

Abstract
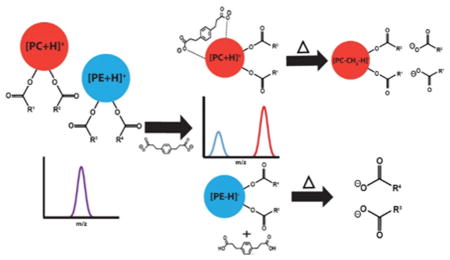
The [M + H]+ cations formed upon electrospray ionization of the glycerophospholipids phosphatidylcholine (PC) and phosphatidylethanolamine (PE) show distinct reactivities upon gas-phase reactions with doubly deprotonated 1,4-phenylenedipropionic acid (PDPA). PC cations undergo charge inversion via adduct formation with subsequent methyl cation and proton transfer to the acid to yield [PC − CH3]− anions. These demethylated PC anions fragment upon ion trap collision-induced dissociation (CID) to yield products that reveal fatty acid chain lengths and degrees of unsaturation. PE cations, on the other hand, undergo charge inversion via double proton transfer to the two carboxylate moieties in doubly deprotonated PDPA to yield [PE − H]− anions. These anions also fragment upon ion trap CID to yield product ions indicative of chain lengths and degrees of unsaturation in the fatty acyl moieties. Advantage is taken of this distinct reactivity to separate isomeric and isobaric PC and PE cations present in mass spectra of lipid mixtures. A cation precursor ion population containing a mixture of PE and PC cations is mass-selected and subjected to ion/ion charge inversion reactions that result in separation of PC and PE anions into different mass-to-charge ratios. Mass selection and subsequent ion trap CID of the lipid anions allows for the characterization of the isomeric lipids within each subclass. The charge inversion approach described here is demonstrated to provide increased signal-to-noise ratios for detection of PCs and PEs relative to the standard negative ionization approach as well as improved mixture analysis performance.
Graphical abstract
The analysis of complex lipid mixtures represents an expanding area of research in the field of mass spectrometry (MS). Soft ionization techniques, such as electrospray ionization (ESI)1 and matrix-assisted laser desorption/ionization (MALDI),2 as well as tandem MS (MS/MS) have played key roles in enabling mass spectrometry to be employed in the study of cellular lipids in biological systems.3,4 Lipids can be classified as either nonpolar (e.g., cholesterol and triacylglycerols) or polar (e.g., glycerophospholipids, sphingolipids, and glycolipids). Phospholipids are the predominant lipid class in eukaroyotic cells and account for approximately 60 mol % of the total lipid.4 The abundant glycerophospholipids contain a phosphate group at the sn-3 position and fatty acid chains attached at the sn-1 and sn-2 positions on the glycerol backbone. The chemical moiety connected to the phosphate ultimately determines the phospholipid subclass. Subclasses such as phosphatidylethanolamines (PEs) and phosphatidylcholines (PCs) compose 75 mol % of phospholipids in eukaryotic membranes, where the PCs and PEs are typically present in a 3:2 ratio, respectively.4 Alteration of PE and PC lipid abundance can provide insight on the malignancy and the metastatic potential of several cancer types.5
Lipid extracts can be admitted to a (tandem) mass spectrometer either via direct infusion or liquid chromatography. Direct infusion MS, or shotgun lipidomics, is now widely deployed for qualitative and quantitative analysis of lipid extracts.6 This strategy is reliant on distinction of the ionized lipids based on either m/z ratio or MS/MS behavior. In this workflow, the unambiguous identification of isomeric or isobaric lipid ions can present an analytical challenge. Although high mass resolution can, in principle, enable the distinction of isobaric ions,7 isomer differentiation relies on MS/MS. For example, upon ESI in positive ion mode, PEs and PCs form abundant [M + H]+ ions that when subjected to collision-induced dissociation (CID) yield characteristic product ions at m/z 184 and [M + H − 141]+, respectively.8,9 These transitions are widely used in precursor ion and neutral loss scans to provide a sum composition identification of lipids (i.e., lipid class with total number of acyl chain carbons and number of degrees of unsaturation) in each class with PE and PC isomers detected in separate spectral acquisitions.10,11 This strategy does not provide acyl chain composition information, which is usually determined from CID spectra acquired in negative ion mode. Although [M − H]− anions from PE lipids can be formed by deprotonation,12 the quaternary ammonium moiety of PC requires the formation of an [M + X]− adduct ion (where X = Cl−, HCO2−, or CH3CO2−) to be observed in negative mode.13,14
Derivatization strategies can be used to derive subclass information as well as fatty acyl chain composition using a single ionization polarity. For example, Reid et al. have described strategies for the functional group specific derivatization of lipids for shotgun lipidomic applications15,16 that result in the conversion of initially isomeric subclass components (e.g., PE versus PC lipids) to different products with distinct masses. This approach, used in conjunction with high resolution mass analysis, essentially acts as a solution-phase, chemically selective separation method that facilitates confident lipid identification. In this work, we use selective gas-phase ion/ion chemistries to separate lipids that are either isomeric or isobaric.
