

ABSTRACT
AIDS virus infections are rarely controlled by cell-mediated immunity, in part due to viral immune evasion and immunodeficiency resulting from CD4+ T-cell infection. One likely aspect of this failure is that antiviral cellular immune responses are either absent or present at low levels during the initial establishment of infection. To test whether an extensive, timely, and effective response could reduce the establishment of infection from a high-dose inoculum, we adoptively transferred large numbers of T cells that were molecularly engineered with anti-simian immunodeficiency virus (anti-SIV) activity into rhesus macaques 3 days following an intrarectal SIV inoculation. To measure in vivo antiviral activity, we assessed the number of viruses transmitted using SIVmac239X, a molecularly tagged viral stock containing 10 genotypic variants, at a dose calculated to transmit 12 founder viruses. Single-genome sequencing of plasma virus revealed that the two animals receiving T cells expressing SIV-specific T-cell receptors (TCRs) had significantly fewer viral genotypes than the two control animals receiving non-SIV-specific T cells (means of 4.0 versus 7.5 transmitted viral genotypes; P = 0.044). Accounting for the likelihood of transmission of multiple viruses of a particular genotype, the calculated means of the total number of founder viruses transmitted were 4.5 and 14.5 in the experimental and control groups, respectively (P = 0.021). Thus, a large antiviral T-cell response timed with virus exposure can limit viral transmission. The presence of strong, preexisting T-cell responses, including those induced by vaccines, might help prevent the establishment of infection at the lower-exposure doses in humans that typically transmit only a single virus.
IMPORTANCE The establishment of AIDS virus infection in an individual is essentially a race between the spreading virus and host immune defenses. Cell-mediated immune responses induced by infection or vaccination are important contributors in limiting viral replication. However, in human immunodeficiency virus (HIV)/SIV infection, the virus usually wins the race, irreversibly crippling the immune system before an effective cellular immune response is developed and active. We found that providing an accelerated response by adoptively transferring large numbers of antiviral T cells shortly after a high-dose mucosal inoculation, while not preventing infection altogether, limited the number of individual viruses transmitted. Thus, the presence of strong, preexisting T-cell responses, including those induced by vaccines, might prevent infection in humans, where the virus exposure is considerably lower.
INTRODUCTION
The CD8+ T cell plays an important role in control of AIDS viruses, i.e., human immunodeficiency virus (HIV) and simian immunodeficiency virus (SIV) (1–10). However, in most cases, CD8+ T-cell responses, whether consisting of responses to infection developed de novo or to prior vaccination, are unable to prevent development of persistent, disseminated infection. This could reflect an intrinsic inability of the generated CD8+ T-cell responses to control AIDS virus infection: certain CD8+ T-cell responses may lack the necessary qualitative effector function profiles against the virus. Additionally, quantitative (11) or spatiotemporal (12) limitations could prevent effective antiviral CD8+ T-cell responses from acting at foci of viral infection and replication, being too little, too late, and/or in the wrong place to affect the establishment of infection.
To test whether the presence of a large, potent virus-specific T-cell response just after viral inoculation, in an otherwise SIV-naive animal, can help limit the establishment of infection, we used a T-cell engineering/adoptive-transfer approach. Adoptive transfer of antigen-specific T cells has been used to examine cell-mediated immune activity against cancer and various pathogens, including viruses (13–15). Adoptive transfer has been most successfully applied in clinical anticancer trials where autologous CD8+ T cells are engineered to express tumor-specific T-cell receptors (TCRs) or, more recently, antitumor chimeric antigen receptors and then infused into cancer patients to produce encouraging antitumor effects (16–20).
This powerful approach has been used less extensively to study the efficacy of anti-AIDS virus immune responses using SIV infection in a rhesus macaques model (12, 21–26). A key challenge for these types of adoptive-transfer experiments is producing sufficient numbers of autologous cells for infusion. Vaccinated rhesus macaques typically have relatively low (∼1% to 4%) levels of virus-specific cells, and even infected animals can exhibit low frequencies of virus-specific CD8+ T cells. Thus, in experiments relying on native virus-specific T cells for adoptive transfer, these relatively low numbers need to be greatly expanded over months in culture, during which time their properties can change, with some clones within the population being lost to senescence or anergy. Also, conventional approaches designed to examine the impact of accelerated timing of the antiviral cellular response by infusion paradoxically require autologous virus-specific T cells to be derived from an immunologically naive animal before infection. To overcome these barriers, we engineered large numbers of T cells obtained from naive animals to express well-characterized, highly effective anti-SIV TCRs specific for immunodominant epitopes (27). By infusing these anti-SIV T cells into animals shortly after they received a large intrarectal challenge inoculum, we successfully reduced the number of founder viruses establishing disseminated infection.
MATERIALS AND METHODS
Rhesus macaque care.
Four Mamu A*01 Indian rhesus macaques (Macaca mulatta) were cared for and housed at the National Cancer Institute (NCI) in Bethesda, MD. Infusions, blood draws, and tissue biopsy procedures were carried out using Animal Care and Use Committee-approved protocols and procedures, including the use of IACUC-approved individual housing to reduce the risk of viral cross contamination. Steps were taken to ensure the welfare of the macaques and to minimize their potential pain or suffering. The NCI and the Frederick National Laboratory are accredited by AAALAC International and follow the Public Health Service Policy for the Care and Use of Laboratory Animals. Animal care was provided in accordance with the procedures outlined in the “Guide for Care and Use of Laboratory Animals” (28).
Cell culture.
T cells were cultured in RPMI 1640 media supplemented with 10% (vol/vol) fetal bovine serum (Gemini Bio Products), 10 mM HEPES buffer, 2 mM glutamine (Life Technologies), interleukin-2 (IL-2; NIH AIDS reagent repository) (50 IU/ml), penicillin (Life Technologies) (100 μg/ml), and streptomycin (Life Technologies) (100 μg/ml). Cells were expanded through biweekly stimulation with anti-CD3 (clone SP34-2; BD Biosciences) (30 ng/ml), IL-2 (50 IU/ml), irradiated human peripheral blood mononucleated cells (PBMC), and an Epstein-Barr virus-transformed B-cell line (TM B-LCL) (12, 29, 30).
Retroviral vectors.
Vector constructs containing rhesus macaque CM9- or SL8-specific TCRs and C-terminally truncated human nerve growth factor receptor (tNGFR) genes were previously described (27, 31). The C-C chemokine receptor 7 (CCR7) and l-selectin (CD62L) genes were isolated by PCR-mediated cDNA cloning from the RNA of rhesus macaque PBMC and were used to construct an MSGV1 vector that expresses a single transcript containing tNGFR/P2A/CCR7/T2A/CD62L coding sequence. Retroviral vectors were generated by transfecting vector constructs into Phoenix-RD114 packaging cells (32).
TCR and homing marker transduction.
Retroviral vector-containing supernatants were loaded onto RetroNectin-coated 24-well plates (treated with 20 μg/ml) and centrifuged at 2,000 × g for 2 h at 32°C. Half of the vector supernatant was removed, and 1 × 106 stimulated T cells were added per well. The cells were then centrifuged at 2,000 × g for 1 h at 32°C followed by incubation at 37°C for 48 h before flow cytometric analysis for tetramer binding or tNGFR expression was performed to determine transduction efficiency. Transduced cells were sorted by the use of paramagnetic beads and LS columns (Miltenyi Biotech, Inc.) using phycoerythrin (PE beads) with either specific tetramers (CM9 peptide major histocompatibility complex [MHC] class I/tetramer [Beckman Coulter] and SL8 peptide/MHC tetramer [NIH Tetramer Core Facility]) or nerve growth factor receptor (NGFR) (ME20.4-1.H4; Miltenyi Biotech) antibody.
ERK1/2 phosphorylation analysis.
Levels of total and phosphorylated extracellular signal-regulated kinase 1 (ERK1)/ERK2 (pERK1/2) in CD8+ T cells (1 ×106 cells in serum-free media) were measured following stimulation with 2 μg/ml of human exodus CCL21 (ProSpec) for 2, 8, and 15 min. Cells were harvested by centrifugation at 10,000 × g for 1 min. The cell pellets were lysed with Pierce immunoprecipitation (IP) lysis buffer containing 1× Halt protease and phosphatase inhibitor cocktail (Thermo Scientific) on ice for 30 min. The nuclear fraction was separated from the cytosolic lysate by centrifugation for 10 min at 10,000 × g, and the supernatant was transferred to a new tube. Prior to SDS fractionation, LDS sample loading buffer (4×) was added to the lysate at a final concentration of 1× and samples were boiled for 5 min.
