fig. 3D). Understanding the context in which PRC2 can act as an oncogene or tumor suppressor in lymphomagenesis and leukemogenesis will undoubtedly be an active area of future research.
Over the past decade, a series of studies has implicated PRC1 in HSC self-renewal and differentiation, which require the core subunits of the Bmi1 and Cbx family of proteins (Lessard and Sauvageau 2003; Park et al. 2003; Klauke et al. 2013). In contrast to PRC2, somatic mutations in the PRC1 components have not been reported for hematopoietic malignancies. However, overexpression of BMI1, an integral subunit that stimulates the ubiquitinase activity of PRC1 toward H2AK119, has been observed in myeloid malignancies and has emerged as a useful indicator for the prognosis of MDS, CMML, and AML (Mihara et al. 2006; Chowdhury et al. 2007; Mohty et al. 2007). However, it remains unclear whether H2AK119 monoubiquitination is concomitantly increased with BMI1 overexpression and whether this histone modification contributes to myeloid malignancies. In Drosophila, H2AK119 monoubiquitination can be removed by the Polycomb-repressive deubiquitinase (PR-DUB) complex, which is comprised of the BRCA1-associated protein 1 (BAP1) homolog, Calypso, and additional sex combs (Asx) (Scheuermann et al. 2010). A mammalian PR-DUB complex containing BAP1 and ASX-like 1 (ASXL1) or ASXL2 was also biochemically isolated and demonstrated to harbor deubiquitinase activity in vitro (Scheuermann et al. 2010; Dey et al. 2012). ASXL1 is frequently mutated in a wide range of myeloid malignancies, most commonly in AML, CMML, MDS, and MPN. The mutations of ASXL1 are associated with a dismal overall prognosis in patients with MDS and AML (Abdel-Wahab et al. 2011, 2012; Bejar et al. 2011; Patel et al. 2012). However, and in contrast to what has been observed in Drosophila, ASXL1 inactivating mutations did not cause significant changes in the H2AK119 monoubiquitination levels in myeloid hematopoietic cells but did promote myeloid transformation through the loss of PRC2-medidated H3K27 methylation, leading to derepression of PRC2 target genes (Fig. 3E; Abdel-Wahab et al. 2012; Inoue et al. 2013).
BAP1 is best known for being recurrently mutated in numerous cancers such as mesothelioma, renal cell carcinoma, and metastatic uveal melanoma (Harbour et al. 2010; Bott et al. 2011; Pena-Llopis et al. 2012). Consistent with a physical association with Asxl1, Bap1 loss of function also leads to myeloid transformation in mice (Dey et al. 2012; LaFave et al. 2015). In contrast to the reduction of H3K27me3 seen with Asxl1 loss, Bap1 deletion leads to increased Ezh2 expression, higher levels of H3K27me3, and enhanced repression of PRC2 target genes in hematopoietic cells. Thus, Ezh2 deletion or inhibition was shown to abrogate myeloid malignancy and mesothelioma induced by Bap1 loss (LaFave et al. 2015). These findings raise the possibility that myeloid transformation resulting from ASXL1 and BAP1 loss could be independent of the function of the BAP1–ASXL1 complex (Fig. 3E). EZH2 dependence for BAP1 mutant malignancies appears to be context-dependent, as BAP1 mutant uveal melanoma cells do not exhibit sensitivity toward EZH2 inhibition, which is in contrast to findings observed in hematopoietic and mesothelioma cells (LaFave et al. 2016; Schoumacher et al. 2016). Future work to dissect the molecular mechanisms and potential cross-talk for the PRC1 and PRC2 complexes in their hematopoietic context and in MDS and other malignancies is another important area requiring investigation.
The nuclear receptor-binding SET domain (NSD) family of H3K36 methyltransferases in hematological malignancies
The NSD family of histone methyltransferases comprises three proteins: NSD1, NSD2 (also known as multiple myeloma SET domain [MMSET] and Wolf-Hirschhorn syndrome candidate 1 [WHSC1]), and NSD3 (also known as WHSC1L1). Although members of the NSD family were initially shown to have enzymatic activity toward multiple residues on histones, subsequent studies demonstrated that these enzymes preferentially implement histone H3K36me2 (Li et al. 2009b; Kuo et al. 2011; Qiao et al. 2011). NSD1 and NSD2 have each been linked to human developmental overgrowth syndromes. The NSD2 gene is among a group of genes deleted in Wolf-Hirschhorn syndrome, and loss of NSD2 is thought to be responsible for a subset of the clinical features characteristic of this syndrome (Andersen et al. 2014). Deletion or loss-of-function mutations involving NSD1 results in Sotos syndrome, an autosomal dominant overgrowth syndrome characterized by a distinctive facial appearance, delayed development, and learning disabilities (Douglas et al. 2003; Turkmen et al. 2003).
The roles of members of the NSD family in normal hematopoiesis have not been investigated. However, alterations of these genes are recurrently observed in hematological malignancies, implying critical roles in normal hematopoiesis. A recurring t(5;11)(q35;p15.5) chromosomal translocation fuses NSD1 to nucleoporin-98 (NUP98) to generate a NUP98–NSD1 chimera in 5% of human AMLs (Cerveira et al. 2003). Transplantation studies in mice demonstrated that expression of the NUP98–NSD1 chimera sustains self-renewal of myeloid progenitors and is sufficient to induce AML in vivo by preventing the EZH2-dependent silencing of the Hoxa9 and Meis1 proto-oncogenes (Wang et al. 2007). NSD2 is commonly overexpressed in 15%–20% of multiple myeloma cases through a chromosomal translocation t(4;14)(p16.3;q32) that juxtaposes the NSD2 gene with the IgH locus (Chesi et al. 1998). Aberrant up-regulation of NSD2 and a subsequent increase of H3K36me2 lead to an altered localization and overall decrease in the levels of H3K27me3 (Kuo et al. 2011; Martinez-Garcia et al. 2011; Popovic et al. 2014). Recent global profiling of histone modifications by mass spectrometry from 115 cancer cell lines identified a cluster of 13 cell lines exhibiting a signature of high H3K36me2. These were divided between multiple myeloma and ALL cell lines, with all but one of the multiple myeloma cell lines having the t4;14 rearrangement with the IgH promoter, while the other multiple myeloma cell line and all of the ALL lines had an activating mutation in the SET domain of NSD2 (E1099K). This activating mutation was found in 14% of the t(12;21) ETV6-RUNX1-containing pediatric ALLs. NSD2 knockdown selectively inhibited the proliferation and xenograft growth of ALL lines harboring the E1099K mutation in NSD2, indicating its requirement for NSD2 mutant ALL (Jaffe et al. 2013). In addition to the therapeutic targeting of NSD2's catalytic activity, an essential function for the second plant homeodomain (PHD) finger in targeting NSD2 to chromatin provides another potential therapeutic strategy (Huang et al. 2013b). Last, the oncogenic potential of NSD3 was first suggested by the observation that it was overexpressed in breast cancer and was later found as a fusion partner of the NUT oncogene in midline carcinoma (Filippakopoulos et al. 2010). In addition, rare cases of AML and MDS have NUP98–NSD3 fusions, but the functional importance of these fusions remains to be demonstrated (Rosati et al. 2002; Taketani et al. 2009).
DNA methylation in hematopoiesis and hematological cancers
Long before the methylation of lysine residues in histones were recognized as important epigenetic marks, cytosine methylation at the C5 position of mammalian genomic DNA, usually in the context of CpG dinucleotides, was known to have a critical role in the epigenetic processes of genomic imprinting and X inactivation (Yang et al. 2015). The regulation of CpG methylation has since been established as being critical for stem cells and their differentiation potential, while aberrant DNA methylation is pervasive in cancer, including in blood malignancies. Cytosine methylation is carried out by a family of DNA methyltransferases, including DNMT1, DNMT3A, and DNMT3B (Okano et al. 1998). DNMT1 is considered as the maintenance DNA methyltransferase that can bind hemimethylated DNA during cell division, resulting in the inheritance of the methylated cytosine state in the daughter strand. DNMT3A and DNMT3B function as de novo DNA methyltransferases, which can methylate the unmethylated cytosines during embryogenesis (Fig. 4A; Stein et al. 1982; Okano et al. 1998; Jones and Liang 2009).
Figure 4.

Misregulation of DNA methylation in hematological malignancies. (A) Dynamics of DNA methylation and demethylation mediated by DNA methyltransferase (DNMT3A, DNMT3B, and DNMT1) and Tet (ten-eleven translocation; TET1, TET2, and TET3) proteins. De novo DNA methyltransferases (DNMT3A and DNMT3B) add a methyl group to the fifth position of cytosine of CpG dinucleotides to form 5-methylcytosine (5mC) during embryonic development. During cell division, the maintenance DNA methyltransferase DNMT1 is targeted to hemimethylated CpG and adds a methyl group to the newly replicated DNA to faithfully maintain the DNA methylation pattern in daughter cells. DNA demethylation is achieved through active and passive pathways. In the active demethylation pathway, 5mC is successively oxidized by members of the TET family to 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC), and 5-carboxylcytosine (5caC). 5caC is further converted to unmethylated cytosine via TDG (thymine DNA glycosylase)-mediated excision and subsequent replacement with unmethylated cytosine by the base excision repair (BER) mechanism. In addition, 5mC and its oxidized derivatives can be replaced with unmethylated cytosine during DNA replication (passive demethylation) if the maintenance DNA methylation is inhibited during DNA replication. (B). Inactivating mutations in DNMT3A and TET2 (red star) are likely to occur in preleukemic cells (multipotent HSCs or progenitors), which increase their self-renewal property and block differentiation. The acquisition of mutations in additional factors (green star) such as NPM and JAK eventually leads to the development of hematological malignancies.
DNA methylation is dynamically regulated during hematopoietic differentiation, and each DNA methyltransferase plays crucial roles in physiological hematopoiesis (Tadokoro et al. 2007; Broske et al. 2009; Trowbridge et al. 2009; Ji et al. 2010; Bock et al. 2012; Challen et al. 2012). Deletion of Dnmt1 in HSCs demonstrates its requirement for HSC self-renewal and differentiation (Broske et al. 2009; Trowbridge et al. 2009). Hematopoietic-specific disruption of Dnmt3a in mice leads to both increased and decreased DNA methylation at individual loci, and these together contribute to persistent self-renewal and a differentiation block of HSCs (Challen et al. 2012). Dnmt3b knockout alone has a negligible impact on HSC function; however, its deletion along with Dnmt3a further enhances the self-renewal versus differentiation of HSCs (Challen et al. 2014).
