

Abstract
The sex chromosomes have special significance in the history of genetics. The chromosomal basis of inheritance was firmly established when Calvin Bridges demonstrated that exceptions to Mendel’s laws of segregation were accompanied at the cytological level by exceptional sex chromosome segregation. The morphological differences between X and Y exploited in Bridges’ experiments arose as a consequence of the evolution of the sex chromosomes. Originally a homologous chromosome pair, the degeneration of the Y chromosome has been accompanied by a requirement for increased expression of the single X chromosome in males. Drosophila has been a model for the study of this dosage compensation and has brought key strengths, including classical genetics, the exceptional cytology of polytene chromosomes, and more recently, comprehensive genomics. The impact of these studies goes beyond sex chromosome regulation, providing valuable insights into mechanisms for the establishment and maintenance of chromatin domains, and for the coordinate regulation of transcription.
Keywords: FlyBook, Drosophila, X chromosome, dosage dependence, genetic screens, male-specific lethal
WITH respect to their sex chromosome constitution, Drosophila females are XX and males are XY. Such a chromosomal difference between the sexes is found in numerous diploid multi-cellular organisms where one sex carries an identical pair of sex chromosomes while, in the other sex, one of these chromosomes has undergone extensive structural modifications. In Drosophila, the Y chromosome has lost most of its genetic content and is retained in the karyotype only because of its specific genetic function in spermatogenesis. As a consequence of the difference in sex chromosomes, genes present on the X chromosome are represented in different doses in females and males, potentially leading to a disparity in the level of their gene products and their stoichiometry relative to autosomal proteins in the two sexes. This problem is preempted by the function of a regulatory mechanism that is sensitive to the ratio of X chromosomes to autosomes (X:A), and has evolved in order to compensate for the dosage differences in X-linked genes between males and females (Figure 1).
Figure 1.

Sex-specific regulation of dosage compensation in Drosophila by the X chromosome to autosome ratio. In diploid females (left), the ratio is 1.0 (XX:2A), which represents a basal or “balanced” state of gene expression. The ratio of 1.0 triggers expression of SXL, which positively regulates female-specific splicing and transcriptional regulators (TRA and DSXF) while repressing the translation of MSL2. Lack of MSL2 prevents inappropriate MSL-complex formation in females. In diploid males (right), the X:A ratio is 0.5 (XY:2A) leading to selective pressure to upregulate X-linked genes. SXL is not expressed, therefore MSL2 is translated, the full MSL complex forms, associates with X-linked genes, and increases their transcription. The lack of SXL also results in male-specific differentiation through expression of DSXM (see Cline and Meyer 1996).
The occurrence of dosage compensation was first reported, three-quarters of a century ago, by Herman Muller (Muller 1932). Muller realized that, within each sex, dosage variation of some partial-loss-of-function mutant alleles that are present on the X chromosome leads to a phenotype that is proportional to the dose. Yet, males that carry one dose and females that carry two doses of the alleles have identical phenotypes (Figure 2) (Muller 1948).
Figure 2.

The wa mutant is a partial loss-of-function allele that allows the deposition of some eye pigments in the eye. The greater the number of wa alleles in the genotype of either females (top row of karyotypes) or males (bottom row of karyotypes) the greater the amount of pigmentation. Surprisingly, females with two doses and males with a single dose of the allele have the same eye color. From Muller 1948; reprinted with permission from The Harvey Society.
The exceptional cytology of polytene chromosomes allowed remarkable early progress in the study of dosage compensation. Polytene chromosomes are produced in larval salivary gland cells, and consist of up to 1024 copies of actively transcribed chromosomal DNA produced by endoreplication in the absence of cell division. These chromosomal copies are lined up in register, and allow interphase chromosomes to be visible under simple light microscopy. Some 25 years after Muller’s discovery, Theodosius Dobzhansky noted that the polytenic X in salivary glands of male larvae is wider and more diffuse than each X in females (Dobzhansky 1957). The significance of this observation was provided by George Rudkin who had determined that the DNA content of the male X is equal to that of each X in females (Aronson et al. 1954). These facts led to the hypothesis that the difference in morphology of the X chromosomes in the two sexes reflects a difference in levels of activity. This hypothesis was substantiated by A. S. Mukherjee in Wolfgang Beermann’s laboratory using transcription autoradiography. Considered, at that time, a state-of-the-art technique in molecular genetics, transcription autoradiography consisted of treating salivary glands with a short pulse of tritiated uridine, covering the spread polytene chromosomes with a photographic film and, following exposure and development of the film, counting the silver grains present over the chromosomes of interest. The results indicated that the levels of nascent transcripts on the male X chromosome and on the paired X chromosomes of females were similar and that the mechanism of compensation, responsible for correcting the difference in gene dosage between the sexes, operated at the level of transcription. As measurements were relative within each nucleus, whether the mechanism relied on increasing the level of transcription in males or reducing it in females could not be determined with certainty (Mukherjee and Beermann 1965).
The discovery of dosage compensation in Drosophila pre-dated the study of the analogous phenomenon in mammals, where the facultative heterochromatization of one of the two X chromosomes in females involves the spread of noncoding RNAs (ncRNAs), as a harbinger of covalent modifications to both DNA and histones (reviewed in Dixon-McDougall and Brown 2016). Similarly it led to the discovery of dosage compensation in Caenorhabditis elegans, where the limited downregulation of both X chromosomes in hermaphrodites involves a subset of proteins and factors that are normally engaged in chromosome condensation during cell division (reviewed in Meyer 2010). In mammals, Susumo Ohno made the landmark discovery that in regenerating female rat liver cells one of the two X chromosomes was heterochromatic, i.e., highly condensed (Ohno et al. 1959). Mary Lyon, in her seminal paper, suggested that the condensed X could be either paternal or maternal in origin and that it is inactivated (Lyon 1961). It was only a year later, in a much longer paper discussing X chromosome gene activity, that she put forward the hypothesis that X inactivation in females is the basis for dosage compensation in mammals (Lyon 1962). Surprisingly, she did not cite any of the Drosophila observations that had led to the realization that such a regulatory mechanism exists in the first place. In contrast, when Barbara Meyer reported the existence of dosage compensation in C. elegans, she discussed her discovery in the perspective of the already extensive literature on the Drosophila mechanism (Meyer and Casson 1986).
The Evolution of Sex Chromosomes Drove the Need for Dosage Compensation
Since the discovery of sex chromosomes, the selective forces that have led to their structural and functional differentiation have been the objects of speculation and of some limited experimentation. The most widely accepted view is that sex determination arose from a simple mating-type system based on a pair of sex-determining alleles: individuals of aa genotype could only “mate” with heterozygous Aa individuals. In such a scheme, the A allele is present only in one mating type, while the a allele is present in both. The evolutionary forces thought to transform such a simple genetic sex-determining mechanism into the existence of dimorphic sex chromosomes most likely involve: (1) the occurrence of mutations on the A-bearing chromosome that are favorable to the Aa mating type and unfavorable to aa individuals; (2) selection for a loss of recombination between the A- and a-bearing chromosomes, mediated for example by inversions, that would result in the linkage of the favorable mutations with the A allele; and (3) the retention in the population of chromosomes carrying randomly occurring mutations because of their association with the mutations favorable to the Aa genotype. As most randomly occurring mutations lead to loss-of-function alleles and are deleterious, the lack of recombination between the A- and a-bearing chromosomes would lead to a progressive, functional degeneration of the A-bearing chromosome. During the course of this process, the genes present in two active doses in aa individuals would be represented by a single active dose in Aa individuals providing the selective pressure for the evolution of a mechanism that would remedy this inequality of gene products between the two mating types or sexes (Lucchesi 1978; Charlesworth 1996).
Different species in the genus Drosophila allow a unique insight into the concomitant evolution of sex chromosomes and dosage compensation. The karyotype of all the members of the genus consists of different arrangements of six chromosomal arms and two “dot” chromosomes (Figure 3). Although numerous inversions and other rearrangements of the chromosomal arms differentiate the species, the genic content of the arms has been substantially maintained across the genus. In Drosophila melanogaster, the ancestral A element is the X chromosome while the fusion of the B and C arms and the D and E arms form the major autosomes. In D. pseudoobscura a fusion of the ancestral A and D arms gave rise to a metacentric X chromosome with both arms exhibiting dosage compensation (Abraham and Lucchesi 1974). The karyotypic configuration of the D. pseudoobscura species is reproduced in females of D. miranda, but the males of this species have an odd number of chromosomes, with one member of a pair of rods (chromosome 2R of D. melanogaster or the ancestral C element) apparently degenerating due to its fusion to the Y chromosome. This chromosome arm is thus present in two doses in females and only a single dose in males, and is thereby considered to be a second X chromosome (X2 or neo-X). In males, the single X2 chromosome pairs with the Y which, instead of being wholly heterochromatic as in D. pseudoobscura, exhibits a number of euchromatic regions that are homologous to the X2 chromosome. These regions are destined to become inactivated and heterochromatic as they accumulate loss-of-function mutations and transposable elements (Steinemann et al. 1993). As evidence that D. miranda provides a window on the evolution of dosage compensation, the X2 chromosome exhibits regions where the activity level in males was less than in females and others with equal levels of gene activity in the two sexes, i.e., exhibiting dosage compensation (Strobel et al. 1978). Concordant results were obtained with respect to the association of the male-specific lethal (MSL) proteins (described below) along the X2 chromosome (Bone and Kuroda 1996; Marin et al. 1996) and the acquisition of chromatin entry sites [CESs, also known as high affinity sites (HASs), described below] to which the complex responsible for dosage compensation is targeted (Alekseyenko et al. 2013).
Figure 3.

Overview of selected Drosophila karyotypes. (A) Diagram of the six chromosomal elements present in various configurations in different species of the genus Drosophila. (B) Male of D. melanogaster. (C) Male of D. pseudoobscura. One of the autosomal elements is fused to the ancestral X, yielding a metacentric X chromosome. (D) Female of D. pseudoobscura or D. miranda. (E) Male of D. miranda. A different autosomal element is fused to the Y chromosome and is in the process of becoming inactivated and heterochromatic. From Lucchesi 1978 reprinted with permission from John Wiley and Sons (New York).