Samples normalized using ß-actin were loaded on a 4% to 20% gradient Tris–glycine polyacrylamide gel, and, after separation, proteins were transferred onto a polyvinylidene difluoride (PVDF) membrane (Millipore Immobilon-P; IPVH00010) using a semidry electroblotter (Ellard Instrumentation) for 1 to 1.5 h. Detection of ERK1/2 and pERK1/2 proteins was performed using polyclonal rabbit antibodies ab17942 (Abcam) and ab4819 (Abcam), respectively, each used at a 1:1,000 dilution, followed by the use of anti-rabbit secondary antibody (A6154; Sigma) at a 1:10,000 dilution. Detection of ß-actin protein was performed using monoclonal antibody (MA5-11116; Thermo Scientific Pierce) diluted 1:1,000, followed by anti-mouse secondary antibody (A4416; Sigma) at a 1:10,000 dilution. Immunoblots were analyzed using a VersaDoc 3000 imaging system (Bio-Rad Laboratories), and band intensities were quantified using Quantity One version 4.5.0 software (Bio-Rad Laboratories).
In vitro chemotaxis assay.
Chemotaxis was measured using transwell inserts (5 μm pore size) on a 24-well plate (Costar). The upper and lower chambers were filled with serum-free media. Cells (200,000) were added to the upper chamber, the cells were allowed to settle for 1 h, and then 1 μg/ml SLC/CCL21 (ProSpec) was added to the lower chamber and cell migration was measured by counting the cells in the lower chamber 3 h after the addition of CCL21. Specific cell migration was calculated by subtracting the number of cells migrating in the absence of chemokine from the number of cells migrating in the presence of chemokine and calculating the ratio of the number of cells in the lower chamber to the number of input cells. Images of wells were taken using an inverted light microscope.
SIVmac239X inoculation.
Macaques were inoculated with 9 × 105 infectious units (TZM-bl blue-cell-forming units) of SIVmac239X intrarectally as previously described (33). The virus stock contains 9 isogenic-sequence-discriminable molecularly tagged variants (variants A to I) plus wild-type (WT) SIVmac239 (10 in total) within a single inoculum stock, with equal proportions of all genotypes represented.
Adoptive T-cell transfer.
Expanded transduced T cells from each animal were pooled and washed, and half the cells were labeled with 5 μM CellTrace Violet (CTV; Life Technologies). Cells were resuspended in 50 ml saline solution supplemented with 2% autologous serum and infused into the femoral vein (2 ml/min) (12, 21). Macaques were subcutaneously administered IL-2 (104 IU/kg of body weight) daily for 10 days following infusion. Numbers of infused cells and weights of animals were as follows: experimental animal 1 (Exp-1), 9 × 108 CM9-6 TCR CD8+ cells, 8.24 kg; Exp-2, 6 × 109 cells (3.5 × 109 CM9-6 TCR CD4+ CCR7/CD62L cells, 1.5 × 109 CM9-6 TCR CD4+ cells, 8.8 × 108 CM9-6 TCR CD4+ cells, and 4.1 × 108 SL8 TCR CD4+ cells), 9.72 kg; control animal 1 (Ctr-1), 2 × 108 tNGFR cells, 7.27 kg; Ctr-2, 1 × 108 tNGFR cells, 5.40 kg.
Flow cytometry.
The antibodies and tetramers used were as follows: CD3 (SP34-2), CD45 (HI30), CD4 (OKT4), CD8 (SK1), CCR7 (3D12), CD62L (SK11), CD28 (CD28.2), CD95 (DX2) (BD Biosciences), and α4-β7 (11718; NIH) (34), NGFR; and ME20.4-1.H4 (Miltenyi Biotech), CM9 peptide MHC class I/tetramer (Beckman Coulter), and SL8 peptide/MHC tetramer (NIH Tetramer Core Facility). Stimulated cells were stained for CD107a (H4A3) and for the intracellular cytokines gamma interferon (IFN-γ) (B27) and macrophage inflammatory protein 1 beta (MIP-1β) (D21-1351; BD Biosciences). More than 100,000 events were acquired on a BD LSRII analyzer (BD Biosciences). Complete blood cell counts were monitored using BD TruCount tubes (BD Biosciences) on a BD FACSVerse flow cytometer (BD Biosciences). Data analysis was performed using FCS Express 4 (De Novo Software).
Quantitation of plasma IFN-γ.
Plasma IFN-γ levels were assayed using a monkey IFN-γ enzyme-linked immunosorbent assay (ELISA) kit (Mabtech) according to the manufacturer's protocol. Detection limits of the assay were 4 to 400 pg/ml.
Virus quantitative real-time PCR.
Viral load was determined from plasma by quantitative PCR (qPCR) as previously described (12, 35). Results are reported as numbers of copies per milliliter, with a detection limit of 30 copies/ml. Cell-associated viral DNA analysis was performed on PBMC as previously described (12, 35). Results are reported as nominal SIV gag DNA copy numbers per 100,000 cell equivalents, as determined by copy numbers of the CCR5 gene.
Sequence analysis of SIV variants.
Single-genome analysis of virions in plasma was performed as previously described (33) on a 300-bp portion of SIVmac239 integrase gene, which contained the engineered genotypes. The number of variants detected systemically was determined from blood plasma at 10 days postchallenge by measuring the number of sequences corresponding to the SIVmac239A to -I and wild-type (WT) genotypes. For the experimental macaques, Exp-1 and -2, we analyzed 121 and 128 sequences, respectively. For the control macaques, Ctr-1 and -2, we analyzed 61 and 109 sequences, respectively.
Statistical methods.
Data in this study were analyzed with linear mixed-effects hierarchical models, analysis of variance (ANOVA), and t tests. Because the animals were inoculated only once prior to infusion, we used a one-tailed t test to compare the means of the numbers of genotypes or founder viruses detected between the two groups with a null hypothesis that the expected mean for the control group was equal to or greater than that of the experimental group. Probability values of less than 0.05 (P < 0.05) were considered to represent statistical significance, and values that were less than 0.10 but greater than 0.05 (0.05 < P < 0.10) were considered to represent marginal statistical significance. All statistical analyses and simulation computations were performed with the R statistical language and environment (36).
Monte Carlo simulation analysis of independent founder events.
Given the limited number (n = 10) of discriminating genotypes in SIVmac239X, there is a near certainty of multiple infections occurring in an individual by one or more of the viral genotypes as the numbers of founder events increase, resulting in an undercount of the founder viruses. To more accurately account for the number of unique genotypes present, we used a computer-based Monte Carlo simulation approach to determine the distribution of independent founder virus transmission based on the observed number of genotypes measured. The simulation assumes a viral inoculum of a population of 10,000 total viral particles with 1,000 particles of each of the 10 variants, corresponding to SIVmac239A to -I and the wild type (WT). From this population, a random number generator (Monte Carlo approach, with potential values 1 to 10) identifies a variant that is drawn from the population representing a successful infecting variant. For each simulation representing an inoculated macaque, the number of sequential draws required to observe a given number of unique variants is determined. This simulation was performed on 1,000 theoretical macaques to account for each of the 10 possible numbers of variants detected, representing founder viruses. In this way, distributions of the numbers of draws required to identify from 1 to 10 variants were generated and then analyzed to produce median, mean, minimal, and maximal estimates of the number of founder viruses present in the macaques given the number of genotypes observed experimentally.
RESULTS
To test whether the presence of SIV-specific effector T cells soon after virus exposure can limit the number of transmitted viral variants that contribute to disseminated systemic infection, we intrarectally inoculated Mamu A0*1 Indian rhesus macaques with SIV and subsequently infused large numbers of antiviral T cells 3 days later. A 3-day lag between inoculation and infusion was chosen to allow establishment of viral foci in mucosal tissues which can serve as targets for the infused cells before establishment of disseminated systemic infection (37) (B. F. Keele, unpublished data).
To generate SIV-specific T cells, we isolated naive T cells (CD3+, CD28+, and CD95−) from PBMC for transduction with retroviral vectors (gating scheme presented in Fig. 1A). Naive cells were selected because, in a comparative study by Hinrichs et al., antitumor TCR-transduced CD8+ T cells generated from a naive population exhibited adoptive antitumor function that was superior to that seen with central memory cells, which are themselves thought to function better than effector memory cells (23, 38). T cells from the two experimental animals, Exp-1 and Exp-2, were transduced with retroviral vectors expressing well-characterized SIV-specific Mamu A0*1-restricted TCR genes that can successfully transfer the ability to suppress SIV replication to CD8+ T cells in vitro (27). CD8+ T cells from Exp-1 were transduced with Gag CM9-6 TCR only, while CD8+ T cells from Exp-2 were transduced with either Gag CM9-6 or Tat SL8-42 TCR. To provide SIV-specific CD4+ T cells to enhance CD8+ antiviral activity in vivo by providing CD4+ T helper function, as suggested by mouse and human studies (39, 40), or by direct antiviral activity on infected cells (41), naive CD4+ T cells from Exp-2 were also transduced with the Gag CM9-6 TCR. Due to the lack of an appropriate SIV-specific MHC class II-restricted TCR, we used the Gag CM9-6 MHC class I-restricted TCR to transfer specific antiviral effector function to the CD4+ T cells as demonstrated previously (reference 42 and unpublished results). In lieu of TCR vectors, T cells from the two control animals, Ctr-1 and Ctr-2, were transduced with a vector encoding an inert truncated form of the nerve growth factor receptor (tNGFR) that allows tracking of their distribution postinfusion by this normally nonlymphoid marker (31, 43). Transduced cells were sorted to greater than 80% purity prior to expansion. The experimental and control animal transductant cultures functionally responded to autologous PMBC pulsed with cognate peptide, producing a characteristic Th1 effector profile, including IFN-γ and MIP-1β intracellular cytokine and CD107a degranulation responses (Fig. 1B).
