

Abstract
Background
Alendronate is an inhibitor of osteoclast-mediated bone resorption, but its clinical utility is limited due to gastrointestinal complications including bleeding erosions.
Aims
We studied whether potent vasodilators hydrogen sulfide (H2S) and carbon monoxide (CO) can protect against alendronate-induced gastric lesions in rats exposed to mild stress.
Methods
Three series (A, B, and C) of Wistar rats received alendronate (150–700 mg/kg i.g., series A) with or without NaHS (5 mg/kg), H2S donor or CORM-2 (5 mg/kg) releasing CO administered i.g. 30 min before alendronate administration (series B) in rats exposed for 3 days before alendronate administration to mild stress (series C). The area of gastric lesions was assessed by planimetry, the gastric blood flow (GBF) was determined by H2-gas clearance technique, and H2S production via CSE/CBS/3-MST activity and the gastric expression of HO-1, HO-2, HIF-1α, NF-κB, iNOS, COX-2, IL-1β, TNF-α, GPx-1 and SOD-2 were analyzed by qPCR or Western blot.
Results
Alendronate dose-dependently produced gastric mucosal lesions and significantly decreased GBF, and these effects were exacerbated by mild stress. NaHS and CORM-2 significantly reduced the alendronate-induced gastric lesions in non-stressed and stressed animals, but only NaHS but not CORM-2 raised H2S production. NaHS and CORM-2 inhibited gastric expression of HIF-1α protein and HO-1, HIF-1α, NF-κB, COX-2, iNOS, IL-1β, TNF-α mRNAs but failed to affect those of HO-2, GPx-1, and SOD-2.
Conclusion
Both H2S and CO released from their donors, NaHS and CORM-2, protect gastric mucosa compromised by stress against alendronate-induced gastric damage via mechanism involving downregulation of HIF-1α, NF-κB and proinflammatory factors COX-2, iNOS, IL-1β, and TNF-α.
Keywords: Alendronate, Gastroprotection, Hydrogen sulfide, Carbon monoxide, Gastric blood flow, Hypoxia-inducible factor-1α
Introduction
Bisphosphonates (BPs) are chemically non-metabolized analogs of inorganic pyrophosphate (PPi), where two P–O bonds were replaced by two C–P structures. This modification determines their resistance to chemical and enzymatic degradation [1–3]. BPs are among the most widely and effectively agents used in the therapy of diseases related to bone remodeling, e.g., osteoporosis and Paget’s disease [4–6]. There are two major classes of BPs-non-nitrogen-containing BPs (NBP, e.g., etidronic acid and clodronic acid) [7, 8] and nitrogen-containing BPs (e.g., alendronic acid, zoledronic acid, and pamidronic acid) [9–12]. Different structures of these two classes of BPs determine their pharmacokinetics and primary mechanism of action. Previous studies revealed that oral administration of BPs, such as alendronate, can cause gastrointestinal adverse effects ranging from mild acid reflux, nausea, vomiting, and diarrhea to serious complications such as gastric erosions and even gastric ulcers [13, 14].
Two enzyme systems are involved in the production of gaseous cellular messenger molecules, carbon monoxide (CO) and hydrogen sulfide (H2S). The heme oxygenase (HO) system is composed by microsomal enzymes, which catalyze α-specific oxidative cleavage of heme to CO, biliverdin and free iron in a multistep, energy-required reaction [15]. The HO-1 isoform with widespread tissue distribution, including the lung, liver, and kidney, has been shown to be overexpressed by a number of stressors [16]. Both HO-1 expression and activity have been also identified in the gastric mucosa [17–19]. Constitutive HO-2 is expressed at high level in the brain and testes [20]. The endogenous H2S biosynthesis depends upon the activity of cystathionine-β-synthase (CBS)/cystathionine-γ-lyase (CSE) system, which has been shown to regulate homocysteine and cysteine metabolism [21, 22]. In addition, H2S may be synthesized by 3-mercaptopyruvate sulfotransferase (3-MST) [23]. Interestingly, increased activity of key H2S enzymatic CSE/CBS pathway seems to afford gastroprotection against stress-induced ulcerogenesis [24] and has been shown to promote ulcer healing in rats [25]. Both H2S and CO were reported to contribute to a modulation of a variety of physiological processes, including vasodilatation, neurotransmission, platelet activation, and aggregation, and have recently emerged as important signaling molecules in gastrointestinal (GI) tract [26–29].
Recent studies revealed that H2S released from its donor, NaHS or synthesized from l-cysteine, exerts gastroprotection against non-topical vascular injury induced by ischemia/reperfusion and topical corrosive irritants such as ethanol [30, 31]. Our group demonstrated that H2S affords gastroprotection against gastric bleeding erosions induced by water immersion and restraint stress (WRS) [24]. Furthermore, CO administered exogenously or that generated endogenously due to the enhanced HO-1 activity has been shown to prevent gastric mucosa against damage induced by ethanol or nonsteroidal anti-inflammatory drugs (NSAIDs) such as aspirin [19, 32].
GI tract is one of the most susceptible systems to stressful stimuli [33]. Local mechanisms for multiple superficial stress-induced lesions are mucosal ischemia, fall in gastric microcirculation, inhibition of gastric mucosal prostaglandins synthesis, and limited mucosal ability to remove back-diffusing protons [34–37]. Oral NBP therapy is likely to be at high risk of developing gastric ulcers, especially in gastric mucosa compromised by NSAIDs, but whether mild stress can affect the complications in gastric mucosa associated with a single BPs treatment has not been so far examined. It is also unknown whether H2S and CO contribute to the mechanism of gastroprotection against those two concomitant deleterious factors, for instance concomitantly observed in BPs patients who suffered from stress episodes [38].
In this study, we selected mild cold stress, which by itself does not result in gastric lesions [39] to determine the susceptibility of gastric mucosa to a subsequent alendronate treatment, and we compared their synergistic injurious action in the rat stomach compared to alendronate alone under non-stressed conditions. Our second goal was to investigate the mechanism of the potential protective action of H2S released from NaHS, or CO released from its donor, tricarbonyldichlororuthenium (II) dimer (CORM-2) against alendronate ulcerogenesis in gastric mucosa with or without exposure to mild stress with special reference to the alteration in blood flow (GBF) and changes in activity of endogenous H2S and CO biosynthetic enzymatic pathways involved in a potential protective action of these two gaseous mediators.
At molecular level, we have investigated whether H2S and CO released from their donors NaHS and CORM-2, respectively, can modulate hypoxia in gastric mucosa via regulation of mRNA and protein expression for hypoxia-inducible factor 1α (HIF-1α), and we examined a potent anti-inflammatory and anti-oxidative actions of CO and H2S by determination of gastric mucosal mRNA expression for heme oxygenase-1 (HO-1), HO-2, and proinflammatory factors inducible nitric oxide synthase (iNOS), cyclooxygenase-2 (COX-2), interleukin-1 β (IL-1β), tumor necrosis factor-α (TNF-α) and expression of anti-oxidative enzymes, glutathione peroxidase (GPx)-1, and superoxide dismutase (SOD)-2. We also determined the relation of HIF-1α with inflammatory factors by determination of mRNA expression for nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) in gastric mucosa of rats pretreated with vehicle or H2S and CO donors.
Materials and Methods
Animals
One hundred and forty male Wistar rats with average weight 220–300 g were used in this study. Rats were fasted for 24 h with free access to drinking water before chemicals and drugs application. The study was approved by the Institutional Animal Care and Use Committee of Jagiellonian University Medical College in Cracow and run in accordance with the statements of the Helsinki Declaration regarding handling of experimental animals.
