


Abstract
Abasic (AP) sites are major DNA lesions and are highly mutagenic. AP site-induced mutagenesis largely depends on translesion synthesis. We have examined the role of DNA polymerase η (Polη) in translesion synthesis of AP sites by replicating a plasmid containing a site-specific AP site in yeast cells. In wild-type cells, AP site bypass resulted in preferred C insertion (62%) over A insertion (21%), as well as −1 deletion (3%), and complex event (14%) containing multiple mutations. In cells lacking Polη (rad30), Rev1, Polζ (rev3), and both Polη and Polζ, translesion synthesis was reduced to 30%, 30%, 15% and 3% of the wild-type level, respectively. C insertion opposite the AP site was reduced in rad30 mutant cells and was abolished in cells lacking Rev1 or Polζ, but significant A insertion was still detected in these mutant cells. While purified yeast Polα effectively inserted an A opposite the AP site in vitro, purified yeast Polδ was much less effective in A insertion opposite the lesion due to its 3′→5′ proofreading exonuclease activity. Purified yeast Polη performed extension synthesis from the primer 3′ A opposite the lesion. These results show that Polη is involved in translesion synthesis of AP sites in yeast cells, and suggest that an important role of Polη is to catalyze extension following A insertion opposite the lesion. Consistent with these conclusions, rad30 mutant cells were sensitive to methyl methanesulfonate (MMS), and rev1 rad30 or rev3 rad30 double mutant cells were synergistically more sensitive to MMS than the respective single mutant strains.
INTRODUCTION
Apurinic/apyrimidinic (AP) sites, also referred to as abasic sites, are a major type of spontaneous DNA lesions. Some environmental agents, such as methyl methanesulfonate (MMS), can also induce AP sites in DNA. AP sites are non-coding. Therefore, copying an AP site by any DNA polymerase is error prone. Consequently, AP sites are highly mutagenic. In Escherichia coli, translesion synthesis of an AP site results in preferential insertion of an A opposite the lesion, leading to the ‘A rule’ hypothesis (1). Such a strong bias opposite an AP site does not appear to be the case in mammals. Using plasmid mutagenesis systems, insertions of A, C, T and G opposite AP sites have all been reported in cultured mammalian cells (2–7). In yeast cells, whereas Lawrence and colleagues (8) observed predominant C insertion opposite AP sites, Haracska et al. (9) reported predominant A insertion.
Translesion synthesis, also referred to as lesion bypass, is the cellular process that directly copies damaged sites of the template during DNA synthesis. It consists of nucleotide insertion opposite the lesion and extension synthesis from opposite the lesion. According to this definition, a DNA polymerase that performs the insertion step, the extension step, or both, qualifies as a translesion polymerase. Extensive in vitro and some in vivo studies have indicated that Polζ and the Y family polymerases are important translesion polymerases in eukaryotes [reviewed in (10–14)]. Polζ belongs to the same B family of DNA polymerases as the replicative Polα, Polδ, and Polε (15,16). In the yeast Saccharomyces cerevisiae, the Y family consists of Polη and Rev1 (16). Mammals contain two additional members of the Y family polymerases: Polκ and Polι (10,16).
Rev1 possesses a dCMP transferase and is efficient in inserting a C opposite an AP site in vitro, but it cannot catalyze extension synthesis from opposite the lesion (17,18). The combination of Rev1 and Polζ, however, results in bypass of the AP site in vitro (17). Genetic experiments have shown that both Rev1 and Polζ are required for AP site-induced mutagenesis in yeast cells (9,19,20). Polζ is believed to function in extension synthesis during AP site bypass (9,17,21). The role of Rev1 in AP site bypass, however, is controversial. Whereas Lawrence and colleagues (19) concluded that Rev1 acts catalytically by inserting C opposite AP site, Haracska et al. (9) concluded that Rev1 predominantly plays a non-catalytic structural role.
Polη was originally discovered as an important translesion polymerase in response to UV-induced TT dimers in an error-free manner (22,23). However, subsequent studies showed that yeast Polη is also responsive to template AP sites by catalysis of G and less frequently A insertions opposite the lesion in vitro (21). Translesion synthesis of AP sites was later extended to the human Polη in vitro, although it prefers A to G for insertion opposite the lesion (24,25). Furthermore, we observed that sequential actions of Polη-catalyzed nucleotide insertion and Polζ-catalyzed extension resulted in AP site bypass in vitro (21). Thus, we proposed that Polη may be involved in error-prone translesion synthesis of some other DNA lesions such as AP sites (21). Indeed, we found recently that Polη is involved in error-prone translesion synthesis of benzo[a]pyrene DNA adducts in yeast cells (26).
To better understand mechanisms of translesion synthesis across AP sites in eukaryotes, we have performed in vitro biochemical and in vivo genetic experiments to examine the role of Polη in AP site bypass in yeast cells. In this report, we (i) describe a site-specific translesion synthesis assay in yeast cells; (ii) demonstrate that yeast Polη is involved in translesion synthesis of AP sites in vivo; and (iii) present evidence suggesting that an important role of Polη in AP site bypass is to catalyze extension synthesis following A insertion opposite the lesion.
MATERIALS AND METHODS
Materials
T4 DNA ligase, the T4 gene 32 protein, T4 polynucleotide kinase and yeast Polη were obtained from Enzymax (Lexington, KY). MMS was purchased from Sigma (St Louis, MO). Yeast Polζ, and the catalytic subunits of yeast Polα and Polδ were purified as previously described (27–29). A 17mer DNA oligonucleotide containing a site-specific tetrahydrofuran (AP site analog) was synthesized by automated DNA phosphoramidite methods by Operon (Alameda, CA). Its sequence is 5′-CGACTXGAAGGATCCGC-3′, where X designates the AP site analog. Other damaged and undamaged DNA oligonucleotides as indicated were synthesized by Integrated DNA Technologies (Coralville, IA).
Yeast strains. The yeast strains used are the wild-type BY4741 (MATa his3 leu2 met15 ura3) and its isogenic BY4741Δrad30 (rad30 deletion mutant), BY4741Δrev1 (rev1 deletion mutant), BY4741Δrev3 (rev3 deletion mutant), BY4741Δrev1Δrad30 (rev1 rad30 double deletion mutant) and BY4741Δrev3Δrad30 (rev3 rad30 double deletion mutant). BY4741 was purchased from ATCC (Manassas, VA). BY4741Δrad30 (lacking Polη) was purchased from Research Genetics (Huntsville, AL). BY4741Δrev3 (lacking Polζ) and BY4741Δrev3Δrad30 were constructed as previously described (26). BY4741Δrev1 (lacking Rev1) was constructed by transforming BY4741 cells with a linearized rev1 deletion plasmid construct. The rev1 deletion clone was confirmed by a functional assay demonstrating reduced UV-resistance and loss of UV-induced mutagenesis. The rev1 deletion strain was further tested for complementation of UV resistance and UV-induced mutagenesis by a plasmid carrying the wild-type REV1 gene. BY4741Δrev1Δrad30 was similarly constructed by transforming BY4741Δrad30 cells with the linearized rev1 deletion plasmid construct and similarly confirmed for its rev1 phenotype as described above.
Construction of plasmid containing a site-specific AP site
The plasmid vector used is pELUf1 (Figure 1A). The strategy for constructing pELUf1 containing a site-specific AP site is shown schematically in Figure 1B. Briefly, E.coli DH5αF′IQ cells containing plasmid pELUf1 were infected with the helper phage M13KO7, and a single-stranded pELUf1 was prepared. The single-stranded pELUf1 was annealed to a 20mer DNA oligonucleotide, 5′-GTGCCCTCCATGGAAAAATC-3′, at its unique NcoI restriction site within the vector URA3 gene, and digested with the NcoI restriction endonuclease. Then, the linearized single-stranded pELUf1 was annealed with a 46mer DNA scaffold, 5′- CTGUGCCCUCCAUGGCGGAUTCUTCUAGUCGGAAAAAUCAGTCAAG-3′, and the 5′-phosphorylated 17mer oligonucleotide containing a site-specific AP site. While the mid-region of the scaffold is complementary to the damaged oligonucleotide, its ends are complementary to the single-stranded pELUf1 ends. Ligation of the damaged oligonucleotide into the pELUf1 vector was performed with T4 DNA ligase at 16°C for 20 h, and DNA was precipitated in ethanol. Finally, the complementary strand of pELUf1 was synthesized with T4 DNA polymerase in the presence of T4 gene 32 protein and 0.5 mM each of dATP, dCTP, dGTP and dUTP, using the scaffold as the primer. The resulting construct, pELUf1–AP, was a double-stranded plasmid containing a site-specific AP site, in which the undamaged strand contained U in place of T. Furthermore, insertion of the damaged oligonucleotide inactivated the vector URA3 gene. Formation of double-stranded plasmid pELUf1–AP was confirmed by electrophoresis on a 1% agarose gel. Construction of pELUf1–AP was performed by Enzymax.
