

Abstract
Most fluorescence microscopes are inefficient, collecting only a small fraction of the emitted light at any instant. Besides wasting valuable signal, this inefficiency also reduces spatial resolution and causes imaging volumes to exhibit significant resolution anisotropy. We describe microscopic and computational techniques that address these problems by simultaneously capturing and subsequently fusing and deconvolving multiple specimen views. Unlike previous methods that serially capture multiple views, our approach improves spatial resolution without introducing any additional illumination dose or compromising temporal resolution relative to conventional imaging. When applying our methods to single-view wide-field or dual-view light-sheet microscopy, we achieve a twofold improvement in volumetric resolution (~235 nm × 235 nm × 340 nm) as demonstrated on a variety of samples including microtubules in Toxoplasma gondii, SpoVM in sporulating Bacillus subtilis, and multiple protein distributions and organelles in eukaryotic cells. In every case, spatial resolution is improved with no drawback by harnessing previously unused fluorescence.
1. INTRODUCTION
Fluorescence microscopy is a powerful imaging tool in biology due to its high sensitivity and specificity [1,2]. However, most implementations remain inefficient: even if a high numerical aperture (NA) lens is used for imaging, most of the emitted fluorescence falls outside the angular acceptance cone of the lens. Besides wasting valuable signal, imaging only a portion of the fluorescence through a single lens also reduces spatial resolution and introduces significant resolution anisotropy (i.e., axial resolution along the detection axis is twofold to threefold poorer than lateral resolution in the focal plane), as any conventional lens collects fewer spatial frequencies along its detection axis than perpendicular to it.
A partial solution to these problems ensues if multiple views of the specimen can be acquired along different detection axes. Although any single view is plagued by an anisotropic point spread function (PSF), fusing all views together and using joint deconvolution to extract the best resolution from each view [3–6] can boost spatial resolution and improve resolution isotropy. Unfortunately, in almost every implementation of multiview imaging we are aware of, views are captured serially. Serial acquisition always introduces a significant cost relative to single-view imaging, since temporal resolution and illumination dose (and thus photobleaching and photodamage) are worsened with each additional view.
A better alternative would be to acquire multiple specimen views simultaneously (subsequently fusing and deconvolving them), thereby boosting spatial resolution without compromising acquisition speed or introducing additional illumination relative to conventional imaging. Here we report such an imaging configuration by extending single-view wide-field microscopy and dual-view light-sheet microscopy [6] to a triple-view microscopy configuration. In both cases volumetric resolution is improved at least twofold to 235 nm × 235 nm × 340 nm by simultaneous acquisition along three detection axes in wide-field mode and two detection axes in light-sheet mode. Key to the success of our approach is the development of improved acquisition, registration, and deconvolution methods that are well adapted to the challenges of simultaneous multiview imaging. The effectiveness of these methods is demonstrated by obtaining high-resolution volumetric (3D) images and time-lapse (4D) image series on a range of fixed and live cellular samples, including microtubules in Toxoplasma gondii, SpoVM in sporulating Bacillus subtilis, and multiple protein distributions and organelles in eukaryotic cells. In all cases, lateral or axial resolution was improved by harnessing previously unused fluorescence.
2. RESULTS
Wide-field microscopy remains a workhorse in biological imaging due to its technical simplicity, speed, and robust performance. When interrogating thin, semi-transparent samples, 3D wide-field imaging can also be substantially improved if deconvolution is properly applied [7]. Nevertheless, substantial resolution anisotropy arises because the single objective typically employed in wide-field imaging collects only a cone of fluorescence emission. For example, if a 1.2 numerical aperture (NA) water-immersion lens is used to image into aqueous specimens, at best only 28% of the full 4 pi solid angle is obtained, resulting in an axial resolution ~3-fold worse than lateral resolution [Figs. 1(c) and 1(d), Figs. S1 and S2; measured lateral resolution before deconvolution, 346 ± 14 nm; axial resolution, 920 ± 56 nm; N = 10; FWHM values from 100 nm fluorescent beads]. We reasoned that simultaneous collection with more detection lenses would cover a larger fraction of the full 4 pi solid angle, thereby better sampling Fourier space [Fig. 1(c)], so we modified a previous, dual-objective setup [6] for simultaneous triple-view wide-field imaging [Fig. 1(a), Section 4, Figs. S3 and S4]. Our microscope places the sample on a conventional glass coverslip, excites the sample with wide-field illumination, and simultaneously collects emissions from two upper, 0.8 NA water lenses [objectives “A” and “B” in Fig. 1(a)] and one lower, 1.2 NA water lens [objective “C” in Fig. 1(a)], imaging them onto three scientific complementary metal-oxide semiconductor (sCMOS) cameras. PSF measurements from each lens (Fig. S2) showed the expected anisotropy characteristic of single-objective imaging, yet by registering all three views and performing joint Richardson–Lucy deconvolution (Section 4, Fig. S5), we were able to obtain a sharper PSF with ~1.5-fold improvement in axial resolution relative to the result obtained when performing single-view deconvolution only [Fig. 1(e), Fig. S2; triple-view axial resolution, 341 ± 13 nm; single-view axial resolution, 539 ± 30 nm]. Lateral resolution was equivalent when considering all three views or only the single, high NA lens (triple-view, 237 ± 13 nm; single-view, 233 ± 7 nm). Here, the lateral (x, y) and axial (z) directions are defined from the bottom objective’s (C) perspective, as shown in Fig. 1(a).
Fig. 1.
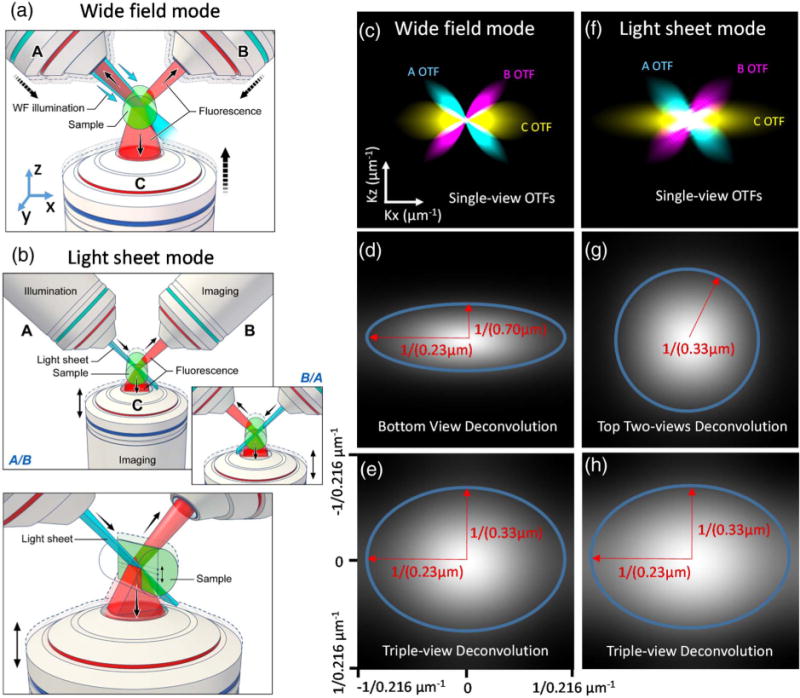
Schematic representation of triple-view wide-field and light-sheet microscopy. (a) In triple-view wide-field microscopy, wide-field illumination is introduced to the sample via one of the two upper objectives (A or B), and all three objectives (A, B, C) simultaneously collect fluorescence emissions from the sample volume. (b) In triple-view light-sheet microscopy, planar illumination is alternately introduced by either of the upper objectives (A/B), with concurrent collection from the other upper objective (B/A) and lower objective (C). Note that A/B are stationary, while C is swept vertically to collect fluorescence from the inclined illuminated plane. Inset shows alternating illumination provided by B; lower panel shows perspective view. (c) Optical transfer functions (OTFs) for each objective in wide-field mode, assuming 0.8 NA for A/B and 1.2 NA for C. A cross section along K x/kz directions is shown to highlight resolution anisotropy between lateral and axial directions. Comparative OTFs are also shown after deconvolution of (d) C alone and (e) all three views. (f) OTFs for each objective in light-sheet mode, assuming the same NAs as in (c). Comparative OTFs are also shown after joint deconvolution of (g) A, B and (h) all three views. In both wide-field and light-sheet microscopy, deconvolution of all three views improves resolution. Blue ellipses and red arrows in (d), (g), (e), and (h) indicate lateral and axial diffraction limits.