FIG 1.

Characterization of SIV-specific TCR transductants. (A) Plots of flow cytometric sorting (presented left to right) for naive T (TN) cells from rhesus macaque PBMC for lymphocytes based on size and granularity determined by side scatter and forward scatter (SSC and FSC, respectively), for naive T cells by CD3+/CD28+/CD95−, and for CD8+ and CD4+ T-cell populations are displayed. (B) Results of flow cytometric analysis of TCR-transduced Exp-2 cells for induction of surface CD107a and intracellular induction of IFN-γ and MIP-1β expression 6 h after coculture with PBMC pulsed with cognate peptides are displayed. Data are representative of results of two independent experiments. The data shown are from transduced Exp-2 cells; Exp-1 exhibited a similar profile (data not shown).
Several studies in humans indicate that a substantial fraction of infused T cells can be found in lung rather than lymphoid tissues (44–47). Indeed, our prior adoptive-transfer studies and those of others performed using rhesus macaques showed extensive localization of infused cells in the lung with little or no detectable persistence in blood or lymph nodes (12, 21, 25). To redirect the infused cells to immune-relevant sites, we supertransduced half of the virus-specific Exp-2 Gag CM9-6 CD4+ T cells with a retroviral vector encoding lymphoid homing proteins rhesus C-C chemokine receptor 7 (CCR7) and l-selectin (CD62L). Additionally, the vector expresses tNGFR to clearly distinguish our infused homing marker transductants from native CCR7- and CD62L-expressing T cells in the Exp-2 samples.
To confirm the function of these homing proteins, we generated and tested Exp-2 CCR7/CD62L CD8+ transductants, termed R7/62L cells, for in vitro receptor-specific signaling and migration. Immunoblotting of samples stimulated with CCL21, the CCR7 ligand, revealed that R7/62L cells signaled with a maximally 8-fold-higher level of ERK1/2 phosphorylation than their unmodified CD8 counterparts after a 2-min treatment (Fig. 2A). Phosphorylation levels in the presence of CCL21 decreased at the latter time points, as reported by others (48), confirming physiological signaling kinetics. The response of the transductants was also considerably higher than that of the positive-control TM B-LCL human B-lymphoblastoid line (Fig. 2A). The R7/62L cell line also exhibited specific migration in a transwell assay (Fig. 2B), with 43% of the cells migrating to the ligand chamber 3 h after CCL21 addition, similarly to the migration of the TM B-LCL positive control (Fig. 2B). In contrast, the unmodified CD8+ T cells did not exhibit any specific migration. Taken together, these results indicated that the CCR7 transductants are functional in vitro.
FIG 2.

Functional testing of CCR7/CD62L transductants. (A) The induction of MAPK/ERK pathway within CCR7/CD62L T-cell transductants (R7/62L) in response to the CCR7 ligand (CCL21)is presented. (Top) ERK1/2 and phospho-ERK immunoblots of cell lysates are presented, with the minutes of CCL21 exposure denoted above each sample, molecular mass standards indicated at left, and bands identified at right. (Bottom) A graph of the measured band intensities is presented with minutes of CCL21 exposure denoted under each sample. (B) A micrograph of CCL21-induced cell migration assayed by transwell CCR7-mediated chemotaxis is presented above a graphic summary of cell counts measured in the transwell assay. Data are representative of results of two independent experiments. (C) Upper row: a flow cytometric analysis for CD62L and IFN-γ of rhesus PBMC left unstimulated or stimulated with anti-CD3 is presented. Lower group: results of flow cytometric analysis of R7/62L cells for CD62L and IFN-γ are presented. Data are representative of results of two independent experiments.
Upon antigenic stimulation, the ectodomain of the CD62L adhesion molecule is cleaved from the surface of T cells by the ADAM17 protease (49). To determine whether our transduced cells appropriately carry out this physiological reaction, we employed an established assay that measures the loss of surface expression on T cells due to ADAM17 protease cleavage that occurs upon antigen stimulation (50). After 6 h of stimulation with the SIV Gag CM9 peptide, flow cytometry analysis of CM9/CCR7/CD62L transduced T cells revealed specific downregulation of the transduced CD62L surface expression on the activated cells, i.e., those with an induced IFN-γ expression response (Fig. 2C). Similar results were observed in a PBMC control, where CD62L loss strongly correlated with induced IFN-γ expression (Fig. 2C), thereby confirming the appropriate activation-induced shedding of CD62L by the transductants.
Even though the TCR-engineered CD8+ T cells produced typical effector intracellular cytokine responses just after transduction and tetramer sorting, because these cultures were extensively expanded through 6 or 7 cycles of anti-CD3 stimulation over 12 weeks, we confirmed our original antigen response results obtained with the expanded SIV-specific T-cell lines, 2 weeks prior to infusion. The activation response results for engineered cells from Exp-1 and Exp-2 revealed that the CD8+ CM9-6 transductants produced IFN-γ and MIP-1β as well as CD107a degranulation responses to SIV Gag CM9 peptide-pulsed antigen-presenting cells (APC) (Fig. 3) at levels similar to those observed just after transduction (Fig. 1B and data not shown).
FIG 3.
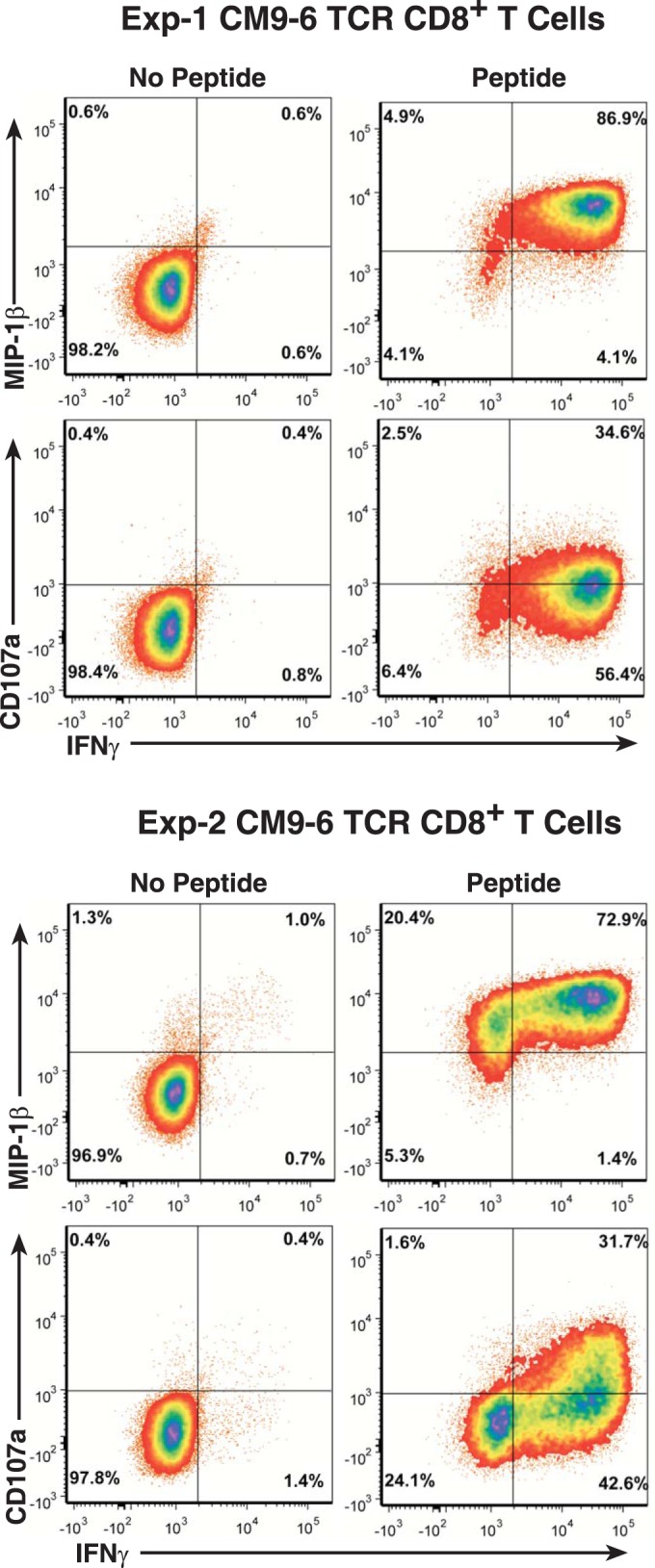
Analysis of in vitro effector response in TCR-transduced T cells prior to infusion. Results of flow cytometric analyses of induced IFN-γ, MIP1-β, and CD107a responses to SIV Gag CM9 peptide-pulsed APC by the expanded TCR-transduced T cells from Exp-1 and Exp-2 2 weeks prior to infusion are presented.