Studies have revealed that the normal distribution of methylated cytosines is disrupted in hematological malignancies, and even subtypes of AML have different DNA methylation patterns (Figueroa et al. 2010). Although a few mutations in DNMT1 and DNMT3B have been reported, cancer genome deep sequencing efforts have identified an overwhelming prevalence of heterozygous mutations of DNMT3A (Ley et al. 2010; Yamashita et al. 2010). DNMT3A mutations have also been detected in patients with MDS and MPN and are associated with an increased risk of progression to AML (Stegelmann et al. 2011; Walter et al. 2011). DNMT3A mutations can be truncating or missense mutations. The most common mutation is the substitution of Arg882 within the catalytic domain to histidine, although substitutions of R882 with other amino acids are also observed (Ley et al. 2010; Yamashita et al. 2010). R882 mutations can induce hypomethylation by disrupting DNMT3A oligomerization (Kim et al. 2013). Two independent studies identified the existence of DNMT3A mutations in preleukemic HSCs in patients with AML. These studies indicated that DNMT3A loss-of-function mutations may confer a self-renewal advantage to HSCs and therefore lead to the development of leukemia after an acquisition of mutations in additional factors over time (Fig. 4B; Corces-Zimmerman et al. 2014; Shlush et al. 2014).
DNA methylation was long considered to be an irreversible epigenetic modification that could be removed only by the passive mechanism of cell division. This view was reversed by the discovery of the TET (ten-eleven translocation) family of dioxygenases that use oxygen, Fe(II), and a-ketoglutarate as substrates in a sequential enzymatic reaction to convert 5-methylcytosine (5mC) into 5-hydroxymethylcytosine (5hmC) and subsequently into 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC) (Iyer et al. 2009; Tahiliani et al. 2009). 5caC is recognized and replaced with a nonmethylated cytosine residue by TDG (thymine DNA glycosylase)-mediated base excision repair during the final step of cytosine demethylation (Fig. 4A; He et al. 2011; Ito et al. 2011). In addition, oxidized derivatives of 5mC by Tet proteins also facilitate passive demethylation by inhibiting DNA binding of DNMT1 during cell division (Fig. 4A; Valinluck and Sowers 2007; Hashimoto et al. 2012). In accordance with the pervasive aberrant cytosine methylation in blood cancers, misregulation of the TET family of proteins is now implicated in oncogenesis. Indeed, TET1 was first identified as an MLL fusion partner in rare cases of AML and ALL that have a t(10,11)(q22;q23) translocation (Ono et al. 2002; Lorsbach et al. 2003; Tahiliani et al. 2009). The chimeric protein lacks the hydroxylase activity, as the cys-rich domain of TET1 that is essential for enzymatic activity is deleted (Tahiliani et al. 2009). Interestingly, studies demonstrated that TET1 is overexpressed in MLL-rearranged leukemia and that TET1 function is required for up-regulating the expression of MLL chimera oncogenic targets such as HOXA9, MEIS1, and PBX3 (Huang et al. 2013a). Reconciling this requirement for TET1 in MLL-rearranged leukemias and the molecular consequence of the MLL-TET1 rearrangement and its interplay with endogenous TET1 has yet to be determined.
Among the TET family of DNA demethylases, TET2 is mutated most frequently in hematopoietic malignancies, whereas alterations in TET1 and TET3 are very rare (Abdel-Wahab et al. 2009). TET2 mutations have been observed in both myeloid and lymphoid malignancies, suggesting that mutations in TET2 occur in early hematopoietic progenitors (Quivoron et al. 2011). TET2 mutations identified in patients with hematological cancers are usually monoallelic and inactivating, indicating that it acts as an haploinsufficient tumor suppressor during hematopoietic transformation (Abdel-Wahab et al. 2009; Jankowska et al. 2009).
In vivo murine studies have clearly demonstrated that loss of TET2 function strongly correlates with hematopoietic transformation. Mice lacking TET2 are viable and fertile and appear to develop normally but develop myeloid malignancies as they age and die from myeloid disorders resembling CMML, MDS, and MPN (Abdel-Wahab et al. 2011; Ko et al. 2011; Moran-Crusio et al. 2011; Quivoron et al. 2011). Further cellular studies revealed that HSCs from TET2−/− mice have an increased ability for self-renewal and expansion and exhibit a competitive advantage over wild-type HSCs for repopulating hematopoietic lineages. TET2 deficiency impedes the overall differentiation potential of HSCs, with a bias toward the expansion of monocyte and macrophage lineages, which is consistent with the myeloid transformation observed in aged mice (Abdel-Wahab et al. 2011; Ko et al. 2011; Moran-Crusio et al. 2011). Despite the prevalent occurrence of TET2 inactivating mutations in lymphoid malignancies, mice rarely develop lymphoma in the absence of TET2. Additionally, the latency for developing myeloid leukemia in TET2-deficient mice is very long, and many elderly individuals with an age-related accumulation of TET2 mutations do not necessarily develop leukemia (Quivoron et al. 2011; Busque et al. 2012). Collectively, these lines of evidence suggest that TET2 mutations alone are not sufficient to initiate lymphoid transformation, and other cooperative oncogenic events are required to drive lymphomagenesis and shorten the latency of myeloid leukemogenesis. Indeed, alterations in multiple genes have been found to coexist with TET2 mutations, such as JAK2, KRAS, NRAS, and SRSF2 and the epigenetic regulators EZH2, DNMT3A, and ASXL1 (Fig. 4B; Tefferi 2011; Shih et al. 2012). The cooperative effects of TET2 loss with other oncogenic alterations on hematopoietic transformation have been confirmed in mice with the combined loss of Tet2 and the mutation in Flt3 (Shih et al. 2015). Therefore, further studies focused on the cooperative effects between TET2 mutations and alterations in other genes should yield significant insights into how genetic and epigenetic alterations contribute to hematopoietic transformation and may provide new therapeutic targets for leukemia with TET2 mutations.
Conclusions and future directions
Despite intensive basic research in the field of epigenetics and chromatin biology, we have a rudimentary understanding of how epigenetic modifiers carry out their function, including the degree to which chromatin modification-dependent and -independent functions contribute to transcriptional regulation during development. Epigenetic modifiers have emerged recently as potential drivers of hematological malignancies through the sequencing of blood cancer. However, the molecular basis for hematopoietic transformation by many of these chromatin modifiers is still largely unclear. For example, the target genes whose expression is misregulated by a given mutant epigenetic modifier are unknown. Future studies need to identify the key deregulated genes and determine how their misexpression contributes to the pathogenesis of hematological malignancies. Also, mutations in many of the epigenetic modifiers individually are generally insufficient for driving hematopoietic transformation, with disease emergence requiring cooperation with alterations in additional regulatory factors. Studies in mice that conditionally inactivate or delete these genes alone and in combination with other chromatin regulatory factors will hopefully provide a more in-depth understanding of the role of these alterations during hematopoietic transformation. In addition to the mouse models of these cancers, hematological cancers are also being modeled in zebrafish (Etchin et al. 2011). These transparent fish are particularly suitable for carrying out genetic and chemical screens that can identify regulators of oncogenesis. Last, although many chromatin modifiers such as MLL3/MLL4/UTX in the COMPASS-like complexes, DNMT3A, and TET2 play a similar tumor suppressor role in different types of cancer; mutations in some of the other epigenetic modifiers may have a tissue-specific role in cancer development. For example, EZH2 mutations are reported to exist in both B-cell lymphoma and T-ALL. However, the impact of these mutations on the biochemical functions of EZH2 in cancer development is distinct. B-cell lymphoma-associated mutations of EZH2 are hyperactivating and lead to increased H3K27 methyltransferase activity, while EZH2 function can be attenuated through inactivating mutations in a subset of T-ALL. Thus, it is of great importance to determine the precise biochemical consequence of mutations in chromatin modifiers in the context of specific hematopoietic cancers. Since many of the chromatin modifiers that are mutated in hematopoietic malignancies are also mutated in solid tumors, the identification of the molecular basis of hematological cancers could also be informative for developing novel therapeutic strategies for other cancers with mutations in these epigenetic modifiers.
Acknowledgments
We are grateful to Dr. E. Smith, Dr. A. Behdad, Dr. A. Piunti, and Dr. P. Ntziachristos for helpful discussions, critical reading of this manuscript, and valuable comments. We also thank M. Miller for preparation of the figures, and L. Shilatifard for editorial assistance. We apologize to colleagues whose studies were not cited in this review due to space limitations. Studies in the Shilatifard laboratory are supported by the National Institutes of Health (R35CA197569).
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.284109.116.