Experimental Design, Chemicals, and Drugs Treatment
Three series of animals (A, B, and C) were randomly selected into experimental groups (6–8 rats each). In series A, rats were administered with single dose of alendronate applied i.g. in a doses ranging from 150 mg/kg up to 700 mg/kg (Sigma-Aldrich, Schnelldorf, Germany) according to the procedure described previously by Ohashi et al. [40]. Alendronate was dissolved in saline and adjusted to pH = 7.0 by adding NaOH (0.1 M) or HCl (0.1 M) [41]. In series B, rats were pretreated with or without NaHS (5 mg/kg), H2S donor, or CORM-2 (5 mg/kg) releasing CO administered i.g. 30 min before alendronate administration (series B). In series C, rats were initially exposed daily to mild stress for 3 days in total before alendronate administration and two hours later after last exposure to stress, they received the pretreatment with NaHS and CORM-2 before subsequent alendronate administration, similarly as animals in series B. In series C, animals were exposed daily for 3 days in total to mild stress by water immersion and restraint in cold water at 23 °C for 30 min (WRS). This short time period of WRS by itself does not result in macroscopic gastric lesions as described previously [39]. At the day 3, the non-stressed control rats (series B) and stressed rats (series C) were administered with vehicle (saline) or alendronate applied i.g. in a single dose of 300 mg/kg, which was established in experiments with dose-dependent damaging effect of this compound. Alendronate at the dose 300 mg/kg was further considered as effective dose-inducing gastric damage and employed in majority of our experimentations. In separate groups of rats (series B and C), NaHS or CORM-2 was administered i.g. in a dose of 5 mg/kg each, which has been shown previously to exert gastroprotective effect against gastric mucosal lesions induced by various damaging agents [19, 24, 31]. Alendronate, NaHS, and CORM-2 were purchased from Sigma-Aldrich (Schnelldorf, Germany) and administered i.g. using orogastric tube as described previously [42].
Determination of Gastric Damage, Examination of GBF, and Gastric Biopsy Sampling for H2S Production and Molecular Assessment
Three days after alendronate administration, the animals of all series A–C were anesthetized with pentobarbital (60 mg/kg i.p.), the abdomen was opened, and the stomach was exposed to measure GBF by H2-gas clearance technique as described previously [43]. The GBF was measured in the oxyntic part of the gastric mucosa not involving mucosal lesions. Average values of three measurements were determined and expressed as a percentage of the change of the GBF value obtained in intact gastric mucosa.
The area of gastric lesions was determined with computerized planimetry (Morphomat, Carl Zeiss, Berlin, Germany) by a person who did not know which experimental group the animals belonged to [19, 43]. Gastric mucosal biopsies (about 200 mg) were taken for the determination of H2S biosynthesis, and the remaining part of stomach was scrapped off on ice, snap-frozen in liquid nitrogen, and stored at−80 °C for further analysis.
Determination of H2S Production Via CSE/CBS and 3-MST Pathways in Gastric Mucosa
The ability of gastric mucosa to produce H2S via CSE/CBS or 3-MST pathway was measured in homogenized tissue in the presence of exogenous substrates using previously described zinc (Zn)-trapping assay [24, 44]. Briefly, gastric mucosa was quickly isolated from uninjured sites, snap-frozen, and stored at −80 °C. The gastric tissue was homogenized in ice-cold 50 mM potassium phosphate buffer, pH = 8.0 (12 % w/v). l-cysteine (10 mM) and pyridoxal-5′-phosphate (P5P) or α-ketoglutarate (α-KG) was added to the tissue homogenate, and the vials including inner tubes with Zn acetate were then incubated in a shaking water bath at 37 °C for 90 min. Trichloroacetic acid (TCA; 50 %; 0.5 ml) was then injected into the reaction mixture through the serum cap. The mixture was left to stand for another 60 min in 50 °C to allow for the trapping of evolved H2S by the Zn acetate. N,N-dimethyl-p-phenylenediamine sulfate (20 mM; 50 μl) in 7.2 M HCl and FeCl3 (30 mM; 50 μl) in 1.2 M HCl were added to the inner tubes. After 20 min, absorbance at 670 nm was measured with a microplate reader (Tecan Sunrise, Männedorf, Switzerland). The calibration curve of absorbance vs. H2S concentration was obtained by using NaHS solution of varying concentrations.
Determination of mRNA Expression for HO-1, HO-2, HIF-1α, NF-κB, COX-2, iNOS, TNF-α, IL-1β, SOD-2, and GPx-1 in Rat Gastric Mucosa by Real-Time Polymerase Chain Reaction (qPCR)
Expression of mRNA in gastric mucosa was determined by real-time PCR as described previously [19]. Briefly, RNA was isolated from gastric mucosal biopsies stored at −80 °C using GeneMATRIX Universal RNA Purification Kit (EURx, Gdansk, Poland) and reversed transcription to cDNA was performed using High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific, Life Technologies, MA, USA). Expression for HO-1, HO-2, HIF-1α, NF-κB, COX-2, iNOS, TNF-α, IL-1β, SOD-2, and GPx-1 was determined by real-time PCR using specific primers, SG qPCR Master Mix (2x) including SYBR-Green (EURx, Gdansk, Poland) and appropriate thermal cycler (7900HT Fast Real-Time PCR System, Thermo Fisher Scientific, Life Technologies, MA, USA). The nucleotide sequences of the primers used in PCR are presented in Table 1. The β-actin gene was used as an internal control. Data were analyzed using 2−ΔCt method [45].
Table 1.
Forward and reverse primers used in the assessment of mRNA expression for β-actin, HO-1, HO-2, HIF-1α, COX-2, iNOS, TNF-α, IL-1β, SOD-2, and GPx-1 by real-time PCR
| Gene | Forward primer | Reverse primer |
|---|---|---|
| β-actin | 5′-GATCAAGATCATTGCTCCTCCTG-3′ | 5′-AGGGTGTAAAACGCAGCTCA-3′ |
| HO-1 | 5′-GTCCCAGGATTTGTCCGAGG-3′ | 5′-GGAGGCCATCACCAGCTTAAA-3′ |
| HO-2 | 5′-CCGGGCAGAAAATACCCAGT-3′ | 5′-ATCAGTGCTTCCTTCCGGTG-3′ |
| HIF-1α | 5′-ATCCATTTTCAGCTCAGGACAC-3′ | 5′-GGTAGGTTTCTGTAACTGGGTCTG-3′ |
| NF-κB | 5′-TTCAACATGGCAGACGACGA-3′ | 5′-CCATCTGTTGACAGTGGTATATCTG-3′ |
| COX-2 | 5′-ATCAGAACCGCATTGCCTCT-3′ | 5′-GCCAGCAATCTGTCTGGTGA-3′ |
| I NOS | 5′-TGGTGAGGGGACTGGACTTT-3′ | 5′-CTCCGTGGGGCTTGTAGTTG-3′ |
| IL-1β | 5′-GCTATGGCAACTGTCCCTGA-3′ | 5′-AGTCAAGGGCTTGGAAGCAA-3′ |
| TNF-α | 5′-TGGGCTCCCTCTCATCAGTT-3′ | 5′-TCCGCTTGGTGGTTTGCTAC-3′ |
| SOD-2 | 5′-GTGGAGAACCCAAAGGAGAGT-3′ | 5′-GGTCCTGATTAGAGCAGGCG-3′ |
| GPx-1 | 5′-CATTGAGAATGTCGCGTCCC-3′ | 5′-TTGCCATTCTCCTGATGTCCG-3′ |
Protein Expression of HIF-1α in Gastric Mucosa Determined by Western Blot
Western blot analysis was used to determine gastric expression of HIF-1α in gastric mucosal samples as described previously [19]. Rabbit polyclonal anti-HIF-1α (Proteintech, Manchester, UK) in dilution of 1:250 in 5 % milk (incubation in 4 °C, overnight) and rabbit policlonal anti-β-actin (Proteintech) in dilution of 1:2000 in 5 % milk (incubation in room temperature, 3 h) were used as primary antibodies. Protein expression was visualized using a horseradish peroxidase-conjugated secondary goat anti-rabbit IgG antibodies (Santa Cruz Biotechnology, Texas, USA) in dilution of 1:2000 in 5 % milk (incubation time 1 h, room temperature). Chemiluminescence was measured using C-DiGit® Blot Scanner (LI-COR, NE, USA). The intensity of the bands was determined and analyzed using Image Studio 4.0 software (LI-COR, NE, USA). The expression of each sample was normalized to the expression of β-actin protein.
Statistical Analysis
Results are expressed as mean ± SEM. Statistical comparison was made by Mann–Whitney U test or Student’s T test. ANOVA with Tukey post hoc or Kruskal–Wallis with Dunn’s post hoc test was used where appropriate. Difference with p < 0.05 was considered significant.