SIVmac239X inoculation and adoptive transfer of SIV-specific T cells.
To measure the ability of the adoptively transferred T cells to reduce the number of transmitted viral variants contributing to disseminated systemic infection, we used SIVmac239X, a “synthetic swarm” virus stock which contains 10 different molecularly tagged genotypes (51). Enumerating how many of the 10 SIVmac239X variants are present in plasma at peak viremia by single-genome sequencing reflects the observable number of distinct transmitted/founder viruses from the inoculum that ultimately contributed to the disseminated infection. A reduction in the number of transmitted/founder variants measured using this system has served as a surrogate measure of vaccine efficacy (52). For this experiment, we chose a dose of SIVmac239X for the intrarectal inoculations intended to transfer ∼12 transmitted/founder viruses to each individual to provide a wide dynamic range of transmitted/founder virus detection and ensure infection in a single dose (33). Given this large inoculum, we deemed it unlikely that the infused cells could prevent infection altogether.
All four rhesus macaques were inoculated intrarectally with SIVmac239X and then infused with the autologous engineered cells 3 days later. Prior to infusion, half of each category of cells was labeled with CellTrace Violet (CTV) fluorescent dye to enable tracking of infused cells in the animals. The two control macaques, Ctr-1 and -2, were infused with the corresponding non-SIV-specific tNGFR-expressing CD8+ T cells. Exp-1 was infused with SIV Gag CM9-specific CD8+ T cells, and Exp-2 was infused with a mixture of SIV Gag CM9- and SIV Tat SL8-specific CD8+ and SIV Gag CM9-specific CD4+ SIV-specific T cells with or without the lymph node homing markers CCR7 and CD62L.
Persistence of infused cells.
On the basis of our previous adoptive transfers, extensively expanded CD8+ T cells typically localize to and persist in the lung (12, 21). The persistence of transduced T cells in the animals was examined by flow cytometry of purified PBMC and of bronchoalveolar lavage (BAL) fluid samples. Infused cells from Exp-2 and Ctr-1 were detectable in PBMC samples for 168 h postinfusion, while infused cells were undetectable in Exp-1 and Ctr-2 PBMC samples after 96 h and 48 h, respectively (Fig. 4A). As seen in our previous studies (12, 21), the highest frequencies of infused cells were found in the BAL fluid samples in all of the animals, being detectable at our last sampled time point, 29 days postinfusion (Fig. 4 and data not shown).
FIG 4.
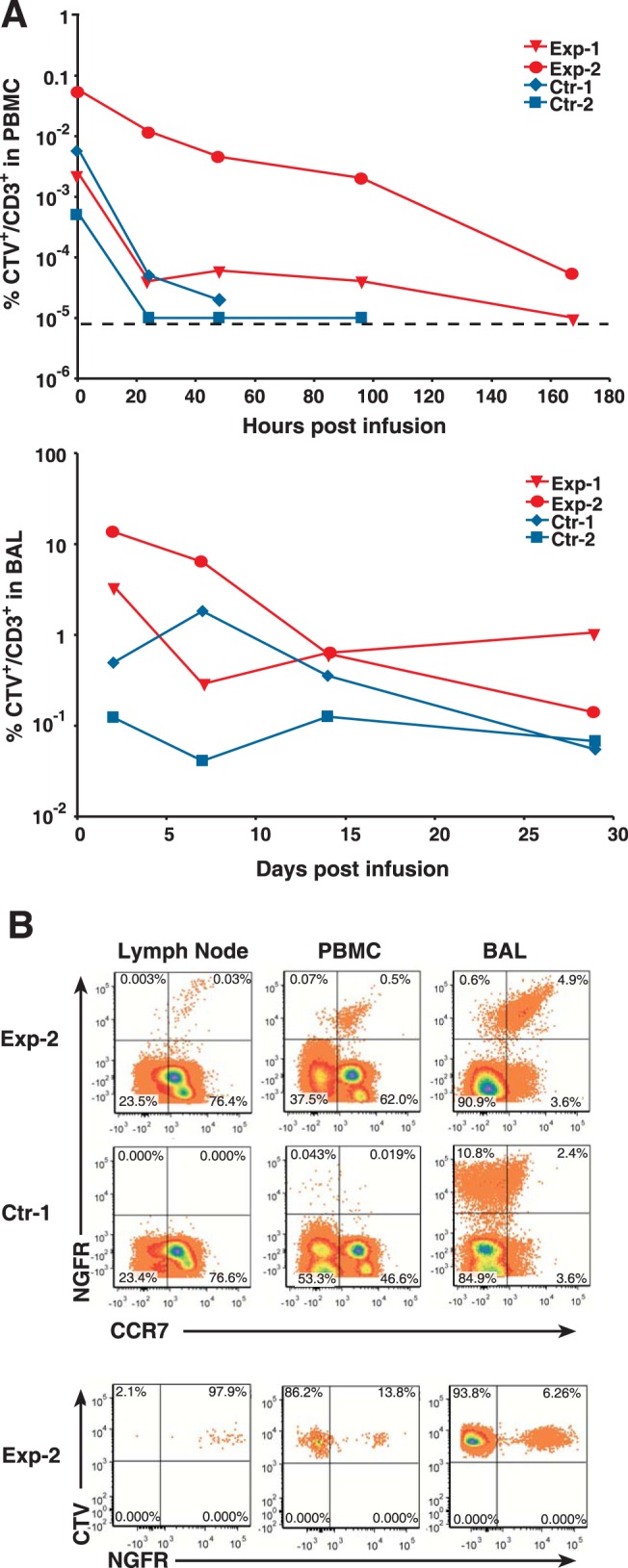
Persistence of infused T cells. (A) Graphs of the percentages of infused T cells (CTV+ CD3+) in blood (PBMC, top) or bronchiolar alveolar lavage fluid (BAL, bottom) samples measured by flow cytometry are presented. A dashed line indicates the level of detection in the flow cytometry assay. (B) Results of flow cytometry analysis for (top) CCR7 and NGFR expressing cells and (bottom) CTV+-gated NGFR expressing infused cells in blood and tissues 48 h postinfusion are presented.
To examine the localization of the Exp-2 CCR7/CD62L transduced cells, we analyzed cells harvested from a 48-h lymph node biopsy specimen from Exp-2. Since our CCR7/CD62L vector also expresses tNGFR, we can distinguish our engineered transductants from the endogenous CCR7-expressing T cells. NGFR and CCR7 flow cytometry analysis of Exp-2 samples gated for CD3+ T cells revealed that 0.03% of the CCR7+ T cells in the lymph node sample were the homing transductants (NGFR+ CCR7+) (Fig. 4B), while PBMC and BAL fluid samples had frequencies of 0.5% and 4.9%, respectively. In contrast, samples from the Ctr-1 animal, which received only tNGFR CD8+ transductants, showed no NGFR-positive cells in the lymph node and few (0.06%) in the PBMC analyses (Fig. 4A). However, the Ctr-1 BAL fluid sample had a higher frequency of infused cells (13%) than the corresponding Exp-2 sample (5%).
To determine if transduction with CCR7/CD62L increased trafficking of infused cells to lymph node, we analyzed the percentage of infused cells that expressed the CCR7/CD62L vector in these samples. Because half of the infused cells were CTV labeled and the CCR7/CD62L vector expresses tNGFR, we could detect the number of infused cells with added homing markers (tNGFR+) as a proportion of the total infused cells (CTV+) found in the lymph node. In the lymph node sample, nearly all (98%) of the infused cells present were the CCR7/CD62L transductants, indicating strong preferential homing of these infused cells to this site. In contrast, the vast majority (94%) of the infused cells found in the BAL fluid samples and 86% of the cells in the PBMC were those without the CCR7/CD62L homing proteins (Fig. 4B), suggesting preferential trafficking away from these sites. Overall, our results are consistent with highly enhanced trafficking to lymph node induced by CCR7/CD62L transduction.