Figure 1.
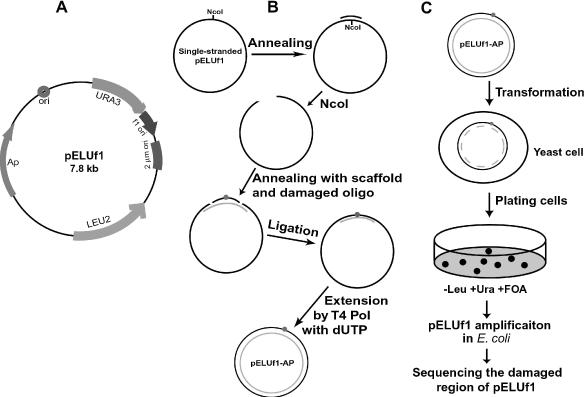
Schematic presentation of an in vivo translesion synthesis assay in yeast. (A) The plasmid used for construction of site-specifically damaged DNA vector. (B) Construction of pELUf1–AP containing a site-specific AP site. The AP site (indicated by a red dot) was carried on a 17mer oligonucleotide. Insertion of the damaged oligonucleotide into the unique NcoI site of the vector inactivated the URA3 gene, thus, allowing yeast cells containing replicated pELUf1–AP to grow on plates containing 5-FOA. The undamaged strand (shown in orange) of pELUf1–AP contained U in place of T. (C) In vivo translesion synthesis assay using the site-specifically damaged pELUf1–AP plasmid. Transformation efficiency was measured in the same experiment by using undamaged pELUf1 and plates lacking both leucine and 5-FOA.
In vivo translesion synthesis assays in yeast
Undamaged pELUf1 or site-specifically damaged pELUf1–AP plasmid (2 μg) was transformed into yeast cells of various strains by the lithium acetate method essentially as described in (30). Immediately after transformation, yeast cells were collected by centrifugation (20 s at 5000 r.p.m.) in a microcentrifuge. Cells transformed with the undamaged pELUf1 were resuspended in 1 ml of sterile water. Aliquots of 0.5–1 μl were diluted and plated onto YNB minimal agar plates (0.17% yeast nitrogen base, 0.49% ammonium sulfate, 2% glucose and 2% agar) lacking leucine to score for transformation efficiency (colonies containing replicated pELUf1). Cells transformed with the site-specifically damaged pELUf1–AP were resuspended in 400 μl of sterile water and were directly plated onto two YNB minimal agar plates lacking leucine but supplemented with 5 mM 5-fluoroorotic acid (5-FOA), 150 μM methionine, and 380 μM uracil to score for colonies containing replicated pELUf1–AP. Cells transformed by the undesired background plasmid pELUf1 without the damaged oligonucleotide insert remained the URA3 wild type and thus cannot grow on plates containing 5-FOA. After incubation at 30°C for 3–4 days, yeast colonies were counted. Translesion synthesis was calculated as transformants per microgram of the damaged plasmid per 106 transformable cells with the undamaged plasmid (i.e. transformants per microgram of the damaged plasmid × 106/transformation efficiency expressed as transformants per microgram of the undamaged plasmid). Relative translesion synthesis was obtained by comparing translesion synthesis in various mutant strains to that in the wild-type cells.
Replicated pELUf1–AP plasmid clones were individually recovered from yeast colonies on the 5-FOA plates by a zymolyase method essentially as described in (31) and amplified in E.coli DH5α cells. Each pELUf1–AP plasmid clone was analyzed by digestion with the BamHI restriction endonuclease. A BamHI restriction site was designed into the damaged 17mer oligonucleotide during construction of pELUf1–AP. The BamHI restriction analysis further eliminated undesired background transformants by the undamaged pELUf1. These background transformants escaped selection by the 5-FOA plates because they contained mutations somewhere in the vector URA3 gene. Plasmid clones that did not contain this added BamHI restriction site were excluded from further analysis and calculation. Finally, the precise specificity of translesion synthesis opposite the site-specific AP site was determined by DNA sequencing.
DNA polymerase assays
A standard DNA polymerase reaction mixture (10 μl) contained 25 mM KH2PO4 (pH 7.0), 5 mM MgCl2, 5 mM dithiothreitol, 100 μg/ml bovine serum albumin, 10% glycerol, 50 μM of dNTPs (dATP, dCTP, dTTP and dGTP individually or together as indicated), 50 fmol of an indicated DNA substrate containing a 32P-labeled primer and a purified DNA polymerase as indicated. After incubation at 30oC for 10 min, reactions were terminated with 7 μl of a stop solution (20 mM EDTA, 95% formamide, 0.05% bromophenol blue and 0.05% xylene cyanol). The reaction products were resolved on a 20% polyacrylamide gel containing 8 M urea and visualized by autoradiography. DNA synthesis products were quantitated by scanning densitometry using the SigmaGel software (Sigma) for analysis.
MMS sensitivity
Yeast cells grown at 30°C to stationary phase in minimum media were harvested by centrifugation, washed with sterile water and resuspended in 100 mM potassium phosphate (pH 7.0) to 4 OD600 cells per milliliter. Cells were then treated with various doses of MMS as indicated at 30°C for 30 min with shaking, followed by centrifugation and washing with sterile water. After dilution, cells were plated in minimum medium plates. Surviving colonies were counted after incubation at 30°C for 2–4 days. Cell survival of each strain was expressed relative to that of untreated cells of the corresponding strain.
RESULTS
An in vivo translesion synthesis assay in yeast
To investigate mechanisms of AP site bypass in cells, we established a plasmid-based in vivo translesion synthesis assay in the yeast S.cerevisiae model system. The vector pELUf1 contains the yeast LEU2 gene for plasmid selection, the yeast URA3 gene for accepting a site-specifically damaged oligonucleotide and the f1 replication origin for production of single-stranded phagemid (Figure 1A). A 17mer oligonucleotide containing a site-specific AP site was ligated into the unique NcoI site of the single-stranded pELUf1 vector, with the help of a scaffold oligonucleotide. The resulting pELUf1–AP was then converted into double-stranded plasmid with T4 DNA polymerase, using the scaffold as the primer and dUTP instead of dTTP. The resulting undamaged second strand of pELUf1–AP contained U in place of T (Figure 1B).
The site-specifically damaged pELUf1–AP was transformed into yeast cells (Figure 1C). Using double-stranded DNA ensured efficient transformation. High levels of U in DNA will lead to extensive DNA fragmentation initiated by uracil-DNA glycosylase (UDG) (32–34). Thus, the U-containing DNA strand was quickly destroyed by the combined actions of UDG and AP endonucleases as the plasmid entered into cells, converting the double-stranded plasmid back into single-stranded DNA. Consequently, plasmid propagation in cells could only be achieved by replicating the damaged DNA strand involving translesion synthesis. Transformed cells were plated on minimal media plates containing uracil and 5-FOA but lacking leucine. While plates without leucine allowed selection for the replicated plasmid, 5-FOA was used to select for ura3 mutant cells. Since insertion of the damaged oligonucleotide had inactivated the URA3 gene, cells containing plasmid pELUf1–AP from translesion synthesis were able to grow on the 5-FOA plate. In contrast, cells transformed by the undesired intact pELUf1 without the oligonucleotide insert remained the URA3 wild type and were therefore unable to grow on the 5-FOA plate. Transformation efficiency was determined by using undamaged and double-stranded pELUf1 plasmid in the same experiment. After correcting for differences in transformation efficiency, translesion synthesis efficiency in various cells relative to that in the wild-type cells was calculated. Replicated plasmid clones were individually isolated from yeast colonies and subsequently amplified in E.coli. To further ensure that the plasmid clones were indeed derived from translesion synthesis in yeast cells, a BamHI site was designed into the damaged oligonucleotide, whereas its complementary scaffold DNA contained a mismatch eliminating this restriction site on the undamaged strand. After amplification in E.coli, each plasmid clone was digested with BamHI. Clones without this BamHI site were excluded from further analysis. Each clone was sequenced to identify the nucleotide inserted opposite the AP site (Figure 1C).