References
- Abdel-Wahab O, Mullally A, Hedvat C, Garcia-Manero G, Patel J, Wadleigh M, Malinge S, Yao J, Kilpivaara O, Bhat R, et al. 2009. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood 114: 144–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdel-Wahab O, Pardanani A, Patel J, Wadleigh M, Lasho T, Heguy A, Beran M, Gilliland DG, Levine RL, Tefferi A. 2011. Concomitant analysis of EZH2 and ASXL1 mutations in myelofibrosis, chronic myelomonocytic leukemia and blast-phase myeloproliferative neoplasms. Leukemia 25: 1200–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdel-Wahab O, Adli M, LaFave LM, Gao J, Hricik T, Shih AH, Pandey S, Patel JP, Chung YR, Koche R, et al. 2012. ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression. Cancer Cell 22: 180–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agger K, Cloos PA, Christensen J, Pasini D, Rose S, Rappsilber J, Issaeva I, Canaani E, Salcini AE, Helin K. 2007. UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature 449: 731–734. [DOI] [PubMed] [Google Scholar]
- Akasaka T, Tsuji K, Kawahira H, Kanno M, Harigaya K, Hu L, Ebihara Y, Nakahata T, Tetsu O, Taniguchi M, et al. 1997. The role of mel-18, a mammalian Polycomb group gene, during IL-7-dependent proliferation of lymphocyte precursors. Immunity 7: 135–146. [DOI] [PubMed] [Google Scholar]
- Andersen EF, Carey JC, Earl DL, Corzo D, Suttie M, Hammond P, South ST. 2014. Deletions involving genes WHSC1 and LETM1 may be necessary, but are not sufficient to cause Wolf-Hirschhorn syndrome. Eur J Hum Genet 22: 464–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aranda S, Mas G, Di Croce L. 2015. Regulation of gene transcription by Polycomb proteins. Sci Adv 1: e1500737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron MH, Isern J, Fraser ST. 2012. The embryonic origins of erythropoiesis in mammals. Blood 119: 4828–4837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beguelin W, Popovic R, Teater M, Jiang Y, Bunting KL, Rosen M, Shen H, Yang SN, Wang L, Ezponda T, et al. 2013. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell 23: 677–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bejar R, Stevenson K, Abdel-Wahab O, Galili N, Nilsson B, Garcia-Manero G, Kantarjian H, Raza A, Levine RL, Neuberg D, et al. 2011. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med 364: 2496–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benyoucef A, Palii CG, Wang C, Porter CJ, Chu A, Dai F, Tremblay V, Rakopoulos P, Singh K, Huang S, et al. 2016. UTX inhibition as selective epigenetic therapy against TAL1-driven T-cell acute lymphoblastic leukemia. Genes Dev 30: 508–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg T, Thoene S, Yap D, Wee T, Schoeler N, Rosten P, Lim E, Bilenky M, Mungall AJ, Oellerich T, et al. 2014. A transgenic mouse model demonstrating the oncogenic role of mutations in the polycomb-group gene EZH2 in lymphomagenesis. Blood 123: 3914–3924. [DOI] [PubMed] [Google Scholar]
- Berger SL, Kouzarides T, Shiekhattar R, Shilatifard A. 2009. An operational definition of epigenetics. Genes Dev 23: 781–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernt KM, Zhu N, Sinha AU, Vempati S, Faber J, Krivtsov AV, Feng Z, Punt N, Daigle A, Bullinger L, et al. 2011. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell 20: 66–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackledge NP, Farcas AM, Kondo T, King HW, McGouran JF, Hanssen LL, Ito S, Cooper S, Kondo K, Koseki Y, et al. 2014. Variant PRC1 complex-dependent H2A ubiquitylation drives PRC2 recruitment and polycomb domain formation. Cell 157: 1445–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock C, Beerman I, Lien WH, Smith ZD, Gu H, Boyle P, Gnirke A, Fuchs E, Rossi DJ, Meissner A. 2012. DNA methylation dynamics during in vivo differentiation of blood and skin stem cells. Mol Cell 47: 633–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borkin D, He S, Miao H, Kempinska K, Pollock J, Chase J, Purohit T, Malik B, Zhao T, Wang J, et al. 2015. Pharmacologic inhibition of the Menin–MLL interaction blocks progression of MLL leukemia in vivo. Cancer Cell 27: 589–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bott M, Brevet M, Taylor BS, Shimizu S, Ito T, Wang L, Creaney J, Lake RA, Zakowski MF, Reva B, et al. 2011. The nuclear deubiquitinase BAP1 is commonly inactivated by somatic mutations and 3p21.1 losses in malignant pleural mesothelioma. Nat Genet 43: 668–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer LA, Plath K, Zeitlinger J, Brambrink T, Medeiros LA, Lee TI, Levine SS, Wernig M, Tajonar A, Ray MK, et al. 2006. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 441: 349–353. [DOI] [PubMed] [Google Scholar]
- Bracken AP, Pasini D, Capra M, Prosperini E, Colli E, Helin K. 2003. EZH2 is downstream of the pRB–E2F pathway, essential for proliferation and amplified in cancer. EMBO J 22: 5323–5335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brien GL, Valerio DG, Armstrong SA. 2016. Exploiting the epigenome to control cancer-promoting gene-expression programs. Cancer Cell 29: 464–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broske AM, Vockentanz L, Kharazi S, Huska MR, Mancini E, Scheller M, Kuhl C, Enns A, Prinz M, Jaenisch R, et al. 2009. DNA methylation protects hematopoietic stem cell multipotency from myeloerythroid restriction. Nat Genet 41: 1207–1215. [DOI] [PubMed] [Google Scholar]
- Buske C, Humphries RK. 2000. Homeobox genes in leukemogenesis. Int J Hematol 71: 301–308. [PubMed] [Google Scholar]
- Busque L, Patel JP, Figueroa ME, Vasanthakumar A, Provost S, Hamilou Z, Mollica L, Li J, Viale A, Heguy A, et al. 2012. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat Genet 44: 1179–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caganova M, Carrisi C, Varano G, Mainoldi F, Zanardi F, Germain PL, George L, Alberghini F, Ferrarini L, Talukder AK, et al. 2013. Germinal center dysregulation by histone methyltransferase EZH2 promotes lymphomagenesis. J Clin Invest 123: 5009–5022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai SF, Chen CW, Armstrong SA. 2015. Drugging chromatin in cancer: recent advances and novel approaches. Mol Cell 60: 561–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cales C, Roman-Trufero M, Pavon L, Serrano I, Melgar T, Endoh M, Perez C, Koseki H, Vidal M. 2008. Inactivation of the polycomb group protein Ring1B unveils an antiproliferative role in hematopoietic cell expansion and cooperation with tumorigenesis associated with Ink4a deletion. Mol Cell Biol 28: 1018–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caligiuri MA, Schichman SA, Strout MP, Mrozek K, Baer MR, Frankel SR, Barcos M, Herzig GP, Croce CM, Bloomfield CD. 1994. Molecular rearrangement of the ALL-1 gene in acute myeloid leukemia without cytogenetic evidence of 11q23 chromosomal translocations. Cancer Res 54: 370–373. [PubMed] [Google Scholar]
- Caligiuri MA, Strout MP, Lawrence D, Arthur DC, Baer MR, Yu F, Knuutila S, Mrozek K, Oberkircher AR, Marcucci G, et al. 1998. Rearrangement of ALL1 (MLL) in acute myeloid leukemia with normal cytogenetics. Cancer Res 58: 55–59. [PubMed] [Google Scholar]
- Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, Jones RS, Zhang Y. 2002. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 298: 1039–1043. [DOI] [PubMed] [Google Scholar]
- Cao F, Townsend EC, Karatas H, Xu J, Li L, Lee S, Liu L, Chen Y, Ouillette P, Zhu J, et al. 2014. Targeting MLL1 H3K4 methyltransferase activity in mixed-lineage leukemia. Mol Cell 53: 247–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caslini C, Yang Z, El-Osta M, Milne TA, Slany RK, Hess JL. 2007. Interaction of MLL amino terminal sequences with menin is required for transformation. Cancer Res 67: 7275–7283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerveira N, Correia C, Doria S, Bizarro S, Rocha P, Gomes P, Torres L, Norton L, Borges BS, Castedo S, et al. 2003. Frequency of NUP98–NSD1 fusion transcript in childhood acute myeloid leukaemia. Leukemia 17: 2244–2247. [DOI] [PubMed] [Google Scholar]
- Challen GA, Sun D, Jeong M, Luo M, Jelinek J, Berg JS, Bock C, Vasanthakumar A, Gu H, Xi Y, et al. 2012. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet 44: 23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Challen GA, Sun D, Mayle A, Jeong M, Luo M, Rodriguez B, Mallaney C, Celik H, Yang L, Xia Z, et al. 2014. Dnmt3a and Dnmt3b have overlapping and distinct functions in hematopoietic stem cells. Cell Stem Cell 15: 350–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang MJ, Wu H, Achille NJ, Reisenauer MR, Chou CW, Zeleznik-Le NJ, Hemenway CS, Zhang W. 2010. Histone H3 lysine 79 methyltransferase Dot1 is required for immortalization by MLL oncogenes. Cancer Res 70: 10234–10242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Liu Y, Rappaport AR, Kitzing T, Schultz N, Zhao Z, Shroff AS, Dickins RA, Vakoc CR, Bradner JE, et al. 2014. MLL3 is a haploinsufficient 7q tumor suppressor in acute myeloid leukemia. Cancer Cell 25: 652–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesi M, Nardini E, Lim RS, Smith KD, Kuehl WM, Bergsagel PL. 1998. The t(4;14) translocation in myeloma dysregulates both FGFR3 and a novel gene, MMSET, resulting in IgH/MMSET hybrid transcripts. Blood 92: 3025–3034. [PubMed] [Google Scholar]
- Cho YW, Hong T, Hong S, Guo H, Yu H, Kim D, Guszczynski T, Dressler GR, Copeland TD, Kalkum M, et al. 2007. PTIP associates with MLL3- and MLL4-containing histone H3 lysine 4 methyltransferase complex. J Biol Chem 282: 20395–20406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury M, Mihara K, Yasunaga S, Ohtaki M, Takihara Y, Kimura A. 2007. Expression of Polycomb-group (PcG) protein BMI-1 predicts prognosis in patients with acute myeloid leukemia. Leukemia 21: 1116–1122. [DOI] [PubMed] [Google Scholar]