We recently demonstrated the gas-phase transformation of PC cations into demethylated PC anions via ion/ion chemistry.17 Long-lived complexes comprised of PC cations and dicarboxylate dianions upon collisional activation transfer a methyl cation and proton from the PC analyte to the reagent. Dissociation of the demethylated PC anions generates structurally informative fragment anions that provide chain length and degree of unsaturation information for the fatty acid chains.17 In this study, we demonstrate gas-phase separation of PE and PC lipids via charge inversion ion/ion chemistry. Ion/ion reactions are performed between doubly deprotonated 1,4-phenylenedipropionic acid (PDPA) and the positive ions from nanoelectrospray (nESI) of PE and PC mixtures. The product ion spectrum following the ion/ion reaction displays singly deprotonated PE anions (i.e., [PE − H]−) and reagent-adducted PC anions (i.e., [PC + PDPA − H]−). Following the ion/ion reaction, ion trap collisional activation of the product anions generates structural information on the fatty acid chains as carboxylate anions. This approach is demonstrated to be able to identify multiple isomeric and isobaric components present in a single peak in the positive ion mass spectrum without recourse to condensed-phase separations or derivatization. The positive ion charge inversion approach is also compared with the conventional direct negative ionization approach and is shown to have an advantages in the reduction of chemical noise in lipid mixture mass analysis and for minimizing mixture complexities within an isolated precursor ion population.
EXPERIMENTAL SECTION
Materials
Methanol, chloroform, and ammonium hydroxide were purchased from Mallinckrodt (Phillipsburg, NJ). Acetonitrile, ammonium acetate, and 1,4-phenylenedipropionic acid (PDPA) were purchased from Sigma-Aldrich (St. Louis, MO). Polar liver extract (bovine), 1-heptadecanoyl-2-(9Z-tetradecenoyl)-sn-glycero-3-phosphocholine, and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine were purchased from Avanti Polar Lipids, Inc. (Alabaster, AL). An equimolar solution of the PC and PE standards was made in 50/50 (v/v) chloroform/methanol with 10 mM ammonium acetate. Solutions of the liver polar extract were prepared at concentrations of 50, 10, 5, and 0.5 μM (assuming an average lipid molecular weight of 760 g/mol) in a solution of 50/50 (v/v) chloroform/methanol with 10 mM ammonium acetate. The reagent anion, doubly deprotonated PDPA, was generated from a solution of PDPA at a concentration of ~220 μM in a solution of 49.5/49.5/1 (v/v/v) acetonitrile/methanol/ammonium hydroxide.
Nomenclature
Where possible, we adopt the shorthand nomenclature recommendations of Liebisch et al.18 Briefly, phosphatidylcholine and phosphatidylethanolamine are abbreviated as PC and PE, respectively. Lipid acyl chains are represented by the number of carbons and degree(s) of unsaturation (e.g., 18:1). Where the acyl chains of a glycerophospholipid are identified but their relative positions are not otherwise defined, the acyl chains are separated by an “_” (e.g., PC 16:0_18:1). Where the relative positions of two acyl chains have been determined, they are reported as separated with a “/” with the substituent at sn-1 on the left and sn-2 on the right (e.g., PC 16:0/18:1).
Mass Spectrometry
All experiments were performed on a TripleTOF 5600 triple quadrupole/time-of-flight mass spectrometer (SCIEX, Concord, ON, Canada), which has been modified to perform ion/ion reactions. The modifications are highly analogous to those described for a SCIEX Q-STAR quadrupole/time-of-flight instrument.19 The essential hardware modification is the ability to apply AC voltages (50–300 kHz, 50–150 V p-p) to the end plates of the q2 reaction cell to enable mutual storage of oppositely charged ions. Alternately pulsed nESI emitters sequentially inject anions and cations (250 ms ion accumulation times were used for each polarity), which are mass selected in transit in Q1 prior to their entry into the q2 reaction cell. Doubly deprotonated PDPA anions are first ionized, isolated in transit in Q1, and accumulated in the q2 reaction cell. Next, the singly charged lipid cations were ionized and transferred to the q2 cell to undergo a mutual storage reaction with the PDPA dianions for 50 ms. For the lipid extract, positively charged ions were monoisotopically isolated in transit in Q1 prior to accumulation in q2. Product anions resulting from the ion/ion reaction were isolated in q2 by ejecting unwanted ions via application of a high amplitude excitation at their resonant frequency. The precursor ions were subjected to single frequency resonance excitation (q = 0.23) to generate the product ion spectrum via CID. In some of the experiments with [PC + PDPA − H]− precursors, a second resonance excitation period was employed to excite the [PC − CH3]− first generation product ion. This was effected by changing the RF amplitude of q2 to bring the [PC − CH3]− to a q-value of 0.23 and using the same resonance excitation waveform that was used with the precursor ion.