We found that the resolution gain enabled by simultaneous triple-view imaging was even more striking when viewing biological specimens. First, we imaged GFP-tagged SpoVM in live Bacillus subtilis bacterial cells. SpoVM is a small protein expressed when B. subtilis undergoes spore formation (sporulation) and localizes to the developing forespore surface [8] to direct assembly of a spherical protein shell, termed the “spore coat” that encases the forespore [9]. Conventional wide-field imaging through objective C revealed the expected distribution of SpoVM around the forespore periphery [Figs. 2(a) and 2(b)], yet axial cuts through the center of the forespore periphery were distorted due to the axial diffraction limit [Fig. 2(b)]. Deconvolution sharpened both lateral and axial views [Fig. 2(e)], but did not address the underlying anisotropy, as the distribution of SpoVM still appeared stretched in z. Conversely, wide-field views simultaneously acquired from objectives A and B were severely blurred in the lateral direction, but better resolved the forespore structure along the axial dimension [Figs. 2(c) and 2(d)]. Using bead-based registration in conjunction with joint deconvolution in order to optimally fuse all three views [Fig. 2(f), Visualization 1 and Visualization 2, Section 4] produced the best images, maintaining the sharp lateral resolution inherent to the lower view while extracting the higher spatial frequencies from the top two views to produce an axial resolution that much better depicted the circular shape of the forespore. We observed the same effect when imaging fixed Toxoplasma gondii ectopically expressing EGFP-Tgβ1-tubulin driven by the T. gondii α1 tubulin promoter [10] [Figs. 2(g)–2(l), Visualization 3]. T. gondii has several tubulin-containing cytoskeletal structures that are highly reproducible, including a truncated cone at the apical end (“conoid”) [11], a pair of intraconoid microtubules, and an array of 22 cortical microtubules [11,12]. Microtubules are also assembled into the mitotic spindle during parasite replication. Nascent daughter buds are formed close to the poles of the mitotic spindle, with each pole resembling a “comet tail” that marks the basal end of the daughters growing within the mother parasite [13]. Individual parasites within an intracellular eight-parasite vacuole were visible in the raw data collected by objective C, but out-of-focus haze obscured the cortical microtubules, and poor axial resolution distorted the cross-sectional parasite shapes [Figs. 2(g) and 2(j)]. Deconvolving the lower view alone reduced haze and improved observation of the cortical microtubules and daughter buds with half-spindle comet tails [red and green arrows, respectively, Fig. 2(h)], but still resulted in significant axial distortion [Fig. 2(k)]. Triple-view deconvolution resulted in high-resolution imaging along both lateral [Fig. 2(i)] and axial [Fig. 2(l)] directions.
Fig. 2.
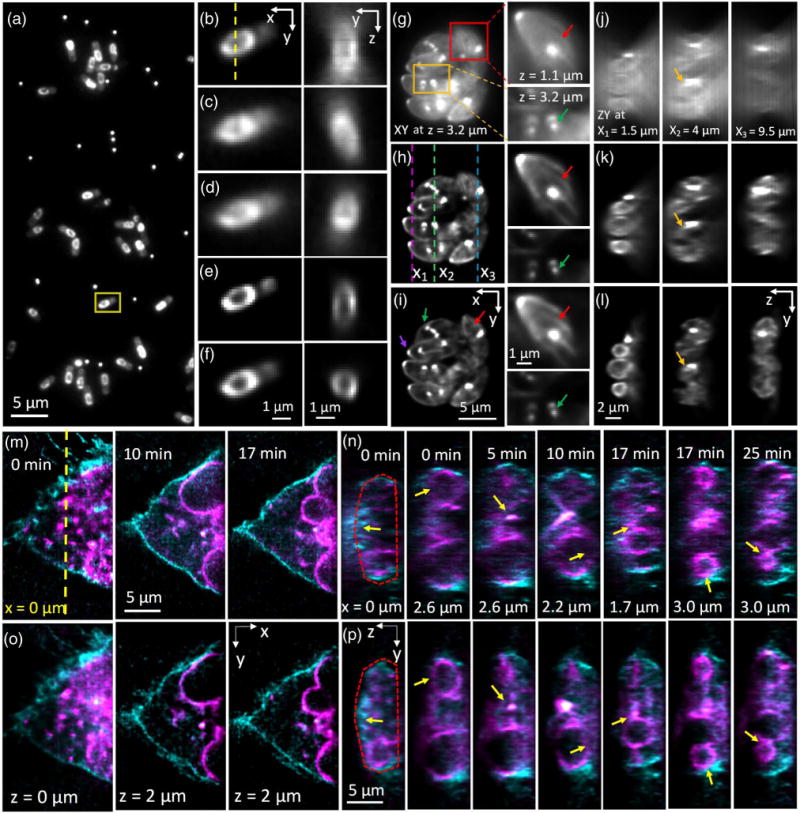
Simultaneous triple-view wide-field imaging improves axial resolution relative to single-view wide-field microscopy. (a) Single-view (perspective from objective C) lateral slice, showing many Bacillus subtilis cells expressing SpoVM-GFP. Also evident are bright puncta, from 100 nm yellow–green fluorescent beads used in aiding registration of all three views. Higher magnification views (all from the perspective of objective C) of the yellow rectangular region in (a) are shown in (b) (captured via objective C), (c) (captured via objective A), (d) (captured via objective B), (e) (deconvolved view captured from objective C), and (f) (triple-view deconvolution). Also shown in (b)–(f) are corresponding axial cuts through the dotted line marked in (b), highlighting the center of the daughter spore. The triple-view deconvolution best captures the circular cross section of the spore. See also Visualization 1 and Visualization 2. (g)–(i) Single lateral slices from imaging volume of fixed Toxoplasma gondii expressing EGFP-Tgβ1-tubulin, as viewed (g) in objective C and after (h) single-view and (i) triple-view deconvolution. Higher magnification views of the red and orange boxed regions at the indicated axial location are shown on the right. The apical ends of the parasites, marked by the conoid (purple arrows), are oriented away from the center of the vacuole. The presence of the mitotic spindle (green arrows) indicates that these parasites are actively replicating, with nascent daughter buds found at the mitotic spindle poles. Red arrows indicate cortical microtubules. (j)–(l) Corresponding axial slices marked as magenta (left), green (middle), and blue (right) dotted lines in (h) (separated by 2.5 and 5.5 μm in the x direction). Triple-view deconvolution (l) preserves the axial shape of the parasites (example indicated by yellow arrows), which is otherwise distorted in single-view imaging (j),(k). See also Visualization 3. (m)–(p) Dual-color, live imaging of U2OS cells expressing Lck-tGFP (cyan), labeling the plasma membrane, and ERGIC3-mCherry (magenta), labeling dilated and vesiculated ER. (m) and (o) highlight lateral sections through the 4D image acquisition at indicated times, after (m) single- and (o) triple-view deconvolution, with relative axial position and time indicated. (n),(p) Corresponding axial cuts at indicated x position [origin is indicated as yellow dotted line in (m)] and time. Yellow arrows indicate features such as nuclear periphery, cell membrane (red dotted lines), or vesicles that are badly distorted after single-view deconvolution, but better resolved after triple-view deconvolution. See also Visualization 4 (comparative time-lapse of axial cuts through sample). 100 iterations were used for single-view deconvolution, and 200 iterations for triple-view deconvolution.
We also used our triple-view wide-field microscope to perform extended two-color, 4D imaging of membranous structures in a live human osteosarcoma cell line (U2OS) over 100 volumes [Figs. 2(m)–2(p), Visualization 4]. We visualized the dynamics of the plasma membrane with Lck-tGFP [cyan in Figs. 2(m)–2(p)]; Lck is a member of the Src family of protein tyrosine kinases that is palmitoylated at its N terminus, and eventually targeted to the plasma membrane. We marked the endomembranous trafficking system using the ER-Golgi intermediate compartment marker ERGIC3 tagged with mCherry [magenta in Figs. 2(m)–2(p)], thus observing the dilated and vesiculated endoplasmic reticulum. Single-view (via objective C) deconvolution resolved lateral membranes well [Fig. 2(m)], but failed to properly resolve the dorsal plasma membrane [Fig. 2(n)]. Additionally, the curvature of membrane-enclosed structures such as vesicles and the nuclear periphery were artificially lengthened [Fig. 2(n)]. These imaging artifacts were significantly improved after triple-view imaging and deconvolution [Figs. 2(o) and 2(p)].
Although simultaneous wide-field imaging has been demonstrated before on fluorescent beads, data from each view were combined by averaging [14], therefore degrading lateral resolution relative to our deconvolution-based approach. Our implementation does not suffer from this problem as we extract the best resolution from each view, and, to our knowledge, it is the first demonstration of the concept on biological specimens. Nevertheless, any wide-field microscope bleaches fluorophores and damages the specimen outside the focal plane. Furthermore, in very densely labeled samples, out-of-focus background (and the associated shot noise) swamps in-focus signal to the extent that recovery of the high-quality in-focus signal is impossible, regardless of deconvolution. Using a thin sheet of light to excite only the focal plane alleviates these problems, so we next adapted our triple-view wide-field microscope for triple-view imaging with light-sheet illumination [Fig. 1(b), Figs. S3 and S6, Section 4].
We previously developed dual-view selective plane illumination microscopy (diSPIM [6,15]), an implementation of light-sheet microscopy that uses two 0.8 NA objectives to excite and detect fluorescence in an alternating duty cycle, subsequently registering and deconvolving the data to achieve isotropic resolution down to ~330 nm (a ~5 fold improvement in axial resolution compared to earlier single-view implementations [16]). Despite the advantages of diSPIM, resolution is still limited by the NA of the upper lenses [Figs. 1(f) and 1(g)], and fluorescence emitted in the direction of the coverslip is not captured. We reasoned that, as with the triple-view wide-field microscope, simultaneously imaging this otherwise neglected signal with a higher NA lens [Figs. 1(b) and 1(f)] could in principle boost the resolution of the diSPIM system [Figs. 1(g) and 1(h)]. The challenge in this case is that each light sheet (which is supplied via the upper lenses, and held stationary) is inclined at a 45 deg angle relative to the lower objective lens and thus extends well beyond its depth of focus. Therefore, during each light-sheet exposure we swept the lower, 1.2 NA objective’s plane of focus through the sample [Fig. 1(b)], imaging this previously unused fluorescence onto the lower sCMOS camera. To aid in discriminating out-of-focus light that would otherwise contaminate this lower view, we synchronized the rolling shutter [17] of the lower sCMOS with the axial sweep of the 1.2 NA objective (Section 4, Figs. S7 and S8). This combination of lens motion and rolling shutter thus effectively allows for imaging with a tilted focal plane. The sample was then laterally translated through the light sheets, allowing for construction of three imaging volumes, as viewed by the three lenses.