Polη is involved in translesion synthesis of AP sites in yeast cells
To determine whether Polη is involved in AP site bypass in cells, we performed in vivo translesion synthesis assays in yeast strains proficient or deficient in Polη. Since Polζ and Rev1 are known to be required for translesion synthesis of AP sites in cells (9,19,20), we also performed in vivo translesion synthesis assays in mutant cells lacking Polζ (rev3 deletion mutant) or Rev1 for comparison. As expected, translesion synthesis of the site-specific AP site was reduced to 30 and 15% of the wild-type level in rev1 and rev3 mutant cells, respectively (Figure 2). In rad30 mutant cells lacking Polη, translesion synthesis was reduced to 30% of the wild-type level (Figure 2). These results show that Polη is involved in translesion synthesis of AP sites in yeast.
Figure 2.
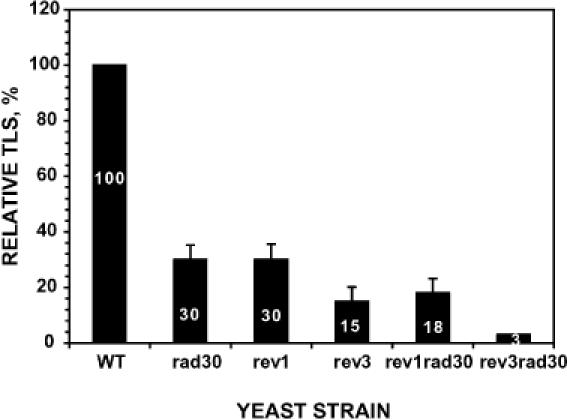
Relative frequencies of AP site translesion synthesis (TLS) in various yeast strains. Using plasmid pELUf1–AP containing a site-specific AP site, in vivo translesion synthesis assays were performed as described in Materials and methods. Relative TLS was obtained by comparing translesion synthesis in various mutant strains to that in the wild-type cells. Slightly different transformation efficiencies as determined with the undamaged pELUf1 have been corrected in the calculation. Standard deviations are shown as error bars. WT, wild type; rad30, lacking Polη; rev1, lacking Rev1; rev3, lacking Polζ; rev1 rad30, lacking both Rev1 and Polη; rev3rad30, lacking both Polζ and Polη.
To gain insights into the genetic relationship between Polη and Rev1 or Polζ in AP site bypass, we performed translesion synthesis assays in yeast cells lacking both Polη and Rev1 or both Polη and Polζ. If Polη functions in the same bypass pathway with Rev1 and Polζ, translesion synthesis in the double mutant would not be further reduced as compared with the single mutants. If the Polη function in AP site bypass involves another mechanism different from that of Rev1 and Polζ, translesion synthesis would be more deficient in the double mutant than in the respective single mutants. As shown in Figure 2, translesion synthesis in the rev1 rad30 double mutant cells was reduced further, as compared to the respective single mutant strains. In the rev3 rad30 double mutant cells, translesion synthesis was severely deficient, retaining only 3% of that in wild-type cells (Figure 2). These results suggest that the Polη function in AP site bypass involves another mechanism different from the bypass mechanism mediated by the Polζ mutagenesis pathway.
Specificity of in vivo translesion synthesis opposite AP sites
To further understand the role of Polη in AP site bypass, we recovered the replicated plasmids from yeast clones and individually amplified them in E.coli for DNA sequencing. Three types of translesion products were observed from various yeast strains: (i) simple translesion synthesis in that 1 nt was inserted, or a −1 deletion was produced, opposite the AP site; (ii) following translesion synthesis opposite the AP site, another mutation was generated downstream of the lesion; and (iii) complex translesion synthesis in that multiple mutations were generated on both sides of the AP site (Figure 3). Translesion synthesis with downstream mutations was rare. Only 2 such clones out of 60 from cells lacking Polη, 1 out of 29 clones from wild-type cells and 1 out of 27 clones from the rev1 mutant cells were recovered.
Figure 3.

Types of AP site bypass products recovered from various yeast strains. The damaged 17mer oligonucleotide contained in plasmid pELUf1–AP is shown as the template for translesion synthesis, with the AP site designated by an X. Simple translesion synthesis with C, A or G insertion, or −1 deletion opposite the AP site are indicated above the damaged template. Complex TLS and TLS with downstream mutations (rare bypass events) are indicated below the damaged template.
T insertion opposite the AP site was not observed among all yeast strains examined. In wild-type cells (Table 1), the majority of translesion synthesis (62%) resulted from C insertion opposite the AP site. Less frequently (21%) A was inserted opposite the lesion. A small fraction (14%) of the bypassed products were derived from complex translesion synthesis, while −1 deletion occurred only as a very minor event (3%). G was not recovered opposite the AP site. In rad30, rev1, rev3, rev1 rad30 and rev3 rad30 mutant cells, the spectra of translesion synthesis were significantly altered (Table 1). In cells lacking either Rev1 or Polζ, C insertion opposite the AP site was not detected (Table 1). In rad30 mutant cells lacking Polη, A insertion was favored over C insertion opposite the AP site (Table 1). Furthermore, G insertion opposite the lesion became detectable in rev3 (lacking Polζ) or rad30 mutant cells (Table 1).
Table 1. Specificity of translesion synthesis opposite the AP site in various yeast strains.
| Straina | Clones sequencedb | TLSc | Complex TLSe | |||
|---|---|---|---|---|---|---|
| A | C | G | Δd | |||
| WT | 29 | 6 (21%) | 18 (62%) | — | 1 (3%) | 4 (14%) |
| rad30 | 60 | 25 (42%) | 18 (30%) | 1 (2%) | 3 (5%) | 13 (22%) |
| rev1 | 27 | 13 (48%) | — | — | 5 (19%) | 9 (33%) |
| rev3 | 34 | 22 (65%) | — | 1 (3%) | 4 (12%) | 7 (21%) |
| rev1 rad30 | 22 | 4 (18%) | — | — | 3 (14%) | 15 (68%) |
| rev3 rad30 | 2 | 2 (100%) | — | — | — | — |
aWT, wild type; rad30, lacking Polη; rev1, lacking Rev1; rev3, lacking Polζ; rev1 rad30, lacking both Rev1 and Polη; rev3 rad30, lacking both Polζ and Polη.
bNumber of independent clones sequenced following in vivo translesion synthesis assays using pELUf1–AP plasmid containing a site-specific AP site. Only two clones were recovered from the rev3 rad30 double mutant cells.
cNumber of various translesion synthesis products are shown. The percentage of a particular type of TLS among all of the bypassed products in a strain is shown in the parenthesis.
dThe −1 deletion.
eOnly one type of complex TLS was observed, which contained mutations on both sides of the AP site. The precise sequence of the complex TLS product is shown in Figure 3.
When the altered bypass in the mutant strains was directly compared to AP site bypass in wild-type cells, expressed as translesion synthesis relative to that in wild-type cells, the effects of Polη, Rev1 and Polζ became more clear (Table 2). For AP site bypass with C incorporation, while Rev1 and Polζ were indispensable, Polη had a strong stimulatory effect (Table 2). In contrast, AP site bypass with A incorporation remained as the most frequent mechanism in cells lacking Polη, Rev1 or Polζ, although such bypass frequency was reduced by approximately 2-fold in these mutant cells (Table 2). This type of translesion synthesis became largely abolished only when both the Polη and Rev1 or both the Polη and Polζ activities were eliminated (Table 2). AP site bypass with −1 deletion was not significantly affected in cells lacking Polη, Rev1 or Polζ, but was abolished in cells lacking both Polη and Polζ (Table 2). The complex translesion product was significantly reduced in cells lacking Polζ and abolished in cells lacking both Polζ and Polη (Table 2). These results suggest that Polη is generally involved in AP site bypass in yeast cells but it acts in a mechanism(s) different from the Polζ mutagenesis pathway that involves Rev1 and Polζ.
Table 2. Changes in translesion synthesis specificity in mutant cells relative to that in wild-type cells.
| Straina | TLSb | Complex TLSd | Total | |||
|---|---|---|---|---|---|---|
| A | C | G | Δc | |||
| WT | 0.21 | 0.62 | — | 0.03 | 0.14 | 1 |
| rad30 | 0.13 | 0.09 | 0.006 | 0.02 | 0.07 | 0.30 |
| rev1 | 0.14 | — | — | 0.06 | 0.10 | 0.30 |
| rev3 | 0.10 | — | 0.005 | 0.02 | 0.03 | 0.15 |
| rev1 rad30 | 0.03 | — | — | 0.03 | 0.12 | 0.18 |
| rev3 rad30 | 0.03 | — | — | — | — | 0.03 |
aWT, wild type; rad30, lacking Polη; rev1, lacking Rev1; rev3, lacking Polζ; rev1 rad30, lacking both Rev1 and Polη; rev3 rad30, lacking both Polζ and Polη.
bTranslesion synthesis in various mutant strains is expressed relative to that in the wild-type cells. Calculation was based on Figure 2 and Table 1.
cThe −1 deletion.
dOnly one type of complex TLS was observed, which contained mutations on both sides of the AP site. The precise sequence of the complex TLS product is shown in Figure 3.