RESULTS AND DISCUSSION
Separation of Synthetic PC and PE Lipids
Scheme 1 summarizes the reaction behavior of singly charged PC and PE cations with doubly deprotonated PDPA. We note that we are using PE to represent all forms of phosphoethanolamines (i.e., primary amine, monomethylamine, and dimethylamine). All forms generate [M − H]− ions upon direct negative ionization and via charge inversion of [M + H]+ with PDPA dianions (see Supporting Information Figures S-1 and S-2 for the charge inversion and CID behaviors of monomethyl and dimethyl PE, respectively). Furthermore, PCs or PEs with ether-linked chains as well as lipids with oxidized side-chains generate ions based on the headgroup identity. For both PCs and PEs, the doubly charged reagent anion gives rise to analyte ion charge inversion but via two distinct pathways: adduct formation for PCs and double proton transfer for PEs. The process of charge inversion proceeds through a long-lived complex comprised of the analyte and reagent ions, namely, [lipid + PDPA − H]−.18,20 In some cases, the noncovalent interactions within the complex are strong enough to result in an adducted analyte ion. In other cases, the noncovalent interactions within the complex are not strong enough to generate adduct ions. Instead, multiple protons are transferred within the complex followed by separation of the reagent and analyte. To achieve charge inversion via multiple proton transfer, the number of protons transferred must exceed the number required to neutralize the analyte ion. In the case of PCs, the major product is the [PC + PDPA − H]− anion, whereas for PEs, the [PE − H]− anion is generated.
Scheme 1.

Charge Inversion Reactions of (a) [PC + H]+ with [PDPA − 2H]2− Generating [PC + PDPA − H]− and (b) [PE + H]+ with [PDPA − 2H]2− Generating [PE − H]− and Neutral PDPA
The reaction phenomenology is illustrated with an equimolar (1 μM) two-component mixture of two isomeric synthetic PE and PC lipids (Figure 1(a)), (PC 17:0/14:1) and (PE 16:0/18:1), upon reaction with PDPA anions. Figure 1(b) shows the isolated cation population comprised of the mixture of [(PC 17:0/14:1) + H]+ and [(PE 16:0/18:1) + H]+ cations (theoretical m/z 718.538). Figure 1(c) shows the post-ion/ion reaction spectrum generated by storage of the mixture of isomeric cations in the presence of the PDPA dianions. The product ion spectrum following the ion/ion reactions shows the generation of a singly deprotonated PE (i.e., [(PE 16:0/18:1) − H]−) and a PDPA-adducted PC (i.e., ([(PC 17:0/14:1) + PDPA − H]−). Energetic transfer conditions to the time-of-flight analyzer produce a demethylated PC anion (i.e., [(PC 17:0/14:1) − CH3]−), which is derived from CID of PDPA-adducted PC anions (vide infra). Note that previous work has shown that no [PC − H]− ions are generated from ion/ion reactions with dianions of dicarboxylic acids.17 Hence, the ion/ion reaction leads to charge inversion of both cations and a separation of the two isomers on the m/z scale. When run separately, ion/ion reactions of [(PE 16:0/18:1) + H]+ and PDPA dianions exhibit charge inversion exclusively via double proton transfer (Figure S-3), whereas charge inversion ion/ion reactions of [(PC 17:0/14:1) + H]+ with PDPA dianions exhibit a dominant [(PC 17:0/14:1) + PDPA − H]− product and no evidence for [PC 17:0/14:1 − H]− (Figure S-4). Ultimately, the differences in the polar head groups of the lipids allow for gas-phase separation of isobaric or isomeric PE and PC lipids via distinct charge inversion processes. It is difficult to determine absolute charge inversion efficiency because the responses of the ion detector in the two polarity modes cannot be assumed to be equal. However, estimates made in previous work using a hybrid linear ion trap/triple quadrupole instrument (Sciex QTRAP 4000) suggested that the charge inversion efficiencies for PCs are roughly 50%.18
Figure 1.

(a) Structures of the ionized lipids [(PC 17:0/14:1) + H]+ and [(PE 16:0/18:1) + H]+. (b) Positive mode ionization and isolation of isomeric ion mixture (1 μM each). (c) Spectrum resulting from the ion/ion reaction between the isomeric ions [(PC 17:0/14:1) + H]+ and [(PE 16:0/18:1) + H]+ and the reagent dianion [PDPA − 2H]2−. Measured m/z values for the labeled peaks are provided in (b) and (c).