Discovery of the First msl Genes
Reasoning that if the mechanism operates in males, mutations that interfered with dosage compensation should be male-lethal, while the reverse would be true if the mechanism operates in females, John Belote set out to screen the major autosomes for sex-specific mutations (Belote and Lucchesi 1980a). Using the genetic scheme described in Box 1, MSL mutants were obtained by identifying three genes: msl1, msl2, and maleless (mle). The latter gene, mle, had already been discovered in natural populations by K. Oishi’s laboratory (Fukunaga et al. 1975), which later reported the existence of a fourth gene (msl3) first found by T. K. Watanabe (Uchida et al. 1981). Linking male-specific lethality to dosage compensation was accomplished for three of the four genes by measuring a male-specific reduction in the level of several X-linked enzymes in mutant males (the mutations allow survival up to the third larval instar) and, in the case of an mle mutant, by demonstrating a male-specific reduction in the level of X-chromosome transcripts (Belote and Lucchesi 1980b; Breen and Lucchesi 1986). Note that for convenience, all four of these genes as well as males absent on the first (mof) (discussed below) are collectively referred to as the msl genes.
BOX 1.
The scheme developed by John Belote to screen for sex-specific lethals on chromosome 2 is illustrated below. A similar scheme was used for chromosome 3.
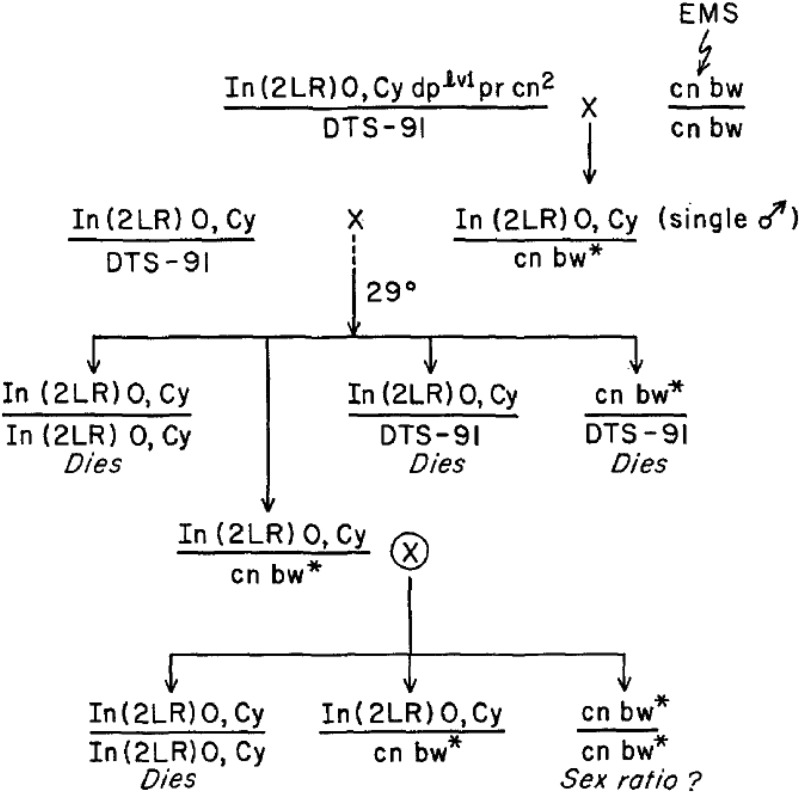
The chromosome marked with Cy is a balancer that is lethal in homozygous condition, and DTS-91 is a dominant temperature-sensitive lethal mutation present on chromosome 2. The use of these chromosomes allows the recovery of fourth-generation progenies that consist of males and female individuals carrying an EMS-treated chromosome over the Cy balancer and individuals homozygous for the treated chromosome to be examined for the absence of one of the two sexes.
For approximately a decade, the research activities of different laboratories focused on four main questions: (1) how do individuals with aneuploid genomes (such as XXX;AA, XX;AAA, or X;AAA) or individuals with partially aneuploid genomes (such as XX;AA) carrying large X-chromosome deficiencies, handle dosage compensation; (2) do X-chromosome genes relocated to the autosomes retain compensation, and do autosomal genes relocated to the X become compensated; (3) what are the interactions between dosage compensation and the sex-differentiation pathway, especially with the master-regulatory gene Sex-lethal (Sxl); and (4) what is the nature of the regulatory sequences that are presumably associated with X-linked genes and that mediate the upregulation of these genes in males (for review see Lucchesi and Manning 1987)? The following decade witnessed the cloning of the msl genes (see below), and the demonstration that their products form a regulatory complex that contains long ncRNAs. These results, added to the discovery by Bryan Turner’s laboratory that the X chromosome in males is highly enriched in a particular histone post-translational modification, H4K16ac (Turner et al. 1992), and to the identification of the MOF acetyltransferase responsible for this modification by Andres Hilfiker (Hilfiker et al. 1997); ushered dosage compensation into the modern molecular biology age as the premier paradigm of the epigenetic regulation of entire chromosomes.
MSL Proteins Bind Along the Length of the Male X Chromosome
The first msl gene to be cloned was mle (Kuroda et al. 1991). The procedure involved the cytological mapping of the gene to a segment missing in two overlapping deficiencies. Within this segment, the breakpoint of a known translocation was associated with a mutant allele of mle (Kernan et al. 1991). Starting with a cloned gene in the vicinity, a “chromosome walk” was carried out by successively screening for overlapping DNA segments in a genomic library until the breakpoint of the translocation was reached. A computer-aided, “conceptual” translation of the sequence that overlapped the break revealed a 140-kDa protein that has two double-stranded RNA binding motifs, appeared to be a member of a particular family of helicases, and was highly homologous to the subsequently identified human RNA helicase A (RHAII). The purified recombinant MLE protein binds single-stranded RNA or DNA and is an ATP-dependent RNA:DNA helicase (Lee et al. 1997).
The msl1 gene was cloned using the same approach (Palmer et al. 1993). The predicted protein did not have any significant resemblance to any of the proteins in the databases available at that time. Two years later, three separate laboratories cloned msl2. This gene had been mapped genetically to chromosome 2, and one cloning strategy relied on a series of Y–2 translocations and on combinations of translocations between chromosomes 2 and 3 to determine its physical location (Bashaw and Baker 1995). Cloning was also achieved by the use of deletions resulting from the imprecise excision of a transposable P element inserted near the msl2 gene, and the selection of a cosmid containing the DNA that was missing in the deletions (Kelley et al. 1995). The third successful attempt at cloning msl2 was based on mapping the physical location of the gene using restriction fragment length polymorphism (RFLP) linkage analysis (Zhou et al. 1995). This method correlates the presence of the phenotype caused by a loss-of-function allele with the presence or absence of restriction enzyme cutting sites, within the segments of a DNA walk. As suspected, because of the presence of a conserved RING finger, human MSL2 is an E3 ubiquitin ligase now known to ubiquitinate p53 (Kruse and Gu 2009) as well as histone H2B (Wu et al. 2011). In flies, MSL2 ubiquitinates itself and the other MSLs and targets them for proteolysis, likely in order to maintain their stoichiometry (Villa et al. 2012). In addition, MSL2 binds DNA through its CXC domain—a stretch of 37 amino acids rich in cysteine (Fauth et al. 2010; Zheng et al. 2014).
The msl3 gene was cloned by the Baker laboratory using a strategy similar to the one used to clone msl2 (Gorman et al. 1995). MSL3 contains a chromodomain that targets the MSLs to active X-chromosome genes by associating with nucleosomes which contain histone H3 methylated at lysine 36 (H3K36me)—a mark deposited concomitantly with transcription (Larschan et al. 2007; Bell et al. 2008; Sural et al. 2008). MSL3 also has affinity for DNA and histone H4 methylated at lysine 20 (Kim et al. 2010; Moore et al. 2010).
Once the msl genes were identified, a major question was whether their gene products might directly regulate the male X chromosome. To address this question, complementary DNA (cDNA) sequences were expressed in Escherichia coli and the resulting protein products were used to generate antisera. In turn, these antisera were used to detect the location of the different msl gene products on polytene chromosomes by indirect immunofluorescence. In all instances, the MSL proteins were present at numerous sites along the entire X chromosome in males and not in females (Figure 4). Importantly, their colocalization suggested that they might form a multi-protein complex. Supporting this contention were the observations that in the absence of MSL1 or MSL2, none of the other proteins associated with the X chromosome (Gorman et al. 1993). Interestingly, in the absence of MSL3 or MLE, a subset of sites is bound by the remaining MSL proteins (Palmer et al. 1994). These sites are now considered nucleation sites for MSL targeting and spreading, which we will discuss in the context of a targeting model below.
Figure 4.

MSL proteins localize to the male X chromosome. MSL3 (red) is present in a reproducible interband pattern on the larval polytene male X chromosome, and not on the autosomes (blue). From Alekseyenko et al. 2006; reprinted with permission from Cold Spring Harbor Laboratory Press (Cold Spring Harbor, NY).
MOF Catalyzes Site-Specific Histone H4 Acetylation on the Male X
Several facts were known in the late 1980s regarding the molecular biology of chromatin: nucleosomes are not only involved in DNA packaging but also play an important role in regulating gene function; nucleosomal histones can be covalently modified and one modification in particular, acetylation, is associated with active transcription; in yeast, among the four acetylated lysines that occur on the terminal tail of histone H4, lysine 16 was uniquely necessary for gene expression. To determine the role of this lysine’s acetylation and that of the other three—lysines 5, 8, and 12—Bryan Turner’s group generated antisera that were able to recognize, individually, each of the acetylated lysines. This was accomplished by injecting rabbits with synthetic peptides acetylated at all four lysines, then testing the antisera present in each rabbit for their in vitro specificity with monoacetylated peptides (Turner and Fellows 1989). To attempt to discern some possible differences in the regulatory functions of nucleosomes containing H4 acetylated at different lysines, Turner and his collaborators immunostained Drosophila polytene chromosomes. Histone H4 acetylated at lysines 5 or 8 was distributed throughout the karyotype and H4 acetylated at lysine 12 was present in β-heterochromatin, in both sexes. Surprisingly, H4 acetylated at lysine 16 (H4K16ac) was found almost exclusively along the X chromosome in males (Turner et al. 1992). This mark colocalizes with the msl gene products on the X chromosome, and is present only if the msl genes are active and dosage compensation is normal (Bone et al. 1994). However, understanding the mechanism for enrichment of H4K16ac on the male X remained unknown until the discovery of the MOF acetyltransferase, the principal enzymatic function underlying dosage compensation.