In vitro activities of Polη in response to a template AP site
Our genetic assay is an endpoint measurement that includes the two distinct steps of in vivo translesion synthesis: nucleotide insertion and extension. Hence, the genetic assay cannot assign the observed effect to either the insertion or extension step, or both. To better define the roles of Polζ and Polη in AP site bypass, we examined in vitro activities of both polymerases in response to an AP site. As shown in Figure 4A, the AP site strongly blocked yeast Polζ in vitro (Figure 4A, lane 1). Nevertheless, weak insertion activity was detected with purified Polζ, which inserted a G opposite the AP site (Figure 4A, lane 5). In contrast, purified yeast Polη effectively inserted a G (Figure 4B, lane 5) and less frequently an A (Figure 4B, lane 2) opposite the AP site in the same sequence context as that used for the in vivo translesion synthesis assays (Figure 4B). Subsequent extension synthesis, however, was inefficient (Figure 4B, lane 1). The Polη results are similar to our earlier observation with another AP site in a different sequence context where the template base immediately 5′ to the lesion was an A (21). Since G insertion opposite the AP site was not detected during in vivo translesion synthesis in wild-type cells (Table 1), we conclude that neither Polη nor Polζ is significantly involved in catalyzing nucleotide insertion opposite the AP site in yeast cells. Apparently, the intrinsic G insertion activity of Polη was not employed to a significant extent for in vivo bypass of AP sites even in the absence of Rev1 or Polζ (Tables 1 and 2).
Figure 4.
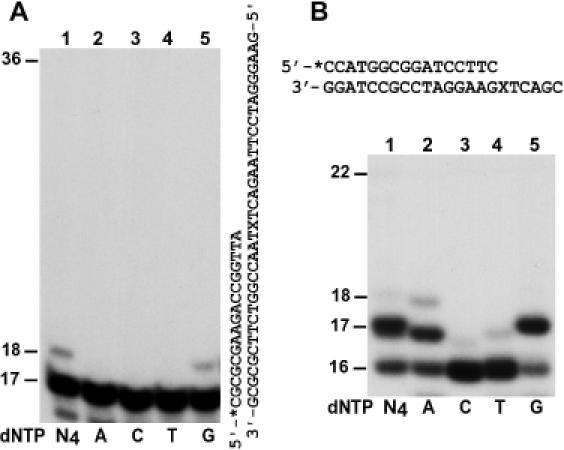
Nucleotide insertion opposite a template AP site by purified yeast Polη and Polζ. (A) A 32P-labeled 17mer primer was annealed to the damaged template with the primer 3′ end terminating right before the AP site as shown on the right. DNA synthesis assays were performed in the presence of a single dATP (A), dCTP (C), dTTP (T) or dGTP (G), or all four dNTPs (N4), using 14 ng (69 fmol) of purified yeast Polζ. Products of DNA synthesis were separated by 20% denaturing polyacrylamide gel and visualized by autoradiography of the gel. DNA size markers in nucleotides are indicated on the left. (B) A 32P-labeled 16mer primer was annealed to the damaged template with the primer 3′ end terminating right before the AP site as shown on the top. DNA synthesis assays were performed with 10 ng (140 fmol) of purified yeast Polη. X, the AP site.
To examine a possible role of Polη in extension synthesis, we separately annealed four 32P-labeled 17mer primers to the damaged template, terminating with an A, C, T or G, respectively, opposite the lesion (Figure 5A). DNA synthesis assays were then performed with purified yeast Polη. For comparison, similar experiments were also performed with an undamaged template G opposite the primer 3′ end (Figure 5A). As expected, Polη was capable of extension synthesis from matched and mismatched primer 3′ end with varying efficiencies (Figure 5A, lanes 1 to 4). The full-length extension products (22mer DNA band) were generated from the T–G (primer–template) and G–G mismatches (Figure 5A, lanes 3 and 4). However, extension of the A–G mismatch yielded predominantly 1 nt shorter products (21mer DNA band) (Figure 5A, lane 1), indicating that extension was mainly mediated by a −1 deletion mechanism through Polη-catalyzed realignment of the primer 3′ A with the next template T prior to extension synthesis. In response to a template AP site, extension synthesis was observed with Polη from the primer 3′ A opposite the lesion, generating both full-length and −1 deletion products in similar quantities (Figure 5A, lane 5). Extension was ineffective from the primer 3′ G opposite the AP site (Figure 5A, lane 8), and was not detected from the primer 3′ C or T (Figure 5A, lanes 6 and 7). To determine whether extension from A opposite the AP site requires a template T immediately 5′ to the lesion, we performed the extension assay again with another DNA substrate containing a template G immediately 5′ to the AP site. As shown in Figure 5B (lanes 3 to 7), extension by Polη was also observed from A opposite the AP site, generating predominantly full-length products. Furthermore, the extension activity of Polη was comparable to that of purified yeast Polζ (Figure 5B, lane 2). Since Polζ is inefficient in copying the last template base even from undamaged templates (27,29), the 35mer extension product of Polζ (Figure 5B, lane 2) most likely resulted from a normal extension mechanism rather than a −1 deletion mechanism. These results show that Polη possesses a significant biochemical activity of extension synthesis from a primer 3′ A opposite an AP site.
Figure 5.
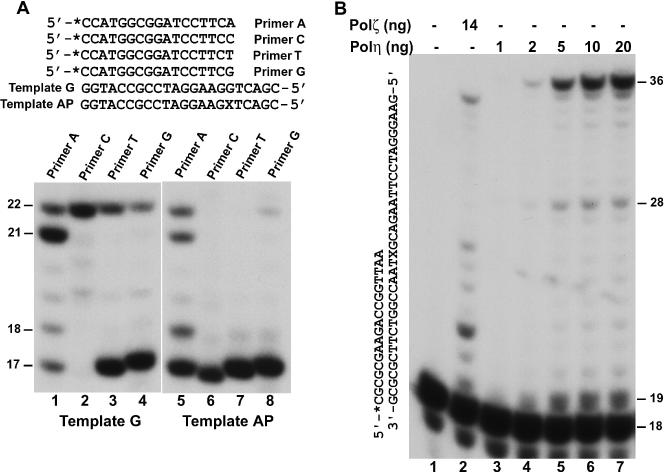
Extension synthesis from opposite the AP site by yeast Polη. (A) Four 32P-labeled 17mer primers were separately annealed to the damaged template (Template AP) or an undamaged template (Template G) with the primer 3′ A, C, T or G, respectively, terminating opposite the AP site or a template G as shown on the top. DNA synthesis assays were performed with 2.5 ng (35 fmol) (lanes 1 to 4) or 10 ng (140 fmol) (lanes 5 to 8) of purified yeast Polη. (B) A 32P-labeled 18mer primers were annealed to the damaged template with the primer 3′ A terminating opposite the AP site as shown on the right. DNA synthesis assays were performed with 14 ng (69 fmol) of purified yeast Polζ (lane 2) or increasing concentrations of purified yeast Polη (lanes 3 to 7). Products of DNA synthesis were separated by 20% denaturing polyacrylamide gel and visualized by autoradiography of the gel. DNA size markers in nucleotides are indicated on the sides. X, the AP site.
Response of the Polδ proofreading exonuclease activity to the primer 3′ A opposite an AP site
Polζ and Rev1 are unable to insert A opposite an AP site (Figure 4) (17,18,35), and cells lacking Polη could still significantly insert A opposite the lesion (Tables 1 and 2). Thus, it is possible that a replicative polymerase such as Polα or Polδ may be important for A insertion opposite AP sites in yeast cells. To test this possibility, we annealed a 16mer primer to the damaged template, terminating right before the AP site (Figure 6). DNA synthesis assays were then performed using the purified catalytic subunits of yeast Polα and Polδ. As shown in Figure 6 (lanes 2 to 6), Polα efficiently inserted an A opposite the AP site, but was unable to extend it. In contrast, Polδ was very inefficient in A insertion opposite the AP site (Figure 6, lanes 8 to 12), and extension from opposite the lesion was not detected.