Results
Figure 1 shows that pretreatment with alendronate (150–700 mg/kg i.g.) dose-dependently increased the mean area of gastric lesions and significantly decreased GBF (p < 0.05). The lower doses 37.5 mg/kg or 75 mg/kg of alendronate failed to induce the gross mucosal lesions, and these results were omitted from the presentation. With increasing the dose of alendronate up to 150 mg/kg, the gastric mucosal lesions were detected and the GBF was significantly decreased (p < 0.05) compared with vehicle (saline) control rats (Fig. 1). The mean area of these gastric lesions was further significantly increased (p < 0.05), and GBF level was significantly decreased (p < 0.05) when alendronate was administered in the dose of 300 mg/kg i.g. as compared with the lower dose of 150 mg/kg of alendronate. Intragastric administration of alendronate in higher dosages of 500 and 700 mg/kg significantly increased the mean lesions area (p < 0.05) and produced a significant fall in GBF as compared with the values observed in animals treated with the dose of 150 mg/kg (p < 0.05) but failed to show significant difference as compared with the dose of 300 mg/kg i.g. Therefore, the dose of 300 mg/kg i.g. of alendronate was selected in further experiments and considered as the truly effective and reproducible dose-inducing gastric damage.
Fig. 1.

Mean lesion area and gastric blood flow (GBF) level in gastric mucosa after alendronate administration (150–700 mg/kg i.g.). Results are mean ± SEM of 6–8 animals for each experimental group. Single asterisk indicates a significant change (p < 0.05) as compared with vehicle (saline). Asterisk and cross indicate a significant change (p < 0.05) as compared with vehicle-control rats and with the group with alendronate administered i.g. in the dose of 150 mg/kg
As shown in Fig. 2, the pretreatment with CORM-2 or NaHS administered in the dose of 5 mg/kg i.g. each, which was shown in our previous studies to reduce gastric lesions by stress and ethanol [19, 24, 31], significantly decreased alendronate-induced increase in mean lesion area (p < 0.05) and significantly increased GBF as compared with vehicle-control rats (p < 0.05). Preexposure of rats for 3 days with mild stress significantly increased alendronate-induced gastric damage (p < 0.05) and significantly decreased the GBF (p < 0.05) as compared with those in vehicle-treated animals. NaHS and CORM-2 administered in the dose of 5 mg/kg significantly decreased the mean lesion area of alendronate-treated rats (p < 0.05) and significantly increased the GBF in gastric mucosa of non-stressed and stressed animals treated with this agent as compared with respective values in vehicle controls (p < 0.05).
Fig. 2.

Mean lesion area of gastric lesions induced by alendronate (300 mg/kg i.g.) and accompanying changes in the gastric blood flow (GBF) in gastric mucosa administered with NaHS (5 mg/kg i.g.) or CORM-2 (5 mg/kg i.g.) with or without exposure to mild stress (WRS). Results are mean ± SEM of 6–8 animals for each experimental group. Asterisk or cross indicate significant change (p < 0.05) as compared with non-stressed vehicle control. Asterisk and cross indicate significant (p < 0.05) difference as compared with vehicle-control gastric mucosa compromised by mild WRS
The macroscopic appearance of rats gastric mucosa pretreated with alendronate (300 mg/kg i.g.) with or without preexposure to mild WRS is presented in Fig. 3a, b, respectively. Alendronate-induced superficial gastric damage, observed as a gray necrotic layer of desquamated epithelium, predominantly located in antral part of gastric mucosa (Fig. 3a). In rats preexposed to mild WRS, alendronate caused numerous gastric hemorrhagic dot-like erosions and band-like lesions which were observed in oxyntic part of the stomach in spite of necrotic damage caused by this agent in antrum (Fig. 3b). Figure 3 (panels C and D) shows the representative gross gastric mucosal injury induced by alendronate in rats compromised by mild WRS and pretreated with CORM-2 (5 mg/kg i.g., Fig. 3c) or NaHS (5 mg/kg i.g., Figure 3d). CORM-2 and NaHS had evidently reduced gross mucosal lesions in rats preexposed to mild WRS followed by administration of alendronate (Fig. 3b, c, d).
Fig. 3.
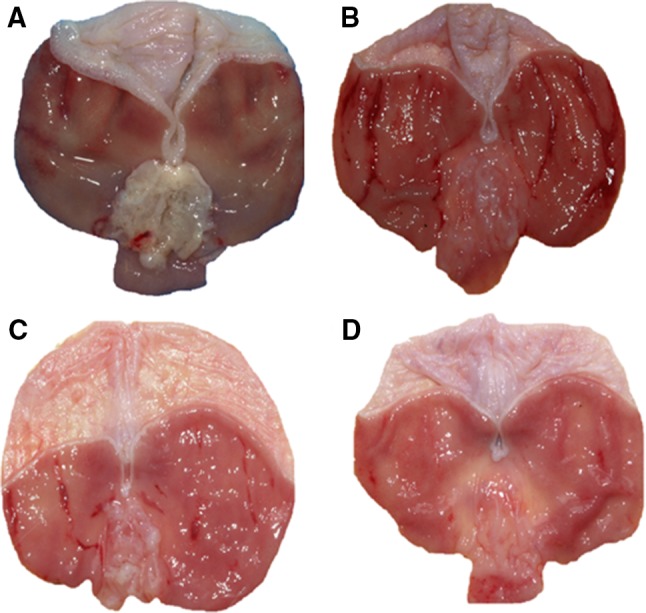
a–d The representative appearance of alendronate-induced gastric lesions in the rat stomach compromised or not with mild water immersion and restraint stress (WRS) with or without i.g. pretreatment with CORM-2 (5 mg/kg) or NaHS (5 mg/kg). Note, the presence of superficial gastric lesions mainly localized in antral part of gastric mucosa in rat stomach exposed to alendronate (300 mg/kg i.g.) (a). In gastric mucosa compromised by mild WRS and administered with alendronate (300 mg/kg i.g.) additionally to damage in antrum, the numerous dot-like and band-like lesions are visible in oxyntic mucosa (b). In rats pretreated with CORM-2 (c) or NaHS (d), the total area of alendronate-induced gastric lesions compromised by mild WRS is markedly reduced (b, c, d)
Production of H2S in gastric mucosa assessed by activity of CSE/CBS was not significantly affected by mild WRS alone as compared to intact rats (Fig. 4a). In rats treated with alendronate (300 mg/kg i.g.) and previously exposed or not to mild WRS, the H2S production analyzed by CSE/CBS pathway was significantly decreased (p < 0.05) as compared with intact rats. The pretreatment with NaHS but not CORM-2 significantly increased (p < 0.05) the gastric mucosal production of H2S determined by CSE/CBS activity as compared with that in gastric mucosa of rats compromised by mild WRS and subsequently treated with the combination of vehicle and alendronate (Fig. 4a). Similarly, H2S production assessed via 3-MST activity was significantly decreased either in non-stressed or in stressed animals and administered with alendronate (300 mg/kg i.g.) (p < 0.05) as compared with intact animals (Fig. 4b). Again, pretreatment with NaHS but not CORM-2 reversed this effect significantly increasing H2S production by 3-MST as compared with vehicle-control gastric mucosa compromised by mild WRS and administered with alendronate (p < 0.05) (Fig. 4b).
Fig. 4.

a, b The effect of mild WRS and alendronate (300 mg/kg i.g.) alone and the combination of mild WRS and alendronate (300 mg/kg i.g.) with or without pretreatment with CORM-2 (5 mg/kg i.g.) or NaHS (5 mg/kg i.g.) on H2S production in gastric mucosa determined by CSE/CBS (a) or 3-MST (b) activity in gastric mucosa. Results are mean ± SEM of 4–6 rats per each group. Significant change (p < 0.05) as compared with the respective values in intact rats was indicated by asterisk. Cross indicates significant change (p < 0.05) comparing to the values obtained in vehicle-control gastric mucosa
Figure 5 (panels A and B) shows mRNA expression of HO-1 (panel A) and HO-2 (panel B) in intact gastric mucosa and in that pretreated with vehicle, NaHS or CORM-2 in rats compromised by mild WRS with or without alendronate treatment. Alendronate administered alone significantly increased mRNA expression for HO-1 as compared with intact rats (p < 0.05) while mild WRS remained without significant effect. Preexposure of gastric mucosa to mild WRS significantly increased alendronate-induced upregulation of HO-1 mRNA expression (p < 0.05). NaHS (5 mg/kg i.g.) and CORM-2 (5 mg/kg i.g.) significantly decreased HO-1 mRNA expression as compared with vehicle-control gastric mucosa in rats compromised by mild WRS for 3 days before the administration of alendronate (p < 0.05) (Fig. 5a). As shown in Fig. 5b, mild WRS and alendronate significantly decreased mRNA expression for HO-2 (p < 0.05), and this effect was not significantly altered when mild WRS was combined with alendronate as compared with animals preexposed to mild WRS alone or administered with alendronate. Neither CORM-2 nor NaHS had influenced the downregulation of HO-2 mRNA expression induced by the combination of alendronate and mild WRS (Fig. 5b).