MS/MS of Charge-Inverted Product Anions
It is well-established that dissociation of deprotonated PE and demethylated PC anions generate product ions carrying structural information relating to the fatty acyl chains (i.e., number of carbons and degree(s) of unsaturation) carried by each phospholipid.21 In Figure 2, ion trap CID is performed on the mass-selected products of the ion/ion reaction associated with Figure 1 to generate structural information on both the PC 17:0/14:1 and PE 16:0/18:1 lipids. Collisional activation of the [(PC 17:0/14:1) + PDPA − H]− anion (Figure 2(a)) produces an abundant [(PC 17:0/14:1) − CH3]− product ion. The presence of the demethylated PC anion is consistent with a proton and methyl cation transfer from the PC lipid to the reagent PDPA. The absence of [PDPA − H]− signal indicates that transfer of a methyl cation and proton is a dominant pathway upon collisional activation, whereas proton transfer without methyl cation transfer, resulting in neutralization of the lipid, is a far less favorable pathway. Collisional activation of the [(PC 17:0/14:1) − CH3]− anion generates a spectrum dominated by fatty acid (FA) carboxylate fragment anions associated with the 17:0 and 14:1 chains (Figure 2(b)). The presence of the carboxylate fragment anions allows for facile determination of the carbon number and the degree of unsaturation in each fatty acid chain. Consistent with prior observations,22 the greater abundance of the [14:1 − H]− product ion relative to that of the [17:0 − H]− product ion is indicative of the 17:0 and 14:1 chains occupying the sn-1 and sn-2 positions, respectively. Results obtained from the ion trap CID of the [(PE 16:0/18:1) − H]− anion derived from the ion/ion reaction are displayed in Figure 2(c). Dominant contributions from the fatty acid carboxylate anions [16:0 − H]− and [18:1 − H]− are noted. The relative abundances of the FA anions are consistent with the 16:0 and 18:1 chains occupying the sn-1 and sn-2 positions, respectively.
Figure 2.

Ion trap CID of product ions generated via ion/ion reaction in the experiment of Figure 1. Activation of (a) [(PC 17:0/14:1) + PDPA − H]−, (b) [(PC 17:0/14:1) − CH3]−, and (c) [(PE 16:0/18:1) − H]−.
Application of Direct Ionization and Charge Inversion to a Lipid Extract
When an analyte of interest can readily form ions of either polarity, as is the case with PEs and PCs, charge inversion reactions can exploit this property to minimize “chemical noise”. This has been demonstrated previously with amino acids present in precipitated blood plasma.23 The charge inversion reaction, therefore, might be anticipated to provide an advantage in the analysis of complex lipid mixtures, particularly where lipids of interest are present at low abundance. A liver polar extract, reported by the supplier to be comprised of a high weight percentage of PC and PE lipids (42 and 26 wt/wt %) among other phospholipids (e.g., phosphatidylinositols, ~10%), and a low percentage of neutral lipids, was prepared at various concentrations in methanol/chloroform in the presence of ammonium acetate. Data were generated from these mixtures using direct negative ionization, direct positive ionization, and the charge inversion approach. As similar conditions as possible were used for each comparison (e.g., the same solution in the same nanospray tip at each concentration and the same ionization time (250 ms)). The major difference was the addition of the steps for anion accumulation (250 ms) and ion/ion reaction (50 ms) for the charge inversion experiment. Results obtained for a 50 μM total lipid concentration obtained from (a) direct negative ionization, (b) direct positive ionization, and (c) charge inversion of the population of ions in (b) via reaction with PDPA dianions are shown in Figure 3.
Figure 3.

Nano-ESI mass spectra of a bovine liver extract (50 μM total lipid concentration) obtained (a) via direct negative ionization, (b) direct positive ionization, and (c) charge inversion of the ions of (b) using [PDPA − 2H]2−. Nominal masses of ions discussed in the manuscript are indicated in the spectra. The tinted regions indicate likely groupings of PCs (light blue) and PEs (pink). In the negative ion spectra, contributions from [PC − CH3]− anions can fall among the deprotonated PE anions.