Figure 6.
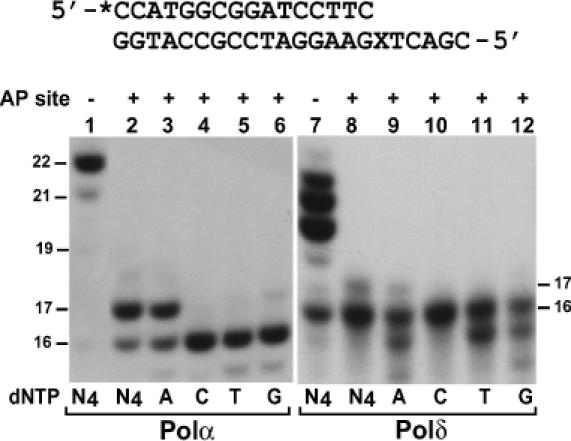
Response of yeast Polα and Polδ to a template AP site. A 32P-labeled 16mer primer was annealed to the damaged template with the primer 3′ end terminating right before the AP site as shown on the top. DNA synthesis assays were performed in the presence of a single dATP (A), dCTP (C), dTTP (T) or dGTP (G) or all four dNTPs (N4) with 12 ng (72 fmol) of the purified catalytic subunit of yeast Polα (lanes 2 to 6) or 15 ng (120 fmol) of the purified catalytic subunit of yeast Polδ (lanes 8 to 12) using the damaged template (AP site, +) as indicated. Control experiments were also performed with the undamaged template containing a G in place of the AP site (AP site, −) (lanes 1 and 7). Products of DNA synthesis were separated by 20% denaturing polyacrylamide gel and visualized by autoradiography of the gel. DNA size markers in nucleotides are indicated on the sides. X, the AP site.
Nucleotide insertion by Polδ opposite the AP site was further reduced in the presence of dATP alone (Figure 6, compare lanes 8 and 9). Moreover, nuclease activities were apparent when the translesion synthesis reactions were performed using dATP, dTTP or dGTP alone (Figure 6, lanes 9, 11 and 12). Therefore, it is possible that A insertion opposite the AP site may be greatly suppressed by the 3′→5′ proofreading exonuclease activity of Polδ. To test this possibility, we determined whether A opposite the AP site is subject to removal by the proofreading exonuclease activity of Polδ. We labeled a 17mer primer with 32P at its 5′ end and annealed it to the damaged template with the primer 3′A terminating opposite the AP site (Figure 7). Purified yeast Polδ was then incubated with this DNA substrate in the absence of deoxyribonucleoside triphosphates. DNA substrates containing a C–G (primer–template) base pair or an A–G mismatch at the primer 3′ end were used as controls. As shown in Figure 7, the 3′→5′ proofreading exonuclease of Polδ was active in removing the A–G mismatch (60% removal), but inactive against the C–G base pair (compare lanes 7 and 8) at the primer 3′ end. The primer 3′ A opposite the AP site was also actively removed by the 3′→5′ proofreading exonuclease of Polδ (68% removal) to a similar extent as the A–G mismatch (Figure 7, compare lanes 2 and 6). The primer 3′ C opposite the AP site, however, was more resistant to the 3′→5′ proofreading exonuclease of Polδ (38% removal) (Figure 7, lane 4). These results show that, while Polα is capable of efficiently inserting an A opposite the AP site, Polδ is much less effective in A insertion opposite the lesion due to its 3′→5′ proofreading exonuclease activity.
Figure 7.
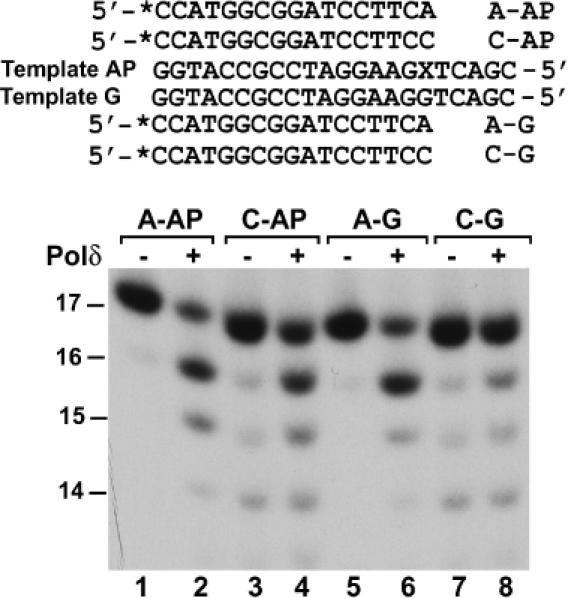
Proofreading exonuclease activity of Polδ against an A opposite the AP site. Two 32P-labeled 17mer primers were separately annealed to the damaged template (Template AP) with the primer 3′ A or C, respectively, terminating opposite the AP site as shown on the top. As controls, the two primers were also separately annealed to an undamaged template (Template G), forming an A–G mismatch or a C–G base pair, respectively, at the primer 3′ end. The four types of DNA substrates as indicated were incubated with 15 ng (120 fmol) of the purified catalytic subunit of yeast Polδ (lanes 2, 4, 6 and 8) at 30°C for 30 min under the DNA polymerase assay conditions in the absence of dNTPs. Lanes 1, 3, 5 and 7, reactions without Polδ. Reaction products were separated by 20% denaturing polyacrylamide gel and visualized by autoradiography of the gel. DNA size markers in nucleotides are indicated on the left. X, the AP site.
Cellular resistance to MMS conferred by Polη, Polζ and Rev1
Genetic studies have demonstrated that defects in translesion synthesis result in cellular sensitivity to DNA-damaging agents. For example, Polη, Polζ and Rev1 play major roles in translesion synthesis in response to UV radiation (22,23,36,37). Consequently, their corresponding mutant cells are hypersensitive to UV radiation (36–40). Based on our results that Polη plays a significant role in the bypass of AP sites in yeast cells, we predicted that rad30 mutant cells lacking Polη would be sensitive to AP site-inducing agents. Even though there is not a known agent that specifically induces AP site in DNA, MMS is probably the most effective agent for AP site induction. As is the case of UV radiation, MMS induces multiple types of DNA lesions. Nevertheless, AP sites are induced by MMS as a major type of lesion, since it reacts with DNA forming mainly unstable N3-methyladenine and N7-methyl guanine adducts, which readily depurinate to AP sites (41,42). To test our prediction, we determined cellular sensitivity to MMS treatment in the presence or absence of Polη. For comparison, rev1 and rev3 mutant cells were also included in these experiments. As expected, Rev1 and rev3 mutant cells were sensitive to MMS treatment as compared to the wild-type cells (Figure 8). Furthermore, the survival curve of rev1 and rev3 mutant cells were essentially identical (Figure 8), consistent with the notion that Rev1 and Polζ function in the same Polζ mutagenesis pathway for AP site bypass. Similarly, rad30 mutant cells were also sensitive to MMS treatment (Figure 8). In the absence of both Rev1 and Polη, the rev1 rad30 double mutant cells became much more sensitive to MMS than either of the single mutant strains (Figure 8). In the absence of both Polζ and Polη, the rev3 rad30 double mutant cells were much more sensitive to MMS than either of the single mutant strains, and exhibited more sensitivity than the rev1 rad30 double mutant (Figure 8). These results show that Polη, like Polζ and Rev1, confers cellular resistance to MMS and that the function of Polη in response to MMS does not completely overlap with the Polζ mutagenesis pathway. These results further support our conclusion that Polη plays a significant role in AP site bypass in yeast cells.
Figure 8.