Fig. 5.

a, b Expression of mRNA for HO-1 (a) and HO-2 (b) in gastric mucosa of intact rats or those exposed to mild water immersion and restraint stress (WRS) alone or administered with alendronate (300 mg/kg i.g.) alone or in combination with mild WRS with or without pretreatment with CORM-2 (5 mg/kg i.g.) or NaHS (5 mg/kg i.g.). Results are expressed as the HO-1 and HO-2 mRNA expression normalized to β-actin. Results are mean ± SEM of 6–9 determinations per group. Asterisk indicates a significant change (p < 0.05) as compared with the respective values in intact gastric mucosa. Double asterisk indicates significant change (p < 0.05) as compared with intact rats and those administered with alendronate (300 mg/kg i.g.) alone. Cross indicates a significant change (p < 0.05) comparing to values obtained in vehicle-control group compromised by mild WRS and treated with alendronate
Expression of mRNA and protein for HIF-1α in gastric mucosa is presented in Fig. 6a, b, respectively. Both mild WRS and alendronate failed to affect mRNA and protein expression of HIF-1α, but when alendronate was combined with mild WRS, a significant increase in gastric mRNA and protein expression of HIF-1α was observed as compared with intact rats and those preexposed to mild WRS only or administered with alendronate alone (p < 0.05). The pretreatment with CORM-2 (5 mg/kg i.g.) or NaHS (5 mg/kg i.g.) significantly decreased the mRNA and protein expression of HIF-1α as compared with vehicle-control gastric mucosa compromised by mild WRS and administered with alendronate (p < 0.05).
Fig. 6.

a, b Expression of mRNA and protein for HIF-1α in gastric mucosa of intact rats or those exposed to mild water immersion and restraint stress (WRS) alone or administered with alendronate (300 mg/kg i.g.) alone or those exposed to the combination of mild WRS and alendronate (300 mg/kg i.g.) with or without pretreatment with CORM-2 (5 mg/kg i.g.) or NaHS (5 mg/kg i.g.). Results are expressed as the HIF-1α mRNA or protein expression of HIF-1α normalized to β-actin. Results are mean ± SEM of 4–8 determinations per group. Asterisk indicates a significant change (p < 0.05) as compared with the respective values in intact gastric mucosa. Cross indicates a significant change (p < 0.05) as compared with the values obtained in vehicle-control gastric mucosa compromised by mild WRS and administered with alendronate (300 mg/kg i.g.). Representative protein expression bands for each experimental group are presented at the bottom of (b)
Figure 7 shows the effect of alendronate with or without the pretreatment with NaHS or CORM-2 on mRNA expression of NF-κB in non-stressed rats and those preexposed to mild WRS. Alendronate combined with mild WRS significantly increased mRNA expression for NF-κB as compared with intact gastric mucosa (p < 0.05) (Fig. 7). CORM-2 (5 mg/kg i.g.) or NaHS (5 mg/kg i.g.) significantly decreased the mRNA expression of NF-κB as compared with vehicle-control gastric mucosa compromised by mild WRS and administered with alendronate (p < 0.05) (Fig. 7).
Fig. 7.

Expression of mRNA for NF-κB in gastric mucosa of intact rats or those exposed to mild water immersion and restraint stress (WRS) alone or administered with alendronate (300 mg/kg i.g.) alone or those exposed to the combination of mild WRS and alendronate (300 mg/kg i.g.) with or without pretreatment with CORM-2 (5 mg/kg i.g.) or NaHS (5 mg/kg i.g.). Results are expressed as the NF-κB mRNA expression normalized to β-actin. Results are mean ± SEM of 4-8 determinations per group. Asterisk indicates a significant change (p < 0.05) as compared with the respective values in intact gastric mucosa. Cross indicates a significant change (p < 0.05) as compared with the values obtained in vehicle-control gastric mucosa compromised by mild WRS and administered with alendronate (300 mg/kg i.g.)
Figure 8 (panels A and B) shows mRNA expression for iNOS (panel A) and COX-2 (panel B) in non-stressed rats and those preexposed to mild WRS and treated with alendronate. When alendronate was combined or not with mild WRS, mRNA expression for iNOS was significantly increased, as compared with intact gastric mucosa (p < 0.05) (Fig. 8a). The expression of COX-2 mRNA was significantly increased in gastric mucosa compromised by mild WRS and administered with alendronate comparing to that in intact gastric mucosa (p < 0.05) (Fig. 8b). CORM-2 (5 mg/kg i.g.) or NaHS (5 mg/kg i.g.) significantly decreased mRNA expression of iNOS and COX-2 as compared with vehicle-control gastric mucosa of rats compromised by mild WRS and administered with alendronate (p < 0.05) (Fig. 8a, b).
Fig. 8.

a, b Expression of mRNA for iNOS (a) and COX-2 (b) in gastric mucosa of intact rats or those exposed to mild water immersion and restraint stress (WRS) alone or administered with alendronate (300 mg/kg i.g.) alone or those exposed to the combination of mild WRS and alendronate (300 mg/kg i.g.) with or without pretreatment with CORM-2 (5 mg/kg i.g.) or NaHS (5 mg/kg i.g.). Results are expressed as the iNOS and COX-2 mRNA expression normalized to β-actin. Results are mean ± SEM of 4-8 determinations per group. Asterisk indicates a significant change (p < 0.05) as compared with the respective values in intact gastric mucosa. Cross indicates significant change (p < 0.05) as compared with the values obtained vehicle-control gastric mucosa compromised by mild WRS and administered with alendronate (300 mg/kg i.g.)
Figure 9 (panels A and B) presents data on mRNA expression for IL-1β (panel A) and TNF-α (panel B) in non-stressed and stressed rats treated with alendronate. The significant increase in mRNA expression for IL-1β and TNF-α was observed in gastric mucosa of rats exposed to mild WRS (vehicle-control) and treated with alendronate as compared with intact gastric mucosa (p < 0.05) (Fig. 9a, b). The pretreatment with CORM-2 (5 mg/kg i.g.) or NaHS (5 mg/kg i.g.) significantly decreased gastric mucosal mRNA expression of these cytokines as compared with vehicle controls (p < 0.05).
Fig. 9.

a, b The expression of mRNA for TNF-α (a) and IL-1β (b) in gastric mucosa of intact rats or those exposed to mild water immersion and restraint stress (WRS) alone or those exposed to the combination of mild WRS and alendronate (300 mg/kg i.g.) with or without pretreatment with CORM-2 (5 mg/kg i.g.) or NaHS (5 mg/kg i.g.). Results are expressed as the TNF-α and IL-1β mRNA expression normalized to β-actin. Results are mean ± SEM of 4–8 determinations per group. Asterisk indicates a significant change (p < 0.05) as compared with the respective values in intact gastric mucosa. Cross indicates significant change (p < 0.05) as compared with the values obtained vehicle-control gastric mucosa compromised by mild WRS and administered with alendronate (300 mg/kg i.g.)
As shown in Fig. 10 (panels A and B), the mRNA expression for GPx-1 and SOD-2 was significantly decreased in alendronate-treated rats preexposed to mild WRS as compared with that recorded in intact gastric mucosa (p < 0.05). The expression of GPx (panel A) and SOD-2 (panel B) mRNAs was significantly decreased in gastric mucosa compromised by mild WRS and further pretreated with vehicle (control) and administered with alendronate as compared with intact rats (p < 0.05). The pretreatment with NaHS (5 mg/kg i.g.) and CORM-2 (5 mg/kg i.g.) did not affect mRNA expression for GPx and SOD-2 downregulated in vehicle-control group of rats exposed to the combination of mild WRS and alendronate (panels A and B).
Fig. 10.