- Corces-Zimmerman MR, Hong WJ, Weissman IL, Medeiros BC, Majeti R. 2014. Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proc Natl Acad Sci 111: 2548–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Core N, Bel S, Gaunt SJ, Aurrand-Lions M, Pearce J, Fisher A, Djabali M. 1997. Altered cellular proliferation and mesoderm patterning in Polycomb-M33-deficient mice. Development 124: 721–729. [DOI] [PubMed] [Google Scholar]
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. 2010. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci 107: 21931–21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen SM, Mayle A, Rossi L, Goodell MA. 2014. Hematopoietic stem cell development: an epigenetic journey. Curr Topics Dev Biol 107: 39–75. [DOI] [PubMed] [Google Scholar]
- Dalla-Favera R, Bregni M, Erikson J, Patterson D, Gallo RC, Croce CM. 1982. Human c-myc onc gene is located on the region of chromosome 8 that is translocated in Burkitt lymphoma cells. Proc Natl Acad Sci 79: 7824–7827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danis E, Yamauchi T, Echanique K, Zhang X, Haladyna JN, Riedel SS, Zhu N, Xie H, Orkin SH, Armstrong SA, et al. 2016. Ezh2 controls an early hematopoietic program and growth and survival signaling in early T cell precursor acute lymphoblastic leukemia. Cell Rep 14: 1953–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Mar Lorente M, Marcos-Gutierrez C, Perez C, Schoorlemmer J, Ramirez A, Magin T, Vidal M. 2000. Loss- and gain-of-function mutations show a polycomb group function for Ring1A in mice. Development 127: 5093–5100. [DOI] [PubMed] [Google Scholar]
- Deshpande AJ, Chen L, Fazio M, Sinha AU, Bernt KM, Banka D, Dias S, Chang J, Olhava EJ, Daigle SR, et al. 2013. Leukemic transformation by the MLL-AF6 fusion oncogene requires the H3K79 methyltransferase Dot1l. Blood 121: 2533–2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey A, Seshasayee D, Noubade R, French DM, Liu J, Chaurushiya MS, Kirkpatrick DS, Pham VC, Lill JR, Bakalarski CE, et al. 2012. Loss of the tumor suppressor BAP1 causes myeloid transformation. Science 337: 1541–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding X, Lin Q, Ensenat-Waser R, Rose-John S, Zenke M. 2012. Polycomb group protein Bmi1 promotes hematopoietic cell development from embryonic stem cells. Stem Cells Dev 21: 121–132. [DOI] [PubMed] [Google Scholar]
- Dorsett D. 1999. Distant liaisons: long-range enhancer-promoter interactions in Drosophila. Curr Opin Genet Dev 9: 505–514. [DOI] [PubMed] [Google Scholar]
- Dou Y, Milne TA, Ruthenburg AJ, Lee S, Lee JW, Verdine GL, Allis CD, Roeder RG. 2006. Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat Struct Mol Biol 13: 713–719. [DOI] [PubMed] [Google Scholar]
- Douglas J, Hanks S, Temple IK, Davies S, Murray A, Upadhyaya M, Tomkins S, Hughes HE, Cole TR, Rahman N. 2003. NSD1 mutations are the major cause of Sotos syndrome and occur in some cases of Weaver syndrome but are rare in other overgrowth phenotypes. Am J Hum Genet 72: 132–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doulatov S, Notta F, Laurenti E, Dick JE. 2012. Hematopoiesis: a human perspective. Cell Stem Cell 10: 120–136. [DOI] [PubMed] [Google Scholar]
- Ernst P, Mabon M, Davidson AJ, Zon LI, Korsmeyer SJ. 2004. An Mll-dependent Hox program drives hematopoietic progenitor expansion. Curr Biol 14: 2063–2069. [DOI] [PubMed] [Google Scholar]
- Ernst T, Chase AJ, Score J, Hidalgo-Curtis CE, Bryant C, Jones AV, Waghorn K, Zoi K, Ross FM, Reiter A, et al. 2010. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet 42: 722–726. [DOI] [PubMed] [Google Scholar]
- Etchin J, Kanki JP, Look AT. 2011. Zebrafish as a model for the study of human cancer. Methods Cell Biol 105: 309–337. [DOI] [PubMed] [Google Scholar]
- Feng Y, Yang Y, Ortega MM, Copeland JN, Zhang M, Jacob JB, Fields TA, Vivian JL, Fields PE. 2010. Early mammalian erythropoiesis requires the Dot1L methyltransferase. Blood 116: 4483–4491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa ME, Lugthart S, Li Y, Erpelinck-Verschueren C, Deng X, Christos PJ, Schifano E, Booth J, van Putten W, Skrabanek L, et al. 2010. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell 17: 13–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, et al. 2010. Selective inhibition of BET bromodomains. Nature 468: 1067–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischle W, Wang Y, Jacobs SA, Kim Y, Allis CD, Khorasanizadeh S. 2003. Molecular basis for the discrimination of repressive methyl-lysine marks in histone H3 by Polycomb and HP1 chromodomains. Genes Dev 17: 1870–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan T, Jude CD, Zaffuto K, Ernst P. 2010. Developmentally induced Mll1 loss reveals defects in postnatal haematopoiesis. Leukemia 24: 1732–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z, Zhang J, Bonasio R, Strino F, Sawai A, Parisi F, Kluger Y, Reinberg D. 2012. PCGF homologs, CBX proteins, and RYBP define functionally distinct PRC1 family complexes. Mol Cell 45: 344–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green MR, Gentles AJ, Nair RV, Irish JM, Kihira S, Liu CL, Kela I, Hopmans ES, Myklebust JH, Ji H, et al. 2013. Hierarchy in somatic mutations arising during genomic evolution and progression of follicular lymphoma. Blood 121: 1604–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green MR, Kihira S, Liu CL, Nair RV, Salari R, Gentles AJ, Irish J, Stehr H, Vicente-Duenas C, Romero-Camarero I, et al. 2015. Mutations in early follicular lymphoma progenitors are associated with suppressed antigen presentation. Proc Natl Acad Sci 112: E1116–E1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grembecka J, He SH, Shi AB, Purohit T, Muntean AG, Sorenson RJ, Showalter HD, Murai MJ, Belcher AM, Hartley T, et al. 2012. Menin–MLL inhibitors reverse oncogenic activity of MLL fusion proteins in leukemia. Nat Chem Biol 8: 277–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groschel S, Sanders MA, Hoogenboezem R, de Wit E, Bouwman BA, Erpelinck C, van der Velden VH, Havermans M, Avellino R, van Lom K, et al. 2014. A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell 157: 369–381. [DOI] [PubMed] [Google Scholar]
- Grossmann V, Kohlmann A, Zenger M, Schindela S, Eder C, Weissmann S, Schnittger S, Kern W, Muller MC, Hochhaus A, et al. 2011. A deep-sequencing study of chronic myeloid leukemia patients in blast crisis (BC-CML) detects mutations in 76.9% of cases. Leukemia 25: 557–560. [DOI] [PubMed] [Google Scholar]
- Guglielmelli P, Biamonte F, Score J, Hidalgo-Curtis C, Cervantes F, Maffioli M, Fanelli T, Ernst T, Winkelman N, Jones AV, et al. 2011. EZH2 mutational status predicts poor survival in myelofibrosis. Blood 118: 5227–5234. [DOI] [PubMed] [Google Scholar]
- Harbour JW, Onken MD, Roberson ED, Duan S, Cao L, Worley LA, Council ML, Matatall KA, Helms C, Bowcock AM. 2010. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science 330: 1410–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto H, Liu Y, Upadhyay AK, Chang Y, Howerton SB, Vertino PM, Zhang X, Cheng X. 2012. Recognition and potential mechanisms for replication and erasure of cytosine hydroxymethylation. Nucleic Acids Res 40: 4841–4849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, Ding J, Jia Y, Chen Z, Li L, et al. 2011. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 333: 1303–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, et al. 2007. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet 39: 311–318. [DOI] [PubMed] [Google Scholar]
- Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, et al. 2009. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 459: 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herranz D, Ambesi-Impiombato A, Palomero T, Schnell SA, Belver L, Wendorff AA, Xu L, Castillo-Martin M, Llobet-Navas D, Cordon-Cardo C, et al. 2014. A NOTCH1-driven MYC enhancer promotes T cell development, transformation and acute lymphoblastic leukemia. Nat Med 20: 1130–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera-Merchan A, Arranz L, Ligos JM, de Molina A, Dominguez O, Gonzalez S. 2012. Ectopic expression of the histone methyltransferase Ezh2 in haematopoietic stem cells causes myeloproliferative disease. Nat Commun 3: 623. [DOI] [PubMed] [Google Scholar]
- Herz HM, Mohan M, Garruss AS, Liang K, Takahashi YH, Mickey K, Voets O, Verrijzer CP, Shilatifard A. 2012. Enhancer-associated H3K4 monomethylation by Trithorax-related, the Drosophila homolog of mammalian Mll3/Mll4. Genes Dev 26: 2604–2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herz HM, Hu D, Shilatifard A. 2014. Enhancer malfunction in cancer. Mol Cell 53: 859–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess JL, Yu BD, Li B, Hanson R, Korsmeyer SJ. 1997. Defects in yolk sac hematopoiesis in Mll-null embryos. Blood 90: 1799–1806. [PubMed] [Google Scholar]
- Hidalgo I, Herrera-Merchan A, Ligos JM, Carramolino L, Nunez J, Martinez F, Dominguez O, Torres M, Gonzalez S. 2012. Ezh1 is required for hematopoietic stem cell maintenance and prevents senescence-like cell cycle arrest. Cell Stem Cell 11: 649–662. [DOI] [PubMed] [Google Scholar]
- Hsieh JJ, Cheng EH, Korsmeyer SJ. 2003. Taspase1: a threonine aspartase required for cleavage of MLL and proper HOX gene expression. Cell 115: 293–303. [DOI] [PubMed] [Google Scholar]
- Hu D, Gao X, Morgan MA, Herz HM, Smith ER, Shilatifard A. 2013a. The MLL3/MLL4 branches of the COMPASS family function as major histone H3K4 monomethylases at enhancers. Mol Cell Biol 33: 4745–4754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu D, Smith ER, Garruss AS, Mohaghegh N, Varberg JM, Lin C, Jackson J, Gao X, Saraf A, Florens L, et al. 2013b. The little elongation complex functions at initiation and elongation phases of snRNA gene transcription. Mol Cell 51: 493–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Jiang X, Li Z, Li Y, Song CX, He C, Sun M, Chen P, Gurbuxani S, Wang J, et al. 2013a. TET1 plays an essential oncogenic role in MLL-rearranged leukemia. Proc Natl Acad Sci 110: 11994–11999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z, Wu H, Chuai S, Xu F, Yan F, Englund N, Wang Z, Zhang H, Fang M, Wang Y, et al. 2013b. NSD2 is recruited through its PHD domain to oncogenic gene loci to drive multiple myeloma. Cancer Res 73: 6277–6288. [DOI] [PubMed] [Google Scholar]