The spectra of Figure 3 are clearly distinct. The negative ion data generated directly (Figure 3(a)) presumably include a mixture of deprotonated PEs and [PC + CH3COO−]− ions, along with anions of any other mixture components that readily form under negative nESI conditions. There are also likely to be contributions from [PC − CH3]− ions from fragmentation of [PC + CH3COO−]− ions upon transfer to the time-of-flight analyzer, as was noted when the PE/PC mixture solution described above was subjected to direct negative ionization (see Figure S-5). All monoisotopic [PE − H]−, [PC − CH3]−, and [PC + CH3COO−]− ions (i.e., the sum of the mass numbers of the individual atoms of the most abundant isotopes, which corresponds to the numbers to the left of the decimal point for the measured masses) are even nominal mass. Oxidized and ether linked forms of PEs and PCs are also of even nominal mass. All major odd mass monoisotopic mass anions are therefore immediately identifiable as arising from other lipid classes or contaminant ions. For example, the prominent peaks at nominal m/z 885 and m/z 887 in Figure 3(a) are consistent with the phosphatidylinositols PI 38:4 and PI 38:3, respectively. The positive ion spectrum, on the other hand, presumably reflects protonated PEs, PC cations, as well as cations from any other components in the mixture that ionize well under positive ion nESI conditions. Figure 3(c) was derived via charge inversion of the positive ions in Figure 3(b) and reflects the ions that can undergo efficient charge inversion and clearly shows products that are spread over a wider m/z range than those derived from direct positive ionization and direct negative ionization. The ions of m/z greater than ~930 are likely to be predominantly [PC + PDPA − H]− ions, as confirmed by MS/MS experiments (vide infra), whereas the lower m/z ions are likely to be comprised of PE anions generated via double proton transfer, as confirmed by MS/MS (vide infra), and [PC − CH3]− ions from fragmentation of the higher m/z [M + PDPA − H]− species upon transfer to the time-of-flight analyzer. On the basis of the separated groups of peaks in Figure 3(c), it is possible to tentatively assign each grouping of peaks as comprised of either PE/[PC − CH3]− or PC species. For example, the grouping of peaks with the most abundant component at nominal m/z 744 in Figure 3(c) is assigned as PE/[PC − CH3]− group 1, the grouping of peaks with the most abundant component at nominal m/z 770 is assigned as PE/[PC − CH3]− group 2, and so forth. The likely locations of these groupings, assigned based on Figure 3(c), are identified in the mass spectra of Figure 3(a and b). The PC group 2 ions at nominal m/z 1008 ([M + PDPA − H]−) correspond to the ions at m/z 788 (i.e., the corresponding [M + H]+ ions) in Figure 3(b) and the ions at m/z 846 (i.e., the corresponding [M + CH3COO−]− ions) in Figure 3(a). There are no contributions from [PC − CH3] ions in the direct positive ion mass spectrum of Figure 3(b). Hence, the labeled groupings are either PE or PC in Figure 3(b).
Figure 4 compares mass spectra derived from the same three types of experiments for a 100-fold dilution (same solvent composition and 10 mM ammonium acetate) of the 50 μM lipid solution (i.e., a 0.5 μM total lipid solution). Data for 5-and 10-fold dilutions are provided in Figures S-6 and S-7, respectively. The 100-fold dilution is the lowest concentration at which ions from most of the lipid groupings can still be detected using the direct negative ionization mode (Figure 4(a)). Evidence for the PC1 and PC4 groups has essentially fallen into the noise at this concentration. The most abundant peaks in the spectrum arise from contaminants in the solvent and/or the added ammonium acetate as they are present in the absence of the lipid sample (data not shown) and are of odd nominal mass. Contaminant peaks are also prominent in the direct positive ionization spectrum (Figure 4(b)). Evidence for ions from all of the major lipid groups, however, is still apparent at this concentration. The charge inversion spectrum (Figure 4(c)) looks very similar to that of Figure 3(c), in terms of the locations and relative abundances of all of the peaks with the notable exception of the appearance of the prominent peak at nominal m/z 857. This peak arises from attachment of a PDPA dianion to a contaminant peak in the positive ion spectrum at nominal m/z 637, which falls below the scale shown in Figure 4(b). Upon CID, the strongly dominant fragment ion from this odd nominal mass ion was observed to be the [PDPA − H]− anion, which arises from proton transfer (data not shown). There is very little evidence that any of the other peaks in the positive ion spectrum that do not fall into the lipid groupings of Figure 3 undergo charge inversion. Such ions are presumably neutralized by the ion/ion reaction. The data of Figures 3 and 4 demonstrate that the ion/ion charge inversion approach leads to lower spectral congestion than either of the direct ionization approaches due both to chemical noise reduction and a greater mass dispersion between PE and PC ions. Figure 4 further demonstrates that significantly greater signal-to-noise ratios for lipid ions are obtained at the lowest lipid mixture concentrations examined in this work using the charge inversion approach.
Figure 4.

Nano-ESI mass spectra of a bovine liver extract (0.5 μM total lipid concentration) obtained via (a) direct negative ionization, (b) direct positive ionization, and (c) charge inversion of the ions of (b) using [PDPA − 2H]2−. In the negative ion spectra, contributions from [PC − CH3]− anions can fall among the deprotonated PE anions.