When imaging 100 nm fluorescent beads, registering and combining all three views via joint deconvolution (Section 4) resulted in ~1.4-fold improvement in lateral resolution relative to diSPIM, as expected given the greater NA of the lower objective [Figs. 1(f)–1(h), Section 4, Figs. S5 and S9; triple-view lateral resolution, 235 ± 15 nm; diSPIM lateral resolution, 306 ± 7 nm]. Axial resolution remained the same as in diSPIM (triple-view, 324 ± 19 nm; diSPIM, 335 ± 8 nm).
The resolution enhancement achieved via triple-view light-sheet imaging also extended to whole, fixed cells (Fig. 3). When imaging immunolabeled microtubules, raw lateral and axial views of the sample from the upper [Fig. 3(a), 3(h), and 3(i)] and lower objectives [Figs. 3(b) and 3(j)] revealed the expected resolution anisotropy along these directions. Notably, images from the lower view [Fig. 3(b)] were comparable in quality to those achieved via the top view [Fig. 3(a)], demonstrating the effectiveness of our partially confocal slit method in imaging such tilted focal planes. After appropriate registration and deconvolution of the three views, microtubules were better resolved laterally in the triple-view deconvolution [Fig. 3(d) and lower panels in Figs. 3(e)–3(g)] compared to the dual-view deconvolution result [Fig. 3(c) and upper panels in Figs. 3(e) and 3(g)]. The axial resolution after triple-view deconvolution remained the same as in dual-view deconvolution [Figs. 3(h)–3(l)].
Fig. 3.
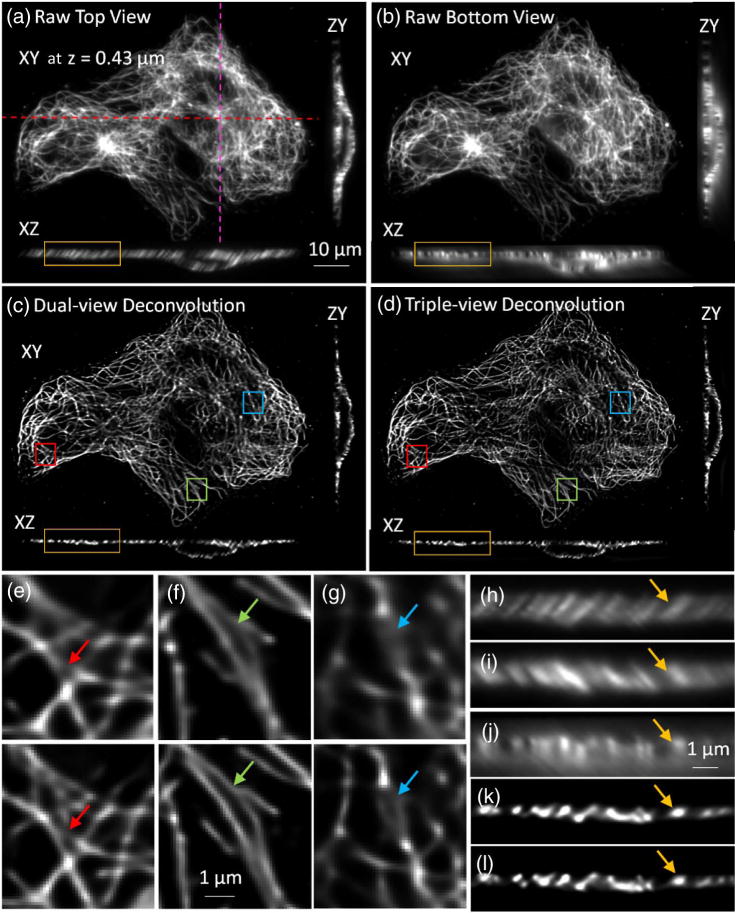
Improvements in lateral resolution after triple-view light-sheet imaging. (a) Fixed and immunolabeled microtubules in U2OS cells, as viewed through top (“A”) objective. Lateral plane 0.43 μm above the coverslip surface, and axial cuts at indicated dotted lines are shown. (b) Same sample and illumination as in (a), but viewed through lower, higher NA objective (“C”), simultaneously acquired with view captured by “A.” (c) Corresponding dual-view (top two views) deconvolution and (d) triple-view deconvolution images are also shown. Higher magnification lateral views corresponding to deconvolved dual- [upper panel, (e), (f), and (g)] and triple-view [lower panel, (e), (f), and (g)] deconvolution are also shown, as are higher magnification axial views [(h) from (a); (j) from (b); (k) from (c); (l) from (d)]. For completeness, a higher magnification axial view from objective B view is also shown (i). As arrows in (e)–(l) highlight, triple-view deconvolution improves lateral resolution without compromising axial resolution inherent to dual-view deconvolution. Sixty iterations were performed for dual-view deconvolution; 180 iterations for triple-view deconvolution.
In addition to boosting the lateral resolution on fixed cells, our method also improves resolution when volumetrically imaging live cells over many time points (Fig. 4). For example, the formation of dynamic structures, such as ruffles and filopodia, driven by the actin cytoskeleton over the entire volume of live macrophages, was more clearly resolved using our triple-view light-sheet system [Figs. 4(a)–4(c), Visualization 5 and Visualization 6] than in diSPIM over 150 volumes in a 40 min timecourse. Due to the 3D, dynamic nature of these actin-based protrusions, they are difficult to image with conventional imaging techniques. We also performed two-color 4D live imaging of GFP-tagged H-Ras and mCherry-Rab8 over 140 volumes, highlighting plasma membrane dynamics and vesicular trafficking from the trans Golgi network in live U2OS cells [Fig. 4(d), Visualization 7 and Visualization 8]. Triple-view light-sheet imaging better resolved filopodia at the rear of the cell [Figs. 4(e) and 4(f)], the reticular Rab network near the base of the cell [Figs. 4(g) and 4(h)], and moving vesicles [Figs. 4(i)–4(l)]. As a final example, we dye-loaded mitochondria with MitoTracker Red (CMXRos) in U2OS cells [Fig. 4(m), Visualization 9]. Triple-view imaging revealed “bead-on-a-string” labeling of mitochondria [Figs. 4(n) and 4(o)], whereas diSPIM imaging resulted in images that appeared artificially homogenous [Fig. 4(n)]. Transient fluctuations in mitochondrial membrane potential [18] were also easily resolved in triple-view light-sheet imaging. We note that in all experiments, bleaching was either minor [Figs. 4(a)–4(c) and Figs. 4(m)–4(o)] or absent [Figs. 4(d)–4(h)].
Fig. 4.
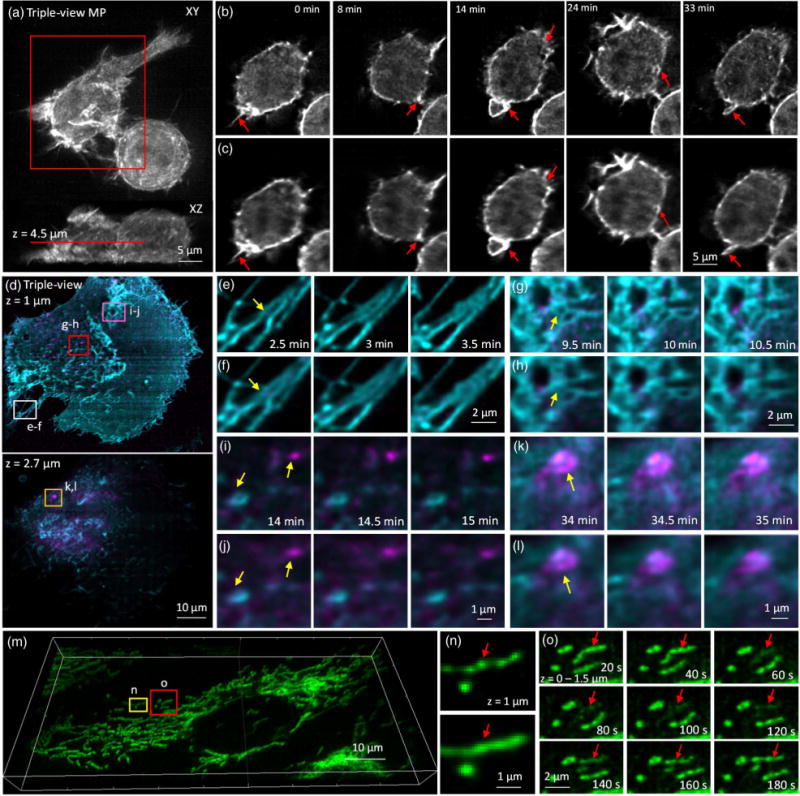
Triple-view light-sheet microscopy improves lateral resolution relative to dual-view light-sheet microscopy. (a) Maximum intensity projection of F-tractin-EGFP in RAW 264.7 macrophage, after triple-view fusion and deconvolution. (b) Higher magnification lateral views of red rectangular region in (a), 4.5 μm from the coverslip surface. Comparative images from dual-view imaging (c) are also shown. Red arrows mark actin protrusions, forming membrane ruffles and filopodia, which are better resolved in triple- than dual-view imaging. See also Visualization 5 and Visualization 6. (d) Two-color imaging of GFP-Ras and mCherry-Rab8 in U2OS cells after triple-view imaging. Lateral planes 1 μm (upper) and 2.7 μm (lower) from the coverslip are shown. Comparative images highlight resolution improvement (yellow arrows) of triple-view [(e), (g), (i), and (k)] relative to dual-view [(f), (h), (j), and (l)] light-sheet imaging, especially in fine structures such as filopodia [(e) and (f)], reticular structures within the plasma membrane [(g) and (h)], and intracellular vesicles and endosomes [(I),(j) and (k),(l)]. Higher magnification images [(e)–(l)] correspond to rectangular regions in (d); see also Visualization 7 and Visualization 8. (m) Mitochondria in U2OS cells were labeled with Mitotracker Red and imaged in triple-view light-sheet microscopy. Imaris rendering is shown. (n) Comparison between triple-view (upper) and dual-view (lower) imaging shows that the nonhomogenous, “beads on a string” staining of mitochondria is better resolved in triple-view imaging. Images are maximum intensity projections over 1.5 μm, and correspond to yellow rectangular region in (m). (o) Higher magnification views of red boxed region in (m) (maximum intensity projections over 1.5 μm) highlighting loss and gain in mitochondrial membrane potential (red arrows) over time. See also Visualization 9. For dual-view deconvolution, 60 iterations were used; for triple-view deconvolution, 180 iterations were used for (a) and (b), 120 iterations for the green channel in (d)–(l), and 100 iterations for the red channel in (d)–(o).