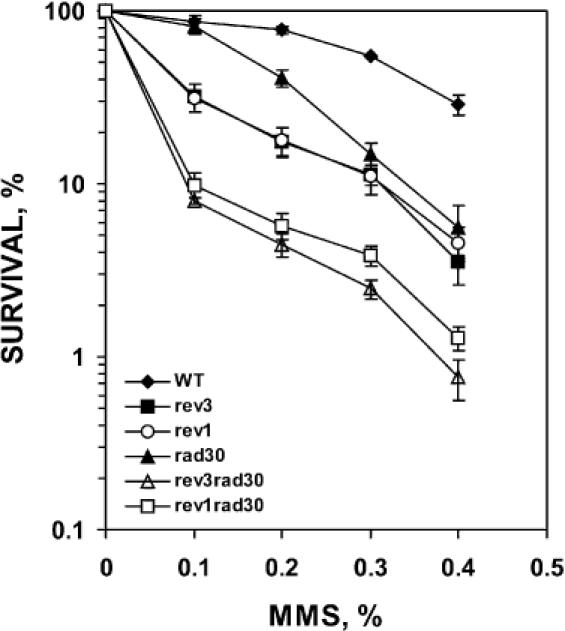
MMS sensitivity of various yeast strains. Yeast cells of various strains were treated with increasing concentrations of MMS as indicated at 30°C for 30 min and the cell survival determined as described in Materials and methods. Survival rates, with the standard deviations shown, are expressed relative to those of untreated cells. Results are the average of three experiments. WT, wild type (closed diamond); rev3, lacking Polζ (closed square); rev1, lacking Rev1 (open circle); rad30, lacking Polη (closed triangle); rev3 rad30, lacking both Polζ and Polη (open triangle); rev1 rad30, lacking both Rev1 and Polη (open square).
DISCUSSION
In this study, we have examined the role of Polη in translesion synthesis of AP sites in yeast cells. In vivo translesion synthesis was directly examined through replication of a plasmid containing a site-specific AP site. A similar assay in yeast cells was originally developed by Lawrence and colleagues (43) using single-stranded plasmid DNA containing a site-specific lesion. Owing to the extremely low transformation efficiency of single-stranded plasmid in yeast cells, the in vivo assay was later modified by using a double-stranded plasmid containing a site-specific lesion located in a small gap (8). This in vivo translesion synthesis assay essentially measures a gap filling reaction in yeast. Nevertheless, the gap filling reaction did reflect translesion synthesis as indicated by a genetic requirement for Polζ and Rev1 (19). In our assay system, a significant modification was made in that the site-specifically damaged and single-stranded plasmid was converted into the double-stranded form prior to transformation, replacing T with U during in vitro synthesis of the complementary strand. Upon entering into cells, the complementary strand was degraded as a result of replacement of T by U, thus, converting the plasmid DNA back into single-stranded form containing a site-specific lesion. An advantage of our system is that replication of the damaged plasmid specifically reflects translesion synthesis without the interference by DNA repair, a template switching mechanism, recombination or selective replication of the undamaged complementary strand. Our experiments were designed such that if the complementary strand were replicated, a T would have been inserted at the position corresponding to the opposite of the original AP site. This was not observed, thus, supporting our expectation that the complementary strand cannot be replicated due to its degradation inside cells. Like earlier systems (19,44), our in vivo translesion synthesis assays also reflected a genetic requirement for Polζ and Rev1 that are known to be required for translesion synthesis of AP sites in cells.
Using our in vivo translesion synthesis assay, we found that Polη makes a significant contribution to AP site bypass in yeast cells. Without Polη, translesion synthesis of AP sites is significantly impaired. Furthermore, the specificity of nucleotide insertion opposite the AP site is altered in rad30 mutant cells lacking Polη. Yeast Polη is capable of inserting a G and less frequently an A opposite an AP site in vitro, regardless of whether a template T or a template A is located immediately 5′ to the lesion (21) (Figure 4B). Thus, if Polη participates in AP site bypass by catalyzing nucleotide insertion opposite the lesion, then, significant G insertion opposite the AP site should be recovered from the replicated plasmids in wild-type cells. This was not the case. Only C and A insertions were detected among the bypassed products in wild-type cells, accounting for 62 and 21%, respectively, of the total bypassed products. Even in the rev1 or rev3 mutant cells that lack the Polζ mutagenesis pathway, G was still not significantly inserted opposite the AP site. Similarly, G insertion opposite the AP site was not observed in a study by Nelson et al. (19). Therefore, although Polη possesses an intrinsic biochemical activity of G insertion opposite AP sites, this activity is not significantly recruited for translesion synthesis of AP sites in vivo. This underscores the importance of performing in vivo genetic experiments to validate the in vitro biochemical results obtained from the various translesion polymerases. Nevertheless, it is possible that Polη may play a minor role in catalyzing G and A insertions opposite AP sites, but G was less effectively extended than A by Polζ (9). In such a case, Polη would make a small contribution to the detected A insertion during AP site bypass in yeast cells.
Then, what is the major role of Polη in translesion synthesis of AP sites in yeast cells? Our genetic and biochemical results together suggest that an important role of Polη in AP site bypass is to catalyze extension synthesis from the primer 3′ A opposite the lesion. This activity was detected by in vitro biochemistry, and was supported by in vivo results that A insertion opposite the AP site was reduced approximately 2-fold in rad30 mutant cells as compared to the wild-type cells (Table 2). We found that extension from opposite the AP site by Polη was most efficient from the primer 3′ A, much less efficient from the primer 3′ G, and not detected from the primer 3′ C or T (Figure 5A). These biochemical properties are also observed with the human Polη (45). Additionally, our genetic experiments suggest that Polη facilitates AP site bypass with C insertion in yeast cells (Table 2). Since Polη is unable to perform effective C insertion or extension synthesis from C opposite the AP site, this polymerase may somehow facilitate Rev1-catalyzed C insertion opposite the lesion. Most recently, it was reported that mouse Polη physically interacts with mouse Rev1 at its C-terminal region (46). Whether there exists a similar interaction between the yeast proteins and whether such an interaction is involved in the putative stimulatory action of Polη on the Rev1 dCMP transferase remain to be determined.
Rev1 efficiently inserts a C opposite a template AP site in vitro (17,18,35). In vivo, C insertion opposite the AP site was abolished in rev1 mutant cells. Therefore, in wild-type cells, C insertion opposite the AP site is catalyzed by the Rev1 dCMP transferase. The subsequent extension synthesis, however, requires another polymerase. In vitro, Polζ is capable of extension synthesis from opposite an AP site (9,17,21). Thus, it is believed that Polζ is an important extension polymerase during translesion synthesis (9,17,21). Consistent with this notion, C insertion was also abolished in rev3 mutant cells, and A insertion was reduced by approximately 2-fold opposite the AP site. In cells lacking Polζ, the significant residual extension activity from 3′ A opposite the AP site probably resulted from Polη activity. Supporting this interpretation, AP site bypass with A insertion opposite the lesion was nearly abolished in cells lacking both Polζ and Polη.
In mutant cells lacking Polη, Polζ or Rev1, A was still significantly inserted opposite the AP site. Thus, a replicative polymerase is likely responsible for the A insertion opposite the AP site in yeast cells. Indeed, opposite an AP site, Polα possesses a significant A insertion activity and Polδ has a weak A insertion capacity in vitro (Figure 7) (9). Since A insertion by Polδ is greatly limited by its 3′→5′ proofreading exonuclease activity, Polα likely plays a significant role in A insertion opposite the AP site in yeast cells. The rare G insertions in the rad30 and rev3 mutant cells (Table 2) were likely catalyzed by Polζ and Polη, respectively. The precise mechanism of complex translesion synthesis involving base substitutions and deletions on both sides of the AP site is not clear. Nevertheless, Polζ and Polη may be involved, as indicated by a reduction of this type of bypass products in the respective mutant strains (Table 2).
Taken together both in vitro and in vivo results, we propose a model for translesion synthesis of AP sites in yeast cells as shown in Figure 9. In our model, most AP sites in DNA block the replicative polymerases before the lesion. Thus, a major mechanism of translesion synthesis is mediated by C insertion opposite the AP site catalyzed by Rev1 and somehow facilitated by Polη. Then, translesion synthesis is completed by Polζ-catalyzed extension (Figure 9). There is another parallel mechanism for translesion synthesis of AP sites, in which Polα and Polδ insert A opposite the lesion. Polη may also contribute to a small fraction of A insertions opposite the lesion. Subsequent extension synthesis is then catalyzed independently by Polζ and Polη (Figure 9). Rev1 probably plays an accessory role for Polζ-catalyzed extension from the primer 3′ A opposite the AP site (Figure 9). The precise role of Rev1, however, is not known. Supporting a role of Rev1 in Polζ-catalyzed extension, A insertion was reduced by approximately 2-fold in the rev1 mutant cells and was largely abolished in rev1 rad30 double mutant cells (Table 2), suggesting a synergistic effect of Rev1 and Polη in extension synthesis from A-terminated primers opposite AP sites. In the rev3 rad30 double mutant cells, extensions in the two paralleled bypass mechanisms are completely blocked, leading to the most severe defect in AP site bypass, as indicated by results from both the in vivo translesion synthesis experiments and the cellular sensitivity to MMS.
Figure 9.