Decreased virus transmission in animals infused with engineered SIV-specific T cells.
Single-genome sequence analysis of viral RNA from plasma at or just after peak viremia (10 days postinfection) detected 7 and 8 of the SIVmac239X genotypes in Ctr-1 and -2, respectively. In contrast, only 3 and 5 genotypes were detected in Exp-1 and Exp-2, respectively. The difference between the mean numbers of genotypes detected between control and experimental groups (3.5) is statistically significant (P = 0.044) (Fig. 5).
FIG 5.
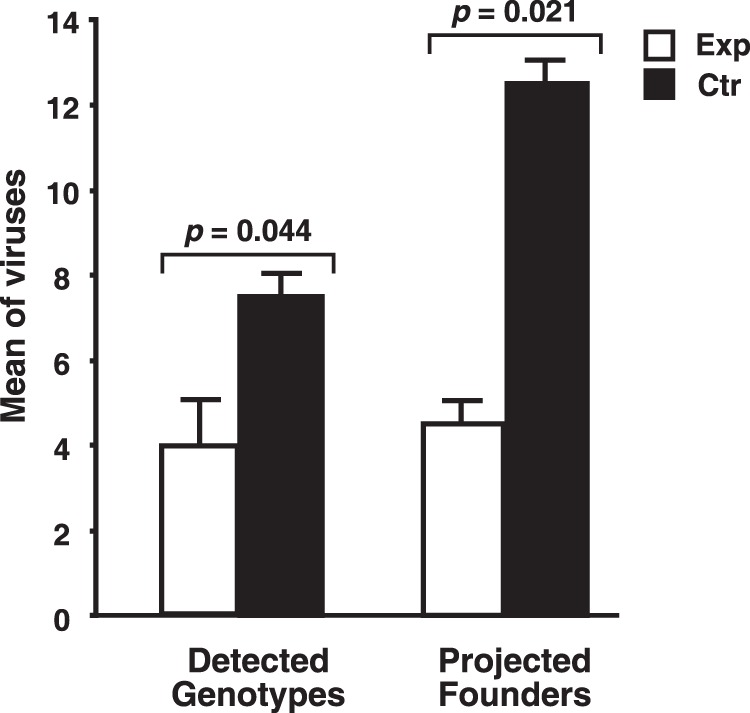
Analysis of founder virus genotypes. A graph of both the observed and projected values representing the means of the numbers of genotypes detected by single-genome sequencing at peak plasma viremia are presented with standard error bars. Statistical probabilities of a null hypothesis calculated by a single-tailed Student's t test are displayed above the bar graphs.
However, note that the numbers presented above represent the minimum number of founder viruses present, because, as the number of genotypes detected increases, the likelihood that one or more of the variants arose from multiple infection events also increases. This results in an undercount of the actual number of founder viruses transmitted by the inoculum. To account for this phenomenon, we developed an iterative Monte Carlo simulation method that empirically predicts the expected number of independent infection events, i.e., founder virus transmission, given the number of genotypes observed as the median from 103 independent trials for each of the possible numbers of genotypes that can be detected, i.e., from 1 to 10 (Table 1) (W. G. Alvord, V. I. Ayala, B. F. Keele, and D. E. Ott, unpublished data). On the basis of this analysis, we found that Exp-1 likely had 3 unique founding infection events and that Exp-2 had 6 whereas Ctr-1 had 14 and Ctr-2 had 11. After adjustment, the means (4.5 founders for experimental animals and 12.5 for the control animals) are still statistically significantly different (P = 0.021) (Fig. 5). Taken together, these data indicate that the infused SIV-specific CD8+ T cells decreased the number of transmitted/founder viruses that ultimately contributed to the establishment of viremia in the experimental animals.
TABLE 1.
Monte Carlo simulation-derived projected founder viruses
| No. of genotypes observed | Median no. of projected founder virus genotypes | Minimuma | Maximumb |
|---|---|---|---|
| 1 | 1 | 1 | 1 |
| 2 | 2 | 2 | 5 |
| 3 | 3 | 3 | 7 |
| 4 | 5 | 4 | 10 |
| 5 | 6 | 5 | 14 |
| 6 | 8 | 6 | 18 |
| 7 | 11 | 7 | 24 |
| 8 | 14 | 8 | 34 |
| 9 | 18 | 9 | 47 |
| 10 | 28 | 10 | 77 |
Data represent the minimum number of genotypes required to observe all 10 genotypes.
Data represent the maximum number of genotypes required to observe all 10 genotypes.
Elevated plasma IFN-γ levels.
One parameter that is used to measure the in vivo activity of infused antigen-specific CD8+ T cells is that of specific increases in plasma cytokine numbers (53), in this case, IFN-γ, which is expressed by our TCR transductants. ELISA analysis of plasma samples from both Exp-1 and -2 detected a 10-fold increase in plasma IFN-γ levels 10 days postinfusion in both animals (Fig. 6). The IFN-γ levels fell to baseline at the 17-day time point for Exp-1, while Exp-2's levels, while peaking lower than those seen with Exp-1, remained clearly elevated for 51 days. In contrast, there was no appreciable increase in IFN-γ levels in the parallel plasma samples from either of the control animals, which received only tNGFR-marked cells. Thus, the increased IFN-γ levels are consistent with the timing of the infused antigen-specific CD8+ T cells and with a functional response to virus-infected cells that reduced the numbers of founder viruses in the experimental animals.
FIG 6.
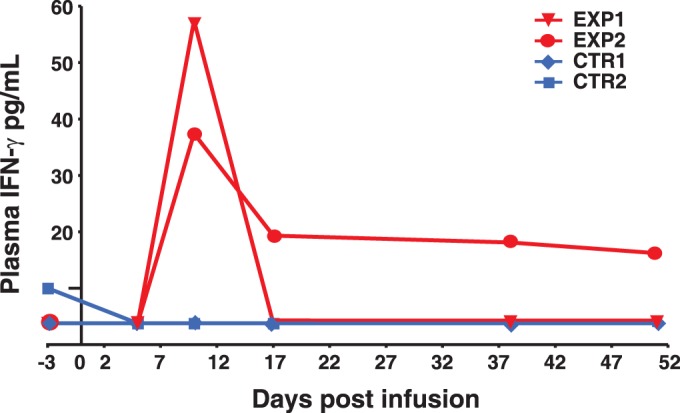
Induction of plasma IFN-γ. A graph of postinfusion plasma IFN-γ detected by ELISA measured 0 to 51 days postinfection is presented.
No effect on viremia.
Despite the reduction in the number of transmitted/founder viruses contributing to viremia, the levels of plasma viral RNA, PBMC-associated viral DNA, and CD4+ T cells were similar among the animals through the acute stage and into the chronic stage, with no clear difference in these infection parameters among the animals (Fig. 7).
FIG 7.

Blood infection parameters. Graphs of plasma viral RNA loads (top), cell-associated viral DNA loads (middle), and CD4+ T-cell counts (bottom) in blood from infected individuals are presented. Dashed lines indicate the level of detection for the PCR assays. Eq, equivalent.
DISCUSSION
The nearly 70% reduction in the levels of founder viruses observed here demonstrates that adoptive transfer of a large antiviral T-cell response rapidly after exposure to virus can effectively reduce the number of distinct infection events that contribute to establishment of viremia in an SIV/rhesus macaque system. This finding indicates that a robust, timed T-cell response can interfere with the establishment of infection that successfully contributes to viremia and disseminated viral replication.
It is important that we used a large-to-overwhelming inoculum to ensure establishment of infection with just a single intrarectal inoculation. Indeed, the 12.5 calculated founder viruses that we observe in the control animals is consistent with the expected transmission of 12 founder viruses based on the inoculation dose used in this study (33). Notably, this dose is considerably higher than the doses used in typical contemporary rhesus macaque vaccine studies which seek to replicate human mucosal transmission of HIV in which only a single transmitted/founder virus is observed in nearly 80% of cases (54). To model this, animals are repeatedly mucosally challenged with titers of inocula intended to infect only a fraction of unvaccinated animals per challenge, typically transmitting one or a few distinct viral variants (52, 55). With the high-dose inoculation used in this study, it is unlikely that the infused cells should have completely eliminated founder virus transmission.
While exhibiting fewer transmitted/founder viruses, the experimental animals receiving antiviral T cells had viremic parameters similar to those seen with the control animals. This was expected, because the transfer of only one transmitted founder virus appears to be sufficient to produce high levels of viremia. Indeed, Liu et al. observed similar levels of viremia in rhesus macaques inoculated with viral doses spanning 3 orders of magnitude (55). Additionally, it is not clear how many of the infused cells persisted beyond the first couple of weeks. Thus, transmission of fewer founder viruses did not alter the disease course after the establishment of infection, as expected.