As described above, the systematic search for sex-specific mutations had focused on the two large autosomes; chromosome 4 was not considered because its small size reduced the probability that it may carry the appropriate genes, and the X chromosome had been ignored because of the operational difficulty of determining whether a male-lethal mutation could be made homozygous and tested for viability in females. Andres Hilfiker circumvented this problem by using an ingenious genetic scheme that allowed the generation of females homozygous for a mutated X chromosome by using the mutation mei-S332 which causes a high frequency of nondisjunctions in the second meiotic division in the female germline (see Box 2). Such females would survive only if the X-linked lethality were male specific. One mutation that conformed to these phenotypic characteristics was isolated and shown to cause the absence of H4K16ac on the X chromosome of dying male larvae. Using RFLP mapping, the mutation was localized to a particular gene on the X chromosome that encodes a protein with substantial similarity to several histone acetyl transferases (Hilfiker et al. 1997). In fact, the mof gene product was shown to be the enzyme responsible for the acetylation of histone H4 at lysine 16 (Smith et al. 2000). The main function of all the other dosage-compensation regulatory proteins in Drosophila is likely to be the correct targeting of this histone acetyltransferase activity to active genes on the male X chromosome.
BOX 2.
The scheme developed by Andres Hilfiker to screen for male-specific lethals on the X chromosome.

Since lethal mutations on the X chromosome affect males whether they are male specific or not, the screen takes advantage of the mei-S332 mutation which causes a high frequency of nondisjunction in the second meiotic division in females. This results in eggs bearing two sister chromosomes that include the EMS-exposed X chromosomes to be tested for male-specific lethality. Fertilized by a Y chromosome, such zygotes develop into females. Therefore, the occurrence of y marked females paralleled by the absence of y marked males is indicative of the presence of an X-linked msl mutation.
Additional features of the screen: F1 females need not be collected as virgins since all of their brothers bear an FM6 chromosome. Diagnostic nondisjunctional females could be present in the F2 generation; however, the low fecundity caused by the mei-S332 mutation makes it profitable to transfer the progeny of the single F1 females and screen in the F3 generation, when a reasonable number of flies have been generated.
In addition to its role in dosage compensation, MOF associates with gene promoters throughout the genome in both males and females (Raja et al. 2010) although it activates only a subset of these genes (Feller et al. 2012). This nondosage-compensation function appears to be dispensable since females lacking MOF are viable (Hilfiker et al. 1997). The characterization of mof in Drosophila led to the identification of its human ortholog (Neal et al. 2000) which, similarly to the Drosophila enzyme, is responsible for the majority of H4K16 acetylation (Smith et al. 2005).
Discovery of the roX RNAs leads to the spreading model for X-chromosome targeting of dosage compensation
The noncoding-RNA components involved in dosage compensation were discovered fortuitously during investigations of the molecular genetic basis of brain function in flies. The laboratory of Ron Davis had used an enhancer-detector transposable element marked with LacZ to establish a collection of cell-specific markers throughout the adult Drosophila brain. The transposable element was induced to reinsert randomly throughout the genome and was activated by neighboring enhancers or neighboring genes (Han et al. 1996). A particular line of flies was obtained with LacZ expression in a brain region of females. A search for genes adjacent to the site of insertion of the transposable element revealed a transcript that was expressed ubiquitously, but only in males. The transcript was spliced and polyadenylated, lacked an open reading frame of any significance, and was found to decorate the X chromosome exclusively (Meller et al. 1997). The gene was named RNA on X 1 (roX1).
At the same time, Hubert Amrein in Richard Axel’s laboratory was initiating studies of sex-specific behavior in Drosophila by preparing cDNA from male and female brains separately and screening libraries for genes exhibiting sex-specific expression. These studies lead to the identification of two separate sets of male-specific transcripts encoded by roX1 and by a new gene—roX2 (Amrein and Axel 1997). Although very different in size and sequence, roX2 RNA shared the same characteristics with roX1: splicing, polyadenylation, no open reading frame, and association with the X chromosome in all tissues examined. Subsequent genetic studies demonstrated that single roX1 or roX2 mutants were viable, but double mutants were MSL; indicating that they play redundant, but essential, functions in dosage compensation (Meller and Rattner 2002).
roX genes are located on the X chromosome in all Drosophila species examined, strongly suggesting a functional importance to their X-linkage (Park et al. 2007; Quinn et al. 2016). However, roX transgenes inserted on autosomes can functionally complement roX1 roX2 double mutants. In this genotype, transgenic roX RNA, as well as the MSL complex, bind not only the male X, but also significant genomic stretches surrounding the autosomal location of the roX transgene (Figure 5) (Park et al. 2002). This result, along with the partial MSL binding pattern seen in msl3 and mle mutants (Palmer et al. 1994), support a model in which the MSL complex first assembles on a subset of nucleation sites and then spreads in cis in a reproducible pattern along the length of the X (Figure 6) (Kelley et al. 1999).
Figure 5.

MSL proteins spread from roX transgenes inserted on autosomes. (Top) Diagram of a roX1 roX2 double-mutant X chromosome (X) and an autosome (A) with a 5-kb wild-type roX+ transgene insertion. (Bottom) Anti-MSL1 staining of male polytene chromosomes carrying the roX+ transgene insertion on an autosome. MSL staining (red) is seen on the roX1 roX2 mutant X chromosome, but also up to 1 Mb in cis to the transgenic insertion (adapted from Park et al. 2002).
Figure 6.

Model for MSL targeting of the X chromosome. Initial assembly occurs on the sites of roX RNA synthesis and may nucleate at CESs before spreading in cis to produce the wild-type MSL pattern on the X chromosome. Assembly is limited to CES in the absence of MSL3 protein (adapted from Kelley et al. 1999).
Sex-Specific Biogenesis of the MSL Complex
Identification of the MSL complex
Given the colocalization of the MSL proteins on the male X chromosome, do they form a multi-subunit complex? The first evidence of a physical association of the MSL proteins was the co-immunoprecipitation of MSL1 and MSL2 (Kelley et al. 1995), followed by the co-immunoprecipitation of these two subunits with MSL3 (Copps et al. 1998). The reason for the absence of MLE from these precipitates can be explained by the fact that its association with the X chromosome in males was RNase sensitive (Richter et al. 1996); MLE might, therefore, be lost during the preparation of nuclear extracts. It is worthwhile to note that this observation led Richter and collaborators to suggest that a hypothetical RNA may be involved in dosage compensation. The roX RNAs were discovered during the following year and their colocalization with the MSL proteins along the X chromosome in males was evidenced by in situ hybridization with antisense, labeled probes (Meller et al. 2000). Using “RNA-friendly” conditions which included the use of a broad-spectrum RNase inhibitor and low salt during extraction, a complex containing all five MSL proteins was eventually isolated and shown to acetylate the H4K16 of nucleosomes (Smith et al. 2000). Recent proteomic analysis using formaldehyde cross-linking to stabilize the complex on DNA recovers MLE, JIL-1 kinase, and roX RNA efficiently (Alekseyenko et al. 2014). JIL-1 enrichment on the male X chromosome and its link to the MSL complex was discovered previously by the Johansen laboratory (Wang et al. 2001). Ongoing structural and functional studies continue to make valuable progress in understanding all of the potential components and organization of the MSL complex (recently reviewed in Keller and Akhtar 2015).
How the complex is assembled
The roX1 RNA is transcribed early during embryogenesis in both males and females; roX2 RNA is transcribed later and only in males. Of all the MSL proteins present at the time of roX RNA synthesis, MLE appears to be necessary for the stabilization of roX1 (Meller 2003) and, by inference, of roX2 RNA. Unless they associate with subunits of the MSL complex, the roX RNAs are unstable. In male embryos, although roX1, roX2, and four MSL proteins are present, formation of the complex must await the synthesis of MSL2; the only MSL protein missing from the maternal contribution. Although very different in size (3.7 kb for roX1 vs. 600 nt for roX2) and with very different primary sequences, the two roX RNAs are functionally redundant (Meller and Rattner 2002). This characteristic depends on the presence of short conserved sequences—the roX boxes—in the 3′ end of the RNAs, and on the formation of stem loops (SLroX1 or SLroX2) containing one of the roX boxes (Figure 7) (Stuckenholz et al. 2003; Park et al. 2007; Kelley et al. 2008; Park et al. 2008). Using one of several currently available techniques to determine the secondary structure of RNA, a number of additional smaller helices can be resolved, two of them located 3′ of SLroX1 and a total of eight in roX2 (Ilik et al. 2013). MLE is necessary for the assembly of the MSL complex since mutations that inactivate its ATPase or helicase functions lead to the association of only MSL1 and MSL2 at the X-chromosome entry sites (Lee et al. 1997; Gu et al. 1998; Morra et al. 2008). MLE’s role in dosage compensation may be to incorporate the roX RNAs into the MSL complex. A second subunit that appears to be required for the stable integration of roX RNA into the complex is MSL2 (Hallacli et al. 2012). Using methodology that allows the mapping of sites of RNA-protein contact at the single nucleotide level, Ilik and colleagues showed that MLE and MSL2 bind roX1 and roX2 at discrete stem loop domains (Ilik et al. 2013). The mechanistic basis and functional consequence of this binding were elucidated by Peter Becker and colleagues, who demonstrated that MLE binds to the major roX stem loop and remodels its topology to allow binding by MSL2 (Maenner et al. 2013). The methods used to determine changes in the stem loop made use of specific RNases that digest double-stranded RNA regions or single-stranded RNAs at specific residues, and by using chemicals that modify bases in single-stranded RNAs; single-stranded RNA regions could also be recovered by hybridization with labeled nucleotides. Two MSL2 subunits bind to a dimer of two MSL1 molecules, which interacts directly with MSL3 and MOF (Hallacli et al. 2012).