Product Ion Spectra of Lipid Mixture Anions
The negative ion mass spectra derived via either negative nano-ESI or charge inversion of cations are comprised of mixtures of deprotonated PEs, adducts of PCs, and [PC − CH3]− ions. The latter ions, which are generated from the PC adduct anions, can overlap in mass with deprotonated PEs that are one methylene group smaller than the PE that is isomeric with the intact PC. The presence of [PC − CH3]− ions can make ambiguous the interpretation of product ion spectra when mass selection is done in the negative ion mode. This ambiguity can be avoided with the charge inversion approach by making the initial mass-selection step in the positive ion mode. Any [PC − CH3]− ions generated during the ion/ion reaction or any ion transfer steps will be 14 Da lower in mass than the [PE − H]− ions. By isolating ions in the positive ion mode, the potential overlap in mass between PE and [PC − CH3]− anions is avoided.
A subset of ions derived from a 10 μM total lipid concentration mixture was subjected to both the direct negative ionization approach and to positive ionization followed by charge inversion. A few illustrative examples are provided here. Figure 5 gives an example in which a precursor ion population is dominated by a single lipid component. Figure 5(a) shows the position/ion reaction product ion spectrum of the m/z 812 precursor ion (1–2 m/z wide isolation) with doubly deprotonated PDPA. No products from double proton transfer are observed, but an abundant PDPA adduct is noted along with a combined proton transfer and methyl cation transfer product. Isolation of the PDPA adduct followed by sequential activation of the adduct and the first generation product from the transfer of a methyl cation and proton to the PDPA dianion yielded the product ion spectrum of Figure 5(b). In this case, only two prominent products are noted that correspond to the FAs 20:3 and 18:0. The mass of the [PC − CH3]− ion (monoisotopic theoretical mass = 796.586 Da, measured mass = 796.568 Da) as well as all CID products are consistent with a PC of empirical formula C46H87NO8P. All evidence points to (PC 20:3_18:0). The relative abundances of the FAs suggests that the 18:0 FA is predominantly present as the sn-1 species and that the 20:3 FA is predominantly present at the sn-2 position. That is, the lipid ions at m/z 812 are predominantly comprised of (PC 18:0/20:3). The measured masses of the precursor ion and product ions all fall within ±20 mDa (i.e., the precision of the mass measurements made over the course of these studies) of the theoretical masses of the assigned species. (Mass measurement accuracies of <5 ppm have been reported with this mass spectrometer platform using a postprocessing algorithm that was not used here.24 Mass accuracies of 20–25 ppm were reported for the raw data, which is consistent with these results.) Figure 5(c) shows the CID spectrum of the ions that corresponds to acetate adduct species generated directly in the negative mode (i.e., neutral lipid mass of 811 Da plus acetate adduct of mass 59 to give a precursor anion of m/z 870). Although the signal-to-noise ratio of this spectrum is lower than that of Figure 5(b), which is likely to be largely due to somewhat lower ionization response for the PC in the negative mode, the information content is essentially identical. Neutral lipids at nominal mass 785 Da provide another example of highly analogous behavior for the ions derived via charge inversion of cations and direct anion formation (see Figure S-8).
Figure 5.

(a) Post-ion/ion reaction product ion spectrum from the m/z 812 precursor cation from a bovine liver extract with [PDPA − 2H]2−. (b) CID product ion spectrum of the [PC + PDPA − H]− complex with subsequent CID of the first generation [PC − CH3]− product. (c) CID product ion spectrum of the [PC + CH3COO−]− adduct formed via direct negative ionization.
An example in which a mixture of isomeric/isobaric species is present within a nominal m/z unit is provided here using the data in Figure 6. Figure 6(a) shows the post-ion/ion product ion spectrum generated from the reaction of the nominal m/z 744 cations (isolation window of 1–2 m/z units) with PDPA dianions. This spectrum reveals the presence of isomeric or isobaric components in the population in that products generated by double proton transfer (nominal m/z 742), PDPA attachment (nominal m/z 964), and combined proton transfer and methyl cation transfer (nominal m/z 728) are observed. In this case, the major charge inversion product results from double proton transfer, which is consistent with the behavior of PE. Figure 6(b) shows the ion trap CID spectrum from the nominal m/z 742 ion, which shows three major products that are consistent with fatty acids 18:0, 18:1, and 18:2. The precursor ion falls within the mass tolerance for a PE with the empirical formula C41H77NO8P (theoretical monoisotopic mass = 742.539 Da, measured isotopic mass = 742.522 Da). The product ions suggest that a mixture of PEs of the form (PE 18:2_18:0) and (PE 18:1/18:1) are the major lipid components in the precursor ion population. The product ion spectrum obtained from the nominal m/z 742 anion derived from direct negative ionization of the lipid mixture is essentially identical (Figure S-9). The most abundant charge inversion product ion formed by PDPA attachment (measured monoisotopic mass = 964.629 Da) is consistent with species formed by PDPA attachment to a PC cation isomer of the protonated PE identified above (monoisotopic mass of C41H79NO8P + C12H12O4 = 964.628 Da). Figure 6(c) shows the product ion spectrum derived from these precursor ions. The first generation product generated from combined proton and methyl cation transfer is also consistent in mass with an isomeric PC (theoretical = 728.523; measured = 728.543). The most abundant low m/z products are consistent with 15:0, 18:2, and 18:1 FAs with the latter two species being the most abundant. The abundant 18-carbon FAs suggest the presence of (PC 18:2_15:0) and (PC 18:1_15:1) lipids, although no clear signal due to [15:1 − H]− ions is present in Figure 6(c).