3. DISCUSSION
The technology described here improves wide-field microscopy (by providing better axial resolution, to ~340 nm) and diSPIM (by improving lateral resolution, to ~235 nm) by collecting “free” light that is neglected in conventional imaging. We note that this lateral resolution is equivalent to that usually reported with confocal microscopy (and higher than that typically achieved with light-sheet microscopy), and the axial resolution is ~2-fold better than that with confocal microscopy. Relative to state-of-the-art multiview light-sheet imaging, our method improves volumetric spatial resolution at least 4x [19], yet requires no custom parts and may be readily assembled with commercially available hardware. As we demonstrate on biological samples (Figs. 2–4) up to 12 μm thick, the resulting images reveal features that are otherwise masked by diffraction.
Earlier multiview approaches for resolution improvement used two opposed objectives for coherent detection of fluorescence emission, coherent superposition of two excitation beams, or both coherent detection and coherent excitation [20]. Since the majority of the available angular aperture is used, these methods enable axial resolution in the ~100 nm range. This improvement is considerably better than what we report here, yet such “4Pi” [21] (or its wide-field alternative, I5M [22]) microscopy relies on high NA lenses (limiting working distance) and is nontrivial to build (mechanisms to actively stabilize the coherence are needed), and the requisite interferometry is very sensitive to refractive index (RI) variations in the sample (often requiring samples to be fixed and/or carefully adjusting the RI in the medium to minimize aberrations). These issues have limited the utility of 4Pi/I5M imaging in live biological imaging. From a practical perspective, we thus suspect that our multiview imaging method—and, in particular, the ability to boost resolution without interference between multiple lenses—is better suited to high-speed, 4D imaging over prolonged durations.
A future implementation that might improve the overall volumetric imaging speed and signal-to-noise ratio (SNR) of the lower view (currently worse than the top views, despite the higher NA; see Supplement 1) in the triple-view light-sheet system would replace the mechanical scan of the lower objective with a passive optic (such as a cubic phase plate [23], multifocus grating [24], or refractive optic [25]) that extends depth-of-field, especially if depth-of-field extension can be established without substantially compromising lateral resolution. Further improving the lower-view SNR might also enable effectively deeper imaging, until depth is limited by either aberrations or scattering—in which case better sample coverage may be required [19]. Additionally, incorporating higher NA objectives, especially in the top view, would produce greater angular coverage, and thus better resolution and SNR in the reconstruction.
The key message of our work is that normally unused light can be simultaneously imaged and fused with the “conventional” view normally captured in fluorescence microscopy, thereby improving 3D spatial resolution without compromising acquisition speed or introducing additional phototoxicity to the sample drawbacks that currently plague other multiview approaches. Although the methods we report here focused on improving wide-field and light-sheet microscopy, we note that simultaneous, multiview capture and image fusion would also improve resolution isotropy in other imaging modalities, such as super-resolution microscopy.
4. METHODS
A. Sample Preparation
For PSF measurements (Figs. S5 and S9), 100 nm yellow–green fluorescent beads (Invitrogen, Cat. # F8803, 1∶2000 dilution in water) were coated on 24 × 50 mm #1.5 coverslips (VWR, 48393241) with 1 mg/ml poly-L lysine. After depositing 50 μL beads on the coverslips, waiting for 10 min, and gently washing the coverslips three to four times in water to remove excess beads from the coverslip, samples were then mounted in the diSPIM chamber and immersed in water.
The Bacillus subtilis strain used in Figs. 2(a)–2(f) is a derivative of B. subtilis PY79, a prototrophic derivative of strain 168 [26]. Strain CVO1195 harbors an ectopic chromosomal copy of spoVM-gfp (driven by its native promoter) integrated at the amyE locus [27]. Cells were grown at 22°C in 2 ml of casein hydrolysate (CH) medium [3] for 16 h. The overnight culture was diluted 1∶20 into 20 ml of fresh CH medium and grown for two additional hours at 37°C, shaking at 225 rpm. Cells were induced to sporulate by the resuspension method in Sterlini and Mandelstam (SM) medium as previously described [28] for 3.5 h. For microscopy, 1 ml of cell culture was centrifuged and resuspended in 100 μl of SM medium to increase cell density. Immediately prior to imaging, 50 μl of poly-L-lysine (1 mg/mL, Sigma-Aldrich, Cat. # P1524) was pipetted onto a cleaned coverslip and smeared across the central region. Next, 100 μl of the bacterial medium was added and allowed to settle for 5 min. 50 μl of a 2000-fold dilution of 100 nm yellow–green bead stock solution was added to the coverslip, and the bacterial/bead slurry allowed it to dry almost completely over 10 min. Finally, the sample was washed three times with resuspension medium A + B (KD Medical, Cat # CUS-0822) and imaged.
For the images in Figs. 2(g)–2(l), T. gondii RHΔhxgprt (Type I) tachyzoites stably expressing an ectopic copy of the T. gondii β1-tubulin isoform fused to EGFP, and driven by the α1-tubulin promoter [10], were used. Parasite cultures were maintained by serial passage in confluent primary human foreskin fibroblast (HFF, ATCC# SCRC-1041) monolayers in Dulbecco’s Modified Eagle’s Medium (DMEM, Gibco) lacking phenol red and antibiotics, supplemented with 4.5 g/L D-glucose, 1% (v/v) heat-inactivated cosmic calf serum (Hyclone), 1 mM sodium pyruvate (Gibco), and 2 mM GlutaMAX-I L-alanyl L-glutamine dipeptide (Gibco), as previously described [13,29]. To prepare the imaging samples, HFFs were seeded onto cleaned, sterile 24 × 50 mm #1.5 glass coverslips to obtain 70%–80% confluency, inoculated with the EGFP-Tgβ1-tubulin-expressing parasites described above, and incubated at 37°C with 5% CO2 for 19–20 h. The T. gondii-infected monolayers were fixed with 3.7% (v/v) freshly made methanol-free formaldehyde in phosphate-buffered saline (PBS) for 10 min at 25°C with gentle shaking, washed three times with PBS containing 0.1 M glycine pH 7.4, and stored at 4°C in PBS containing 0.1 M glycine and 10 mM NaN3. Before imaging, 100 μl of 500x-diluted 100 nm yellow–green beads was incubated with the sample for 10 min. The sample was subsequently washed three times with PBS prior to imaging. Beads were added to bacterial and Toxoplasma samples in order to improve the accuracy of registration (Fig. S13; also see Section 4.F.3).
RAW 264.7 macrophages (ATCC) displayed in Figs. 4(a)–4(c) were grown in RPMI 1640 medium, supplemented with 10% fetal bovine serum (Gibco), at 37°C in a 5% CO2 balanced environment. The probe for actin filaments, F-tractin-EGFP [30], was transfected into macrophages using an Amaxa Nucleofector system (Lonza) one day before imaging, and cells were plated onto #1.5 rectangular glass coverslips with the same procedure mentioned above. For imaging, the cells were maintained in an RPMI 1640 medium, supplemented with 20 mM HEPES pH 7.4, at 37°C via a customized temperature control system (ATC-100, ACUITYnano LLC, Rockville, MD). This compact temperature control system includes a PID (proportional–integral–derivative) controller, external temperature probes, and Peltier and resistive heating elements that were tailored to both the diSPIM imaging chamber and objective lenses.
For the eukaryotic, time-lapse imaging presented in Figs. 2(m)–2(p) and Figs. 4(d)–4(o), we imaged the human osteosarcoma cell line U2OS. Cells were routinely maintained in DMEM supplemented glucose (1 g/L), glutamine (1 mM), pyruvate (1 mM), and 10% FBS (Hyclone) and were grown at 37°C in a 5% CO2 environment. Two days before imaging, the cells were plated onto 24 mm × 50 mm, #1.5 rectangular coverslips that had been acid treated in 1M HCl overnight, rinsed extensively with deionized water, submerged into boiling milliQ water for 5 min, rinsed again with cold milliQ water, and autoclaved. The next day, cells were transfected with organelle markers using Turbofect (Thermo Fisher Scientific). To mark the plasma membrane in Figs. 2(m)–2(p), we used the Lck-tGFP construct (Origene, Cat# RC100017), which encodes for LCK proto-oncogene, Src family tyrosine kinase (LCK), transcript variant 2, mRNA (NM_005356.4). Lck is myristoylated and palmitoylated at its N-terminus and is eventually targeted to the plasma membrane. To mark endoplasmic reticulum derived structures, we overexpressed ER-Golgi Intermediate compartment marker ERGIC3 tagged with mCherry (ERGIC3-mCherry). To mark the plasma membrane and trafficking to and from the trans Golgi network to the plasma membrane in Figs. 4(d)–4(l), we transfected cells with two small GTPases, GFP-tagged H-Ras, and mCherry-tagged Rab8 [31]. Mitochondria in Figs. 4(m)–4(o) were labeled with the membrane potential-sensitive dye, MitoTracker Red (CMXRos, Life Technologies), at a final concentration of 200 nM, by incubating cells with the dye for 15 min. Following incubation, the medium was replaced with fresh, dye-free medium and left in the CO2 incubator for another 20–30 min to buffer the new medium. During live cell imaging, U2OS samples were immersed in low glucose (1 g/L) DMEM supplemented with glutamine, pyruvate, and 10% FBS without phenol red and antibiotics.