a, b The expression of mRNA for GPx-1 and SOD-2 in gastric mucosa of intact rats or those exposed to mild water immersion and restraint stress (WRS) alone or administered with alendronate (300 mg/kg i.g.) alone or those exposed to the combination of mild WRS and alendronate (300 mg/kg i.g.) with or without pretreatment with CORM-2 (5 mg/kg i.g.) or NaHS (5 mg/kg i.g.). Results are expressed as the GPx-1 and SOD-2 mRNA expression normalized to β-actin. Results are mean ± SEM of 4-8 determinations per group. Asterisk indicates a significant change (p < 0.05) as compared with the respective values in intact gastric mucosa
Discussion
The resistance of stomach to gastric mucosal injury is maintained by the mucosal defense mechanisms consisting of protective mucus and alkaline secretion of bicarbonate, the maintenance of gastric microcirculation, the hydrophobicity of mucosal surface, the presence of vasoactive agents such as prostaglandins (PGs), sensory neuropeptides released from primary afferent capsaicin-sensitive sensory neurons, and three interacting gaseous mediators NO, CO, and H2S [28, 31, 46–48].
The use of BPs has been reported to be closely associated with GI adverse events in humans and experimental animals [49]. In our study, we adapted experimental model of alendronate-induced gastric damage [40], and we confirmed that this drug when administered orally in graded concentrations caused macroscopic lesions in the rat stomach. Our results are corroborative with previous observations that various BPs evoke mainly antral lesions accompanied by severe edema, neutrophil accumulation, and epithelial cell loss determined microscopically [50–52].
It has been recognized long time ago that the gastric microcirculation plays a crucial role in gastric mucosal defense [53], and this study provides the evidence that alendronate-induced gastric damage is accompanied by the decrease in GBF. The prominent fall in GBF was observed with the dose of 300 mg/kg of alendronate, which was further used in our subsequent determinations. Thus, we demonstrated that the formation of gastric damage by alendronate is associated with perturbation in the GBF, which could be at variance with previous observations [54]. This discrepancy reflecting alteration in GBF may be due to different dosages of BPs and techniques of assessment of gastric mucosal blood flow used before [54] and in our present study. In the present study, we demonstrated for the first time that preexposure of rats to mild stress exacerbated the alendronate-induced gastric damage and produced greater fall in GBF as compared with those recorded in non-stressed animals exposed to alendronate. Therefore, we propose that this mild stress could weaken the gastric mucosal barrier, thus predisposing gastric mucosa to injury induced by alendronate. Interestingly, the increased susceptibility for gastric damage induced by BPs has been previously observed in rats pretreated with indomethacin [38].
Hypoxia-inducible factor 1 alpha (HIF-1α), the transcription factor, whose expression is oxygen-dependent and increased by prolonged hypoxia, has been shown to contribute to tumor proliferation and angiogenesis [55–57]. In our present study, the alendronate-induced gastric damage in rats preexposed to mild stress resulted in an increase in mRNA and protein expression for HIF-1α; the effect that has not been observed in non-stressed rats administered with alendronate. Thus, we propose that the mechanism of mild stress affecting pathomechanism of alendronate-induced gastric damage may involve hypoxia, which can trigger exacerbation of alendronate-induced ulcerogenesis in the rat stomach. This notion is supported by our finding that expression of NF-κB mRNA as well as HIF-1α was increased in gastric mucosa compromised by mild stress and administered with alendronate. These observations suggest that NF-κB pathway plays a role in injurious action of alendronate due to regulation of hypoxia, which remains corroborative with the conclusion of study by van Uden et al. [58] who demonstrated in vitro that NF-κB is involved in the regulation of HIF-1α expression. Moreover, we observed that expression of mRNA expression for HO-1, possibly enhanced by inflammation and pro-inflammatory factors iNOS, COX-2, and cytokines such as IL-1β and TNF-α, was increased in gastric mucosa compromised by mild stress and administered with alendronate. Additionally, the mRNA expression of anti-oxidative enzymes GPx-1 and SOD-2 was inhibited in alendronate-treated animals with or without preexposure to mild stress. This indicates that mild stress not only exacerbated the alendronate-induced gastric damage possibly mediated by the enhanced expression of pro-inflammatory factors but also profoundly decreased expression of anti-oxidative enzymes GPx-1 and SOD-2. This observation is in keeping with the findings by Nicolau et al. [41] and Silva et al. [59] who also reported increased TNF-α and IL-1β protein concentration in alendronate-treated gastric mucosa. However, in contrast to our findings where a single dose of alendronate was used in majority of our experiments, Silva et al. [59] have demonstrated decreased iNOS and eNOS protein expression after 4 days of subsequent alendronate administrations, suggesting that suppression of major enzymes of NO biosynthesis could be considered as one of the possible mechanism by which this drug evokes its damaging activity. This effect on NO biosynthesis caused by chronic vs single alendronate administration should be further determined but our experimental design with alendronate administered in a large single dose resulted in an excessive production of NO possibly derived from overexpression of iNOS acting as a potentially harmful agent and the proinflammatory factor in the gastric mucosa [60–62]. Nevertheless, the decreased gastric mucosal glutathione (GSH) and the increased malondialdehyde (MDA) contents reported in their study [59] and diminished GPx-1 and SOD-2 mRNA expression demonstrated in our present work clearly indicate that alendronate-evoked gastric lesions are due to a failure of anti-oxidative mechanisms in gastric mucosa exposed to this BPs.
Interestingly, we demonstrated to our best knowledge for the first time that H2S production via CSE/CBS and 3-MST pathways was markedly decreased in gastric mucosa compromised by mild WRS and administered with alendronate, suggesting that this gaseous mediator produced endogenously plays a crucial role in pathogenesis of alendronate-induced gastric lesions. This notion is supported by the fact that the activity of these H2S-keynote enzymes was decreased in vehicle-pretreated control rats administered with alendronate and exposed or not to mild WRS. This effect was accompanied by the decreased mRNA expression for HO-2, which is one of two major enzymes, besides HO-1, producing endogenous CO. We conclude that CO next to H2S can be involved in pathogenesis of damage induced by alendronate and could be also implicated in gastroprotection against alendronate-induced damage of gastric mucosa compromised or not by mild stress.
We found that H2S donor, NaHS, and CO-releasing CORM-2 attenuated gastric mucosal lesions induced by alendronate and that this effect was accompanied by the rise in GBF. Moreover, the preexposure of gastric mucosa to mild stress exaggerated alendronate-induced gastric damage, and the pretreatment with NaHS and CORM-2 also reduced these lesions and elevated GBF. These findings confirm our previous observations that CORM-2 (1 mg/kg i.g.) and NaHS (5 mg/kg i.g.) prevented gastric mucosa against stress-induced gastric damage [24, 63]. In this study, both compounds were administered in the dose of 5 mg/kg i.g., which has been shown in our previous studies to exert gastroprotection against acute WRS- and ethanol-induced gastric damage [19, 24, 31]. Interestingly, at the dose used in our present study, CORM-2 has been shown to increase CO content in gastric mucosa and COHb level in blood [19]. Results of our study with both donors of H2S and CO seem to correspond with the observations of Costa et al. [51] and Nicolau et al. [41] who showed in two independent studies that the treatment with dimanganese decacarbonyl (DMDC) and Lawesson’s reagent, another compounds donating of CO and H2S, respectively, exhibited a protective effect against alendronate-induced gastric lesions in rats. Moreover, our present study demonstrates that NaHS increased H2S production via CSE/CBS and 3-MST activity pathways, which was profoundly decreased in gastric mucosa compromised by mild stress and administered with alendronate. This is in keeping with our previous observation that NaHS increased H2S production via CSE/CBS activity [24] confirming the “reciprocal” interaction of this chemical donor releasing H2S with endogenous biosynthesis of this gaseous molecule endorsing its protective and hyperemic activity against gastric mucosal injury. Therefore, we conclude that the common mechanism of protective action of CORM-2 and NaHS against BPs and other damaging agents can involve an increase in CO and H2S bioavailability in gastric mucosa.