- Inoue D, Kitaura J, Togami K, Nishimura K, Enomoto Y, Uchida T, Kagiyama Y, Kawabata KC, Nakahara F, Izawa K, et al. 2013. Myelodysplastic syndromes are induced by histone methylation-altering ASXL1 mutations. J Clin Invest 123: 4627–4640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, Zhang Y. 2011. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 333: 1300–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwama A, Oguro H, Negishi M, Kato Y, Morita Y, Tsukui H, Ema H, Kamijo T, Katoh-Fukui Y, Koseki H, et al. 2004. Enhanced self-renewal of hematopoietic stem cells mediated by the polycomb gene product Bmi-1. Immunity 21: 843–851. [DOI] [PubMed] [Google Scholar]
- Iyer LM, Tahiliani M, Rao A, Aravind L. 2009. Prediction of novel families of enzymes involved in oxidative and other complex modifications of bases in nucleic acids. Cell Cycle 8: 1698–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe JD, Wang Y, Chan HM, Zhang J, Huether R, Kryukov GV, Bhang HE, Taylor JE, Hu M, Englund NP, et al. 2013. Global chromatin profiling reveals NSD2 mutations in pediatric acute lymphoblastic leukemia. Nat Genet 45: 1386–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowska AM, Szpurka H, Tiu RV, Makishima H, Afable M, Huh J, O'Keefe CL, Ganetzky R, McDevitt MA, Maciejewski JP. 2009. Loss of heterozygosity 4q24 and TET2 mutations associated with myelodysplastic/myeloproliferative neoplasms. Blood 113: 6403–6410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowska AM, Makishima H, Tiu RV, Szpurka H, Huang Y, Traina F, Visconte V, Sugimoto Y, Prince C, O'Keefe C, et al. 2011. Mutational spectrum analysis of chronic myelomonocytic leukemia includes genes associated with epigenetic regulation: UTX, EZH2, and DNMT3A. Blood 118: 3932–3941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji H, Ehrlich LI, Seita J, Murakami P, Doi A, Lindau P, Lee H, Aryee MJ, Irizarry RA, Kim K, et al. 2010. Comprehensive methylome map of lineage commitment from haematopoietic progenitors. Nature 467: 338–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PA, Liang G. 2009. Rethinking how DNA methylation patterns are maintained. Nat Rev Genet 10: 805–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jude CD, Climer L, Xu D, Artinger E, Fisher JK, Ernst P. 2007. Unique and independent roles for MLL in adult hematopoietic stem cells and progenitors. Cell Stem Cell 1: 324–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajiume T, Ninomiya Y, Ishihara H, Kanno R, Kanno M. 2004. Polycomb group gene mel-18 modulates the self-renewal activity and cell cycle status of hematopoietic stem cells. Exp Hematol 32: 571–578. [DOI] [PubMed] [Google Scholar]
- Kalb R, Latwiel S, Baymaz HI, Jansen PW, Muller CW, Vermeulen M, Muller J. 2014. Histone H2A monoubiquitination promotes histone H3 methylation in Polycomb repression. Nat Struct Mol Biol 21: 569–571. [DOI] [PubMed] [Google Scholar]
- Kamminga LM, Bystrykh LV, Boer AC, Houwer S, Douma J, Weersing E, Dontje B, de Haan G. 2006. The polycomb group gene Ezh2 prevents hematopoietic stem cell exhaustion. Blood 107: 2170–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanda H, Nguyen A, Chen L, Okano H, Hariharan IK. 2013. The Drosophila ortholog of MLL3 and MLL4, trithorax related, functions as a negative regulator of tissue growth. Mol Cell Biol 33: 1702–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, et al. 2013. Mutational landscape and significance across 12 major cancer types. Nature 502: 333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karatas H, Townsend EC, Cao F, Chen Y, Bernard D, Liu L, Lei M, Dou Y, Wang S. 2013. High-affinity, small-molecule peptidomimetic inhibitors of MLL1/WDR5 protein–protein interaction. J Am Chem Soc 135: 669–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SJ, Zhao H, Hardikar S, Singh AK, Goodell MA, Chen T. 2013. A DNMT3A mutation common in AML exhibits dominant-negative effects in murine ES cells. Blood 122: 4086–4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klauke K, Radulovic V, Broekhuis M, Weersing E, Zwart E, Olthof S, Ritsema M, Bruggeman S, Wu X, Helin K, et al. 2013. Polycomb Cbx family members mediate the balance between haematopoietic stem cell self-renewal and differentiation. Nat Cell Biol 15: 353–362. [DOI] [PubMed] [Google Scholar]
- Kleer CG, Cao Q, Varambally S, Shen R, Ota I, Tomlins SA, Ghosh D, Sewalt RG, Otte AP, Hayes DF, et al. 2003. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci 100: 11606–11611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutson SK, Wigle TJ, Warholic NM, Sneeringer CJ, Allain CJ, Klaus CR, Sacks JD, Raimondi A, Majer CR, Song J, et al. 2012. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat Chem Biol 8: 890–896. [DOI] [PubMed] [Google Scholar]
- Ko M, Bandukwala HS, An J, Lamperti ED, Thompson EC, Hastie R, Tsangaratou A, Rajewsky K, Koralov SB, Rao A. 2011. Ten-eleven-translocation 2 (TET2) negatively regulates homeostasis and differentiation of hematopoietic stem cells in mice. Proc Natl Acad Sci 108: 14566–14571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo Y, Shen L, Cheng AS, Ahmed S, Boumber Y, Charo C, Yamochi T, Urano T, Furukawa K, Kwabi-Addo B, et al. 2008. Gene silencing in cancer by histone H3 lysine 27 trimethylation independent of promoter DNA methylation. Nat Genet 40: 741–750. [DOI] [PubMed] [Google Scholar]
- Krivtsov AV, Feng Z, Lemieux ME, Faber J, Vempati S, Sinha AU, Xia X, Jesneck J, Bracken AP, Silverman LB, et al. 2008. H3K79 methylation profiles define murine and human MLL-AF4 leukemias. Cancer Cell 14: 355–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroeze LI, Nikoloski G, da Silva-Coelho P, van Hoogen P, Stevens-Linders E, Kuiper RP, Schnittger S, Haferlach T, Pahl HL, van der Reijden BA, et al. 2012. Genetic defects in PRC2 components other than EZH2 are not common in myeloid malignancies. Blood 119: 1318–1319. [DOI] [PubMed] [Google Scholar]
- Krogan NJ, Dover J, Khorrami S, Greenblatt JF, Schneider J, Johnston M, Shilatifard A. 2002. COMPASS, a histone H3 (lysine 4) methyltransferase required for telomeric silencing of gene expression. J Biol Chem 277: 10753–10755. [DOI] [PubMed] [Google Scholar]
- Kroon E, Krosl J, Thorsteinsdottir U, Baban S, Buchberg AM, Sauvageau G. 1998. Hoxa9 transforms primary bone marrow cells through specific collaboration with Meis1a but not Pbx1b. EMBO J 17: 3714–3725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku M, Koche RP, Rheinbay E, Mendenhall EM, Endoh M, Mikkelsen TS, Presser A, Nusbaum C, Xie X, Chi AS, et al. 2008. Genomewide analysis of PRC1 and PRC2 occupancy identifies two classes of bivalent domains. PLoS Genet 4: e1000242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn MW, Hadler MJ, Daigle SR, Koche RP, Krivtsov AV, Olhava EJ, Caligiuri MA, Huang G, Bradner JE, Pollock RM, et al. 2015. MLL partial tandem duplication leukemia cells are sensitive to small molecule DOT1L inhibition. Haematologica 100: e190–e193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo AJ, Cheung P, Chen K, Zee BM, Kioi M, Lauring J, Xi Y, Park BH, Shi X, Garcia BA, et al. 2011. NSD2 links dimethylation of histone H3 at lysine 36 to oncogenic programming. Mol Cell 44: 609–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFave LM, Beguelin W, Koche R, Teater M, Spitzer B, Chramiec A, Papalexi E, Keller MD, Hricik T, Konstantinoff K, et al. 2015. Loss of BAP1 function leads to EZH2-dependent transformation. Nat Med 21: 1344–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFave LM, Beguelin W, Koche R, Teater M, Spitzer B, Chramiec A, Papalexi E, Keller MD, Hricik T, Konstantinoff K, et al. 2016. Reply to ‘Uveal melanoma cells are resistant to EZH2 inhibition regardless of BAP1 status’. Nat Med 22: 578–579. [DOI] [PubMed] [Google Scholar]
- Lan F, Bayliss PE, Rinn JL, Whetstine JR, Wang JK, Chen S, Iwase S, Alpatov R, Issaeva I, Canaani E, et al. 2007. A histone H3 lysine 27 demethylase regulates animal posterior development. Nature 449: 689–694. [DOI] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz G. 2014. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 505: 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MG, Villa R, Trojer P, Norman J, Yan KP, Reinberg D, Di Croce L, Shiekhattar R. 2007. Demethylation of H3K27 regulates polycomb recruitment and H2A ubiquitination. Science 318: 447–450. [DOI] [PubMed] [Google Scholar]
- Lee JE, Wang C, Xu S, Cho YW, Wang L, Feng X, Baldridge A, Sartorelli V, Zhuang L, Peng W, et al. 2013. H3K4 mono- and di-methyltransferase MLL4 is required for enhancer activation during cell differentiation. Elife 2: e01503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SC, Miller S, Hyland C, Kauppi M, Lebois M, Di Rago L, Metcalf D, Kinkel SA, Josefsson EC, Blewitt ME, et al. 2015. Polycomb repressive complex 2 component Suz12 is required for hematopoietic stem cell function and lymphopoiesis. Blood 126: 167–175. [DOI] [PubMed] [Google Scholar]
- Lessard J, Sauvageau G. 2003. Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature 423: 255–260. [DOI] [PubMed] [Google Scholar]
- Lewis EB. 1978. A gene complex controlling segmentation in Drosophila. Nature 276: 565–570. [DOI] [PubMed] [Google Scholar]
- Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, Kandoth C, Payton JE, Baty J, Welch J, et al. 2010. DNMT3A mutations in acute myeloid leukemia. N Engl J Med 363: 2424–2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Clevers H. 2010. Coexistence of quiescent and active adult stem cells in mammals. Science 327: 542–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Gonzalez ME, Toy K, Filzen T, Merajver SD, Kleer CG. 2009a. Targeted overexpression of EZH2 in the mammary gland disrupts ductal morphogenesis and causes epithelial hyperplasia. Am J Pathol 175: 1246–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Trojer P, Xu CF, Cheung P, Kuo A, Drury WJ III, Qiao Q, Neubert TA, Xu RM, Gozani O, et al. 2009b. The target of the NSD family of histone lysine methyltransferases depends on the nature of the substrate. J Biol Chem 284: 34283–34295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C, Smith ER, Takahashi H, Lai KC, Martin-Brown S, Florens L, Washburn MP, Conaway JW, Conaway RC, Shilatifard A. 2010. AFF4, a component of the ELL/P-TEFb elongation complex and a shared subunit of MLL chimeras, can link transcription elongation to leukemia. Mol Cell 37: 429–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindberg EH. 2005. Strategies for biology- and molecular-based treatment of myelodysplastic syndromes. Curr Drug Targets 6: 713–725. [DOI] [PubMed] [Google Scholar]
- Lindqvist CM, Nordlund J, Ekman D, Johansson A, Moghadam BT, Raine A, Overnas E, Dahlberg J, Wahlberg P, Henriksson N, et al. 2015. The mutational landscape in pediatric acute lymphoblastic leukemia deciphered by whole genome sequencing. Hum Mutat 36: 118–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Cao L, Chen J, Song S, Lee IH, Quijano C, Liu H, Keyvanfar K, Chen H, Cao LY, et al. 2009. Bmi1 regulates mitochondrial function and the DNA damage response pathway. Nature 459: 387–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohr JG, Stojanov P, Lawrence MS, Auclair D, Chapuy B, Sougnez C, Cruz-Gordillo P, Knoechel B, Asmann YW, Slager SL, et al. 2012. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc Natl Acad Sci 109: 3879–3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorsbach RB, Moore J, Mathew S, Raimondi SC, Mukatira ST, Downing JR. 2003. TET1, a member of a novel protein family, is fused to MLL in acute myeloid leukemia containing the t(10;11)(q22;q23). Leukemia 17: 637–641. [DOI] [PubMed] [Google Scholar]
- Lux CT, Yoshimoto M, McGrath K, Conway SJ, Palis J, Yoder MC. 2008. All primitive and definitive hematopoietic progenitor cells emerging before E10 in the mouse embryo are products of the yolk sac. Blood 111: 3435–3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majewski IJ, Blewitt ME, de Graaf CA, McManus EJ, Bahlo M, Hilton AA, Hyland CD, Smyth GK, Corbin JE, Metcalf D, et al. 2008. Polycomb repressive complex 2 (PRC2) restricts hematopoietic stem cell activity. PLoS Biol 6: e93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majewski IJ, Ritchie ME, Phipson B, Corbin J, Pakusch M, Ebert A, Busslinger M, Koseki H, Hu Y, Smyth GK, et al. 2010. Opposing roles of polycomb repressive complexes in hematopoietic stem and progenitor cells. Blood 116: 731–739. [DOI] [PubMed] [Google Scholar]
- Mansour MR, Abraham BJ, Anders L, Berezovskaya A, Gutierrez A, Durbin AD, Etchin J, Lawton L, Sallan SE, Silverman LB, et al. 2014. Oncogene regulation. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science 346: 1373–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mar BG, Bullinger L, Basu E, Schlis K, Silverman LB, Dohner K, Armstrong SA. 2012. Sequencing histone-modifying enzymes identifies UTX mutations in acute lymphoblastic leukemia. Leukemia 26: 1881–1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R, Reinberg D. 2011. The Polycomb complex PRC2 and its mark in life. Nature 469: 343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R, Li G, Sarma K, Blais A, Zavadil J, Woodcock CL, Dynlacht BD, Reinberg D. 2008. Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol Cell 32: 503–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez AM, Schuettengruber B, Sakr S, Janic A, Gonzalez C, Cavalli G. 2009. Polyhomeotic has a tumor suppressor activity mediated by repression of Notch signaling. Nat Genet 41: 1076–1082. [DOI] [PubMed] [Google Scholar]
- Martinez-Garcia E, Popovic R, Min DJ, Sweet SM, Thomas PM, Zamdborg L, Heffner A, Will C, Lamy L, Staudt LM, et al. 2011. The MMSET histone methyl transferase switches global histone methylation and alters gene expression in t(4;14) multiple myeloma cells. Blood 117: 211–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCabe MT, Graves AP, Ganji G, Diaz E, Halsey WS, Jiang Y, Smitheman KN, Ott HM, Pappalardi MB, Allen KE, et al. 2012a. Mutation of A677 in histone methyltransferase EZH2 in human B-cell lymphoma promotes hypertrimethylation of histone H3 on lysine 27 (H3K27). Proc Natl Acad Sci 109: 2989–2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, Liu Y, Graves AP, Della Pietra A III, Diaz E, et al. 2012b. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 492: 108–112. [DOI] [PubMed] [Google Scholar]
- McMahon KA, Hiew SY, Hadjur S, Veiga-Fernandes H, Menzel U, Price AJ, Kioussis D, Williams O, Brady HJ. 2007. Mll has a critical role in fetal and adult hematopoietic stem cell self-renewal. Cell Stem Cell 1: 338–345. [DOI] [PubMed] [Google Scholar]
- Medvinsky A, Rybtsov S, Taoudi S. 2011. Embryonic origin of the adult hematopoietic system: advances and questions. Development 138: 1017–1031. [DOI] [PubMed] [Google Scholar]
- Mihara K, Chowdhury M, Nakaju N, Hidani S, Ihara A, Hyodo H, Yasunaga S, Takihara Y, Kimura A. 2006. Bmi-1 is useful as a novel molecular marker for predicting progression of myelodysplastic syndrome and patient prognosis. Blood 107: 305–308. [DOI] [PubMed] [Google Scholar]
- Miller T, Krogan NJ, Dover J, Erdjument-Bromage H, Tempst P, Johnston M, Greenblatt JF, Shilatifard A. 2001. COMPASS: a complex of proteins associated with a trithorax-related SET domain protein. Proc Natl Acad Sci 98: 12902–12907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne TA, Briggs SD, Brock HW, Martin ME, Gibbs D, Allis CD, Hess JL. 2002. MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol Cell 10: 1107–1117. [DOI] [PubMed] [Google Scholar]
- Milne TA, Kim J, Wang GG, Stadler SC, Basrur V, Whitcomb SJ, Wang Z, Ruthenburg AJ, Elenitoba-Johnson KS, Roeder RG, et al. 2010. Multiple interactions recruit MLL1 and MLL1 fusion proteins to the HOXA9 locus in leukemogenesis. Mol Cell 38: 853–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min J, Zhang Y, Xu RM. 2003. Structural basis for specific binding of Polycomb chromodomain to histone H3 methylated at Lys 27. Genes Dev 17: 1823–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra BP, Zaffuto KM, Artinger EL, Org T, Mikkola HK, Cheng C, Djabali M, Ernst P. 2014. The histone methyltransferase activity of MLL1 is dispensable for hematopoiesis and leukemogenesis. Cell Rep 7: 1239–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki-Kashio M, Aoyama K, Sashida G, Oshima M, Tomioka T, Muto T, Wang C, Iwama A. 2015. Ezh2 loss in hematopoietic stem cells predisposes mice to develop heterogeneous malignancies in an Ezh1-dependent manner. Blood 126: 1172–1183. [DOI] [PubMed] [Google Scholar]
- Mohan M, Herz HM, Takahashi YH, Lin C, Lai KC, Zhang Y, Washburn MP, Florens L, Shilatifard A. 2010a. Linking H3K79 trimethylation to Wnt signaling through a novel Dot1-containing complex (DotCom). Genes Dev 24: 574–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohan M, Lin C, Guest E, Shilatifard A. 2010b. Licensed to elongate: a molecular mechanism for MLL-based leukaemogenesis. Nat Rev Cancer 10: 721–728. [DOI] [PubMed] [Google Scholar]
- Mohan M, Herz HM, Smith ER, Zhang Y, Jackson J, Washburn MP, Florens L, Eissenberg JC, Shilatifard A. 2011. The COMPASS family of H3K4 methylases in Drosophila. Mol Cell Biol 31: 4310–4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohty M, Yong AS, Szydlo RM, Apperley JF, Melo JV. 2007. The polycomb group BMI1 gene is a molecular marker for predicting prognosis of chronic myeloid leukemia. Blood 110: 380–383. [DOI] [PubMed] [Google Scholar]
- Moore MA, Metcalf D. 1970. Ontogeny of the haemopoietic system: yolk sac origin of in vivo and in vitro colony forming cells in the developing mouse embryo. Br J Haematol 18: 279–296. [DOI] [PubMed] [Google Scholar]
- Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, Figueroa ME, Vasanthakumar A, Patel J, Zhao X, et al. 2011. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell 20: 11–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan MA, Shilatifard A. 2015. Chromatin signatures of cancer. Genes Dev 29: 238–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin RD, Mendez-Lago M, Mungall AJ, Goya R, Mungall KL, Corbett RD, Johnson NA, Severson TM, Chiu R, Field M, et al. 2011. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature 476: 298–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrozek K. 2008. Cytogenetic, molecular genetic, and clinical characteristics of acute myeloid leukemia with a complex karyotype. Semin Oncol 35: 365–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muto T, Sashida G, Oshima M, Wendt GR, Mochizuki-Kashio M, Nagata Y, Sanada M, Miyagi S, Saraya A, Kamio A, et al. 2013. Concurrent loss of Ezh2 and Tet2 cooperates in the pathogenesis of myelodysplastic disorders. J Exp Med 210: 2627–2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Mori T, Tada S, Krajewski W, Rozovskaia T, Wassell R, Dubois G, Mazo A, Croce CM, Canaani E. 2002. ALL-1 is a histone methyltransferase that assembles a supercomplex of proteins involved in transcriptional regulation. Mol Cell 10: 1119–1128. [DOI] [PubMed] [Google Scholar]