Overall, our infused T cells did not persist at appreciable levels in blood, while the BAL fluid samples contained the largest relative frequency of infused endogenous cells over the follow-up period. Consistent with these findings, preferential retention of infused cells in the lung, often referred to as lung entrapment, has been observed after adoptive transfers of both T-cells and stem cells (12, 21, 24, 56). While this pattern of trafficking and persistence of the infused cells may be related in part to the preinfusion culture conditions, especially in the case of expansion during extended culture, activated T cells have been observed to reside in normal lung in a process that appears to involve LFA-1/ICAM-1 adhesion (44, 46). Thus, pulmonary localization by infused T cells might in part reflect a physiologically relevant process. It is also important that the absolute number of infused cells localized to the lung and the fraction of the infused cells that this frequency represents are not clear from BAL fluid sampling, underscoring some of the challenges in definitive characterization of trafficking of infused cells after adoptive transfer.
The localization and fate of the infused cells are important and yet poorly understood parameters in adoptive-transfer experiments. Here we show that homing to lymph node tissues can be manipulated by T-cell engineering: the CCR7/CD62L transductants preferentially homed to the lymph node while mostly avoiding the lung. These results provide a proof of principle for using this type of molecular engineering to enhance trafficking of infused antiviral T cells to lymphoid organs of interest.
This engineered T-cell approach, applied to studies of T-cell immunology and control of AIDS viruses by cellular immune responses through use of adoptive transfer in nonhuman primates, opens up a variety of possibilities. By producing T cells with defined TCRs having effective anti-SIV activity, rhesus macaque adoptive-transfer experiments can be carried out on naive animals or those that might be missing a particular response. Additionally, transducing T cells with effective TCRs en masse enables adoptive-transfer experiments with large numbers of virus-specific cells, expanded with fewer stimulation cycles and better persistence (results reported here and unpublished data analyzed by V. I. Ayala, M. T. Trivett, S. Jain, and D. E. Ott) than in our prior studies performed with extensively expanded native SIV-specific T-cell clones (12, 21).
The ability to easily and robustly measure the number of viral variants transmitted during infection using a defined system provides a powerful tool for understanding viral transmission. Here, we expand the use of the SIVmac239X 10 variant “synthetic swarm” transmitted/founder virus enumeration strategy (33) to assess the ability of adoptively transferred engineered T cells with demonstrated anti-SIV activity to effect a reduction in the number of transmitted/founder variants contributing to systemic infection after a high-dose intrarectal inoculation. To overcome the near certainty of an undercount of variants due to the occurrence of multiple infection events by 1 or more of the 10 individual genotypes at the inoculum level used, we used iterative Monte Carlo simulations to generate a conversion table that projects the most probable number of founder viruses present for a given number of observed genotypes. Our method presented here extends the utility of this powerful SIVmac293X system by enabling clearer interpretation of experiments with high-dose inoculations and challenges that would otherwise be confounded by multiple infections.
Our finding that the presence of a strong cellular immune response can limit the establishment of distinct transmitted/founder viruses indicates that an actively poised cellular immune response could supplement natural bottlenecks (57), e.g., innate immunity, intrinsic resistance, or mucosal barriers, and could further reduce or prevent infection. By necessity, our experimental design required a high-dose, clearly nonphysiological viral inoculum to ensure the establishment of infection to coordinate with the timing of T-cell infusion. Under conditions more closely approximating typical mucosal transmission of HIV, i.e., low inoculum levels that produce at most only one or two founder viruses (54), potent antiviral cellular immune responses could potentially protect high-risk individuals from infection (58). Thus, the presence of effective antiviral T cells during infection could eliminate nascent foci of infection before they become established.
ACKNOWLEDGMENTS
We thank Kelli Oswald, Rebecca Shoemaker, and Randy Fast for viral load analyses; Vicky Coulter, Adam Wiles, Rodney Wiles, and Don Johnson for sample processing; Carolyn Reid for sequencing the SIVmac239X virus; Abigail Lara and James Thomas for performing Trucount analysis; and Jeff Lifson for helpful input and critical readings of the manuscript. We thank Hans-Peter Kleim for providing the Phoenix RD114 clone 22 packaging cell line. The following reagents were obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: human IL-2 from Hoffmann-La Roche Inc., Nutley, NJ; α4β7 (catalog no. 11718) from A. A. Ansari; and SL8 peptide/MHC tetramer from the NIH Tetramer Facility, Emory University, Atlanta, GA.
This project was funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health, under contract no. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
REFERENCES
- 1.Hel Z, Nacsa J, Tryniszewska E, Tsai W-P, Parks RW, Montefiori DC, Felber BK, Tartaglia J, Pavlakis GN, Franchini G. 2002. Containment of simian immunodeficiency virus infection in vaccinated macaques: correlation with the magnitude of virus-specific pre- and postchallenge CD4+ and CD8+ T cell responses. J Immunol 169:4778–4787. doi: 10.4049/jimmunol.169.9.4778. [DOI] [PubMed] [Google Scholar]
- 2.Hazuda DJ, Young SD, Guare JP, Anthony NJ, Gomez RP, Wai JS, Vacca JP, Handt L, Motzel SL, Klein HJ, Dornadula G, Danovich RM, Witmer MV, Wilson KAA, Tussey L, Schleif WA, Gabryelski LS, Jin L, Miller MD, Casimiro DR, Emini EA, Shiver JW. 2004. Integrase inhibitors and cellular immunity suppress retroviral replication in rhesus macaques. Science 305:528–532. doi: 10.1126/science.1098632. [DOI] [PubMed] [Google Scholar]
- 3.Fernandez CS, Smith MZ, Batten CJ, de Rose R, Reece JC, Rollman E, Venturi V, Davenport MP, Kent SJ. 2007. Vaccine-induced T cells control reversion of AIDS virus immune escape mutants. J Virol 81:4137–4144. doi: 10.1128/JVI.02193-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barouch DH, Kunstman J, Kuroda MJ, Schmitz JE, Santra S, Peyerl FW, Krivulka GR, Beaudry K, Lifton MA, Gorgone DA, Montefiori DC, Lewis MG, Wolinsky SM, Letvin NL. 2002. Eventual AIDS vaccine failure in a rhesus monkey by viral escape from cytotoxic T lymphocytes. Nature 415:335–339. doi: 10.1038/415335a. [DOI] [PubMed] [Google Scholar]
- 5.Ueno T, Idegami Y, Motozono C, Oka S, Takiguchi M. 2007. Altering effects of antigenic variations in HIV-1 on antiviral effectiveness of HIV-specific CTLs. J Immunol 178:5513–5523. doi: 10.4049/jimmunol.178.9.5513. [DOI] [PubMed] [Google Scholar]
- 6.Loffredo JT, Burwitz BJ, Rakasz EG, Spencer SP, Stephany JJ, Vela JPG, Martin SR, Reed J, Piaskowski SM, Furlott J, Weisgrau KL, Rodrigues DS, Soma T, Napoé G, Friedrich TC, Wilson NA, Kallas EG, Watkins DI. 2007. The antiviral efficacy of simian immunodeficiency virus-specific CD8+ T cells is unrelated to epitope specificity and is abrogated by viral escape. J Virol 81:2624–2634. doi: 10.1128/JVI.01912-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Loffredo JT, Sidney J, Wojewoda C, Dodds E, Reynolds MR, Napoé G, Mothé BR, O'Connor DH, Wilson NA, Watkins DI, Sette A. 2004. Identification of seventeen new simian immunodeficiency virus-derived CD8+ T cell epitopes restricted by the high frequency molecule, Mamu-A*02, and potential escape from CTL recognition. J Immunol 173:5064–5076. doi: 10.4049/jimmunol.173.8.5064. [DOI] [PubMed] [Google Scholar]