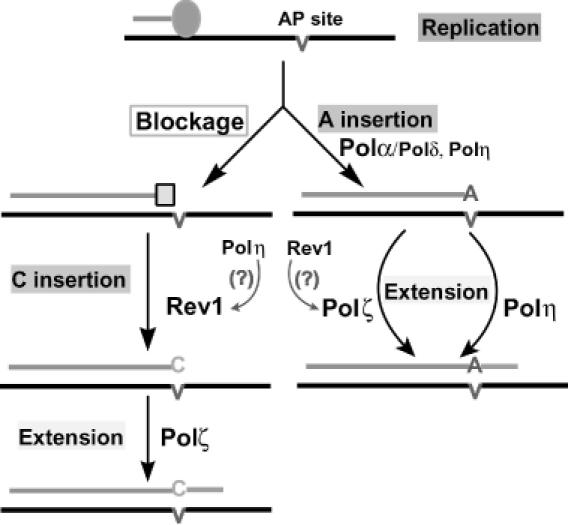
A mechanistic model of AP site bypass in yeast cells. When the replication complex (represented by the filled blue oval) encounters AP sites in the DNA template, two parallel bypass mechanisms can occur. In one mechanism (scheme on the left side), the replicative polymerase is blocked by the lesion and subsequently replaced by Rev1. C insertion is catalyzed by Rev1 and stimulated by Polη. Bypass is completed by Polζ-catalyzed extension from opposite the lesion. In the other mechanism (scheme on the right), A is inserted opposite the AP site by Polα and Polδ. Polη may also catalyze A insertion to a limited extent. Extension is then catalyzed independently by Polζ or Polη. Rev1-catalyzed C insertion may be facilitated by Polη, and Polζ-catalyzed extension may be facilitated by Rev1, although the precise nature of such facilitation is not clear yet.
Lawrence and colleagues (8,19) originally reported that Rev1-catalyzed C insertion opposite the lesion was the predominant mechanism of AP site bypass in yeast cells. However, Haracska et al. (9) recently concluded that Polδ-catalyzed A insertion was the major mechanism of AP site bypass. Our model of AP site bypass could provide an explanation for these conflicting reports. Our studies and those of Lawrence and colleagues with wild-type cells were performed under normal growth conditions, thus yielding similar results of AP site bypass. Haracska et al. (9), however, performed their experiments with the anp1 apn2 double mutant strain under MMS treatment conditions that killed nearly all cells (<0.3% cell survival). The double mutant cells are unable to repair AP sites in DNA. Hence, MMS treatment of these cells would yield excessive unrepaired AP sites in the genome during replication. Excessive AP sites may lead to saturation of the Rev1-catalyzed C insertion mechanism (Figure 9). Consequently, the majority of AP sites would have to be bypassed by the Polα-catalyzed A insertion mechanism (Figure 9), resulting in predominant A insertion opposite AP sites under the experimental conditions of Haracska et al. (9).
As indicated by our results and our model (Figure 9), AP site bypass in yeast cells is a complex cellular process involving different mechanisms and multiple polymerases. Such complexity offers some levels of functional redundancy. Indeed, some residual levels of AP site bypass can still take place in the absence of any single translesion polymerase. In higher eukaryotes, AP site bypass is further complicated by the presence of two more Y family translesion polymerases: Polκ and Polι, which efficiently and predominantly insert an A and a G, respectively, opposite a template AP site in vitro (47–49). Thus, these two polymerases may participate in nucleotide insertion during AP site bypass in mammalian cells. Furthermore, Polκ is able to perform efficient extension synthesis by a −1 deletion mechanism in certain sequence contexts in vitro (47,49), raising the possibility that it may also participate in extension synthesis to some extent from the primer 3′ A opposite the AP site. A better understanding of AP site bypass in higher eukaryotes should be facilitated by genetic analyses as we have performed and the conceptual model we have generated here in the yeast model system.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Yanbin Zhang for technical assistance and initiating the in vivo translesion synthesis assays. We thank Dongyu Guo for purifying yeast DNA polymerases α, δ and ζ. This work was supported by a NIH grant CA92528.
REFERENCES
- 1.Strauss B.S. (1991) The ‘A rule’ of mutagen specificity: a consequence of DNA polymerase bypass of non-instructional lesions? Bioessays, 13, 79–84. [DOI] [PubMed] [Google Scholar]
- 2.Gentil A., Renault,G., Madzak,C., Margot,A., Cabral-Neto,J.B., Vasseur,J.J., Rayner,B., Imbach,J.L. and Sarasin,A. (1990) Mutagenic properties of a unique abasic site in mammalian cells. Biochem. Biophys. Res. Commun., 173, 704–710. [DOI] [PubMed] [Google Scholar]
- 3.Gentil A., Cabral-Neto,J.B., Mariage-Samson,R., Margot,A., Imbach,J.L., Rayner,B. and Sarasin,A. (1992) Mutagenicity of a unique apurinic/apyrimidinic site in mammalian cells. J. Mol. Biol., 227, 981–984. [DOI] [PubMed] [Google Scholar]
- 4.Neto J.B., Gentil,A., Cabral,R.E. and Sarasin,A. (1992) Mutation spectrum of heat-induced abasic sites on a single-stranded shuttle vector replicated in mammalian cells. J. Biol. Chem., 267, 19718–19723. [PubMed] [Google Scholar]
- 5.Cabral Neto J.B., Cabral,R.E., Margot,A., Le Page,F., Sarasin,A. and Gentil,A. (1994) Coding properties of a unique apurinic/apyrimidinic site replicated in mammalian cells. J. Mol. Biol., 240, 416–420. [DOI] [PubMed] [Google Scholar]
- 6.Takeshita M. and Eisenberg,W. (1994) Mechanism of mutation on DNA templates containing synthetic abasic sites: study with a double strand vector. Nucleic Acids Res., 22, 1897–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klinedinst D.K. and Drinkwater,N.R. (1992) Mutagenesis by apurinic sites in normal and ataxia telangiectasia human lymphoblastoid cells. Mol. Carcinog., 6, 32–42. [DOI] [PubMed] [Google Scholar]
- 8.Gibbs P.E. and Lawrence,C.W. (1995) Novel mutagenic properties of abasic sites in Saccharomyces cerevisiae. J. Mol. Biol., 251, 229–236. [DOI] [PubMed] [Google Scholar]
- 9.Haracska L., Unk,I., Johnson,R.E., Johansson,E., Burgers,P.M., Prakash,S. and Prakash,L. (2001) Roles of yeast DNA polymerases δ and ζ and of Rev1 in the bypass of abasic sites. Genes Dev., 15, 945–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ohmori H., Friedberg,E.C., Fuchs,R.P.P., Goodman,M.F., Hanaoka,F., Hinkle,D., Kunkel,T.A., Lawrence,C.W., Livneh,Z., Nohmi,T. et al. (2001) The Y-family of DNA polymerases. Mol. Cell, 8, 7–8. [DOI] [PubMed] [Google Scholar]
- 11.Wang Z. (2001) Translesion synthesis by the UmuC family of DNA polymerases. Mutat. Res., 486, 59–70. [DOI] [PubMed] [Google Scholar]
- 12.Wang Z. (2001) DNA damage-induced mutagenesis: a novel target for cancer prevention. Mol. Interv., 1, 269–281. [PubMed] [Google Scholar]
- 13.Livneh Z. (2001) DNA damage control by novel DNA polymerases: translesion replication and mutagenesis. J. Biol. Chem., 276, 25639–25642. [DOI] [PubMed] [Google Scholar]
- 14.Friedberg E.C., Fischhaber,P.L. and Kisker,C. (2001) Error-prone DNA polymerases: novel structures and the benefits of infidelity. Cell, 107, 9–12. [DOI] [PubMed] [Google Scholar]
- 15.Lin W., Wu,X. and Wang,Z. (1999) A full-length cDNA of hREV3 is predicted to encode DNA polymerase ζ for damage-induced mutagenesis in humans. Mutat. Res., 433, 89–98. [DOI] [PubMed] [Google Scholar]
- 16.Burgers P.M., Koonin,E.V., Bruford,E., Blanco,L., Burtis,K.C., Christman,M.F., Copeland,W.C., Friedberg,E.C., Hanaoka,F., Hinkle,D.C. et al. (2001) Eukaryotic DNA polymerases: proposal for a revised nomenclature. J. Biol. Chem., 276, 43487–43490. [DOI] [PubMed] [Google Scholar]
- 17.Nelson J.R., Lawrence,C.W. and Hinkle,D.C. (1996) Deoxycytidyl transferase activity of yeast REV1 protein. Nature, 382, 729–731. [DOI] [PubMed] [Google Scholar]