For fixed cell imaging in Fig. 3, we cultured the human osteosarcoma cell line U2OS, fixed cells, and immunolabeled microtubules according to previously described methods [32]. For T cell imaging in Fig. S14, stably transfected GFP-Actin E6-1 Jurkat T cells were grown in RPMI 1640 medium with L-glutamine and supplemented with 10% FBS, at 37°C in a 5% CO2 environment. Before imaging, 1 ml of cells was centrifuged at 250 RCF for 5 min, resuspended in the L-15 imaging buffer supplemented with 2% FBS, and plated onto rectangular glass coverslips. Cleaned coverslips (as above) were coated with anti-CD3 antibody (Hit-3a, eBiosciences, San Diego, CA) at 10 μg/ml for 2 h at 37°C prior to imaging.
B. Triple-View Microscopy
A previously published diSPIM frame [6] served as the base for the triple-view microscopy experiments conducted in this work. An XY piezo stage (Physik Instrumente, P-545.2C7, 200 μm × 200 μm) was bolted on the top of a motorized XY stage, which was in turn attached to a modular microscope base (Applied Scientific Instrumentation, RAMM and MS-2000). Rectangular coverslips containing samples were placed in an imaging chamber (Applied Scientific Instrumentation, I-3078-2450), the chamber placed into a stage insert (Applied Scientific Instrumentation, PI545.2C7-3078), and the insert mounted to the piezo stage. The MS-2000 stage was used for coarse sample positioning before imaging, and the piezo stage was used to step the sample through the stationary light sheets to create imaging volumes.
Two 40x, 0.8 NA water-immersion objectives [OBJ A and OBJ B in Figs. 1(a) and 1(b), Nikon Cat. # MRD07420] were held in the conventional, perpendicular diSPIM configuration with a custom objective mount (Applied Scientific Instrumentation, RAO-DUAL-PI). The 60x, 1.2 NA water objective [OBJ C in Figs. 1(a) and 1(b), Olympus UPLSAPO60XWPSF] was mounted in the epi-fluorescence module of the microscope base. Each objective was housed within a piezoelectric objective positioner (PZT, Physik Instrumente, PIFOC-P726), enabling independent axial control of each detection objective.
C. Triple-View Excitation Optics
The excitation optics for triple-view light-sheet microscopy are similar to those in our previous, nonfiber coupled diSPIM [6], except that the light sheets are created with a cylindrical beam expander and a rectangular slit (Fig. S3). After combining a 488 nm laser (Newport, PC14584) and a 561 nm laser (Crystalaser, CL-561-050) with a dichroic mirror (DC, Semrock, Di01-R488-25 × 36), the output beams are passed through an acousto-optic tunable filter (AOTF, Quanta Tech, AOTFnC-400.650-TN) for power and shuttering control. Light sheets are created by a 3x cylindrical beam expander (CL1 and CL2, Thorlabs LJ1821L1-A, f = 50 mm and LJ1629RM, f = 150 mm) and finely adjusted with a mechanical slit (SLIT, Thorlabs, VA100). The light sheets are reimaged to the sample via lens pairs L1 and L2 (Thorlabs, AC254-200-A-ML, f = 200 mm and AC254-250-A-ML, f = 250 mm) and L3 (Thorlabs, AC-254-300-A, f = 300 mm) and OBJ A (Nikon, MRD07420, 40x, 0.8 NA, water immersion, f = 5 mm) for A view excitation, or L4 and OBJ B for B view excitation (L4 and OBJ B has the same specifications with L3 and OBJ A, respectively). The A view and B illumination paths are also independently shuttered/switched via liquid-crystal shutters (SHUTTER, Meadowlark Optics, Cat. # LCS200, with controller D5020). The resultant light sheet had thickness of ~1.5 μm (FHWM at beam waist) and a width of ~80 μm. In all light-sheet imaging experiments, the samples were laterally moved through the excitation via the XY piezo stage mentioned above.
For wide-field triple-view imaging, a galvanometric mirror (GALVO, Thorlabs, GVSM001) was placed at the front focal plane of L1 and reimaged to the back focal planes of OBJ A and OBJ B via lens pair L2/L3 or L2/L4. Scanning this GALVO over an angular range of ±0.2 deg (mechanical) at 1 kHZ translated the light sheet axially at the sample plane to create a wide-field excitation with cross-sectional area ~80 μm × 80 μm. In all wide-field imaging experiments, the samples were held stationary and the OBJ A/B/C plane of focus simultaneously translated through the sample via piezoelectric objective positioners.
D. Triple-View Detection Optics
Triple-view fluorescence (in either wide-field mode or light-sheet mode) was collected via the objectives A–C, transmitted through dichroic mirrors (Chroma, ZT405/488/561/640rpc), filtered through long-pass and notch emission filters (Semrock, LP02-488RU-25 and NF03-561E-25) to reject 488 and 561 nm excitation light, respectively, and imaged with 200 mm tube lenses (Applied Scientific Instrumentation, C60-TUBE_B) for objectives A/B or a 180 mm tube lens (Edmund Cat. # 86-835) for objective C onto three sCMOS cameras (PCO, Edge 5.5). The resulting image pixel sizes for the top views were 6.5 μm/40 = 162.5 nm, and for the bottom view 6.5 μm/60 = 108.3 nm.
E. Triple-View Data Acquisition
In the wide-field triple-view acquisition, all three sCMOS cameras were operated in a hybrid rolling/global shutter mode by illuminating the sample only when all lines were exposed—the same scheme used in previous diSPIM acquisition [6]. Figure S4 illustrates the entire control scheme; all waveforms were produced by using analog outputs of a data acquisition (DAQ) card (National Instruments, PCI 6733). Three pulse trains were used for triggering the three sCMOS cameras, and three analog step-wise waveforms (10 ms at each step or imaging plane) were used to drive the three piezoelectric objective stages. A triangle waveform with 1 kHz frequency was used to rotate the GALVO, thereby creating effectively wide-field illumination during the exposure time of each camera. All analog waveforms were synchronized with an internal digital trigger input (the PFI 0 channel in the DAQ card).
When performing triple-view light-sheet imaging, the two sCMOS cameras corresponding to objectives A/B were also operated in the hybrid rolling/global shutter mode, but the lower camera was operated in a virtual confocal slit mode to obtain partially confocal images during light-sheet illumination introduced from objectives A/B (Figs. S7 and S8) since the resulting light sheets are tilted at 45 deg relative to the lower objective. The PCO Edge sCMOS camera allows users to implement a “light-sheet,” rolling-shutter mode similar to the Hamamatsu Orca Flash 4.0 by (1) decreasing the pixel clock to the slow speed mode, 95.3 MHz; (2) changing the default rolling mode (double side rolling) to single side rolling, i.e., from the top row to the bottom row of the camera chip; (3) setting the output data format to 16 bit; and (4) setting the number of exposure rows and the exposure time for the rows. In this mode, at any instant only certain rows are exposed and most of the detection pixels are masked, blocking out-of-focus light at these masked locations. The image is then built as the virtual slit “rolls” along the camera chip (Fig. S7).
When introducing the stationary light sheet via objective A, the objective C is scanned from the top of the imaging volume to the bottom (near the coverslip), while the virtual slit “rolls” from the top to the center of the camera. Switching the light sheet so that it is introduced via objective B, objective C completes a reverse scan (from the coverslip to the top of the imaging volume), and the virtual slit continues rolling from the center of the chip to the bottom of the camera (Fig. S7). During this single cycle of triple-view excitation, two lower views are simultaneously obtained as a single image, although to minimize downstream registration and computation we only used half of this image (the bottom view corresponding to light-sheet introduction via objective A) when subsequently performing joint deconvolution.
Synchronizing illumination and detection in this way (Figs. S6 and S7) rejected much out-of-focus fluorescence that would otherwise contaminate the lower view (Fig. S8). In all light-sheet experiments (Figs. 3 and 4), the slit width (i.e., the number of exposed rows) was set at 10 pixels (1.08 μm) to achieve the most rejection of out-of-focus light while retaining in-focus signal generated within the light sheet.
Careful synchronization of the lower objective position and the camera rolling slit is critical for optimal use of the rolling shutter. We achieved this synchronization by (1) setting the objective PZT oscillation range to the same as the field of view in the camera in the rolling dimension (e.g., 120 pixels in the acquired images corresponds to 120 × 0.108 μm = 13 μm scanning range); (2) setting the PZT oscillation speed to exactly match the camera rolling speed defined by the product of exposure time for the “slit” and the acquired image size (e.g., a PZT scan of 13 μm in 25 ms implies an exposure time of 25 ms/120 pixels = 208 μs for the “slit”); and (3) tuning the start position of the objective PZT to match the first row of the acquired image. In the experiments, the PZT offset was varied every 500 nm to determine the best synchronization by manual judgment of the acquired image quality. Simplifying the synchronization by using a stationary illumination sheet is one reason we translated the sample through the light sheet with our piezoelectric stage, rather than sweeping the light sheet through the sample, when acquiring volumetric imaging stacks. We note that the stage-scanning method also allows us to achieve a larger imaging area [e.g., ~100 μm laterally in Fig. 4(m)] than in previous diSPIM [6,15]: as the light-sheet beam waist is always located at the center of samples, optimal sectioning is maintained even over a large field of view [33].