Figure 7.

Diagram of the two roX RNAs showing the location of the conserved sequences (roX boxes) and structural features (stem loops). The sequences forming the stem loops are different in the two RNAs except for one copy of the roX box which they share. From Maenner et al. 2013; reprinted with permission from Elsevier (Amsterdam).
The complex does not assemble in females
During the course of his seminal investigations on the role of Sxl in sex determination, Tom Cline suggested that this gene might also act in dosage compensation to limit the occurrence of this regulatory mechanism to males. He hypothesized that a dominant mutation of Sxl that is expressed in males (SxlM) should cause lethality by preventing X-chromosome upregulation, while the female-specific null allele Sxlf would kill females by allowing the hyper-activation of both X chromosomes (Figure 1) (Cline 1979). These contentions were validated using transcription autoradiography (Lucchesi and Skripsky 1981). An insight into the mechanism used by the Sxl gene product to insure the male-specific occurrence of dosage compensation awaited the molecular characterization of the sex-determining pathway and of the MSLs. The SXL protein is produced in females where it controls the splicing of transformer (tra) messenger RNA (mRNA), encoded by the next gene in the sex determination regulatory cascade. TRA protein, along with TRA-2, functions to regulate the sex-specific splicing of doublesex mRNA, resulting in distinct forms of the DSX transcription factor in males and females. SXL protein influences splicing by binding to poly-U rich sequences (reviewed in Cline and Meyer 1996). Similar binding sites were discovered in the 5′ and 3′ untranslated regions of the msl2 gene transcript (Kelley et al. 1995). The causative relation between the presence of the SXL binding sites on the msl2 message and the absence of the MSL2 protein in females was demonstrated by generating transgenic lines expressing an msl2 gene that lacked the SXL binding sites; females of these lines exhibited very reduced viability and their two X chromosomes were decorated by MSL2, MSL1, and MLE in larval salivary gland nuclei, suggesting the hyper-activation of their X chromosomes by the dosage-compensation mechanism (Kelley et al. 1995). In females, transcription of the Sxl gene initiates from an early establishment promoter and then continues from a maintenance promoter that remains active throughout female development. The SXL protein produced by the transient activity of the early promoter was shown to prevent the binding of MSL1 to polytene chromosomes and, by inference, to abrogate the function or presence of MSL2 (McDowell et al. 1996). The regulation of MSL2 synthesis by SXL did not appear to involve a sex-specific splicing event leading to a sequence lacking an open reading frame; rather it pointed to the possibility that SXL interfered with the translation of the msl2 mRNA in females (Kelley et al. 1997). In fact, SXL binding prevents the association of the msl2 transcript with ribosomal subunits (Gebauer et al. 1999; Gebauer et al. 2003).
Dosage Compensation Goes Genomic
The immunostaining of MSL complex on polytene chromosomes, as described above, demonstrated several remarkable features of MSL targeting. First, the exquisite X specificity of MSL binding in wild type males; second, a subset of sites, termed CESs seen in msl mutants; and third, the acquisition of new sites on autosomes upon roX gene transposition. These observations became the basis for a “spreading model” in which the MSL complex is proposed to bind at a limited number of initial sites, including the two roX genes, and then spread in cis to fill its full pattern on the X chromosome (Figure 6) (Kelley et al. 1999). With the development of genome-wide methods, this model and alternatives have been tested by specifically searching for sequences or chromatin features that might explain how the complex binds the X and not the autosomes.
The initial surprise, when chromatin immunoprecipitation (ChIP) coupled with microarray analysis (ChIP-chip) was first applied to this problem, was that the MSL complex is concentrated on X-linked gene bodies rather than promoter or intergenic DNA, where cis-acting regulatory elements are typically thought to be located (Alekseyenko et al. 2006; Gilfillan et al. 2006; Legube et al. 2006). In fact, the complex, as well as the H4K16ac chromatin mark that it catalyzes, occupies active genes with a 3′ bias (Figure 8). Furthermore, no sequence specificity was evident from this broad binding pattern. Rather, binding correlated with gene activity, and subsequent analyses demonstrated that active autosomal genes were likewise bound if inserted on the X chromosome (Gorchakov et al. 2009). Binding in gene bodies coincides with H3K36me3, a general mark on active genes (Larschan et al. 2007; Bell et al. 2008).
Figure 8.

MSL complex associates with the bodies of active X-linked genes with a 3′ bias. The metagene profiles show X-linked genes with the greatest MSL occupancy in red (bound genes, n = 700, the vast majority of which are transcribed), compared to unbound genes in green (n = 702, largely not expressed), or to all genes on the 2L autosomal arm in gray. The average bound profile covers the bodies of active genes on the male X, with a 3′ bias. All transcription units were scaled and aligned at their 5′ and 3′ ends. Chr, chromosome. From Alekseyenko et al. 2006; reprinted with permission from Cold Spring Harbor Laboratory Press (Cold Spring Harbor, NY).
Discovery of the MSL recognition element
If the MSL complex is attracted to active genes, what limits it to the X chromosome? To identify the chromatin entry sites (CESs) implicated in initial recognition of the X, ChIP-chip was performed in msl3 mutants where the entry-site pattern was first discovered on polytene chromosomes (Figure 6). The 150 potential entry sites identified by MSL2 ChIP-chip revealed a common GA-rich MSL recognition element (MRE) (Figure 9) (Alekseyenko et al. 2008). Studies of MSL binding in suboptimal cross-linking conditions identified high affinity sites (HASs) (Straub and Becker 2008), which are largely the same as the CESs. The 21-bp MRE motif is only slightly enriched on the X chromosome (approximately twofold), but this is doubled when considering its preferential location within or 3′ to active genes (greater than fourfold enrichment). When inserted on an autosome, an entry site can direct local MSL spreading to flanking active autosomal genes (Alekseyenko et al. 2008). However, roX genes appear to be the most effective drivers of MSL spreading (Larschan et al. 2007), and may be a special class of entry site because they produce ncRNAs that associate directly with the complex. Biochemical analyses have not yielded evidence for additional ncRNA components of the MSL complex to date (Oh et al. 2003; Alekseyenko et al. 2014).
Figure 9.

Recovery of the CESs and their consensus sequence. Crossing scheme that results in recovery of msl3 mutant male embryos, along with their msl3+ sisters. Since only males express MSL2, ChIP of the total population results in selective recovery of MSL2 sites remaining in the absence of MSL3, also known as the CESs. The Multiple EM for Motif Elicitation algorithm revealed a consensus MRE logo based on the 150 sites recovered (adapted from Alekseyenko et al. 2008).
The molecular necessity of CES has been difficult to assess, as they are assumed to function redundantly along the length of the X. However, evolution of sex chromosomes is an ongoing process as can be seen in the karyotypes of several Drosophila species (Figure 3). In particular, analyses in D. miranda strongly suggest the functional importance of CES as an evolving neo-X chromosome exhibits the recent acquisition of MREs with the analogous consensus sequence and spacing (every ∼50 kb on X) as in D. melanogaster (Alekseyenko et al. 2013). Extensive new analyses of roX RNAs from 35 Drosophilid species, including the genomic mapping of occupancy of four roX RNA orthologs, revealed that one mode for the evolution of new MRE sequences is likely to be from preexisting RNA splicing signals (Quinn et al. 2016). These results provide strong support for the functional significance of CES, and for the spreading model in the establishment of dosage compensation.
Finding a DNA sequence implicated in targeting of the MSL complex solves only part of the puzzle. The direct-binding factor(s) for the MRE motif were key missing links in dosage compensation. An RNA-interference (RNAi) screen coupled with a cell-based assay and genome-wide mapping identified the CG1832 zinc-finger protein as a key factor required for MSL targeting, and a strong candidate for sequence-specific binding of the MRE (Larschan et al. 2012). Subsequent direct interaction of CG1832 protein (renamed chromatin-linked adaptor for MSL proteins, CLAMP) with the MRE sequence was documented in vitro (Soruco et al. 2013). Independently, the CXC motif in MSL2 was discovered to bind and form a cocrystal with the MRE sequence (Zheng et al. 2014), providing a direct DNA contact with the core MSL complex. CLAMP binds the MRE sequence on all chromosomes but displays a clear preference for X (Soruco et al. 2013), thus perhaps its binding is stabilized in conjunction with MSL2.
In spite of all of these advances, understanding the exquisite X specificity of MSL binding is still a major challenge. Current evidence indicates that, as with many transcription factors, the preexisting chromatin environment and flanking sequence composition may help select functional binding sites by the MSL complex. Bioinformatic analysis of chromatin profiles from the Drosophila Model Organism Encyclopedia of DNA Elements (modENCODE) project, combined with experimental results inducing MSL complex in female cells, suggest that an active chromatin context plays a critical role in the initial binding of the MSL complex to the X (Alekseyenko et al. 2012). Furthermore, the GC content of the DNA surrounding functional binding sites is significantly higher than for nonfunctional motifs. However, many unanswered questions remain, including why MREs on autosomes can meet all of the currently known criteria but escape targeting.
Potential role of repetitive sequences on X
The conservation of specific-repeat enrichment on the X, including rapid population of neo-X chromosomes by these repeats, strongly suggests a functional role in dosage compensation (Pardue et al. 1987; Waring and Pollack 1987; DiBartolomeis et al. 1992). Recent genetic studies have raised the possibility that small RNAs derived from repetitive sequences on X play a role in chromosome-specific recognition. First, lowered dosage of the RNAi machinery can enhance lethality in males partially deficient in roX RNA function. Second, lowered dosage of the RNAi machinery can partially rescue females with inappropriate dosage compensation. Third, transgenes expressing hairpin RNAs derived from the X-specific 1.688 satellite sequence produce abundant small interfering RNAs (siRNAs) and increase male survival in mutants with defective X recognition (Menon and Meller 2012; Menon et al. 2014). MLE also exhibits strong affinity for hairpin RNAs in vivo (Cugusi et al. 2016). Unexpectedly, MSL proteins and roX RNAs were also recently implicated in full expression of autosomal heterochromatic genes in males, a sex-specific function separate from X-chromosome dosage compensation (Koya and Meller 2015). Perhaps a common role for ncRNAs in chromatin underlies all of these novel findings.