Figure 6.

(a) Post-ion/ion product ion spectrum from the reaction of the m/z 744 precursor cation from a bovine liver extract with [PDPA − 2H]2−. (b) CID product ion spectrum of the [PE − H]− ion generated by charge inversion. (c) CID product ion spectrum of the PDPA adduct ion generated by charge inversion with subsequent activation of the [PC − CH3]− product. (d) Product ion spectrum from CID of the acetate adducts generated via direct negative ionization.
The acetate adducts of the neutral lipids at nominal mass 743 Da appear at m/z 802 in the mass spectrum generated by direct negative ionization of the 10 μM solution. Figure 6(d) shows the product ion spectrum generated from ion trap CID of the m/z 802 precursor ion population. The product ion spectra of the anionic adducts of the neutral lipids of 743 Da nominal mass generated via charge inversion (Figure 6(c)) and via direct negative ionization (Figure 6(d)) are quite distinct. All of the product ions observed in the FA anion region (i.e., roughly m/z 225–350) in Figure 6(c) are also present in Figure 6(d). This suggests that the same PCs found with the charge inversion method are also indicated by the direct negative ionization approach. However, several additional prominent ions, such as those at nominal m/z 255, 283, and 303 are also apparent. These ions fall within the mass tolerance for [16:0 − H]−, [18:0 − H]− and [20:4 − H]− anions, respectively. It is noteworthy that another additional product ion at m/z 728 corresponds in mass to the [PE − H]− ion of Figure 6(b). This implies that at least some of the precursor ion population is comprised of the acetate adduct of nominal mass 743 Da PE lipids. At least some of the FA anions observed in Figure 6(d) can arise from further fragmentation of these species. It is noteworthy that the most abundant FA anion in Figure 6(d) is absent in Figure 6(b and c) (as well as Figure S-9), which implies the presence of one or more additional components. It possible that the acetate adducts of the isomeric PC/PE components at 743 Da nominal mass are mixed with [M − H]− ions formed directly from deprotonation of molecules of 803 Da nominal mass. The mixture of precursor ions present at the m/z of the acetate adduct of the 743 nominal mass lipids is clearly more complex than the mixture of adduct anions generated by the charge inversion of the m/z 744 cations. A very similar scenario was noted for the lipids at 767 Da nominal mass in which the ions at the m/z of the acetate adduct formed via direct ionization appeared to be a mixture of adducts of the 767 Da PEs and PCs as well as [M − H]− anions of molecules of nominal mass 827 Da (see Figure S-10).
CONCLUSIONS
A key difference between phosphatidylcholines and phosphatidylethanolamines is that the headgroup of the former is comprised of a quaternary ammonium group whereas the latter is characterized by a primary amine. This difference gives rise to distinct reactivities for the corresponding singly charged ions in reaction with doubly deprotonated dicarboxylic acids. PC cations undergo charge inversion by dianion attachment with subsequent proton and methyl cation transfers upon activation of the complex. PE cations, conversely, undergo charge inversion via double proton transfer. The net result is that isomeric PE and PC lipids give rise to charge inversion products of different m/z. In both cases, ion trap CID yields information about fatty acid carbon number and degree of unsaturation.
Deprotonated PEs and anionic adducts of PCs are conventionally generated via direct negative ionization, which also generates anions from any species amenable to anion formation. The approach described here ultimately relies on ionization yields in the positive mode, which particularly favors species with strong basic sites that are readily protonated (e.g., PEs) and species with fixed charge cationic sites (e.g., PCs). The charge inversion reaction inherently selects for species that can readily form either anions or cations, such as PEs and PCs. The use of positive ionization followed by ion/ion charge inversion for the generation of lipid anions from PEs and PCs has three potentially attractive features relative to direct negative ionization. The first is that the charge inversion reactions can eliminate significant fractions of the chemical noise that arises from species that ionize efficiently in only one polarity. The second is that positive ionization can be more efficient than negative ionization, particularly for PCs, which can translate to lower detection limits. The third is that the MS3 approach illustrated here avoids the generation of mixtures of ions comprised of adduct anions of mixture components of lower mass with deprotonated species of higher mass species. By mass selecting a cation prior to charge inversion, the possibility for creating new mixtures of precursor ions, which can occur with direct negative ionization, is eliminated.