It is of interest that both CORM-2 and NaHS downregulated expression of HIF-1α mRNA and protein, which were elevated in gastric mucosa after exposure of rats to combined action of mild stress and alendronate. This confirms that NaHS-releasing H2S and CORM-2-releasing CO can afford gastroprotective effects by counteracting of hypoxia, which is involved in injurious activity and mucosal dysfunction induced by combination of mild stress and alendronate. Indeed, Guillen et al. [64] have demonstrated that CORM-2 dose-dependently inhibited HIF-1α activation in primary chondrocytes, and Flannigan et al. [65] have observed that the H2S-releasing diallyl disulfide while maintaining the HIF-1α expression reduced the severity of experimental colitis in rodents. The level of tissue oxygen seems to be critical because in another study [66] the administration of H2S inhibited HIF-1α stabilization under conditions of hypoxia provided by the 1 and 3 % O2 but not by the level of hypoxia resulting from the 0.1 % O2 content.
We observed that CO and H2S released from their donors, CORM-2, and NaHS, respectively, clearly reduced mRNA expression for HO-1. We propose that HIF-1α and HO-1 expression could act as markers of tissue hypoxia and inflammation because both HIF-1α and HO-1 mRNAs expression were increased in gastric mucosa of rats treated with alendronate and exposed to mild stress. This notion is supported by the fact that application of CORM-2 or NaHS, which have decreased HIF-1α stabilization and HO-1 mRNA expression, greatly reduced hypoxia and inflammation induced by alendronate in gastric mucosa compromised by mild stress. Therefore, it is reasonable to assume that the decrease in stabilization of HIF-1α and the restoration of HO-1 expression could, at least in part, account for the observed anti-inflammatory and anti-oxidizing effects of H2S and CO released from NaHS or CORM-2, respectively.
We observed that CORM-2 and NaHS reduced the mRNA expression for pro-inflammatory factors including iNOS, COX-2, and cytokines such as TNF-α and IL-1β that were all upregulated in gastric mucosa of rats exposed to the combination of mild stress and alendronate. This is in parallel with the results of Nicolau et al. [41] who reported decreased TNF-α and IL-1β levels in alendronate-treated gastric mucosa after H2S donor administration and the study of Costa et al. [51] who revealed the same effect after application of DMDC, a CO donor. Interestingly, CORM-2 and NaHS decreased mRNA expression of NF-κB upregulated in gastric mucosa compromised by mild stress and administered with alendronate. This indicates that H2S and CO can downregulate the NF-κB pathway, thus explaining in part the anti-hypoxic and anti-inflammatory action of these gaseous molecules. In agreement with this notion, Wei et al. [67] have demonstrated that CORM-2 decreased NF-κB activity and TNF-α concentration in rats with ischemia-/reperfusion-induced hepatic injury. Moreover, Chen et al. [68] have shown that NaHS inhibited nuclear translocation of NF-κB and attenuated TNF-α/interferon-γ-induced injury in monolayer culture of Caco-2 cells.
Interestingly, neither CORM-2 nor NaHS affected mRNA expression for GPx-1 or SOD-2, which was downregulated in gastric mucosa compromised by mild WRS and administered with alendronate. This suggests that H2S- and CO-induced gastroprotection against mucosal damage induced by alendronate does not involve the alterations in expression of these enzymes. However, Nicolau et al. [41] and Costa et al. [51] have demonstrated that using different H2S donor, Lawesson’s reagent, and CO-releasing molecule, DMDC, these donors reversed the effect of alendronate on the activity of another oxidative parameters including GSH and MDA in gastric mucosa.
We conclude that endogenous gaseous mediators, CO and H2S, play an important role in the maintenance of gastric integrity and exert a beneficial protective effect against alendronate-induced gastric lesions in the presence of mild stressor. We provided the evidence that the increased bioavailability of these gaseous transmitters released from their donors NaHS and CORM-2 can be responsible for the gastroprotection against alendronate-induced gastric damage with or without exposure to mild stress. Interestingly, the NaHS-induced protection is accompanied by the increase in endogenous H2S biosynthesis, but CORM-2 seems to act independently from endogenous H2S. The common mechanism of CO and H2S gastroprotective action involves an increase in gastric microcirculation, the reduction in hypoxia by attenuation of HIF-1α, and the attenuation of inflammation by downregulation of NF-κB and pro-inflammatory factors, such as iNOS, COX-2, IL-1β, and TNF-α expression in gastric mucosa exposed to alendronate combined with mild stress. Moreover, our study documented that NaHS and CORM-2 increased the gastric bioavailability of two important vasoactive mediators H2S and CO, thus contributing to the restoration of gastric blood flow, which was decreased after alendronate administration. Therefore, this local hyperemic activity of gaseous mediators H2S and CO seems to be primary mechanism by which these two gaseous mediators exhibit gastroprotection. This increase in gastric microcirculation can contribute to limiting of the extent of hypoxia and inflammation induced by this bisphosphonate as manifested in our present study by the decrease in HIF-1α stabilization and the downregulation of expression of “cytoprotective” HO-1.
Acknowledgments
This study was supported by a Grant from the Jagiellonian University Medical College (No. K/DSC/003122) for JS, Department of Physiology, Jagiellonian University Medical College, Cracow, Poland.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Contributor Information
Marcin Magierowski, Email: m.magierowski@uj.edu.pl.
Katarzyna Magierowska, Email: k.jasnos@interia.pl.
Jakub Szmyd, Email: jakub.szmyd@gmail.com.
Marcin Surmiak, Email: marcin.surmiak@uj.edu.pl.
Zbigniew Sliwowski, Email: agazs@poczta.fm.
Slawomir Kwiecien, Email: skwiecien@cm-uj.krakow.pl.
Tomasz Brzozowski, Email: mpbrzozo@cyf-kr.edu.pl.
References
- 1.Fleisch H. Bisphosphonates: mechanisms of action. Endocr Rev. 1998;19:80–100. doi: 10.1210/edrv.19.1.0325. [DOI] [PubMed] [Google Scholar]
- 2.Drake MT, Clarke BL, Khosla S. Bisphosphonates: mechanism of action and role in clinical practice. Mayo Clin Proc. 2008;83:1032–1045. doi: 10.4065/83.9.1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pazianas M, Abrahamsen B, Ferrari S, Russell RG. Eliminating the need for fasting with oral administration of bisphosphonates. Ther Clin Risk Manag. 2013;9:395–402. doi: 10.2147/TCRM.S52291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reid IR, Hosking DJ. Bisphosphonates in Paget’s disease. Bone. 2011;49:89–94. doi: 10.1016/j.bone.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 5.Tella SH, Gallagher JC. Prevention and treatment of postmenopausal osteoporosis. J Steroid Biochem Mol Biol. 2014;142:155–170. doi: 10.1016/j.jsbmb.2013.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cremers SC, Pillai G, Papapoulous SE. Pharmacokinetics/pharmacodynamics of bisphosphonates: use for optimization of intermittent therapy for osteoporosis. Clin Pharmacokinet. 2005;44:551–570. doi: 10.2165/00003088-200544060-00001. [DOI] [PubMed] [Google Scholar]