Linkage and transcription play the pivotal roles in the final MSL binding pattern, irrespective of gene origin and DNA sequence
An alternative to the spreading model, discussed above, is that each MSL binding site is directed by DNA sequence, but that the elements are difficult to identify because they were degenerate, with varying affinities (Fagegaltier and Baker 2004; Oh et al. 2004; Dahlsveen et al. 2006). Although autosomal genes could be bound by MSL complex when a roX gene was inserted in cis on that chromosome (Park et al. 2002; Larschan et al. 2007), proponents of an affinities model argued that this was a highly artificial situation, potentially irrelevant to normal assembly and targeting on X. To distinguish between the two models, Gorchakov et al. (2009) proposed a test with distinct predictions for the fate of autosomal genes inserted on X. The spreading model predicted binding of the inserted autosomal genes, dependent on their transcriptional state. The affinities model predicted that the majority of autosomal genes should be skipped, unless they fortuitously carried a DNA sequence that had sufficient affinity to function within the MSL-rich X chromosome. The result was unambiguous. MSL complex bound, acetylated, and upregulated the transcriptionally active autosomal genes inserted on 14- and 65-kb autosomal transgenes tested at multiple locations on X, without any evidence for skipping. Therefore, a long-sought specific DNA sequence within each X-linked gene is not obligatory for MSL binding. However, the mechanistic basis for spreading remains far from understood.
To make further progress, it will be critical to determine what attracts the MSL complex to active genes. This occurs, at least in part, through association of the MSL3 chromodomain to the H3K36me3 active-chromatin mark. However, this is clearly not the only factor, as binding is diminished but not abolished in a set2 mutant, which lacks detectable H3K36me3 (Larschan et al. 2007). Importantly, roX RNAs are also required for spreading, as MSL complex binding is restricted to CES in their absence (Figueiredo et al. 2012). Thus, one of the most interesting outstanding questions is what targets the roX RNAs to active genes to facilitate spreading.
Transcriptional Regulation on the Male X
There is a large body of evidence that the MSL complex functions by increasing the transcription of X-linked genes. For example, Dobzhansky (1957) proposed that the increased volume of the wild-type male polytene X chromosome compared to single autosomes might reflect its increased activity. Consistent with this observation, Belote and Lucchesi noted a reduced width of the polytene X chromosome in mle mutants (Belote and Lucchesi 1980b). The male-specific lethality of the msl mutants and the X-chromosome binding of the MSL proteins are all consistent with a direct function on the X. The most definitive evidence comes from experiments in which the MSL complex is locally spread on autosomes. When genome-wide RNA expression is measured in transgenic flies with distinct insertion sites, local autosomal gene expression is increased, coincident with the location of MSL spreading specific to each line (Park et al. 2010).
The biochemical mechanisms for increasing gene expression on X must function over a wide dynamic range of transcription levels and differential expression patterns. Historically, experimental analysis of MSL function at the mechanistic level has been very challenging due to the small magnitude of the chromosome-wide effect and the lack of an in vitro system for biochemical analysis. However, with the development of new genomic tools to address transcriptional mechanisms using high throughput sequencing, the field has begun to make critical progress in this area.
The linkage of histone acetylation to transcriptional activity has been firmly established from yeast to human. Acetylation at multiple positions on the N-terminal tails of both H3 and H4 is typically seen on nucleosomes concentrated around the transcription start sites of active genes. Notably, the dosage compensated X chromosomes in Drosophila males display further enrichment for a specific acetylated form, H4K16ac, beyond 5′ ends, across the bodies, with a bias toward the 3′ ends of active genes (Smith et al. 2001). This modification is the specific product of the MOF histone acetyltransferase component of the MSL complex (Hilfiker et al. 1997; Smith et al. 2000). The significance of this distribution has been a major focus in the analysis of how transcriptional output is increased on the male X chromosome.
New genome-wide technologies have allowed experimental analysis of effects of MSL perturbation on the occupancy of RNA polymerase across genes on X and autosomes, and on nascent RNA transcription. Larschan et al. (2011) used global run-on sequencing (GRO-seq) to examine the specific effect of MSL-complex RNAi knockdown on RNA polymerase II (Pol II) on a genome-wide level. In this method, active polymerases are captured on the DNA during the isolation of nuclear extracts, and then mapped along genes via their ability to create a short tag of nascent RNA when provided with a pulse of labeled nucleotides. Results indicated that the MSL complex increases the density of Pol II across the bodies of active X-linked genes compared to autosomal genes, reminiscent of the original assays of nascent RNA linked to polytene chromosomes by Mukherjee and Beermann (1965). Notably, the considerable amount of paused RNA polymerase seen at 5′ ends of both X and autosomal genes did not appear to be strongly affected by MSL function.
In contrast, using ChIP followed by sequencing (ChIP-seq) for Pol II, Conrad et al. (2012) reported an MSL-dependent increase in Pol II occupancy at 5′ ends of X-linked genes, without any further increase over gene bodies. This supported an alternative model in which improved 5′ Pol II recruitment is the key mechanism for dosage compensation in Drosophila, with no role in the elongation phase of transcription (Conrad et al. 2012; Ferrari et al. 2013a; Straub and Becker 2013; Vaquerizas et al. 2013). It should be noted, however, that neither GRO-seq nor ChIP-seq provided the ability to differentiate with certainty between the recruitment, initiation, pausing, and elongation steps in early transcription (Ferrari et al. 2013b).
Further evidence for a role for the MSL complex in transcriptional elongation came from nascent RNA sequencing (Nascent-seq), a method to selectively isolate Pol II and nascent transcripts attached to the DNA template, based on the remarkable stability of transcriptionally engaged polymerase. Direct Nascent-seq documented dosage compensation with a new level of precision by comparing the positions of Pol II on X and autosomal genes at nucleotide resolution. The comparison of X and autosomes from additional GRO-seq and 5′ short RNA sequencing data also complemented the Nascent-seq analyses for assessment of the role of Pol II 5′ recruitment. The composite model from all of these studies suggests that male X-linked transcription is differentially regulated upon release from 5′ pausing, with increasing compensation as polymerase transcribes through X-linked gene bodies (Ferrari et al. 2013b). This “jumpstart and gain” model (Figure 10) still encompasses two interpretations. One possibility is that H4K16 acetylation increases processivity of Pol II which might otherwise terminate prematurely during elongation. Alternatively, increased efficiency of successfully elongating Pol II may result in faster release of paused polymerase molecules waiting to engage in productive elongation. In either case, 5′ replenishment of RNA polymerase does not appear to be the rate-limiting factor for most dosage-compensated genes on X.
Figure 10.

Jumpstart and gain model for dosage compensation. The transcription profile of genes on autosomes (Auto, blue line) compared to dosage compensated genes on X (chrX, red line). Both attract abundant Pol II, producing short transcripts while paused. An increase of mRNA production occurs on X-linked genes due to H4K16ac enriched on gene bodies. The facilitated steps include pause release (jumpstart), and a measureable gain during elongation. From Ferrari et al. 2013b; reprinted with permission from Elsevier (Amsterdam).
If pausing release and the ability to efficiently elongate through the chromatin template are the likely steps affected by MSL function, is the acetylation at Lys16 on histone H4 the sole cause? This is an appealing model, as nucleosomes are known to be a barrier to efficient transcription, and acetylation can oppose the tendency of nucleosomes to cause compaction in vitro (Shogren-Knaak et al. 2006; Robinson et al. 2008). However, to date it has not been possible to separate the role that the MOF acetyltransferase plays in MSL complex integrity and targeting from its role in histone acetylation. Evidence that H4K16 acetylation may not explain all of MSL-dependent dosage compensation comes from studies demonstrating a role for MLE helicase activity and enrichment for Topoisomerase II function on X-linked genes (Cugusi et al. 2013). Furthermore, a role for higher order structure in the X chromosomal “territory” within the nucleus cannot be excluded (Akhtar and Gasser 2007; Grimaud and Becker 2009).
Comparisons of mRNAs from male and female somatic cells demonstrate near perfect dosage compensation of X-linked genes (Gupta et al. 2006), raising the question of how such a precise level of transcriptional control can occur. One possibility is that the transcriptional elongation process is already quite efficient, with a maximum improvement approaching twofold. Alternative models suggest that MOF’s intrinsic ability to strongly augment transcription must be somehow dampened in a precisely quantitative manner by the other MSL subunits (Sun and Birchler 2009; Villa et al. 2012). A related issue is whether MSL-complex activity accounts for the full twofold effect. Although measurements of MSL-dependent regulation suggest a <1.5-fold rather than a perfect 2-fold effect in the transcriptional studies cited above, this could be due to the inability to create a complete knockdown with transient RNAi in cell culture. Alternatively, additional dosage compensation mechanisms may normally contribute. These may include direct repression of X-linked mRNA stability by SXL (Gergen 1987; Kelley et al. 1995); MSL-independent regulation of early embryonic transcription (Lott et al. 2011), but not in all Drosophila species (Lott et al. 2014); and general feedback regulation seen for dosage disturbances on all chromosomes (Stenberg and Larsson 2011; Chen and Oliver 2015). Furthermore, the male germline displays dosage compensation without requiring MSL function (Rastelli and Kuroda 1998; Gupta et al. 2006). Current active areas of research into the mechanisms and targeting of somatic dosage compensation include the three-dimensional structure at the level of MSL-complex subunit interactions, as well as at the level of whole chromosome organization. The molecular mechanism for roX ncRNA function remains one of the most fascinating and challenging current topics.