- Neff T, Armstrong SA. 2013. Recent progress toward epigenetic therapies: the example of mixed lineage leukemia. Blood 121: 4847–4853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Vosberg S, Schlee C, Heesch S, Schwartz S, Gokbuget N, Hoelzer D, Graf A, Krebs S, Bartram I, et al. 2015. Mutational spectrum of adult T-ALL. Oncotarget 6: 2754–2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen AT, Taranova O, He J, Zhang Y. 2011. DOT1L, the H3K79 methyltransferase, is required for MLL-AF9-mediated leukemogenesis. Blood 117: 6912–6922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikoloski G, Langemeijer SM, Kuiper RP, Knops R, Massop M, Tonnissen ER, van der Heijden A, Scheele TN, Vandenberghe P, de Witte T, et al. 2010. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat Genet 42: 665–667. [DOI] [PubMed] [Google Scholar]
- Ntziachristos P, Tsirigos A, Van Vlierberghe P, Nedjic J, Trimarchi T, Flaherty MS, Ferres-Marco D, da Ros V, Tang Z, Siegle J, et al. 2012. Genetic inactivation of the polycomb repressive complex 2 in T cell acute lymphoblastic leukemia. Nat Med 18: 298–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ntziachristos P, Tsirigos A, Welstead GG, Trimarchi T, Bakogianni S, Xu L, Loizou E, Holmfeldt L, Strikoudis A, King B, et al. 2014. Contrasting roles of histone 3 lysine 27 demethylases in acute lymphoblastic leukaemia. Nature 514: 513–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oguro H, Iwama A, Morita Y, Kamijo T, van Lohuizen M, Nakauchi H. 2006. Differential impact of Ink4a and Arf on hematopoietic stem cells and their bone marrow microenvironment in Bmi1-deficient mice. J Exp Med 203: 2247–2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oguro H, Yuan J, Ichikawa H, Ikawa T, Yamazaki S, Kawamoto H, Nakauchi H, Iwama A. 2010. Poised lineage specification in multipotential hematopoietic stem and progenitor cells by the polycomb protein Bmi1. Cell Stem Cell 6: 279–286. [DOI] [PubMed] [Google Scholar]
- Okada Y, Feng Q, Lin Y, Jiang Q, Li Y, Coffield VM, Su L, Xu G, Zhang Y. 2005. hDOT1L links histone methylation to leukemogenesis. Cell 121: 167–178. [DOI] [PubMed] [Google Scholar]
- Okano M, Xie S, Li E. 1998. Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat Genet 19: 219–220. [DOI] [PubMed] [Google Scholar]
- Okosun J, Bodor C, Wang J, Araf S, Yang CY, Pan C, Boller S, Cittaro D, Bozek M, Iqbal S, et al. 2014. Integrated genomic analysis identifies recurrent mutations and evolution patterns driving the initiation and progression of follicular lymphoma. Nat Genet 46: 176–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono R, Taki T, Taketani T, Taniwaki M, Kobayashi H, Hayashi Y. 2002. LCX, leukemia-associated protein with a CXXC domain, is fused to MLL in acute myeloid leukemia with trilineage dysplasia having t(10;11)(q22;q23). Cancer Res 62: 4075–4080. [PubMed] [Google Scholar]
- Orkin SH, Zon LI. 2008. Hematopoiesis: an evolving paradigm for stem cell biology. Cell 132: 631–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega-Molina A, Boss IW, Canela A, Pan H, Jiang Y, Zhao C, Jiang M, Hu D, Agirre X, Niesvizky I, et al. 2015. The histone lysine methyltransferase KMT2D sustains a gene expression program that represses B cell lymphoma development. Nat Med 21: 1199–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park IK, Qian D, Kiel M, Becker MW, Pihalja M, Weissman IL, Morrison SJ, Clarke MF. 2003. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature 423: 302–305. [DOI] [PubMed] [Google Scholar]
- Park SS, Kim JS, Tessarollo L, Owens JD, Peng L, Han SS, Tae Chung S, Torrey TA, Cheung WC, Polakiewicz RD, et al. 2005. Insertion of c-Myc into Igh induces B-cell and plasma-cell neoplasms in mice. Cancer Res 65: 1306–1315. [DOI] [PubMed] [Google Scholar]
- Pasini D, Bracken AP, Jensen MR, Lazzerini Denchi E, Helin K. 2004. Suz12 is essential for mouse development and for EZH2 histone methyltransferase activity. EMBO J 23: 4061–4071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasqualucci L, Dominguez-Sola D, Chiarenza A, Fabbri G, Grunn A, Trifonov V, Kasper LH, Lerach S, Tang H, Ma J, et al. 2011. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature 471: 189–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasqualucci L, Khiabanian H, Fangazio M, Vasishtha M, Messina M, Holmes AB, Ouillette P, Trifonov V, Rossi D, Tabbo F, et al. 2014. Genetics of follicular lymphoma transformation. Cell Rep 6: 130–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel A, Dharmarajan V, Cosgrove MS. 2008. Structure of WDR5 bound to mixed lineage leukemia protein-1 peptide. J Biol Chem 283: 32158–32161. [DOI] [PubMed] [Google Scholar]
- Patel A, Dharmarajan V, Vought VE, Cosgrove MS. 2009. On the mechanism of multiple lysine methylation by the human mixed lineage leukemia protein-1 (MLL1) core complex. J Biol Chem 284: 24242–24256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel JP, Gonen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, Van Vlierberghe P, Dolgalev I, Thomas S, Aminova O, et al. 2012. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med 366: 1079–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pena-Llopis S, Vega-Rubin-de-Celis S, Liao A, Leng N, Pavia-Jimenez A, Wang S, Yamasaki T, Zhrebker L, Sivanand S, Spence P, et al. 2012. BAP1 loss defines a new class of renal cell carcinoma. Nat Genet 44: 751–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piunti A, Shilatifard A. 2016. Epigenetic balance of gene expression by Polycomb and COMPASS families. Science 352: aad9780. [DOI] [PubMed] [Google Scholar]
- Piunti A, Rossi A, Cerutti A, Albert M, Jammula S, Scelfo A, Cedrone L, Fragola G, Olsson L, Koseki H, et al. 2014. Polycomb proteins control proliferation and transformation independently of cell cycle checkpoints by regulating DNA replication. Nat Commun 5: 3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popovic R, Martinez-Garcia E, Giannopoulou EG, Zhang Q, Zhang Q, Ezponda T, Shah MY, Zheng Y, Will CM, Small EC, et al. 2014. Histone methyltransferase MMSET/NSD2 alters EZH2 binding and reprograms the myeloma epigenome through global and focal changes in H3K36 and H3K27 methylation. PLoS Genet 10: e1004566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao Q, Li Y, Chen Z, Wang M, Reinberg D, Xu RM. 2011. The structure of NSD1 reveals an autoregulatory mechanism underlying histone H3K36 methylation. J Biol Chem 286: 8361–8368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quivoron C, Couronne L, Della Valle V, Lopez CK, Plo I, Wagner-Ballon O, Do Cruzeiro M, Delhommeau F, Arnulf B, Stern MH, et al. 2011. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell 20: 25–38. [DOI] [PubMed] [Google Scholar]
- Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. 2011. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 470: 279–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosati R, La Starza R, Veronese A, Aventin A, Schwienbacher C, Vallespi T, Negrini M, Martelli MF, Mecucci C. 2002. NUP98 is fused to the NSD3 gene in acute myeloid leukemia associated with t(8;11)(p11.2;p15). Blood 99: 3857–3860. [DOI] [PubMed] [Google Scholar]
- Saeidi K. 2016. Myeloproliferative neoplasms: current molecular biology and genetics. Crit Rev Oncol Hematol 98: 375–389. [DOI] [PubMed] [Google Scholar]
- Santos MA, Faryabi RB, Ergen AV, Day AM, Malhowski A, Canela A, Onozawa M, Lee JE, Callen E, Gutierrez-Martinez P, et al. 2014. DNA-damage-induced differentiation of leukaemic cells as an anti-cancer barrier. Nature 514: 107–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuermann JC, de Ayala Alonso AG, Oktaba K, Ly-Hartig N, McGinty RK, Fraterman S, Wilm M, Muir TW, Muller J. 2010. Histone H2A deubiquitinase activity of the Polycomb repressive complex PR-DUB. Nature 465: 243–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schichman SA, Caligiuri MA, Gu Y, Strout MP, Canaani E, Bloomfield CD, Croce CM. 1994. ALL-1 partial duplication in acute leukemia. Proc Natl Acad Sci 91: 6236–6239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlesinger Y, Straussman R, Keshet I, Farkash S, Hecht M, Zimmerman J, Eden E, Yakhini Z, Ben-Shushan E, Reubinoff BE, et al. 2007. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat Genet 39: 232–236. [DOI] [PubMed] [Google Scholar]
- Schoumacher M, Le Corre S, Houy A, Mulugeta E, Stern MH, Roman-Roman S, Margueron R. 2016. Uveal melanoma cells are resistant to EZH2 inhibition regardless of BAP1 status. Nat Med 22: 577–578. [DOI] [PubMed] [Google Scholar]
- Schwartz YB, Kahn TG, Nix DA, Li XY, Bourgon R, Biggin M, Pirrotta V. 2006. Genome-wide analysis of Polycomb targets in Drosophila melanogaster. Nat Genet 38: 700–705. [DOI] [PubMed] [Google Scholar]
- Score J, Hidalgo-Curtis C, Jones AV, Winkelmann N, Skinner A, Ward D, Zoi K, Ernst T, Stegelmann F, Dohner K, et al. 2012. Inactivation of polycomb repressive complex 2 components in myeloproliferative and myelodysplastic/myeloproliferative neoplasms. Blood 119: 1208–1213. [DOI] [PubMed] [Google Scholar]
- Shi AB, Murai MJ, He SH, Lund G, Hartley T, Purohit T, Reddy G, Chruszcz M, Grembecka J, Cierpicki T. 2012. Structural insights into inhibition of the bivalent menin–MLL interaction by small molecules in leukemia. Blood 120: 4461–4469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih AH, Abdel-Wahab O, Patel JP, Levine RL. 2012. The role of mutations in epigenetic regulators in myeloid malignancies. Nat Rev Cancer 12: 599–612. [DOI] [PubMed] [Google Scholar]
- Shih AH, Jiang Y, Meydan C, Shank K, Pandey S, Barreyro L, Antony-Debre I, Viale A, Socci N, Sun Y, et al. 2015. Mutational cooperativity linked to combinatorial epigenetic gain of function in acute myeloid leukemia. Cancer Cell 27: 502–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shilatifard A. 1998. Factors regulating the transcriptional elongation activity of RNA polymerase II. FASEB J 12: 1437–1446. [DOI] [PubMed] [Google Scholar]