- 18.Lin W., Xin,H., Zhang,Y., Wu,X., Yuan,F. and Wang,Z. (1999) The human REV1 gene codes for a DNA template-dependent dCMP transferase. Nucleic Acids Res., 27, 4468–4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nelson J.R., Gibbs,P.E., Nowicka,A.M., Hinkle,D.C. and Lawrence,C.W. (2000) Evidence for a second function for Saccharomyces cerevisiae Rev1p. Mol. Microbiol., 37, 549–554. [DOI] [PubMed] [Google Scholar]
- 20.Johnson R.E., Torres-Ramos,C.A., Izumi,T., Mitra,S., Prakash,S. and Prakash,L. (1998) Identification of APN2, the Saccharomyces cerevisiae homolog of the major human AP endonuclease HAP1, and its role in the repair of abasic sites. Genes Dev., 12, 3137–3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yuan F., Zhang,Y., Rajpal,D.K., Wu,X., Guo,D., Wang,M., Taylor,J.-S. and Wang,Z. (2000) Specificity of DNA lesion bypass by the yeast DNA polymerase η. J. Biol. Chem., 275, 8233–8239. [DOI] [PubMed] [Google Scholar]
- 22.Johnson R.E., Prakash,S. and Prakash,L. (1999) Efficient bypass of a thymine-thymine dimer by yeast DNA polymerase, Polη. Science, 283, 1001–1004. [DOI] [PubMed] [Google Scholar]
- 23.Masutani C., Araki,M., Yamada,A., Kusumoto,R., Nogimori,T., Maekawa,T., Iwai,S. and Hanaoka,F. (1999) Xeroderma pigmentosum variant (XP-V) correcting protein from HeLa cells has a thymine dimer bypass DNA polymerase activity. EMBO J., 18, 3491–3501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Y., Yuan,F., Wu,X., Rechkoblit,O., Taylor,J.-S., Geacintov,N.E. and Wang,Z. (2000) Error-prone lesion bypass by human DNA polymerase η. Nucleic Acids Res., 28, 4717–4724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Masutani C., Kusumoto,R., Iwai,S. and Hanaoka,F. (2000) Mechanisms of accurate translesion synthesis by human DNA polymerase η. EMBO J., 19, 3100–3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xie Z., Braithwaite,E., Guo,D., Bo,Z., Geacintov,N.E. and Wang,Z. (2003) Mutagenesis of benzo[a]pyrene diol epoxide in yeast: requirement for DNA polymerase ζ and involvement of DNA polymerase η. Biochemistry, 42, 11253–11262. [DOI] [PubMed] [Google Scholar]
- 27.Guo D., Wu,X., Rajpal,D.K., Taylor,J.-S. and Wang,Z. (2001) Translesion synthesis by yeast DNA polymerase ζ from templates containing lesions of ultraviolet radiation and acetylaminofluorene. Nucleic Acids Res., 29, 2875–2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Y., Wu,X., Guo,D., Rechkoblit,O. and Wang,Z. (2002) Activities of human DNA polyemrase κ in response to the major benzo[a]pyrene DNA adduct: error-free bypass and extension synthesis from opposite the lesion. DNA Repair, 1, 559–569. [DOI] [PubMed] [Google Scholar]
- 29.Guo D., Xie,Z., Shen,H., Bo,Z. and Wang,Z. (2004) Translesion synthesis of acetylaminofluorene-dG adducts by DNA polymerase ζ is stimulated by yeast Rev1 protein. Nucleic Acids Res., 32, 1122–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Becker D.M. and Guarente,L. (1991) High-efficiency transformation of yeast by electroporation. Methods Enzymol., 194, 182–187. [DOI] [PubMed] [Google Scholar]
- 31.Strathern J.N. and Higgins,D.R. (1991) Recovery of plasmids from yeast into Escherichia coli: shuttle vectors. Methods Enzymol., 194, 319–329. [DOI] [PubMed] [Google Scholar]
- 32.Duncan B.K., Rockstroh,P.A. and Warner,H.R. (1978) Escherichia coli K-12 mutants deficient in uracil-DNA glycosylase. J. Bacteriol., 134, 1039–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Warner H.R. and Duncan,B.K. (1978) In vivo synthesis and properties of uracil-containing DNA. Nature, 272, 32–34. [DOI] [PubMed] [Google Scholar]
- 34.Kunkel T.A. (1985) Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc. Natl Acad. Sci. USA, 82, 488–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Y., Wu,X., Rechkoblit,O., Geacintov,N.E., Taylor,J.-S. and Wang,Z. (2002) Response of human REV1 to different DNA damage: preferential dCMP insertion opposite the lesion. Nucleic Acids Res., 30, 1630–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morrison A., Christensen,R.B., Alley,J., Beck,A.K., Bernstine,E.G., Lemontt,J.F. and Lawrence,C.W. (1989) REV3, a Saccharomyces cerevisiae gene whose function is required for induced mutagenesis, is predicted to encode a nonessential DNA polymerase. J. Bacteriol., 171, 5659–5667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Larimer F.W., Perry,J.R. and Hardigree,A.A. (1989) The REV1 gene of Saccharomyces cerevisiae: isolation, sequence, and functional analysis. J. Bacteriol., 171, 230–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McDonald J.P., Levine,A.S. and Woodgate,R. (1997) The Saccharomyces cerevisiae RAD30 gene, a homologue of Escherichia coli dinB and umuC, is DNA damage inducible and functions in a novel error-free postreplication repair mechanism. Genetics, 147, 1557–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roush A.A., Suarez,M., Friedberg,E.C., Radman,M. and Siede,W. (1998) Deletion of the Saccharomyces cerevisiae gene RAD30 encoding an Escherichia coli DinB homolog confers UV radiation sensitivity and altered mutability. Mol. Gen. Genet., 257, 686–692. [DOI] [PubMed] [Google Scholar]
- 40.Maher V.M., Ouellette,L.M., Curren,R.D. and McCormick,J.J. (1976) Frequency of ultraviolet light-induced mutations is higher in exeroderma pimentosum variant cells than in normal human cells. Nature, 261, 593–595. [DOI] [PubMed] [Google Scholar]
- 41.Friedberg E.C., Walker,G.C. and Siede,W. (1995) DNA Repair and Mutagenesis. American Society of Microbiology Press, Washington, DC. [Google Scholar]
- 42.Loeb L.A. and Preston,B.D. (1986) Mutagenesis by apurinic/apyrimidinic sites. Annu. Rev. Genet., 20, 201–230. [DOI] [PubMed] [Google Scholar]
- 43.Gibbs P.E., Kilbey,B.J., Banerjee,S.K. and Lawrence,C.W. (1993) The frequency and accuracy of replication past a thymine-thymine cyclobutane dimer are very different in Saccharomyces cerevisiae and Escherichia coli. J. Bacteriol., 175, 2607–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baynton K., Bresson-Roy,A. and Fuchs,R.P. (1999) Distinct roles for Rev1p and Rev7p during translesion synthesis in Saccharomyces cerevisiae. Mol. Microbiol., 34, 124–133. [DOI] [PubMed] [Google Scholar]
- 45.Haracska L., Washington,M.T., Prakash,S. and Prakash,L. (2001) Inefficient bypass of an abasic site by DNA polymerase η. J. Biol. Chem., 276, 6861–6866. [DOI] [PubMed] [Google Scholar]
- 46.Guo C., Fischhaber,P.L., Luk-Paszyc,M.J., Masuda,Y., Zhou,J., Kamiya,K., Kisker,C. and Friedberg,E.C. (2003) Mouse Rev1 protein interacts with multiple DNA polymerases involved in translesion DNA synthesis. EMBO J., 22, 6621–6630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang Y., Yuan,F., Wu,X., Wang,M., Rechkoblit,O., Taylor,J.-S., Geacintov,N.E. and Wang,Z. (2000) Error-free and error-prone lesion bypass by human DNA polymerase κ in vitro. Nucleic Acids Res., 28, 4138–4146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Y., Yuan,F., Wu,X., Taylor,J.-S. and Wang,Z. (2001) Response of human DNA polymerase ι to DNA lesions. Nucleic Acids Res., 29, 928–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ohashi E., Ogi,T., Kusumoto,R., Iwai,S., Masutani,C., Hanaoka,F. and Ohmori,H. (2000) Error-prone bypass of certain DNA lesions by the human DNA polymerase κ. Genes Dev., 14, 1589–1594. [PMC free article] [PubMed] [Google Scholar]