- 7.Frith JC, Monkkonen J, Auriola S, et al. The molecular mechanism of action of the anti-resorptive and anti-inflammatory drug clodronate: evidence for the formation in vivo of a metabolite that inhibits bone resorption and causes osteoclast and macrophage apoptosis. Arthritis Rheum. 2001;44:2201–2210. doi: 10.1002/1529-0131(200109)44:9<2201::AID-ART374>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 8.Lehenkari PP, Kellinsalmi M, Napankangas JP, et al. Further insight into mechanism of action of clodronate: inhibition of mitochondrial ADP/ATP translocase by a nonhydrolyzable, adenine-containing metabolite. Mol Pharmacol. 2002;61:1255–1262. doi: 10.1124/mol.61.5.1255. [DOI] [PubMed] [Google Scholar]
- 9.Van Beek E, Pieterman E, Cohen L, Löwik C, Papapoulos S. Farnesyl pyrophosphate synthase is the molecular target of nitrogen-containing bisphosphonates. Biochem Biophys Res Commun. 1999;264:108–111. doi: 10.1006/bbrc.1999.1499. [DOI] [PubMed] [Google Scholar]
- 10.Bergstrom JD, Bostedor RG, Masarachia PJ, Reszka AA, Rodan G. Alendronate is a specific, nanomolar inhibitor of farnesyl diphosphate synthase. Arch Biochem Biophys. 2000;373:231–241. doi: 10.1006/abbi.1999.1502. [DOI] [PubMed] [Google Scholar]
- 11.Kimmel DB. Mechanism of action, pharmacokinetic and pharmacodynamic profile and clinical applications of nitrogen-containing bisphosphonates. J Dent Res. 2007;86:1022–1033. doi: 10.1177/154405910708601102. [DOI] [PubMed] [Google Scholar]
- 12.Hayden RS, Vollrats M, Kaplan DL. Effects of clodronate and alendronate on osteoclast and osteoblast co-cultures on silk-hydroxyapatite films. Acta Biomater. 2014;10:486–493. doi: 10.1016/j.actbio.2013.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Graham DY, Malaty HM. Alendronate gastric ulcers. Alimen. Pharmacol Ther. 1999;13:515–519. doi: 10.1046/j.1365-2036.1999.00488.x. [DOI] [PubMed] [Google Scholar]
- 14.Graham DY. What the gastroenterologist should know about the gastrointestinal safety profiles of bisphosphonates. Dig Dis Sci. 2002;47:1665–1678. doi: 10.1023/A:1016495221567. [DOI] [PubMed] [Google Scholar]
- 15.Ryter SW, Choi AM. Heme oxygenase-1/carbon monoxide: from metabolism to molecular therapy. Am J Respir Cell Mol Biol. 2009;41:251–260. doi: 10.1165/rcmb.2009-0170TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim YM, Pae HO, Park JE, et al. Heme oxygenase in the regulation of vascular biology: from molecular mechanisms to therapeutic opportunities. Antioxid Redox Signal. 2011;14:137–167. doi: 10.1089/ars.2010.3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo JS, Cho CH, Wang WP, Shen XZ, Cheng CL, Koo MW. Expression and activities of three inducible enzymes in the healing of gastric ulcers in rats. World J Gastroenterol. 2003;9:1767–1771. doi: 10.3748/wjg.v9.i8.1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Becker JC, Grosser N, Boknik P, Schröder H, Domschke W, Pohle T. Gastroprotection by vitamin C–a heme oxygenase-1-dependent mechanism? Biochem Biophys Res Commun. 2003;312:507–512. doi: 10.1016/j.bbrc.2003.10.146. [DOI] [PubMed] [Google Scholar]
- 19.Magierowska K, Magierowski M, Hubalewska-Mazgaj M, et al. Carbon monoxide (CO) released from tricarbonyldichlororuthenium (II) dimer (CORM-2) in gastroprotection against experimental ethanol-induced gastric damage. PLoS One. 2015;10:e0140493. doi: 10.1371/journal.pone.0140493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Muñoz-Sánchez J, Chánez-Cárdenas ME. A review on heme oxygenase-2: focus on cellular protection and oxygen response. Oxid Med Cell Longey. 2014;2014:604981. doi: 10.1155/2014/604981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stipanuk MH, Beck PW. Characterization of the enzymatic capacity for cysteine desulphydration in liver and kidney of the rat. Biochem J. 1982;206:267–277. doi: 10.1042/bj2060267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Swaroop M, Bradley K, Ohura T, et al. Rat cystathionine beta-synthase. Gene organization and alternative splicing. J Biol Chem. 1992;267:11455–11461. [PubMed] [Google Scholar]
- 23.Shibuya N, Tanaka M, Yoshida M, et al. 3-Mercaptopyruvate sulfurtransferase produces hydrogen sulfide and bound sulfane sulfur in the brain. Antioxid Redox Signal. 2009;11:703–714. doi: 10.1089/ars.2008.2253. [DOI] [PubMed] [Google Scholar]
- 24.Magierowski M, Jasnos K, Kwiecien S, et al. Endogenous prostaglandins and afferent sensory nerves in gastroprotective effect of hydrogen sulfide against stress-induced gastric lesions. PLoS One. 2015;10:e0118972. doi: 10.1371/journal.pone.0118972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wallace JL, Dicay M, McKnight W, et al. Hydrogen sulfide enhances ulcer healing in rats. FASEB J. 2007;21:4070–4076. doi: 10.1096/fj.07-8669com. [DOI] [PubMed] [Google Scholar]
- 26.Chan MV, Wallace JL. Hydrogen sulfide-based therapeutics and gastrointestinal diseases: translating physiology to treatments. Am J Physiol Gastrointest Liver Physiol. 2013;305:G467–G473. doi: 10.1152/ajpgi.00169.2013. [DOI] [PubMed] [Google Scholar]
- 27.Gibbons SJ, Verhulst PJ, Bharucha A, Farrugia G. Review article: carbon monoxide in gastrointestinal physiology and its potential in therapeutics. Aliment Pharmacol Ther. 2013;38:689–702. doi: 10.1111/apt.12467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Farrugia G, Szurszewski JH. Carbon monoxide, hydrogen sulfide, and nitric oxide as signaling molecules in the gastrointestinal tract. Gastroenterology. 2014;147:303–313. doi: 10.1053/j.gastro.2014.04.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jasnos K, Magierowski M, Kwiecien S, Brzozowski T. Carbon monoxide in human physiology-its role in the gastrointestinal tract. Postepy Hig Med Dosw (Online) 2014;68:101–109. doi: 10.5604/17322693.1087527. [DOI] [PubMed] [Google Scholar]
- 30.Magierowski M, Magierowska K, Kwiecien S, Brzozowski T. Gaseous mediators nitric oxide and hydrogen sulfide in the mechanism of gastrointestinal integrity, protection and ulcer healing. Molecules. 2015;20:9099–9123. doi: 10.3390/molecules20059099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mard SA, Neisi N, Solgi G, Hassanpour M, Darbor M, Maleki M. Gastroprotective effect of NaHS against mucosal lesions induced by ischemia-reperfusion injury in rat. Dig Dis Sci. 2012;57:1496–1503. doi: 10.1007/s10620-012-2051-5. [DOI] [PubMed] [Google Scholar]
- 32.Uc X, Zhu BA, Wagner GR, Buettner DJ. Berg. Heme Oxygenase 1 is protective against non-steroidal anti-inflammatory drug-induced gastric ulcers. J Pediatr Gastroenterol Nutr. 2012;54:471–476. doi: 10.1097/MPG.0b013e3182334fdf. [DOI] [PubMed] [Google Scholar]
- 33.Bhatia V, Tandon RK. Stress and the gastrointestinal tract. J Gastroenterol Hepatol. 2005;20:332–339. doi: 10.1111/j.1440-1746.2004.03508.x. [DOI] [PubMed] [Google Scholar]
- 34.Marrone GC, Silen W. Pathogenesis, diagnosis and treatment of acute gastric mucosal lesions. Clin Gastroenterol. 1984;13:635–650. [PubMed] [Google Scholar]
- 35.Konturek PK, Brzozowski T, Konturek SJ, Dembiński A. Role of epidermal growth factor, prostaglandin and sulfhydryls in stress-induced gastric lesions. Gastroenterology. 1990;99:1607–1615. doi: 10.1016/0016-5085(90)90464-c. [DOI] [PubMed] [Google Scholar]
- 36.Yabana T, Yachi A. Stress-induced vascular damage and ulcer. Dig Dis Sci. 1988;33:751–761. doi: 10.1007/BF01540441. [DOI] [PubMed] [Google Scholar]
- 37.Kwiecien S, Pawlik MW, Brzozowski T, Pawlik WW, Konturek SJ. Reactive oxygen metabolite action in experimental, stress model of gastric mucosa damage. Gastroenterol Pol. 2010;17:234–243. [Google Scholar]
- 38.Blank MA, Gibson GW, Myers WR, Dierckman TA, Phipps RJ, Smith PN. Gastric damage in the rat with nitrogen-containing bisphosphonates depends on pH. Aliment Pharmacol Ther. 2000;14:1215–1223. doi: 10.1046/j.1365-2036.2000.00816.x. [DOI] [PubMed] [Google Scholar]