Exemplary control waveforms for implementing dual-color triple-view light-sheet imaging are summarized in Fig. S6, including a 20 Hz triangle waveform to drive the lower-view piezoelectric objective positioner, a step-wise waveform (50 ms/plane, 0.2 Hz volume imaging rate for 100 planes) to drive the XY piezo stage, three external trigger signals for each sCMOS camera that output signals to turn on/off the excitation via the AOTF and liquid-crystal shutters, and two square waveforms to switch the AOTF excitation wavelengths and control the resulting excitation power. Programs controlling waveforms and DAQ were written in Labview (National Instruments), and programs controlling image acquisition (via sCMOS cameras) were written in the Python programming language [34,35]. Software is available upon request from the authors.
F. Triple-View Data Processing
Raw image data from the three views of data are merged to produce a single volumetric view, after processing steps that include background subtraction, interpolation, transformation, registration, and deconvolution. Registration is implemented in the open-source Medical Imaging Processing, Analyzing and Visualization (MIPAV) programming environment [36] (http://mipav.cit.nih.gov/); other processing steps are implemented in Matlab (R2015a) with both CPU (Intel Xeon, ES-2690-v3, 48 threads, 128 GB memory) and GPU (using an Nvidia Quadro K6000 graphics card, 12 GB memory) programming. Details follow.
1. Background Subtraction
Raw light-sheet and wide-field images were preprocessed by subtracting an average of 100 dark (no excitation light) background frames acquired under the same imaging conditions as during the experiment. This process removes any residual room light, but does not alter the underlying noise characteristics of the data nor the final deconvolution outcome.
2. Interpolation and Transformation
Raw triple-view images, either in wide-field or light-sheet mode, are acquired from three volumetric views (via OBJ A/B/C). To merge these three views into a single volumetric view, first one view is chosen as the perspective view, and the other two views are transformed to the common perspective view. Here we choose to visualize samples from the bottom objective’s (C) perspective (with coordinates x, y, and z; Figs. S10 and S11), a traditional view for most microscopy, because the purpose of our triple-view system is to improve the axial resolution of this lower view (in wide-field mode) or its lateral resolution (in light-sheet mode). We thus transformed the two top views’ perspectives to the bottom view’s perspective according to their relative angles (we initially assume a 45 deg relationship between objective A/B’s optical axis and objective C’s optical axis for the initial transformation, but allow a ±5 deg searching range to determine the angle more precisely during fine registration, described in the following section). Considering that the lateral resolution limit of the lower objective ~λ/(2 ∗ NA) is ~218 nm for our 1.2 NA lens at λ = 525 nm, we designed the lower-view magnification so that the raw object space pixel size is 108.3 nm, thereby satisfying the Nyquist sampling criterion. The top views’ object space pixel size is 162.5 nm, and the slice spacing in stack acquisitions varied between 108.3 nm (beads, fixed cells) and 5 × 108.3 nm = 541.5 nm (time-lapse light-sheet imaging) depending on the imaging study. In all cases, appropriate coordinate transformations and linear interpolation are applied to obtain consistent orientations and an isotropic voxel size of 108.3 nm × 108.3 nm × 108.3 nm prior to fine registration. Further details on interpolation and transformation follow.
In conventional diSPIM, we typically define a coordinate system from one of the top view’s perspectives. For example, the light-sheet plane is defined along coordinates xt, yt, where xt corresponds to the propagation direction of the light sheet and yt to the direction along the light-sheet width. The light sheet and the detection objective in conventional diSPIM are co-swept through the sample along the zt direction (perpendicular to the xt, yt plane) to create an imaging volume (Fig. S10). When translating the sample through a fixed light sheet (stage-scanning mode, used in all light-sheet imaging), the sheet and detection plane are maintained at a fixed position and the piezo stage steps the samples in the x direction (x is defined with respect to the lower objective’s coordinate system) to obtain successive imaging planes. These planes appear to spread laterally across the field of view of the upper objectives, as evidenced by the parallelogram (shown along the xt, zt plane) and acquired pixels (red dots) in Fig. S10.
The raw pixel size in the top two views is 162.5 nm along the xt, yt directions. To obtain an isotropic pixel size of 108.3 nm from the bottom view’s perspective, we first digitally upsample the acquired images 1.5x along yt and 1.5/sqrt(2)x along xt, respectively, as shown by the yellow dots and grids in Fig. S10. For imaging beads and fixed cells (Figs. S3 and S9), the spacing between acquisition planes (here denoted by “s,” equivalent to sqrt(2) ∗ zt) was set to 108.3 nm along the x direction, and further interpolation is not required. For time-lapse light-sheet imaging (Fig. 4), the spacing was set to 5 × 108.3 nm along the x direction in order to avoid undue dose to the sample, and interpolation was used to fill in the gaps, as shown by the green diamonds in Fig. S10. The key consideration here is to preserve the high-resolution lateral dimensions of each native view, so interpolation is always performed along the lower-resolution native axial dimension of each view. For example, for this top view illustrated in Fig. S10, this interpolation was performed along the native axial zt direction instead of along the x or z directions. After interpolation, the stage-scanned top views (with coordinates xt, yt, and s) are transformed to the coordinate system of the bottom objective (x, y, z, as shown in Fig. S10) according to
| (1) |
When acquiring lower-view images under light-sheet excitation introduced by a top objective, each image (defined along coordinates xb, yb) is actually a projection of the 45 deg light-sheet plane onto the lateral plane in the bottom view’s perspective (with coordinates x, y, z as before), and thus the z direction is mapped to xb (y is identical to yb, Fig. S11). Since the acquired pixel size in xb, yb is 108.3 nm, no interpolation along xb and yb is required when reassigning the coordinates, as shown by the red dots and grids in Fig. S11. As before, the piezo stage translates the samples in the x direction to obtain successive imaging planes, which results in a shift (i.e., the spacing “s”) in x from xb. Equation (2) summarizes the transformation from the raw stage-scanned data to the conventional perspective of the bottom objective. After this transformation, interpolation is used to fill in pixels at every 108.3 nm along the z direction, as shown by the blue rectangles in Fig. S11. Note that the interpolation is performed along the z instead of x coordinate direction, because this is the bottom view’s native lower-resolution axial dimension:
| (2) |
Finally, when considering wide-field triple-view acquisition, interpolation and transformation are relatively straightforward: we tri-linearly interpolated along each detection objective axis to obtain an isotropic pixel size (108.3 nm), and then rotated the data obtained in the two top views by 45 deg along the yt axis into the bottom-view coordinate system (x, y, z) according to Eq. (3):
| (3) |
3. Registration
After transforming the coordinate system of all three raw views (in triple-view light-sheet imaging) into the coordinate system of the conventional bottom view using the procedure above, the views are coarsely registered. To perform a finer registration, we adopted the same method used in earlier diSPIM registration [6]; i.e., we used an intensity-based method in the open-source MIPAV programming environment to optimize an affine transformation between views, with 12 deg of freedom (translation, rotation, scaling, and skewing). To increase registration accuracy (Fig. S12), we (1) deconvolved each view (with the deconvolution algorithm detailed below) to increase image quality, manually removing regions with poor SNR; (2) registered the deconvolved view B to the deconvolved view A, thus obtaining a registration matrix mapping view B to view A; (3) applied this registration matrix to the raw view B, thus registering it to the raw view A; (4) performed joint deconvolution on the two registered, raw views A and B; (5) registered the jointly deconvolved views A/B to the deconvolved lower view C, thus obtaining a registration matrix mapping views A/B to view C; and, finally, (6) applied both registration matrices (view B to view A, then views A/B to view C) to register all three raw views to the coordinate system of the lower view. For time series imaging we applied this process to the same early time point in each view, obtaining a set of registration matrices that were then applied to all other time points in the 4D dataset.
For wide-field triple-view registration, we followed the same procedure. Registration was made more difficult in some samples by the presence of out-of-focus light. In these cases [Figs. 2(a)–2(l)], 100 nm yellow–green fluorescent beads were added to the samples. Regions of poor SNR and with significant out-of focus contamination were then manually masked, so that the registration mainly relied on the sharp, in-focus bead signals. An example of the improvement offered with bead-based registration is shown in Fig. S13.
4. Joint Deconvolution
As with previous diSPIM deconvolution [6,15], the algorithm we developed to fuse triple-view datasets is based on Richardson–Lucy iterative deconvolution [37,38], which is appropriate for images that are contaminated by Poisson noise. We implemented our method in the Fourier domain and used a graphics processing unit (GPU) to speed up deconvolution of the registered triple-view volumes. Equation (4) shows one iteration of the triple-view joint deconvolution, which involves six convolutions:
| (4) |
Here ⊗ denotes convolution operation; ViewA, ViewB, and ViewC are the measured, interpolated, transformed, registered, and normalized triple-view volumes, where A and B denote the two top views and C denotes the lower view; PSFA, PSFB, and PSFC are PSFs corresponding to A, B, and C views, respectively, whereas , , are the adjoint PSFs; m, n, and k are the dimensions of the PSF; Ek is the estimate from the previous iteration, with k denoting the current iteration number; the initial estimate E0 is the arithmetic fusion of the three view data; EA, EB, and EC are the updated estimates calculated for each view; and the new estimate Ek+1 is the arithmetic average of all three estimates. We found that this additive deconvolution provided better SNR and rejection of out-of-focus light compared to multiplicative deconvolution (i.e., the geometrical average of the three estimates [39]) or alternating deconvolution (i.e., joint deconvolution based on ordered subsets expectation maximization in conventional diSPIM [40]), perhaps due to the mitigation of noise inherent to view C. A comparison of these three methods is presented in Fig. S14.