Dosage Compensation in Drosophila as a Model for Chromatin-Based Regulation
Historically, fruit flies had a tremendous head start in genetic studies, and this history, combined with the outstanding repertoire of visible and chromosomal phenotypes available for study have allowed many “firsts” to occur in this organism. As noted at the beginning of this chapter, sex chromosome dosage compensation was first recognized in Drosophila, decades prior to its discovery in mammals and worms. Subsequently, the fly community enjoyed the polytene chromosomes as a precursor to microarray technology years before genome-wide binding studies were possible. The connection between histone acetylation and gene activity was in part established by beautiful polytene chromosome immunostaining studies, showing increased site-specific acetylation on the male X (Turner et al. 1992). Furthermore, MOF, identified in a particularly clever fly genetic screen (Box 2), was the first histone acetyltransferase to have a clearly defined genetic function (Hilfiker et al. 1997). Finally, the roles of nucleation and spreading and ncRNAs in the establishment of chromatin-based targeting (Kelley et al. 1999) are becoming widely recognized general principles for all large genomes. Again and again the tiny fruit fly endures as a valuable model for all higher organisms.
Acknowledgments
We thank all our wonderful colleagues in the field, past and present. A.H. gives special thanks to Denise Hilfiker-Kleiner for essential support in identification of the mof mutant. In this review we have focused mainly on classical studies in dosage compensation, and apologize for any omissions regarding current areas of investigation. We are also very grateful to the National Institutes of Health, which has supported research in dosage compensation over many decades. Male-specific lethal research in the Kuroda laboratory is supported by National Institutes of Health grant GM-45744. J.C.L. is the Asa Griggs Candler Emeritus Professor of Biology at Emory University.
Footnotes
Communicating editor: B. Oliver
Literature Cited
- Abraham I., Lucchesi J. C., 1974. Dosage compensation of genes on the left and right arms of the X chromosome of Drosophila pseudoobscura and Drosophila willistoni. Genetics 78: 1119–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhtar A., Gasser S. M., 2007. The nuclear envelope and transcriptional control. Nat. Rev. Genet. 8: 507–517. [DOI] [PubMed] [Google Scholar]
- Alekseyenko A. A., Larschan E., Lai W. R., Park P. J., Kuroda M. I., 2006. High-resolution ChIP-chip analysis reveals that the Drosophila MSL complex selectively identifies active genes on the male X chromosome. Genes Dev. 20: 848–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alekseyenko A. A., Peng S., Larschan E., Gorchakov A. A., Lee O. K., et al. , 2008. A sequence motif within chromatin entry sites directs MSL establishment on the Drosophila X chromosome. Cell 134: 599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alekseyenko A. A., Ho J. W., Peng S., Gelbart M., Tolstorukov M. Y., et al. , 2012. Sequence-specific targeting of dosage compensation in Drosophila favors an active chromatin context. PLoS Genet. 8: e1002646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alekseyenko A. A., Ellison C. E., Gorchakov A. A., Zhou Q., Kaiser V. B., et al. , 2013. Conservation and de novo acquisition of dosage compensation on newly evolved sex chromosomes in Drosophila. Genes Dev. 27: 853–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alekseyenko A. A., Gorchakov A. A., Zee B. M., Fuchs S. M., Kharchenko P. V., et al. , 2014. Heterochromatin-associated interactions of Drosophila HP1a with dADD1, HIPP1, and repetitive RNAs. Genes Dev. 28: 1445–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amrein H., Axel R., 1997. Genes expressed in neurons of adult male Drosophila. Cell 88: 459–469. [DOI] [PubMed] [Google Scholar]
- Aronson J. F., Rudkin G. T., Schultz Z., 1954. A comparison of giant X-chromosomes in male and female Drosophila melanogaster by cytophotometry in the ultraviolet. J. Histochem. Cytochem. 2: 458–459. [Google Scholar]
- Bashaw G. J., Baker B. S., 1995. The msl-2 dosage compensation gene of Drosophila encodes a putative DNA-binding protein whose expression is sex specifically regulated by Sex-lethal. Development 121: 3245–3258. [DOI] [PubMed] [Google Scholar]
- Bell O., Conrad T., Kind J., Wirbelauer C., Akhtar A., et al. , 2008. Transcription-coupled methylation of histone H3 at lysine 36 regulates dosage compensation by enhancing recruitment of the MSL complex in Drosophila melanogaster. Mol. Cell. Biol. 28: 3401–3409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belote J. M., Lucchesi J. C., 1980a Male-specific lethal mutations of Drosophila melanogaster. Genetics 96: 165–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belote J. M., Lucchesi J. C., 1980b Control of X chromosome transcription by the maleless gene in Drosophila. Nature 285: 573–575. [DOI] [PubMed] [Google Scholar]
- Bone J. R., Kuroda M. I., 1996. Dosage compensation regulatory proteins and the evolution of sex chromosomes in Drosophila. Genetics 144: 705–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bone J. R., Lavender J., Richman R., Palmer M. J., Turner B. M., et al. , 1994. Acetylated histone H4 on the male X chromosome is associated with dosage compensation in Drosophila. Genes Dev. 8: 96–104. [DOI] [PubMed] [Google Scholar]
- Breen T. R., Lucchesi J. C., 1986. Analysis of the dosage compensation of a specific transcript in Drosophila melanogaster. Genetics 112: 483–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B., 1996. The evolution of chromosomal sex determination and dosage compensation. Curr. Biol. 6: 149–162. [DOI] [PubMed] [Google Scholar]
- Chen Z. X., Oliver B., 2015. X Chromosome and Autosome Dosage Responses in Drosophila melanogaster Heads. G3 (Bethesda) 5: 1057–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline T. W., 1979. A male-specific lethal mutation in Drosophila melanogaster that transforms sex. Dev. Biol. 72: 266–275. [DOI] [PubMed] [Google Scholar]
- Cline T. W., Meyer B. J., 1996. Vive la difference: males vs. females in flies vs. worms. Annu. Rev. Genet. 30: 637–702. [DOI] [PubMed] [Google Scholar]
- Conrad T., Cavalli F. M., Vaquerizas J. M., Luscombe N. M., Akhtar A., 2012. Drosophila dosage compensation involves enhanced Pol II recruitment to male X-linked promoters. Science 337: 742–746. [DOI] [PubMed] [Google Scholar]
- Copps K., Richman R., Lyman L. M., Chang K. A., Rampersad-Ammons J., et al. , 1998. Complex formation by the Drosophila MSL proteins: role of the MSL2 RING finger in protein complex assembly. EMBO J. 17: 5409–5417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cugusi S., Ramos E., Ling H., Yokoyama R., Luk K. M., et al. , 2013. Topoisomerase II plays a role in dosage compensation in Drosophila. Transcription 4: 238–250. [DOI] [PubMed] [Google Scholar]
- Cugusi S., Li Y., Jin P., Lucchesi J. C., 2016. The Drosophila Helicase MLE Targets Hairpin Structures in Genomic Transcripts. PLoS Genet. 12: e1005761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlsveen I. K., Gilfillan G. D., Shelest V. I., Lamm R., Becker P. B., 2006. Targeting determinants of dosage compensation in Drosophila. PLoS Genet. 2: e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiBartolomeis S. M., Tartof K. D., Jackson F. R., 1992. A superfamily of Drosophila satellite related (SR) DNA repeats restricted to the X chromosome euchromatin. Nucleic Acids Res. 20: 1113–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon-McDougall T., Brown C., 2016. The making of a Barr body: the mosaic of factors that eXIST on the mammalian inactive X chromosome. Biochem. Cell Biol. 94: 56–70. [DOI] [PubMed] [Google Scholar]
- Dobzhansky T., 1957. The X-chromosome in the larval salivary glands of hybrids Drosophila insularis × Drosophila tropicalis. Chromosoma 8: 691–698. [DOI] [PubMed] [Google Scholar]
- Fagegaltier D., Baker B. S., 2004. X chromosome sites autonomously recruit the dosage compensation complex in Drosophila males. PLoS Biol. 2: e341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauth T., Muller-Planitz F., Konig C., Straub T., Becker P. B., 2010. The DNA binding CXC domain of MSL2 is required for faithful targeting the Dosage Compensation Complex to the X chromosome. Nucleic Acids Res. 38: 3209–3221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feller C., Prestel M., Hartmann H., Straub T., Soding J., et al. , 2012. The MOF-containing NSL complex associates globally with housekeeping genes, but activates only a defined subset. Nucleic Acids Res. 40: 1509–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari F., Jung Y. L., Kharchenko P. V., Plachetka A., Alekseyenko A. A., et al. , 2013a Comment on “Drosophila dosage compensation involves enhanced Pol II recruitment to male X-linked promoters”. Science 340: 273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari F., Plachetka A., Alekseyenko A. A., Jung Y. L., Ozsolak F., et al. , 2013b “Jump start and gain” model for dosage compensation in Drosophila based on direct sequencing of nascent transcripts. Cell Reports 5: 629–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueiredo M. L., Philip P., Stenberg P., Larsson J., 2012. HP1a recruitment to promoters is independent of H3K9 methylation in Drosophila melanogaster. PLoS Genet. 8: e1003061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukunaga A., Tanaka A., Oishi K., 1975. Maleless, a recessive autosomal mutant of Drosophila melanogaster that specifically kills male zygotes. Genetics 81: 135–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebauer F., Corona D. F., Preiss T., Becker P. B., Hentze M. W., 1999. Translational control of dosage compensation in Drosophila by Sex-lethal: cooperative silencing via the 5′ and 3′ UTRs of msl-2 mRNA is independent of the poly(A) tail. EMBO J. 18: 6146–6154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebauer F., Grskovic M., Hentze M. W., 2003. Drosophila sex-lethal inhibits the stable association of the 40S ribosomal subunit with msl-2 mRNA. Mol. Cell 11: 1397–1404. [DOI] [PubMed] [Google Scholar]