- 39.Brzozowski T, Konturek PC, Konturek SJ, et al. Expression of cyclooxygenase (COX)-1 and COX-2 in adaptive cytoprotection induced by mild stress. J Physiol Paris. 2000;94:83–91. doi: 10.1016/S0928-4257(00)00145-5. [DOI] [PubMed] [Google Scholar]
- 40.Ohashi Y, Aihara E, Takasuka H, Takahashi K, Takeuchi K. Antral ulcers induced by alendronate, a nitrogen-containing bisphosphonate, in rat stomachs–prophylactic effect of rebamipide. J Physiol Pharmacol. 2009;60:85–93. [PubMed] [Google Scholar]
- 41.Nicolau LA, Silva RO, Damasceno SR, et al. The hydrogen sulfide donor, Lawesson’s reagent, prevents alendronate-induced gastric damage in rats. Braz J Med Biol Res. 2013;46:708–714. doi: 10.1590/1414-431X20133030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Konturek SJ, Brzozowski T, Pytko-Polonczyk J, Drozdowicz D. Exogenous and endogenous cholecystokinin protects gastric mucosa against the damage caused by ethanol in rats. Eur J Pharmacol. 1995;273:57–62. doi: 10.1016/0014-2999(94)00674-V. [DOI] [PubMed] [Google Scholar]
- 43.Magierowski M, Jasnos K, Sliwowski Z, et al. Exogenous asymmetric dimethylarginine (ADMA) in pathogenesis of ischemia-reperfusion-induced gastric lesions: interaction with protective nitric oxide (NO) and calcitonin gene-related peptide (CGRP) Int J Mol Sci. 2014;15:4946–4964. doi: 10.3390/ijms15034946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Flannigan KL, Ferraz JG, Wang R, Wallace JL. Enhanced synthesis and diminished degradation of hydrogen sulfide in experimental colitis: a site-specific, pro-resolution mechanism. PLoS One. 2013;8:e71962. doi: 10.1371/journal.pone.0071962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 46.Laine L, Takeuchi K, Tarnawski A. Gastric mucosal defense and cytoprotection: bench to bedside. Gastroenterology. 2008;135:41–60. doi: 10.1053/j.gastro.2008.05.030. [DOI] [PubMed] [Google Scholar]
- 47.Wallace JL. Physiological and pathophysiological roles of hydrogen sulfide in the gastrointestinal tract. Antioxid Redox Signal. 2010;12:1125–1133. doi: 10.1089/ars.2009.2900. [DOI] [PubMed] [Google Scholar]
- 48.Magierowski M, Jasnos K, Kwiecien S, Brzozowski T. Role of hydrogen sulfide in the physiology of gastrointestinal tract and in the mechanism of gastroprotection. Postepy Hig Med Dosw (Online) 2013;67:150–156. doi: 10.5604/17322693.1038356. [DOI] [PubMed] [Google Scholar]
- 49.Lanza FL. Gastrointestinal adverse effects of bisphosphonates: etiology, incidence and prevention. Treat Endocrinol. 2002;1:37–43. doi: 10.2165/00024677-200201010-00004. [DOI] [PubMed] [Google Scholar]
- 50.Amagase K, Inaba A, Senta T, et al. Gastric ulcerogenic and healing impairment effects of risedronate, a nitrogen-containing bisphosphonate in rats. Comparison with alendronate and minodronate. J Physiol Pharmacol. 2011;62:609–618. [PubMed] [Google Scholar]
- 51.Costa NR, Silva RO, Nicolau LA, et al. Role of soluble guanylate cyclase activation in the gastroprotective effect of the HO-1/CO pathway against alendronate-induced gastric damage in rats. Eur J Pharmacol. 2013;700:51–59. doi: 10.1016/j.ejphar.2012.12.007. [DOI] [PubMed] [Google Scholar]
- 52.Carvalho NS, Silva MM, Silva RO, et al. Protective effects of simvastatin against alendronate-induced gastric mucosal injury in rats. Dig Dis Sci. 2016;61:400–409. doi: 10.1007/s10620-015-3890-7. [DOI] [PubMed] [Google Scholar]
- 53.Guth PH. Current concepts in gastric microcirculatory pathophysiology. Yale J Biol Med. 1992;65:677–688. [PMC free article] [PubMed] [Google Scholar]
- 54.Wallace JL, Dicay M, McKnight W, Bastaki S, Blank MA. N-bisphosphonates cause gastric epithelial injury independent of effects on the microcirculation. Aliment Pharmacol Ther. 1999;13:1675–1682. doi: 10.1046/j.1365-2036.1999.00658.x. [DOI] [PubMed] [Google Scholar]
- 55.Wang GL, Jiang BH, Rue EA, Samenza GL. Hypoxia inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA. 1995;92:5510–5514. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jung F, Palmer LA, Zhou N, Johns RA. Hypoxic regulation of inducible nitric oxide synthase via hypoxia inducible factor-1 in cardiac myocytes. Circ Res. 2000;86:319–325. doi: 10.1161/01.RES.86.3.319. [DOI] [PubMed] [Google Scholar]
- 57.Pugh CW, Ratcliffe PJ. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat Med. 2003;9:677–684. doi: 10.1038/nm0603-677. [DOI] [PubMed] [Google Scholar]
- 58.van Uden P, Kenneth NS, Rocha S. Regulation of hypoxia-inducible factor-1alpha by NF-kappaB. Biochem J. 2008;412:477–484. doi: 10.1042/BJ20080476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Silva RO, Lucetti LT, Wong DV, et al. Alendronate induces gastric damage by reducing nitric oxide synthase expression and NO/cGMP/K(ATP) signaling pathway. Nitric Oxide. 2014;40:22–30. doi: 10.1016/j.niox.2014.05.002. [DOI] [PubMed] [Google Scholar]
- 60.Lamarque D, Whittle BJ. Involvement of peroxynitrite in the lipid peroxidation induced by nitric oxide in rat gastric mucosa. Eur J Pharmacol. 1996;313:R5–R7. doi: 10.1016/0014-2999(96)00669-3. [DOI] [PubMed] [Google Scholar]
- 61.Souza MH, Lemos HP, Oliveira RB, Cunha FQ. Gastric damage and granulocyte infiltration induced by indomethacin in tumor necrosis factor receptor 1 (TNF-R1) or inducible nitric oxide synthase (iNOS) deficient mice. Gut. 2004;53:791–796. doi: 10.1136/gut.2002.012930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kwiecien S, Jasnos K, Magierowski M, et al. Lipid peroxidation, reactive oxygen species and antioxidative factors in the pathogenesis of gastric mucosal lesions and mechanism of protection against oxidative stress-induced gastric injury. J Physiol Pharmacol. 2014;65:613–622. [PubMed] [Google Scholar]
- 63.Magierowska K, Magierowski M, Surmiak M, et al. The protective role of carbon monoxide (CO) produced by heme oxygenases and derived from the co-releasing molecule CORM-2 in the pathogenesis of stress-induced gastric lesions: evidence for non-involvement of nitric oxide (NO). Int J Mol Sci 2016;17(4). doi:10.3390/ijms17040442. [DOI] [PMC free article] [PubMed]
- 64.Guillén MI, Megías J, Clérigues V, Gomar F, Alcaraz MJ. The CO-releasing molecule CORM-2 is a novel regulator of the inflammatory process in osteoarthritic chondrocytes. Rheumatology (Oxford) 2008;47:1323–1328. doi: 10.1093/rheumatology/ken264. [DOI] [PubMed] [Google Scholar]
- 65.Flannigan KL, Agbor TA, Motta JP, et al. Proresolution effects of hydrogen sulfide during colitis are mediated through hypoxia-inducible factor-1α. FASEB J. 2015;29:1591–1602. doi: 10.1096/fj.14-266015. [DOI] [PubMed] [Google Scholar]
- 66.Kai S, Tanaka T, Daijo H, et al. Hydrogen sulfide inhibits hypoxia- but not anoxia-induced hypoxia-inducible factor 1 activation in a von hippel-lindau- and mitochondria-dependent manner. Antioxid Redox Signal. 2012;16:203–216. doi: 10.1089/ars.2011.3882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wei Y, Chen P, de Bruyn M, Zhang W, Bremer E, Helfrich W. Carbon monoxide-releasing molecule-2 (CORM-2) attenuates acute hepatic ischemia reperfusion injury in rats. BMC Gastroenterol. 2010;10:42. doi: 10.1186/1471-230X-10-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen SW, Zhu J, Zuo S, et al. Protective effect of hydrogen sulfide on TNF-α and IFN-γ-induced injury of intestinal epithelial barrier function in Caco-2 monolayers. Inflamm Res. 2015;64:789–797. doi: 10.1007/s00011-015-0862-5. [DOI] [PubMed] [Google Scholar]