In previous diSPIM deconvolution, we employed 3D Gaussian PSFs to speed processing (the convolution is then separable in each spatial dimension). However, here we used either theoretical or experimentally measured PSFs in computing the convolution, as the PSFs for views A and B are not separable Gaussian functions when visualized from the bottom view, and the bottom view C’s PSF cannot be simply modeled as a Gaussian PSF in the light-sheet mode (Fig. S15; also see Section 4.G).
Direct, 3D convolution in the spatial domain is computationally taxing, so we used the discrete Fourier transform (DFT) to speed processing. Since the circular convolution of two vectors is equal to the inverse discrete Fourier transform (DFT−1) of the product of the two vectors’ DFTs, the linear convolution operation in the deconvolution algorithm can be calculated according to Eq. (5): (1) pad the PSF with zeros to match the dimensions of the input image E; (2) circularly shift the PSF stack so that the center of the PSF is the first element of the stack; (3) compute the DFT to create the optical transfer function (OTF) of the circularly shifted PSF; (4) multiply the OTF and the DFT of the input image E, and then compute the DFT−1, equivalent to the convolution of E and PSF; (5) compute the DFT−1 of the product of the conjugate OTF and the input image E’s DFT, which is the convolution E with the adjoint PSF; and (6) use these results in Eq. (4) above:
| (5) |
The above calculations [Eqs. (4) and (5)] were implemented in Matlab by employing built-in, multithreaded fast Fourier transform (FFT) algorithms with a dual-CPU (2.6 GHz, 24 cores, 48 threads, and 128 GB memory) and the NVIDIA CUDA fast Fourier transform library (cuFFT) with a GPU (2880 CUDA cores and 12 GB memory). With the dual-CPU processor, it takes 5.2 s/deconvolution iteration when processing a triple-view stack with 350 × 240 × 256 pixels per view, or ~15 min for a full deconvolution (typically 180 iterations for convergence). Using the GPU is preferred, if the GPU memory is large enough to hold all variables (including the three input images ViewA, ViewB, and ViewC; three estimate images EA, EB, and EC; and three OTFs, e.g., 4.2 GB for a triple-view 16-bit stack with size 350 × 240 × 256 pixels/view and double precision in FFT calculations). For example, for the stack size above, GPU processing required only 350 ms for one iteration, a factor of 15-fold improvement when compared to the CPU option. A triple-view, dual-color dataset with 100 time points requires ~3 h to deconvolve with the GPU, versus 48 h with the CPU. However, for larger datasets [e.g., Fig. 4(m), with a stack size of 980 × 450 × 256], there was insufficient memory in our single-GPU card to store all variables simultaneously, and data were transferred to/from GPU memory after each deconvolution iteration. This slowed GPU computation considerably, although it was still faster (by 2–3×) than the CPU-based option.
The number of iterations used in deconvolution is an important parameter since the Richardson–Lucy iteration will lead to excessive noise and artifact amplification if run for too many iterations. Our goal was to iterate until the theoretical resolution limit of each imaging configuration was achieved, but not beyond. To do so, we analyzed the convergence of the lateral and axial resolution (FWHM) in wide-field and light-sheet imaging of 100 nm yellow–green fluorescent beads. As shown in Fig. S5, the results reveal that 180–230 iterations are needed to achieve the lateral resolution limit in wide-field triple-view imaging, compared to 60–100 iterations for only the bottom view’s deconvolution; likewise, 140–180 iterations are needed to achieve the axial resolution limit in light-sheet triple-view imaging, compared to 60–90 iterations for only the two top views’ joint deconvolution. For all biological wide-field imaging shown in Fig. 2, we choose 100 and 200 iterations for bottom-view and triple-view deconvolution, respectively. For the cell light-sheet imaging in Figs. 3 and 4, the iteration number is set at 60 for dual-view deconvolution, whereas the triple-view deconvolution iteration number varied depending on SNR (higher SNR allows more iterations before noise is amplified): we thus used 180 for Figs. 3, 4(a), and 4(b), 120 for the GFP channel in Figs. 4(d)–4(l), and 100 for the mCherry channel in Figs. 4(d)–4(l) and Figs. 4(m)–4(o).
Although in every case we used more iterations in triple-view than in dual-view imaging, we note that the gain in resolution/image quality in triple-view imaging is evident even at early iteration cycles. Conversely, dual-view deconvolution never attained the resolution enhancement observed with triple-view deconvolution, even if we extended the number of iterations used in for dual-view imaging to the number used in triple-view imaging. This can be seen in the difference between the black and red curves in Fig. S5 and is further demonstrated in Fig. S16.
G. PSF Modeling
In order to minimize artifacts in the DFT/DFT−1 induced by zero padding, we first extended our model of the PSF (up to 1024 × 1024 × 512 pixels), and then cropped the PSFs to have the same size as each triple-view input image (e.g., 350 × 240 × 256 pixels). We simulated the three wide-field PSFs with the PSF Generator (ImageJ plugin, http://bigwww.epfl.ch/algorithms/psfgenerator/) using the “Born and Wolf” model with appropriate numerical aperture (0.8 NA for top views and 1.2 for lower view), refractive index of immersion medium (1.33 for water), and wavelength (525 nm as a typical emission wavelength for GFP). We further rotated the top PSFs to the conventional coordinates (visualized from the bottom view) according to Eq. (3).
In the light-sheet mode, the top two PSFs are multiplied by the excitation light sheet. A 3D Gaussian sheet with a 45 deg tilt (with respect to the bottom view) and a 1.5 μm beam waist was created, and then multiplied with the 0.8 NA wide-field PSFs generated above. The bottom-view PSF depends on both the lower objective motion and the confocal slit function. As shown in Fig. S15, the bottom PSF is computed by (1) generating a series of axially bottom-view wide-field PSFs within the rolling slit width, (2) averaging these shifted PSFs, and (3) multiplying the averaged, shifted PSFs with the Gaussian light sheet. The simulated PSFs are noise free, and work well for deconvolution of data with moderate SNR [Figs. 4(d)–4(l)]. However, they do not model objective aberrations, which are especially evident in the high NA bottom view. Thus, we measured the PSFs derived from ten 100 nm yellow–green beads, registered and averaged them to reduce noise, and used the resulting experimentally derived PSF when deconvolving the bottom view. This procedure resulted in slightly better resolution recovery in the triple-view deconvolution of data with high SNR [Figs. 3 and 4(a)–4(c)]. All the PSFs are normalized to the total energy (i.e., divided by sum of each PSF) during the deconvolution.
H. Drift Correction
For some experiments [Figs. 2(d)–2(l)], the sample drifted slightly. We corrected this drift after triple-view deconvolution by registering subsequent time points to the first time point as follows: we (1) computed a 2D rigid registration of the maximum intensity projections (XY) between each successive stack and the first stack using an intensity-based method; (2) applied the resulting transformation matrix to XY planes in each stack; (3) computed a maximum intensity projection (XZ) for each stack; (4) condensed each maximum intensity projection across the X dimension to derive a single axial (XZ) line profile for each stack; (5) determined the axial location of the bottom of the cell, located at the maximal signal intensity of the axial line profile; and (6) offset the Z slices in each time point to appropriately align all XY planes in each stack to the bottom of the cell. All processing steps were implemented in Matlab.
I. Image Intensity Correction
In the triple-view joint deconvolution procedure, all three volumetric views are normalized to their respective peak intensity before deconvolution. In order to restore any bleaching inherent to the raw time-lapse imaging data, an intensity correction was performed by multiplying each deconvolved volume with the corresponding average intensity of the raw data (we choose the raw view A as reference).
Supplementary Material
Acknowledgments
We thank Mark Reinhardt and Martin Missio (PCO) for helping us to implement the confocal slit mode with our sCMOS cameras, Jiali Lei (Florida International University) for help in beads PSF distilling and GPU testing, Alan Hoofring (Medical Arts Design Section, NIH) for help with illustrations, Jianyong Tang (NIAID, NIH) for help in preparing figures/movies produced by Imaris, John Murray (Indiana University) for helpful advice on Toxoplasma imaging, S. Abrahamsson and C. Bargmann (The Rockfeller University) for useful discussions, L. Maldonado-Baez (NINDS, NIH) for providing the mCherry-tagged Rab8 construct, and H. Eden (NIBIB, NIH) for useful comments on the paper.
Funding. National Institute of Biomedical Imaging and Bioengineering (NIBIB); National Cancer Institute (NCI); National Heart, Lung, and Blood Institute (NHLBI); Marine Biological Laboratory (MBL) MBL-UChicago Exploratory Research Fund Research Award; National Institute of General Medical Sciences (NIGMS) (R25GM109439); National Natural Science Foundation of China (NSFC) (61427807, 61271083, 61525106); Colciencias–Fulbright Scholarship; March of Dimes Foundation (MODF) (6-FY15-198).
Footnotes
OCIS codes: (170.2520) Fluorescence microscopy; (110.0110) Imaging systems; (170.3880) Medical and biological imaging
See Supplement 1 for supporting content.