- 8.Jin X, Bauer DE, Tuttleton SE, Lewin S, Gettie A, Blanchard J, Irwin CE, Safrit JT, Mittler J, Weinberger L, Kostrikis LG, Zhang L, Perelson AS, Ho DD. 1999. Dramatic rise in plasma viremia after CD8(+) T cell depletion in simian immunodeficiency virus-infected macaques. J Exp Med 189:991–998. doi: 10.1084/jem.189.6.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Metzner KJ, Jin X, Lee FV, Gettie A, Bauer DE, Di Mascio M, Perelson AS, Marx PA, Ho DD, Kostrikis LG, Connor RI. 2000. Effects of in vivo CD8(+) T cell depletion on virus replication in rhesus macaques immunized with a live, attenuated simian immunodeficiency virus vaccine. J Exp Med 191:1921–1931. doi: 10.1084/jem.191.11.1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lifson JD, Rossio JL, Piatak M, Parks T, Li L, Kiser R, Coalter V, Fisher B, Flynn BM, Czajak S, Hirsch VM, Reimann KA, Schmitz JE, Ghrayeb J, Bischofberger N, Nowak MA, Desrosiers RC, Wodarz D. 2001. Role of CD8(+) lymphocytes in control of simian immunodeficiency virus infection and resistance to rechallenge after transient early antiretroviral treatment. J Virol 75:10187–10199. doi: 10.1128/JVI.75.21.10187-10199.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reynolds MR, Rakasz E, Skinner PJ, White C, Abel K, Ma Z-M, Compton L, Napoé G, Wilson N, Miller CJ, Haase A, Watkins DI. 2005. CD8+ T-lymphocyte response to major immunodominant epitopes after vaginal exposure to simian immunodeficiency virus: too late and too little. J Virol 79:9228–9235. doi: 10.1128/JVI.79.14.9228-9235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Minang JT, Trivett MT, Bolton DL, Trubey CM, Estes JD, Li Y, Smedley J, Pung R, Rosati M, Jalah R, Pavlakis GN, Felber BK, Piatak M, Roederer M, Lifson JD, Ott DE, Ohlen C. 2010. Distribution, persistence, and efficacy of adoptively transferred central and effector memory-derived autologous simian immunodeficiency virus-specific CD8+ T cell clones in rhesus macaques during acute infection. J Immunol 184:315–326. doi: 10.4049/jimmunol.0902410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berger C, Turtle CJ, Jensen MC, Riddell SR. 2009. Adoptive transfer of virus-specific and tumor-specific T cell immunity. Curr Opin Immunol 21:224–232. doi: 10.1016/j.coi.2009.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Greenberg PD, Finch RJ, Gavin MA, Kalos M, Lewinsohn DA, Lonergan M, Lord JD, Nelson BH, Ohlen C, Sing AP, Warren EH, Yee C, Riddell SR. 1998. Genetic modification of T-cell clones for therapy of human viral and malignant diseases. Cancer J Sci Am 4(Suppl 1):S100–S105. [PubMed] [Google Scholar]
- 15.Restifo NP, Dudley ME, Rosenberg SA. 2012. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol 12:269–281. doi: 10.1038/nri3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Phan GQ, Rosenberg SA. 2013. Adoptive cell transfer for patients with metastatic melanoma: the potential and promise of cancer immunotherapy. Cancer Control 20:289–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barrett DM, Grupp SA, June CH. 2015. Chimeric antigen receptor- and TCR-modified T cells enter Main Street and Wall Street. J Immunol 195:755–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang L, Morgan RA. 2012. Genetic engineering with T cell receptors. Adv Drug Deliv Rev 64:756–762. doi: 10.1016/j.addr.2011.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morgan RA, Dudley ME, Yu YY, Zheng Z, Robbins PF, Theoret MR, Wunderlich JR, Hughes MS, Restifo NP, Rosenberg SA. 2003. High efficiency TCR gene transfer into primary human lymphocytes affords avid recognition of melanoma tumor antigen glycoprotein 100 and does not alter the recognition of autologous melanoma antigens. J Immunol 171:3287–3295. doi: 10.4049/jimmunol.171.6.3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, Royal RE, Topalian SL, Kammula US, Restifo NP, Zheng Z, Nahvi A, de Vries CR, Rogers-Freezer LJ, Mavroukakis SA, Rosenberg SA. 2006. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 314:126–129. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bolton DL, Minang JT, Trivett MT, Song K, Tuscher JJ, Li Y, Piatak M Jr, O'Connor D, Lifson JD, Roederer M, Ohlen C. 2010. Trafficking, persistence, and activation state of adoptively transferred allogeneic and autologous simian immunodeficiency virus-specific CD8(+) T cell clones during acute and chronic infection of rhesus macaques. J Immunol 184:303–314. doi: 10.4049/jimmunol.0902413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mee ET, Stebbings R, Hall J, Giles E, Almond N, Rose NJ. 2014. Allogeneic lymphocyte transfer in MHC-identical siblings and MHC-identical unrelated Mauritian cynomolgus macaques. PLoS One 9:e88670. doi: 10.1371/journal.pone.0088670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Berger C, Jensen MC, Lansdorp PM, Gough M, Elliott C, Riddell SR. 2008. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J Clin Invest 118:294–305. doi: 10.1172/JCI32103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Greene JM, Lhost JJ, Hines PJ, Scarlotta M, Harris M, Burwitz BJ, Budde ML, Dudley DM, Pham N, Cain B, Mac Nair CE, Weiker MK, O'Connor SL, Friedrich TC, O'Connor DH. 2013. Adoptive transfer of lymphocytes isolated from simian immunodeficiency virus SIVmac239Deltanef-vaccinated macaques does not affect acute-phase viral loads but may reduce chronic-phase viral loads in major histocompatibility complex-matched recipients. J Virol 87:7382–7392. doi: 10.1128/JVI.00348-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mohns MS, Greene JM, Cain BT, Pham NH, Gostick E, Price DA, O'Connor DH. 2015. Expansion of simian immunodeficiency virus (SIV)-specific CD8 T cell lines from SIV-naive Mauritian cynomolgus macaques for adoptive transfer. J Virol 89:9748–9757. doi: 10.1128/JVI.00993-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Greene JM, Burwitz BJ, Blasky AJ, Mattila TL, Hong JJ, Rakasz EG, Wiseman RW, Hasenkrug KJ, Skinner PJ, O'Connor SL, O'Connor DH. 2008. Allogeneic lymphocytes persist and traffic in feral MHC-matched Mauritian cynomolgus macaques. PLoS One 3:e2384. doi: 10.1371/journal.pone.0002384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barsov EV, Trivett MT, Minang JT, Sun H, Ohlen C, Ott DE. 2011. Transduction of SIV-specific TCR genes into rhesus macaque CD8 T cells conveys the ability to suppress SIV replication. PLoS One 6:e23703. doi: 10.1371/journal.pone.0023703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.National Research Council. 2011. Guide for care and use of laboratory animals. National Academies Press, Washington, DC. [Google Scholar]
- 29.Riddell SR, Greenberg PD. 1990. The use of anti-CD3 and anti-CD28 monoclonal antibodies to clone and expand human antigen-specific T cells. J Immunol Methods 128:189–201. doi: 10.1016/0022-1759(90)90210-M. [DOI] [PubMed] [Google Scholar]
- 30.Berger C, Huang ML, Gough M, Greenberg PD, Riddell SR, Kiem HP. 2001. Nonmyeloablative immunosuppressive regimen prolongs in vivo persistence of gene-modified autologous T cells in a nonhuman primate model. J Virol 75:799–808. doi: 10.1128/JVI.75.2.799-808.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Andersen H, Barsov EV, Trivett MT, Trubey CM, Giavedoni LD, Lifson JD, Ott DE, Ohlen C. 2007. Transduction with human telomerase reverse transcriptase immortalizes a rhesus macaque CD8+ T cell clone with maintenance of surface marker phenotype and function. AIDS Res Hum Retroviruses 23:456–465. doi: 10.1089/aid.2006.0194. [DOI] [PubMed] [Google Scholar]
- 32.Neff T, Peterson LJ, Morris JC, Thompson J, Zhang X, Horn PA, Thomasson BM, Kiem HP. 2004. Efficient gene transfer to hematopoietic repopulating cells using concentrated RD114-pseudotype vectors produced by human packaging cells. Mol Ther 9:157–159. doi: 10.1016/j.ymthe.2003.11.011. [DOI] [PubMed] [Google Scholar]
- 33.Del Prete GQ, Park H, Fennessey CM, Reid C, Lipkey L, Newman L, Oswald K, Kahl C, Piatak M, Quiñones OA, Alvord WG, Smedley J, Estes JD, Lifson JD, Picker LJ, Keele BF. 2014. Molecularly tagged simian immunodeficiency virus SIVmac239 synthetic swarm for tracking independent infection events. J Virol 88:8077–8090. doi: 10.1128/JVI.01026-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lazarovits AI, Moscicki RA, Kurnick JT, Camerini D, Bhan AK, Baird LG, Erikson M, Colvin RB. 1984. Lymphocyte activation antigens. I. A monoclonal antibody, anti-Act I, defines a new late lymphocyte activation antigen. J Immunol 133:1857–1862. [PubMed] [Google Scholar]