- Gergen J. P., 1987. Dosage Compensation in Drosophila: Evidence That daughterless and Sex-lethal Control X Chromosome Activity at the Blastoderm Stage of Embryogenesis. Genetics 117: 477–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilfillan G. D., Straub T., de Wit E., Greil F., Lamm R., et al. , 2006. Chromosome-wide gene-specific targeting of the Drosophila dosage compensation complex. Genes Dev. 20: 858–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorchakov A. A., Alekseyenko A. A., Kharchenko P., Park P. J., Kuroda M. I., 2009. Long-range spreading of dosage compensation in Drosophila captures transcribed autosomal genes inserted on X. Genes Dev. 23: 2266–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorman M., Kuroda M. I., Baker B. S., 1993. Regulation of the sex-specific binding of the maleless dosage compensation protein to the male X chromosome in Drosophila. Cell 72: 39–49. [DOI] [PubMed] [Google Scholar]
- Gorman M., Franke A., Baker B. S., 1995. Molecular characterization of the male-specific lethal-3 gene and investigations of the regulation of dosage compensation in Drosophila. Development 121: 463–475. [DOI] [PubMed] [Google Scholar]
- Grimaud C., Becker P. B., 2009. The dosage compensation complex shapes the conformation of the X chromosome in Drosophila. Genes Dev. 23: 2490–2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W., Szauter P., Lucchesi J. C., 1998. Targeting of MOF, a putative histone acetyl transferase, to the X chromosome of Drosophila melanogaster. Dev. Genet. 22: 56–64. [DOI] [PubMed] [Google Scholar]
- Gupta V., Parisi M., Sturgill D., Nuttall R., Doctolero M., et al. , 2006. Global analysis of X-chromosome dosage compensation. J. Biol. 5: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallacli E., Lipp M., Georgiev P., Spielman C., Cusack S., et al. , 2012. Msl1-mediated dimerization of the dosage compensation complex is essential for male X-chromosome regulation in Drosophila. Mol. Cell 48: 587–600. [DOI] [PubMed] [Google Scholar]
- Han P. L., Meller V., Davis R. L., 1996. The Drosophila brain revisited by enhancer detection. J. Neurobiol. 31: 88–102. [DOI] [PubMed] [Google Scholar]
- Hilfiker A., Hilfiker-Kleiner D., Pannuti A., Lucchesi J. C., 1997. mof, a putative acetyl transferase gene related to the Tip60 and MOZ human genes and to the SAS genes of yeast, is required for dosage compensation in Drosophila. EMBO J. 16: 2054–2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilik I. A., Quinn J. J., Georgiev P., Tavares-Cadete F., Maticzka D., et al. , 2013. Tandem stem-loops in roX RNAs act together to mediate X chromosome dosage compensation in Drosophila. Mol. Cell 51: 156–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller C. I., Akhtar A., 2015. The MSL complex: juggling RNA-protein interactions for dosage compensation and beyond. Curr. Opin. Genet. Dev. 31: 1–11. [DOI] [PubMed] [Google Scholar]
- Kelley R. L., Solovyeva I., Lyman L. M., Richman R., Solovyev V., et al. , 1995. Expression of msl-2 causes assembly of dosage compensation regulators on the X chromosomes and female lethality in Drosophila. Cell 81: 867–877. [DOI] [PubMed] [Google Scholar]
- Kelley R. L., Wang J., Bell L., Kuroda M. I., 1997. Sex lethal controls dosage compensation in Drosophila by a non-splicing mechanism. Nature 387: 195–199. [DOI] [PubMed] [Google Scholar]
- Kelley R. L., Meller V. H., Gordadze P. R., Roman G., Davis R. L., et al. , 1999. Epigenetic spreading of the Drosophila dosage compensation complex from roX RNA genes into flanking chromatin. Cell 98: 513–522. [DOI] [PubMed] [Google Scholar]
- Kelley R. L., Lee O. K., Shim Y. K., 2008. Transcription rate of noncoding roX1 RNA controls local spreading of the Drosophila MSL chromatin remodeling complex. Mech. Dev. 125: 1009–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kernan M. J., Kuroda M. I., Kreber R., Baker B. S., Ganetzky B., 1991. napts, a mutation affecting sodium channel activity in Drosophila, is an allele of mle, a regulator of X chromosome transcription. Cell 66: 949–959. [DOI] [PubMed] [Google Scholar]
- Kim D., Blus B. J., Chandra V., Huang P., Rastinejad F., et al. , 2010. Corecognition of DNA and a methylated histone tail by the MSL3 chromodomain. Nat. Struct. Mol. Biol. 17: 1027–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koya S. K., Meller V. H., 2015. Modulation of Heterochromatin by Male Specific Lethal Proteins and roX RNA in Drosophila melanogaster Males. PLoS One 10: e0140259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse J. P., Gu W., 2009. MSL2 promotes Mdm2-independent cytoplasmic localization of p53. J. Biol. Chem. 284: 3250–3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroda M. I., Kernan M. J., Kreber R., Ganetzky B., Baker B. S., 1991. The maleless protein associates with the X chromosome to regulate dosage compensation in Drosophila. Cell 66: 935–947. [DOI] [PubMed] [Google Scholar]
- Larschan E., Alekseyenko A. A., Gortchakov A. A., Peng S., Li B., et al. , 2007. MSL complex is attracted to genes marked by H3K36 trimethylation using a sequence-independent mechanism. Mol. Cell 28: 121–133. [DOI] [PubMed] [Google Scholar]
- Larschan E., Bishop E. P., Kharchenko P. V., Core L. J., Lis J. T., et al. , 2011. X chromosome dosage compensation via enhanced transcriptional elongation in Drosophila. Nature 471: 115–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larschan E., Soruco M. M., Lee O. K., Peng S., Bishop E., et al. , 2012. Identification of chromatin-associated regulators of MSL complex targeting in Drosophila dosage compensation. PLoS Genet. 8: e1002830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C. G., Chang K. A., Kuroda M. I., Hurwitz J., 1997. The NTPase/helicase activities of Drosophila maleless, an essential factor in dosage compensation. EMBO J. 16: 2671–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legube G., McWeeney S. K., Lercher M. J., Akhtar A., 2006. X-chromosome-wide profiling of MSL-1 distribution and dosage compensation in Drosophila. Genes Dev. 20: 871–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lott S. E., Villalta J. E., Schroth G. P., Luo S., Tonkin L. A., et al. , 2011. Noncanonical compensation of zygotic X transcription in early Drosophila melanogaster development revealed through single-embryo RNA-seq. PLoS Biol. 9: e1000590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lott S. E., Villalta J. E., Zhou Q., Bachtrog D., Eisen M. B., 2014. Sex-specific embryonic gene expression in species with newly evolved sex chromosomes. PLoS Genet. 10: e1004159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucchesi J. C., 1978. Gene dosage compensation and the evolution of sex chromosomes. Science 202: 711–716. [DOI] [PubMed] [Google Scholar]
- Lucchesi J. C., Manning J. E., 1987. Gene dosage compensation in Drosophila melanogaster. Adv. Genet. 24: 371–429. [DOI] [PubMed] [Google Scholar]
- Lucchesi J. C., Skripsky T., 1981. The link between dosage compensation and sex differentiation in Drosophila melanogaster. Chromosoma 82: 217–227. [DOI] [PubMed] [Google Scholar]
- Lyon M. F., 1961. Gene action in the X-chromosome of the mouse (Mus musculus L.). Nature 190: 372–373. [DOI] [PubMed] [Google Scholar]
- Lyon M. F., 1962. Sex chromatin and gene action in the mammalian X-chromosome. Am. J. Hum. Genet. 14: 135–148. [PMC free article] [PubMed] [Google Scholar]
- Maenner S., Muller M., Frohlich J., Langer D., Becker P. B., 2013. ATP-dependent roX RNA remodeling by the helicase maleless enables specific association of MSL proteins. Mol. Cell 51: 174–184. [DOI] [PubMed] [Google Scholar]
- Marin I., Franke A., Bashaw G. J., Baker B. S., 1996. The dosage compensation system of Drosophila is co-opted by newly evolved X chromosomes. Nature 383: 160–163. [DOI] [PubMed] [Google Scholar]
- McDowell K. A., Hilfiker A., Lucchesi J. C., 1996. Dosage compensation in Drosophila: the X chromosome binding of MSL-1 and MSL-2 in female embryos is prevented by the early expression of the Sxl gene. Mech. Dev. 57: 113–119. [DOI] [PubMed] [Google Scholar]
- Meller V. H., 2003. Initiation of dosage compensation in Drosophila embryos depends on expression of the roX RNAs. Mech. Dev. 120: 759–767. [DOI] [PubMed] [Google Scholar]
- Meller V. H., Rattner B. P., 2002. The roX genes encode redundant male-specific lethal transcripts required for targeting of the MSL complex. EMBO J. 21: 1084–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meller V. H., Wu K. H., Roman G., Kuroda M. I., Davis R. L., 1997. roX1 RNA paints the X chromosome of male Drosophila and is regulated by the dosage compensation system. Cell 88: 445–457. [DOI] [PubMed] [Google Scholar]
- Meller V. H., Gordadze P. R., Park Y., Chu X., Stuckenholz C., et al. , 2000. Ordered assembly of roX RNAs into MSL complexes on the dosage-compensated X chromosome in Drosophila. Curr. Biol. 10: 136–143. [DOI] [PubMed] [Google Scholar]
- Menon D. U., Meller V. H., 2012. A role for siRNA in X-chromosome dosage compensation in Drosophila melanogaster. Genetics 191: 1023–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon D. U., Coarfa C., Xiao W., Gunaratne P. H., Meller V. H., 2014. siRNAs from an X-linked satellite repeat promote X-chromosome recognition in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 111: 16460–16465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer B. J., 2010. Targeting X chromosomes for repression. Curr. Opin. Genet. Dev. 20: 179–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer B. J., Casson L. P., 1986. Caenorhabditis elegans compensates for the difference in X chromosome dosage between the sexes by regulating transcript levels. Cell 47: 871–881. [DOI] [PubMed] [Google Scholar]