References
- 1.Fischer RS, Wu Y, Kanchanawong PS, Shroff H, Waterman CM. Microscopy in 3D: a biologist’s toolbox. Trends Cell Biol. 2011;21:682–691. doi: 10.1016/j.tcb.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Winter PW, Shroff H. Faster fluorescence microscopy: advances in high speed biological imaging. Curr Opin Chem Biol. 2014;20:46–53. doi: 10.1016/j.cbpa.2014.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Temerinac-Ott M, Ronneberger O, Ochs P, Driever W, Brox T, Burkhardt H. Multiview deblurring for 3-D images from light-sheet-based fluorescence microscopy. IEEE Trans Image Process. 2012;21:1863–1873. doi: 10.1109/TIP.2011.2181528. [DOI] [PubMed] [Google Scholar]
- 4.Ingaramo M, York AG, Hoogendoorn E, Postma M, Shroff H, Patterson GH. Richardson-Lucy deconvolution as a general tool for combining images with complementary strengths. ChemPhysChem. 2014;15:794–800. doi: 10.1002/cphc.201300831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Krzic U. PhD thesis. University of Heidelberg; 2009. Multiple-view microscopy with light-sheet based fluorescence microscope. [Google Scholar]
- 6.Wu Y, Wawrzusin P, Senseney J, Fischer RS, Christensen R, Santella A, York AG, Winter PW, Waterman CM, Bao Z, Colón-Ramos DA, McAuliffe M, Shroff H. Spatially isotropic four-dimensional imaging with dual-view plane illumination microscopy. Nat Biotechnol. 2013;31:1032–1038. doi: 10.1038/nbt.2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McNally JG, Karpova T, Cooper J, Conchello JA. Three-dimensional imaging by deconvolution microscopy. Methods. 1999;19:373–385. doi: 10.1006/meth.1999.0873. [DOI] [PubMed] [Google Scholar]
- 8.Gill RLJ, Castaing JP, Hsin J, Tan IS, Wang X, Huang KC, Tian F, Ramamurthi KS. Structural basis for the geometry-driven localization of a small protein. Proc Natl Acad Sci USA. 2015;112:E1908–E1915. doi: 10.1073/pnas.1423868112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McKenney PT, Driks A, Eichenberger P. The Bacillus subtilis endospore: assembly and functions of the multilayered coat. Nat Rev Microbiol. 2013;11:33–44. doi: 10.1038/nrmicro2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hu K, Suravajjala S, DiLullo C, Roos DS, Murray JM. Identification of new tubulin isoforms in Toxoplasma gondii. ASCB Annual Meeting. 2003 Poster, Abstract #L460. [Google Scholar]
- 11.Hu K, Roos DS, Murray JM. A novel polymer of tubulin forms the conoid of Toxoplasma gondii. J Cell Biol. 2002;156:1039–1050. doi: 10.1083/jcb.200112086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nichols BA, Chiappino ML. Cytoskeleton of Toxoplasma gondii. J Protozool. 1987;34:217–226. doi: 10.1111/j.1550-7408.1987.tb03162.x. [DOI] [PubMed] [Google Scholar]
- 13.Liu J, He Y, Benmerzouga I, Sullivan WJJ, Morrissette NS, Murray JM, Hu K. An ensemble of specifically targeted proteins stabilizes cortical microtubules in the human parasite Toxoplasma gondii. Mol Biol Cell. 2016;27:549–571. doi: 10.1091/mbc.E15-11-0754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Swoger J, Huisken J, Stelzer EHK. Multiple imaging axis microscopy improves resolution for thick-sample applications. Opt Lett. 2003;28:1654–1656. doi: 10.1364/ol.28.001654. [DOI] [PubMed] [Google Scholar]
- 15.Kumar A, Wu Y, Christensen R, Chandris P, Gandler W, McCreedy E, Bokinsky A, Colon-Ramos D, Bao Z, McAuliffe M, Rondeau G, Shroff H. Dual-view plane illumination microscopy for rapid and spatially isotropic imaging. Nat Protoc. 2014;9:2555–2573. doi: 10.1038/nprot.2014.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu Y, Ghitani A, Christensen R, Santella A, Du Z, Rondeau G, Bao Z, Colon-Ramos D, Shroff H. Inverted selective plane illumination microscopy (iSPIM) enables coupled cell identity lineaging and neurodevelopmental imaging in Caenorhabditis elegans. Proc Natl Acad Sci USA. 2011;108:17708–17713. doi: 10.1073/pnas.1108494108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baumgart E, Kubitscheck U. Scanned light sheet microscopy with confocal slit detection. Opt Express. 2012;20:21805–21814. doi: 10.1364/OE.20.021805. [DOI] [PubMed] [Google Scholar]
- 18.Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, Yuan J, Deeney JT, Corkey BE, Shirihai OS. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chhetri RK, Amat F, Wan Y, Hockendorf B, Lemon WC, Keller P. Whole-animal functional and developmental imaging with isotropic spatial resolution. Nat Methods. 2015;12:1171–1178. doi: 10.1038/nmeth.3632. [DOI] [PubMed] [Google Scholar]
- 20.Gustafsson MGL. Extended resolution fluorescence microscopy. Curr Opin Struct Biol. 1999;9:627–628. doi: 10.1016/s0959-440x(99)00016-0. [DOI] [PubMed] [Google Scholar]
- 21.Hell SW, Stelzer EHK. Properties of a 4Pi confocal fluorescence microscope. J Opt Soc Am A. 1992;9:2159–2166. [Google Scholar]
- 22.Gustafsson MGL, Agard DA, Sedat JW. I5M: 3D widefield light microscopy with better than 100 nm axial resolution. J Microsc. 1999;195:10–16. doi: 10.1046/j.1365-2818.1999.00576.x. [DOI] [PubMed] [Google Scholar]
- 23.Olarte OE, Andilla J, Artigas D, Loza-Alvarez P. Decoupled illumination detection in light sheet microscopy for fast volumetric imaging. Optica. 2015;2:702–705. [Google Scholar]
- 24.Abrahamsson S, Chen J, Hajj B, Stallinga S, Katsov AY, Wisniewski J, Mizuguchi G, Soule P, Mueller F, Darzacq C Dugast, Darzacq X, Wu C, Bargmann CI, Agard DA, Dahan M, Gustafsson MGL. Fast multicolor 3D imaging using aberration-corrected multifocus microscopy. Nat Methods. 2012;10:60–63. doi: 10.1038/nmeth.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abrahamsson S, Usawa S, Gustafsson MGL. A new approach to extended focus for high-speed high-resolution biological microscopy. Proc SPIE. 2006;6090:60900N. [Google Scholar]
- 26.Youngman P, Perkins JB, Losick R. Construction of a cloning site near one end of Tn917 into which foreign DNA may be inserted without affecting transposition in Bacillus subtilis or expression of the transposon-borne erm gene. Plasmid. 1984;12:1–9. doi: 10.1016/0147-619x(84)90061-1. [DOI] [PubMed] [Google Scholar]
- 27.van Ooij C, Losick R. Subcellular localization of a small sporulation protein in Bacillus subtilis. J Bacteriol. 2003;185:1391–1398. doi: 10.1128/JB.185.4.1391-1398.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sterlini JM, Mandelstam J. Commitment to sporulation in Bacillus subtilis and its relationship to development of actinomycin resistance. Biochem J. 1969;113:29–37. doi: 10.1042/bj1130029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leung JM, Rould MA, Konradt C, Hunter CA, Ward GE. Disruption of TgPHIL1 alters specific parameters of Toxoplasma gondii motility as measured in a quantitative, three-dimensional live motility assay. PLoS One. 2014;9:e85763. doi: 10.1371/journal.pone.0085763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schell MJ, Erneux C, Irvine RF. Inositol 1, 4, 5-triphosphate 3-kinase A associates with F-actin and dendritic spines via its N-terminus. J Biol Chem. 2001;276:37537–37546. doi: 10.1074/jbc.M104101200. [DOI] [PubMed] [Google Scholar]
- 31.York AG, Chandris P, Nogare DD, Head J, Wawrzusin P, Fischer RS, Chitnis AB, Shroff H. Instant super-resolution imaging in live cells and embryos via analog image processing. Nat Methods. 2013;10:1122–1126. doi: 10.1038/nmeth.2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Winter PW, York AG, Nogare DD, Ingaramo M, Christensen R, Chitnis AB, Patterson GH, Shroff H. Two-photon instant structured illumination microscopy improves the depth penetration of super-resolution imaging in thick scattering samples. Optica. 2014;1:181–191. doi: 10.1364/OPTICA.1.000181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kumar A, Christensen R, Guo M, Chandris P, Duncan W, Wu Y, Santella A, Moyle M, Winter PW, Colon-Ramos D, Bao Z, Shroff H. Using stage- and slit-scanning to improve contrast and optical sectioning in dual-view inverted light-sheet microscopy (diSPIM) Biol Bull. doi: 10.1086/689589. to be published. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oliphant T. Python for scientific computing. Comput Sci Eng. 2007;9:10–20. [Google Scholar]
- 35.Jones E, Oliphant T, Peterson P. SciPy: Open Source Scientific Tools for Python. 2001 http://www.scipy.org.
- 36.McAuliffe MJ, Lalonde FM, McGarry D, Gandler W, Csaky K, Trus BL. Proceedings of 14th IEEE Symposium on Computer-Based Medical Systems. IEEE; 2001. Medical image processing, analysis, and visualization in clinical research; pp. 381–386. [Google Scholar]
- 37.Richardson WH. Bayesian-based iterative method of image restoration. J Opt Soc Am. 1972;62:55–59. [Google Scholar]
- 38.Lucy LB. An iterative technique for the rectification of observed distributions. Astron J. 1974;79:745–754. [Google Scholar]
- 39.Preibisch S, Amat F, Stamataki E, Sarov M, Singer RH, Myers EW, Tomancak P. Efficient Bayesian-based multiview deconvolution. Nat Methods. 2014;11:645–648. doi: 10.1038/nmeth.2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hudson HM, Larkin RS. Accelerated image reconstruction using ordered subsets of projection data. IEEE Trans Med Imaging. 1994;13:601–609. doi: 10.1109/42.363108. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.