- Shilatifard A. 2012. The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annu Rev Biochem 81: 65–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shilatifard A, Lane WS, Jackson KW, Conaway RC, Conaway JW. 1996. An RNA polymerase II elongation factor encoded by the human ELL gene. Science 271: 1873–1876. [DOI] [PubMed] [Google Scholar]
- Shlush LI, Zandi S, Mitchell A, Chen WC, Brandwein JM, Gupta V, Kennedy JA, Schimmer AD, Schuh AC, Yee KW, et al. 2014. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 506: 328–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon C, Chagraoui J, Krosl J, Gendron P, Wilhelm B, Lemieux S, Boucher G, Chagnon P, Drouin S, Lambert R, et al. 2012. A key role for EZH2 and associated genes in mouse and human adult T-cell acute leukemia. Genes Dev 26: 651–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slack JM. 2002. Conrad Hal Waddington: the last Renaissance biologist? Nat Rev Genet 3: 889–895. [DOI] [PubMed] [Google Scholar]
- Smith E, Shilatifard A. 2013. Transcriptional elongation checkpoint control in development and disease. Genes Dev 27: 1079–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith E, Shilatifard A. 2014. Enhancer biology and enhanceropathies. Nat Struct Mol Biol 21: 210–219. [DOI] [PubMed] [Google Scholar]
- Smith ER, Lin C, Garrett AS, Thornton J, Mohaghegh N, Hu D, Jackson J, Saraf A, Swanson SK, Seidel C, et al. 2011. The little elongation complex regulates small nuclear RNA transcription. Mol Cell 44: 954–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souroullas GP, Jeck WR, Parker JS, Simon JM, Liu JY, Paulk J, Xiong J, Clark KS, Fedoriw Y, Qi J, et al. 2016. An oncogenic Ezh2 mutation induces tumors through global redistribution of histone 3 lysine 27 trimethylation. Nat Med 22: 632–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegelmann F, Bullinger L, Schlenk RF, Paschka P, Griesshammer M, Blersch C, Kuhn S, Schauer S, Dohner H, Dohner K. 2011. DNMT3A mutations in myeloproliferative neoplasms. Leukemia 25: 1217–1219. [DOI] [PubMed] [Google Scholar]
- Steidl U, Steidl C, Ebralidze A, Chapuy B, Han HJ, Will B, Rosenbauer F, Becker A, Wagner K, Koschmieder S, et al. 2007. A distal single nucleotide polymorphism alters long-range regulation of the PU.1 gene in acute myeloid leukemia. J Clin Invest 117: 2611–2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein R, Gruenbaum Y, Pollack Y, Razin A, Cedar H. 1982. Clonal inheritance of the pattern of DNA methylation in mouse cells. Proc Natl Acad Sci 79: 61–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struhl G. 1981. A gene product required for correct initiation of segmental determination in Drosophila. Nature 293: 36–41. [DOI] [PubMed] [Google Scholar]
- Su IH, Basavaraj A, Krutchinsky AN, Hobert O, Ullrich A, Chait BT, Tarakhovsky A. 2003. Ezh2 controls B cell development through histone H3 methylation and Igh rearrangement. Nat Immunol 4: 124–131. [DOI] [PubMed] [Google Scholar]
- Tadokoro Y, Ema H, Okano M, Li E, Nakauchi H. 2007. De novo DNA methyltransferase is essential for self-renewal, but not for differentiation, in hematopoietic stem cells. J Exp Med 204: 715–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, et al. 2009. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324: 930–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taketani T, Taki T, Nakamura H, Taniwaki M, Masuda J, Hayashi Y. 2009. NUP98–NSD3 fusion gene in radiation-associated myelodysplastic syndrome with t(8;11)(p11;p15) and expression pattern of NSD family genes. Cancer Genet Cytogenet 190: 108–112. [DOI] [PubMed] [Google Scholar]
- Tavares L, Dimitrova E, Oxley D, Webster J, Poot R, Demmers J, Bezstarosti K, Taylor S, Ura H, Koide H, et al. 2012. RYBP–PRC1 complexes mediate H2A ubiquitylation at polycomb target sites independently of PRC2 and H3K27me3. Cell 148: 664–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tefferi A. 2011. Mutations galore in myeloproliferative neoplasms: would the real Spartacus please stand up? Leukemia 25: 1059–1063. [DOI] [PubMed] [Google Scholar]
- Tetsu O, Ishihara H, Kanno R, Kamiyasu M, Inoue H, Tokuhisa T, Taniguchi M, Kanno M. 1998. mel-18 negatively regulates cell cycle progression upon B cell antigen receptor stimulation through a cascade leading to c-myc/cdc25. Immunity 9: 439–448. [DOI] [PubMed] [Google Scholar]
- Trowbridge JJ, Snow JW, Kim J, Orkin SH. 2009. DNA methyltransferase 1 is essential for and uniquely regulates hematopoietic stem and progenitor cells. Cell Stem Cell 5: 442–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumes DJ, Onodera A, Suzuki A, Shinoda K, Endo Y, Iwamura C, Hosokawa H, Koseki H, Tokoyoda K, Suzuki Y, et al. 2013. The polycomb protein Ezh2 regulates differentiation and plasticity of CD4+ T helper type 1 and type 2 cells. Immunity 39: 819–832. [DOI] [PubMed] [Google Scholar]
- Turkmen S, Gillessen-Kaesbach G, Meinecke P, Albrecht B, Neumann LM, Hesse V, Palanduz S, Balg S, Majewski F, Fuchs S, et al. 2003. Mutations in NSD1 are responsible for Sotos syndrome, but are not a frequent finding in other overgrowth phenotypes. Eur J Hum Genet 11: 858–865. [DOI] [PubMed] [Google Scholar]
- Valinluck V, Sowers LC. 2007. Endogenous cytosine damage products alter the site selectivity of human DNA maintenance methyltransferase DNMT1. Cancer Res 67: 946–950. [DOI] [PubMed] [Google Scholar]
- van den Boom V, Rozenveld-Geugien M, Bonardi F, Malanga D, van Gosliga D, Heijink AM, Viglietto G, Morrone G, Fusetti F, Vellenga E, et al. 2013. Nonredundant and locus-specific gene repression functions of PRC1 paralog family members in human hematopoietic stem/progenitor cells. Blood 121: 2452–2461. [DOI] [PubMed] [Google Scholar]
- van Haaften G, Dalgliesh GL, Davies H, Chen L, Bignell G, Greenman C, Edkins S, Hardy C, O'Meara S, Teague J, et al. 2009. Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. Nat Genet 41: 521–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, Ghosh D, Pienta KJ, Sewalt RG, Otte AP, et al. 2002. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 419: 624–629. [DOI] [PubMed] [Google Scholar]
- Walter MJ, Ding L, Shen D, Shao J, Grillot M, McLellan M, Fulton R, Schmidt H, Kalicki-Veizer J, O'Laughlin M, et al. 2011. Recurrent DNMT3A mutations in patients with myelodysplastic syndromes. Leukemia 25: 1153–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GG, Cai L, Pasillas MP, Kamps MP. 2007. NUP98–NSD1 links H3K36 methylation to Hox-A gene activation and leukaemogenesis. Nat Cell Biol 9: 804–812. [DOI] [PubMed] [Google Scholar]
- Whitman SP, Liu S, Vukosavljevic T, Rush LJ, Yu L, Liu C, Klisovic MI, Maharry K, Guimond M, Strout MP, et al. 2005. The MLL partial tandem duplication: evidence for recessive gain-of-function in acute myeloid leukemia identifies a novel patient subgroup for molecular-targeted therapy. Blood 106: 345–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods BA, Levine RL. 2015. The role of mutations in epigenetic regulators in myeloid malignancies. Immunol Rev 263: 22–35. [DOI] [PubMed] [Google Scholar]
- Xie H, Xu J, Hsu JH, Nguyen M, Fujiwara Y, Peng C, Orkin SH. 2014. Polycomb repressive complex 2 regulates normal hematopoietic stem cell function in a developmental-stage-specific manner. Cell Stem Cell 14: 68–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita Y, Yuan J, Suetake I, Suzuki H, Ishikawa Y, Choi YL, Ueno T, Soda M, Hamada T, Haruta H, et al. 2010. Array-based genomic resequencing of human leukemia. Oncogene 29: 3723–3731. [DOI] [PubMed] [Google Scholar]
- Yamazaki H, Suzuki M, Otsuki A, Shimizu R, Bresnick EH, Engel JD, Yamamoto M. 2014. A remote GATA2 hematopoietic enhancer drives leukemogenesis in inv(3)(q21;q26) by activating EVI1 expression. Cancer Cell 25: 415–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Rau R, Goodell MA. 2015. DNMT3A in haematological malignancies. Nat Rev Cancer 15: 152–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap DB, Chu J, Berg T, Schapira M, Cheng SW, Moradian A, Morin RD, Mungall AJ, Meissner B, Boyle M, et al. 2011. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood 117: 2451–2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama A, Wang Z, Wysocka J, Sanyal M, Aufiero DJ, Kitabayashi I, Herr W, Cleary ML. 2004. Leukemia proto-oncoprotein MLL forms a SET1-like histone methyltransferase complex with menin to regulate Hox gene expression. Mol Cell Biol 24: 5639–5649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama A, Somervaille TC, Smith KS, Rozenblatt-Rosen O, Meyerson M, Cleary ML. 2005. The menin tumor suppressor protein is an essential oncogenic cofactor for MLL-associated leukemogenesis. Cell 123: 207–218. [DOI] [PubMed] [Google Scholar]
- Zeisig BB, Milne T, Garcia-Cuellar MP, Schreiner S, Martin ME, Fuchs U, Borkhardt A, Chanda SK, Walker J, Soden R, et al. 2004. Hoxa9 and Meis1 are key targets for MLL–ENL-mediated cellular immortalization. Mol Cell Biol 24: 617–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, Easton J, Chen X, Wang J, Rusch M, et al. 2012. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 481: 157–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Grubor V, Love CL, Banerjee A, Richards KL, Mieczkowski PA, Dunphy C, Choi W, Au WY, Srivastava G, et al. 2013. Genetic heterogeneity of diffuse large B-cell lymphoma. Proc Natl Acad Sci 110: 1398–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Kinkel S, Maksimovic J, Bandala-Sanchez E, Tanzer MC, Naselli G, Zhang JG, Zhan Y, Lew AM, Silke J, et al. 2014. The polycomb repressive complex 2 governs life and death of peripheral T cells. Blood 124: 737–749. [DOI] [PubMed] [Google Scholar]
- Zhang J, Dominguez-Sola D, Hussein S, Lee JE, Holmes AB, Bansal M, Vlasevska S, Mo T, Tang H, Basso K, et al. 2015. Disruption of KMT2D perturbs germinal center B cell development and promotes lymphomagenesis. Nat Med 21: 1190–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]