- 35.Cline AN, Bess JW, Piatak M, Lifson JD. 2005. Highly sensitive SIV plasma viral load assay: practical considerations, realistic performance expectations, and application to reverse engineering of vaccines for AIDS. J Med Primatol 34:303–312. doi: 10.1111/j.1600-0684.2005.00128.x. [DOI] [PubMed] [Google Scholar]
- 36.Team RC. 2014. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: http://www.R-project.org/. [Google Scholar]
- 37.Barouch DH, Ghneim K, Bosche WJ, Li Y, Berkemeier B, Hull M, Bhattacharyya S, Cameron M, Liu J, Smith K, Borducchi E, Cabral C, Peter L, Brinkman A, Shetty M, Li H, Gittens C, Baker C, Wagner W, Lewis MG, Colantonio A, Kang HJ, Li W, Lifson JD, Piatak M Jr, Sekaly RP. 2016. Rapid inflammasome activation following mucosal SIV infection of rhesus monkeys. Cell 165:656–667. doi: 10.1016/j.cell.2016.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hinrichs CS, Borman ZA, Cassard L, Gattinoni L, Spolski R, Yu Z, Sanchez-Perez L, Muranski P, Kern SJ, Logun C, Palmer DC, Ji Y, Reger RN, Leonard WJ, Danner RL, Rosenberg SA, Restifo NP. 2009. Adoptively transferred effector cells derived from naive rather than central memory CD8+ T cells mediate superior antitumor immunity. Proc Natl Acad Sci U S A 106:17469–17474. doi: 10.1073/pnas.0907448106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kalams SA, Walker BD. 1998. The critical need for CD4 help in maintaining effective cytotoxic T lymphocyte responses. J Exp Med 188:2199–2204. doi: 10.1084/jem.188.12.2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Planz O, Ehl S, Furrer E, Horvath E, Brundler MA, Hengartner H, Zinkernagel RM. 1997. A critical role for neutralizing-antibody-producing B cells, CD4(+) T cells, and interferons in persistent and acute infections of mice with lymphocytic choriomeningitis virus: implications for adoptive immunotherapy of virus carriers. Proc Natl Acad Sci U S A 94:6874–6879. doi: 10.1073/pnas.94.13.6874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ayala VI, Trivett MT, Coren LV, Jain S, Bohn PS, Wiseman RW, O'Connor DH, Ohlen C, Ott DE. 2016. A novel SIV gag-specific CD4T-cell clone suppresses SIV replication in CD4T cells revealing the interplay between antiviral effector cells and their infected targets. Virology 493:100–112. doi: 10.1016/j.virol.2016.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jain S, Trivett MT, Ayala VI, Ohlen C, Ott DE. 2015. African green monkey TRIM5alpha restriction in simian immunodeficiency virus-specific rhesus macaque effector CD4 T cells enhances their survival and antiviral function. J Virol 89:4449–4456. doi: 10.1128/JVI.03598-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bonini C, Grez M, Traversari C, Ciceri F, Marktel S, Ferrari G, Dinauer M, Sadat M, Aiuti A, Deola S, Radrizzani M, Hagenbeek A, Apperley J, Ebeling S, Martens A, Kolb HJ, Weber M, Lotti F, Grande A, Weissinger E, Bueren JA, Lamana M, Falkenburg JH, Heemskerk MH, Austin T, Kornblau S, Marini F, Benati C, Magnani Z, Cazzaniga S, Toma S, Gallo-Stampino C, Introna M, Slavin S, Greenberg PD, Bregni M, Mavilio F, Bordignon C. 2003. Safety of retroviral gene marking with a truncated NGF receptor. Nat Med 9:367–369. doi: 10.1038/nm0403-367. [DOI] [PubMed] [Google Scholar]
- 44.Dixon AE, Mandac JB, Martin PJ, Hackman RC, Madtes DK, Clark JG. 2000. Adherence of adoptively transferred alloreactive Th1 cells in lung: partial dependence on LFA-1 and ICAM-1. Am J Physiol Lung Cell Mol Physiol 279:L583–L591. [DOI] [PubMed] [Google Scholar]
- 45.Kelsen J, Agnholt J, Falborg L, Nielsen JT, Romer JL, Hoffmann HJ, Dahlerup JF. 2004. Indium-labelled human gut-derived T cells from healthy subjects with strong in vitro adhesion to MAdCAM-1 show no detectable homing to the gut in vivo. Clin Exp Immunol 138:66–74. doi: 10.1111/j.1365-2249.2004.02578.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Galkina E, Thatte J, Dabak V, Williams MB, Ley K, Braciale TJ. 2005. Preferential migration of effector CD8+ T cells into the interstitium of the normal lung. J Clin Invest 115:3473–3483. doi: 10.1172/JCI24482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bernhard H, Neudorfer J, Gebhard K, Conrad H, Hermann C, Nährig J, Fend F, Weber W, Busch DH, Peschel C. 2008. Adoptive transfer of autologous, HER2-specific, cytotoxic T lymphocytes for the treatment of HER2-overexpressing breast cancer. Cancer Immunol Immunother 57:271–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shi GX, Harrison K, Wilson GL, Moratz C, Kehrl JH. 2002. RGS13 regulates germinal center B lymphocytes responsiveness to CXC chemokine ligand (CXCL)12 and CXCL13. J Immunol 169:2507–2515. doi: 10.4049/jimmunol.169.5.2507. [DOI] [PubMed] [Google Scholar]
- 49.Peschon JJ, Slack JL, Reddy P, Stocking KL, Sunnarborg SW, Lee DC, Russell WE, Castner BJ, Johnson RS, Fitzner JN, Boyce RW, Nelson N, Kozlosky CJ, Wolfson MF, Rauch CT, Cerretti DP, Paxton RJ, March CJ, Black RA. 1998. An essential role for ectodomain shedding in mammalian development. Science 282:1281–1284. doi: 10.1126/science.282.5392.1281. [DOI] [PubMed] [Google Scholar]
- 50.Yang S, Liu F, Wang QJ, Rosenberg SA, Morgan RA. 2011. The shedding of CD62L (L-selectin) regulates the acquisition of lytic activity in human tumor reactive T lymphocytes. PLoS One 6:e22560. doi: 10.1371/journal.pone.0022560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Del Prete GQ, Ailers B, Moldt B, Keele BF, Estes JD, Rodriguez A, Sampias M, Oswald K, Fast R, Trubey CM, Chertova E, Smedley J, LaBranche CC, Montefiori DC, Burton DR, Shaw GM, Markowitz M, Piatak M Jr, KewalRamani VN, Bieniasz PD, Lifson JD, Hatziioannou T. 2014. Selection of unadapted, pathogenic SHIVs encoding newly transmitted HIV-1 envelope proteins. Cell Host Microbe 16:412–418. doi: 10.1016/j.chom.2014.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vaccari M, Keele BF, Bosinger SE, Doster MN, Ma Z-M, Pollara J, Hryniewicz A, Ferrari G, Guan Y, Forthal DN, Venzon D, Fenizia C, Morgan T, Montefiori D, Lifson JD, Miller CJ, Silvestri G, Rosati M, Felber BK, Pavlakis GN, Tartaglia J, Franchini G. 2013. Protection afforded by an HIV vaccine candidate in macaques depends on the dose of SIVmac251 at challenge exposure. J Virol 87:3538–3548. doi: 10.1128/JVI.02863-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, June CH. 2011. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med 3:95ra73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Keele BF, Giorgi EE, Salazar-Gonzalez JF, Decker JM, Pham KT, Salazar MG, Sun C, Grayson T, Wang S, Li H, Wei X, Jiang C, Kirchherr JL, Gao F, Anderson JA, Ping LH, Swanstrom R, Tomaras GD, Blattner WA, Goepfert PA, Kilby JM, Saag MS, Delwart EL, Busch MP, Cohen MS, Montefiori DC, Haynes BF, Gaschen B, Athreya GS, Lee HY, Wood N, Seoighe C, Perelson AS, Bhattacharya T, Korber BT, Hahn BH, Shaw GM. 2008. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc Natl Acad Sci U S A 105:7552–7557. doi: 10.1073/pnas.0802203105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu J, Keele BF, Li H, Keating S, Norris PJ, Carville A, Mansfield KG, Tomaras GD, Haynes BF, Kolodkin-Gal D, Letvin NL, Hahn BH, Shaw GM, Barouch DH. 2010. Low-dose mucosal simian immunodeficiency virus infection restricts early replication kinetics and transmitted virus variants in rhesus monkeys. J Virol 84:10406–10412. doi: 10.1128/JVI.01155-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schrepfer S, Deuse T, Reichenspurner H, Fischbein MP, Robbins RC, Pelletier MP. 2007. Stem cell transplantation: the lung barrier. Transplant Proc 39:573–576. doi: 10.1016/j.transproceed.2006.12.019. [DOI] [PubMed] [Google Scholar]
- 57.Keele BF, Estes JD. 2011. Barriers to mucosal transmission of immunodeficiency viruses. Blood 118:839–846. doi: 10.1182/blood-2010-12-325860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Picker LJ, Hansen SG, Lifson JD. 2012. New paradigms for HIV/AIDS vaccine development. Annu Rev Med 63:95–111. doi: 10.1146/annurev-med-042010-085643. [DOI] [PMC free article] [PubMed] [Google Scholar]