- Moore S. A., Ferhatoglu Y., Jia Y., Al-Jiab R. A., Scott M. J., 2010. Structural and biochemical studies on the chromo-barrel domain of male specific lethal 3 (MSL3) reveal a binding preference for mono- or dimethyllysine 20 on histone H4. J. Biol. Chem. 285: 40879–40890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morra R., Smith E. R., Yokoyama R., Lucchesi J. C., 2008. The MLE subunit of the Drosophila MSL complex uses its ATPase activity for dosage compensation and its helicase activity for targeting. Mol. Cell. Biol. 28: 958–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee A. S., Beermann W., 1965. Synthesis of ribonucleic acid by the X-chromosomes of Drosophila melanogaster and the problem of dosage compensation. Nature 207: 785–786. [DOI] [PubMed] [Google Scholar]
- Muller, H. J., 1932 Further studies on the nature and causes of gene mutations. Proceedings of the Sixth International Congress of Genetics, Ithaca, NY. 1: 213–255. [Google Scholar]
- Muller H. J., 1948. Evidence of the precision of genetic adaptation. Harvey Lect Ser 43: 165–229. [Google Scholar]
- Neal K. C., Pannuti A., Smith E. R., Lucchesi J. C., 2000. A new human member of the MYST family of histone acetyl transferases with high sequence similarity to Drosophila MOF. Biochim. Biophys. Acta 1490: 170–174. [DOI] [PubMed] [Google Scholar]
- Oh H., Park Y., Kuroda M. I., 2003. Local spreading of MSL complexes from roX genes on the Drosophila X chromosome. Genes Dev. 17: 1334–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh H., Bone J. R., Kuroda M. I., 2004. Multiple classes of MSL binding sites target dosage compensation to the X chromosome of Drosophila. Curr. Biol. 14: 481–487. [DOI] [PubMed] [Google Scholar]
- Ohno S., Kaplan W. D., Kinosita R., 1959. Formation of the sex chromatin by a single X-chromosome in liver cells of Rattus norvegicus. Exp. Cell Res. 18: 415–418. [DOI] [PubMed] [Google Scholar]
- Palmer M. J., Mergner V. A., Richman R., Manning J. E., Kuroda M. I., et al. , 1993. The male-specific lethal-one (msl-1) gene of Drosophila melanogaster encodes a novel protein that associates with the X chromosome in males. Genetics 134: 545–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer M. J., Richman R., Richter L., Kuroda M. I., 1994. Sex-specific regulation of the male-specific lethal-1 dosage compensation gene in Drosophila. Genes Dev. 8: 698–706. [DOI] [PubMed] [Google Scholar]
- Pardue M. L., Lowenhaupt K., Rich A., Nordheim A., 1987. (dC-dA)n.(dG-dT)n sequences have evolutionarily conserved chromosomal locations in Drosophila with implications for roles in chromosome structure and function. EMBO J. 6: 1781–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S. W., Kang Y., Sypula J. G., Choi J., Oh H., et al. , 2007. An evolutionarily conserved domain of roX2 RNA is sufficient for induction of H4-Lys16 acetylation on the Drosophila X chromosome. Genetics 177: 1429–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S. W., Kuroda M. I., Park Y., 2008. Regulation of histone H4 Lys16 acetylation by predicted alternative secondary structures in roX noncoding RNAs. Mol. Cell. Biol. 28: 4952–4962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S. W., Oh H., Lin Y. R., Park Y., 2010. MSL cis-spreading from roX genes up-regulates the neighboring genes. Biochem. Biophys. Res. Commun. 399: 227–231. [DOI] [PubMed] [Google Scholar]
- Park Y., Kelley R. L., Oh H., Kuroda M. I., Meller V. H., 2002. Extent of chromatin spreading determined by roX RNA recruitment of MSL proteins. Science 298: 1620–1623. [DOI] [PubMed] [Google Scholar]
- Quinn J. J., Zhang Q. C., Georgiev P., Ilik I. A., Akhtar A., et al. , 2016. Rapid evolutionary turnover underlies conserved lncRNA-genome interactions. Genes Dev. 30: 191–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raja S. J., Charapitsa I., Conrad T., Vaquerizas J. M., Gebhardt P., et al. , 2010. The nonspecific lethal complex is a transcriptional regulator in Drosophila. Mol. Cell 38: 827–841. [DOI] [PubMed] [Google Scholar]
- Rastelli L., Kuroda M. I., 1998. An analysis of maleless and histone H4 acetylation in Drosophila melanogaster spermatogenesis. Mech. Dev. 71: 107–117. [DOI] [PubMed] [Google Scholar]
- Richter, L., J. R. Bone, and M. I. Kuroda, 1996 RNA-dependent association of the Drosophila maleless protein with the male X chromosome. Genes Cells. 1: 325–336. [DOI] [PubMed]
- Robinson P. J., An W., Routh A., Martino F., Chapman L., et al. , 2008. 30 nm chromatin fibre decompaction requires both H4–K16 acetylation and linker histone eviction. J. Mol. Biol. 381: 816–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shogren-Knaak M., Ishii H., Sun J. M., Pazin M. J., Davie J. R., et al. , 2006. Histone H4–K16 acetylation controls chromatin structure and protein interactions. Science 311: 844–847. [DOI] [PubMed] [Google Scholar]
- Smith E. R., Pannuti A., Gu W., Steurnagel A., Cook R. G., et al. , 2000. The Drosophila MSL complex acetylates histone H4 at lysine 16, a chromatin modification linked to dosage compensation. Mol. Cell. Biol. 20: 312–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith E. R., Allis C. D., Lucchesi J. C., 2001. Linking global histone acetylation to the transcription enhancement of X-chromosomal genes in Drosophila males. J. Biol. Chem. 276: 31483–31486. [DOI] [PubMed] [Google Scholar]
- Smith E. R., Cayrou C., Huang R., Lane W. S., Cote J., et al. , 2005. A human protein complex homologous to the Drosophila MSL complex is responsible for the majority of histone H4 acetylation at lysine 16. Mol. Cell. Biol. 25: 9175–9188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soruco M. M., Chery J., Bishop E. P., Siggers T., Tolstorukov M. Y., et al. , 2013. The CLAMP protein links the MSL complex to the X chromosome during Drosophila dosage compensation. Genes Dev. 27: 1551–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinemann M., Steinemann S., Lottspeich F., 1993. How Y chromosomes become genetically inert. Proc. Natl. Acad. Sci. USA 90: 5737–5741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenberg P., Larsson J., 2011. Buffering and the evolution of chromosome-wide gene regulation. Chromosoma 120: 213–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub T., Becker P. B., 2008. DNA sequence and the organization of chromosomal domains. Curr. Opin. Genet. Dev. 18: 175–180. [DOI] [PubMed] [Google Scholar]
- Straub T., Becker P. B., 2013. Comment on “Drosophila dosage compensation involves enhanced Pol II recruitment to male X-linked promoters”. Science 340: 273. [DOI] [PubMed] [Google Scholar]
- Strobel E., Pelling C., Arnheim N., 1978. Incomplete dosage compensation in an evolving Drosophila sex chromosome. Proc. Natl. Acad. Sci. USA 75: 931–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuckenholz C., Meller V. H., Kuroda M. I., 2003. Functional redundancy within roX1, a noncoding RNA involved in dosage compensation in Drosophila melanogaster. Genetics 164: 1003–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X., Birchler J. A., 2009. Interaction study of the male specific lethal (MSL) complex and trans-acting dosage effects in metafemales of Drosophila melanogaster. Cytogenet. Genome Res. 124: 298–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sural T. H., Peng S., Li B., Workman J. L., Park P. J., et al. , 2008. The MSL3 chromodomain directs a key targeting step for dosage compensation of the Drosophila melanogaster X chromosome. Nat. Struct. Mol. Biol. 15: 1318–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner, B. M., and G. Fellows, 1989 Specific antibodies reveal ordered and cell-cycle-related use of histone-H4 acetylation sites in mammalian cells. Eur J Biochem. 179: 131–139. [DOI] [PubMed]
- Turner B. M., Birley A. J., Lavender J., 1992. Histone H4 isoforms acetylated at specific lysine residues define individual chromosomes and chromatin domains in Drosophila polytene nuclei. Cell 69: 375–384. [DOI] [PubMed] [Google Scholar]
- Uchida S., Uenoyama T., Oishi K., 1981. Studies on the sex-specific lethals of Drosophila melanogaster. III. A third chromosome male-specific lethal mutant. Jpn. J. Genet. 56: 523–527. [Google Scholar]
- Vaquerizas J. M., Cavalli F. M., Conrad T., Akhtar A., Luscombe N. M., 2013. Response to Comments on “Drosophila Dosage Compensation Involves Enhanced Pol II Recruitment to Male X–Linked Promoters”. Science 340: 273. [DOI] [PubMed] [Google Scholar]
- Villa R., Forne I., Muller M., Imhof A., Straub T., et al. , 2012. MSL2 combines sensor and effector functions in homeostatic control of the Drosophila dosage compensation machinery. Mol. Cell 48: 647–654. [DOI] [PubMed] [Google Scholar]
- Wang Y., Zhang W., Jin Y., Johansen J., Johansen K. M., 2001. The JIL-1 tandem kinase mediates histone H3 phosphorylation and is required for maintenance of chromatin structure in Drosophila. Cell 105: 433–443. [DOI] [PubMed] [Google Scholar]
- Waring G. L., Pollack J. C., 1987. Cloning and characterization of a dispersed, multicopy, X chromosome sequence in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 84: 2843–2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L., Zee B. M., Wang Y., Garcia B. A., Dou Y., 2011. The RING finger protein MSL2 in the MOF complex is an E3 ubiquitin ligase for H2B K34 and is involved in crosstalk with H3 K4 and K79 methylation. Mol. Cell 43: 132–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S., Villa R., Wang J., Feng Y., Becker P. B., et al. , 2014. Structural basis of X chromosome DNA recognition by the MSL2 CXC domain during Drosophila dosage compensation. Genes Dev. 28: 2652–2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou S., Yang Y., Scott M. J., Pannuti A., Fehr K. C., et al. , 1995. Male-specific lethal 2, a dosage compensation gene of Drosophila, undergoes sex-specific regulation and encodes a protein with a RING finger and a metallothionein-like cysteine cluster. EMBO J. 14: 2884–2895. [DOI] [PMC free article] [PubMed] [Google Scholar]