Supplementary Material
Acknowledgments
This research was supported by the National Institutes of Health under Grant GM 45372 and AB Sciex. S.J.B. acknowledges support from the Australian Research Council through the Centres of Excellence (CE0561607) and Discovery (DP120102922, DP150101715) Programs.
Footnotes
The authors declare no competing financial interest.
- Some of the spectra mentioned in the text that support the presentation of this work (PDF)
References
- 1.Yamashita M, Fenn JB. J Phys Chem. 1984;88:4451–4459. [Google Scholar]
- 2.Karas M, Bachman D, Bahr U, Hillenkamp F. Int J Mass Spectrom Ion Processes. 1987;78:53–68. [Google Scholar]
- 3.Han X, Gross RW. Mass Spectrom Rev. 2005;24:367–412. doi: 10.1002/mas.20023. [DOI] [PubMed] [Google Scholar]
- 4.Pulfer M, Murphy RC. Mass Spectrom Rev. 2003;22:332–364. doi: 10.1002/mas.10061. [DOI] [PubMed] [Google Scholar]
- 5.Smith RE, Lespi P, Di Luca M, Bustos C, Marra FA, de Alaniz MJT, Marra CA. Lipids. 2008;43:79–89. doi: 10.1007/s11745-007-3133-6. [DOI] [PubMed] [Google Scholar]
- 6.Han X, Yang K, Gross RW. Mass Spectrom Rev. 2012;31:134–178. doi: 10.1002/mas.20342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schwudke D, Hannich JT, Surendranath V, Grimard V, Moehring T, Burton L, Kurzchalia T, Shevchencko A. Anal Chem. 2007;79:4083–4093. doi: 10.1021/ac062455y. [DOI] [PubMed] [Google Scholar]
- 8.Hsu FF, Turk J. J Am Soc Mass Spectrom. 2003;14:352–363. doi: 10.1016/S1044-0305(03)00064-3. [DOI] [PubMed] [Google Scholar]
- 9.Hsu FF, Turk J. J Mass Spectrom. 2000;35:595–606. doi: 10.1002/(SICI)1096-9888(200005)35:5<595::AID-JMS965>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 10.Cole MJ, Enke CG. Anal Chem. 1991;63:1032–1038. doi: 10.1021/ac00010a020. [DOI] [PubMed] [Google Scholar]
- 11.Brugger B, Erben G, Sandhoff R, Wieland FT, Lehmann WD. Proc Natl Acad Sci U S A. 1997;94:2339–2344. doi: 10.1073/pnas.94.6.2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hsu FF, Turk J. J Am Soc Mass Spectrom. 2000;11:892–899. doi: 10.1016/S1044-0305(00)00159-8. [DOI] [PubMed] [Google Scholar]
- 13.Harrison KA, Murphy RC. J Mass Spectrom. 1995;30:1772–1773. [Google Scholar]
- 14.Ekroos K, Esjing CS, Bahr U, Karas M, Simons K, Shevchenko A. J Lipid Res. 2003;44:2181–2192. doi: 10.1194/jlr.D300020-JLR200. [DOI] [PubMed] [Google Scholar]
- 15.Fhaner CJ, Liu S, Ji H, Simpson RJ, Reid GE. Anal Chem. 2012;84:8917–8926. doi: 10.1021/ac302154g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fhaner CJ, Liu J, Zhou X, Reid GE. Mass Spectrom. 2013;2:S0015. doi: 10.5702/massspectrometry.S0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stutzman JR, Blanksby SJ, McLuckey SA. Anal Chem. 2013;85:3752–3757. doi: 10.1021/ac400190k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liebisch G, Vizcaíno JA, Köfeler H, Trötzmüller M, Griffiths WJ, Schmitz G, Spener F, Wakelam MJO. J Lipid Res. 2013;54:1523–1530. doi: 10.1194/jlr.M033506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xia Y, Chrisman PA, Erickson DE, Liu J, Liang X, Londry FA, Yang MJ, McLuckey SA. Anal Chem. 2006;78:4146–4154. doi: 10.1021/ac0606296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Emory JF, McLuckey SA. Int J Mass Spectrom. 2008;276:102–109. [Google Scholar]
- 21.Murphy RC, Axelsen PH. Mass Spectrom Rev. 2011;30:579–599. doi: 10.1002/mas.20284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ekroos K, Esjng CS, Bahr U, Karas M, Simons K, Shevchenko A. J Lipid Res. 2003;44:2181–2192. doi: 10.1194/jlr.D300020-JLR200. [DOI] [PubMed] [Google Scholar]
- 23.Hassell KM, LeBlanc Y, McLuckey SA. Anal Chem. 2011;83:3252–3255. doi: 10.1021/ac200439k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Andrews GL, Simons BL, Young JB, Hawkridge AM, Muddiman DC. Anal Chem. 2011;83:5442–5446. doi: 10.1021/ac200812d. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.