

Abstract
N‐myc & STAT Interactor, NMI, is a protein that has mostly been studied for its physical interactions with transcription factors that play critical roles in tumor growth, progression and metastasis. NMI is an inducible protein, thus its intracellular levels and location can vary dramatically, influencing a diverse array of cellular functions in a context‐dependent manner. The physical interactions of NMI with its binding partners have been linked to many aspects of tumor biology including DNA damage response, cell death, epithelial‐to‐mesenchymal transition and stemness. Thus, discovering more details about the function(s) of NMI could reveal key insights into how transcription factors like c‐Myc, STATs and BRCA1 are contextually regulated. Although a normal, physiological function of NMI has not yet been discovered, it has potential roles in pathologies ranging from viral infection to cancer. This review provides a timely perspective of the unfolding roles of NMI with specific focus on cancer progression and metastasis.
Keywords: N‐myc interactor, cancer, transcription
Introduction
Multiple studies have been focused on elucidating the roles of transcription factors, but less attention has been given to proteins that indirectly influence specific transcriptional events. These proteins are of critical importance because of their organ‐specific, spatio‐temporal regulatory ability. Understanding these indirect influences may hold the key to revealing intricate, biologically relevant details of signaling pathway regulation.
N‐myc and STAT Interactor, NMI, is one such protein that has been realized as one of the proteins that indirectly influence specific transcriptional events. The NMI gene resides on chromosome 2q23. In humans, NMI protein expression has been detected in all fetal tissues except the brain, and its expression is highest in the adult spleen, liver and kidneys.1 The human NMI gene can code for two distinct mature mRNA transcripts that are translated identically into NMI protein products. The NMI protein is typically localized to the cytoplasm, but it has also been detected in the nucleus by multiple investigators.2, 3 As its name reveals, NMI was first identified as a binding protein partner of oncogenic transcription factors, N‐myc and c‐Myc.1 The synopsis of published literature about NMI indicates that this 38 kDa protein may function as an adapter molecule, capable of different functions depending on cellular context and proteins with which it interacts. The first one hundred amino acids of NMI comprise a coiled‐coil domain that is homologous to the C. elegans protein, CEF59. The second domain consisting of the next one hundred amino acids is referred to as the NMI/IFP35 (NID) domain, due to its homology with IFP35 (interferon inducible protein 35). All predicted domains of NMI are reported to be involved in protein–protein interactions (Fig. 1 a). Several proteins confirmed to interact with NMI are transcription factors that influence (positively or negatively) tumor progression. The studies that substantiate these important protein–protein interactions also support the proposition that NMI may have an important role in cancer (Fig. 1 b).
Figure 1.
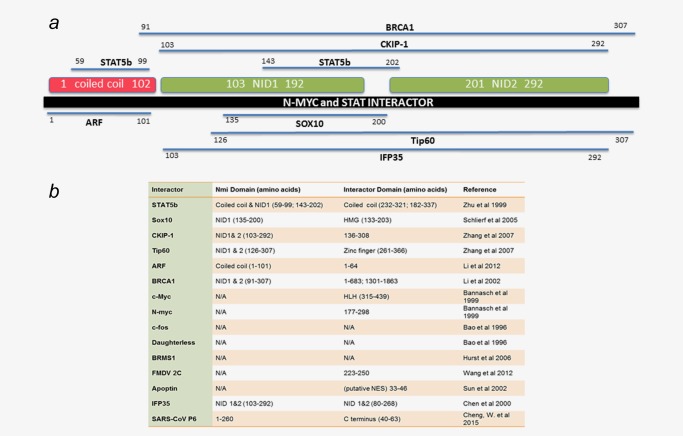
Protein interactors of NMI. ( a ) The validated interacting partners of NMI are depicted along with the functional domains of NMI. Three domains have been characterized within the NMI protein. The first domain comprises the initial one hundred amino acids and resembles a coiled coil structure. The second two domains are named NID domains for their homology to the protein IFP35 and have unknown function. Mapped interactors are shown in their respective binding regions within the NMI protein. ( b ) All interactors of NMI are listed including those which have not been mutated to determine the specific binding region responsible for the interaction. Amino acids determined necessary for binding with each interacting protein are listed as well as their confirmed or speculated domains. As is apparent by the table NMI binds a wide variety of proteins including transcription factors.
Indeed, a number of studies have been aimed at elucidating the role of NMI in cancer as summarized in Table 1. Devine et al. found that NMI protein expression is higher in early stage breast cancer than in later stages, with the lowest expression observed in patients with Stage 4 disease.4 Moreover, NMI mRNA expression is inversely related to the grade of clinical breast cancer specimens, indicating that the loss of NMI correlates with progression to aggressive disease. Surveys of breast tumor specimens from the Cancer Genome Atlas show little copy number variation or mutation of NMI, yet some independent studies have identified single nucleotide polymorphisms (SNPs) in the NMI gene that are present in breast cancer and ovarian cancer patient samples.5, 6 Whether these SNPs cause mutations that alter NMI function was not investigated.
Table 1.
NMI in Diverse Cancer Types
| Cancer type | NMI expression | Findings | Study type | References |
|---|---|---|---|---|
| Breast cancer | Lost with increasing stage | Inhibits EMT, lung metastasis, and tumor growth | Cell lines, xenograft, Patient samples | Fillmore et al., 2009,55 Devine et al., 20144 |
| Glioblastoma multiforme | Increased expression with grade | Promotes growth by regulating G1/S transition | Patient samples, xenograft, cell lines (U251,U87) | Meng et al., 20148 |
| Neuroblastoma | Unknown | Binds N‐myc | Cell line (kelly neuroblastoma) | Bannasch et al., 199978 |
| Colorectal cancer | Unknown | Binds IFP35,induced by IFNα | Cell line (HT‐29) | Zhou et al., 200075 |
| Cervical cancer | Unknown | Induced by IFN γ | Cell line (Hela) | Li et al., 201214 |
| Acute T cell leukemia | Unknown | Induced by IFN γ binds with IFP35 | Cell line (Jurkat) | Chen et al., 2000,76 Nagel et al., 201128 |
| Ovarian cancer | SNPs detected | SNPs are associated with reduced risk of ovarian cancer | Patient samples | Quaye et al., 20095 |
Cancer‐specific studies of NMI are summarized according to what type of cancer was investigated. A variety of cell lines from varying cancer types have been used to determine the functional role of NMI. Relative NMI expression compared to the corresponding normal tissue is listed according to the data presented in each study. Overall findings from each study are summarized including the methods and reagents utilized. It is apparent that in multiple cancer types NMI remains to be inducible by interferon gamma.
In contrast to the findings in epithelial cancers, NMI expression in gliomas indicates poor patient prognosis and has been found by Meng et al. to independently predict overall survival and progression‐free survival.7 SNPs in the NMI gene correlated with susceptibility to glioma. These SNPs were identified in the non‐coding regions of the NMI gene, and a number of SNPs that conferred risk also increased the activity of the NMI promoter.8 Furthermore, NMI mRNA and protein was most highly expressed in Grade 4 glioblastoma multiforme (GBM), the most common advanced stage of glioma.7 Opposing functions of genes in different cancer types has been observed before; and in the case of NMI, the studies of patient specimens show an inverse relationship of the effect of NMI in epithelial cancers versus glial cancers. Taken together, the published literature on NMI suggests that its function can dramatically influence multiple aspects of cancer.
Tumor Initiation and Growth
Several investigations have observed that NMI is involved in signaling mechanisms that contribute to tumor initiation (Fig. 2).
Figure 2.
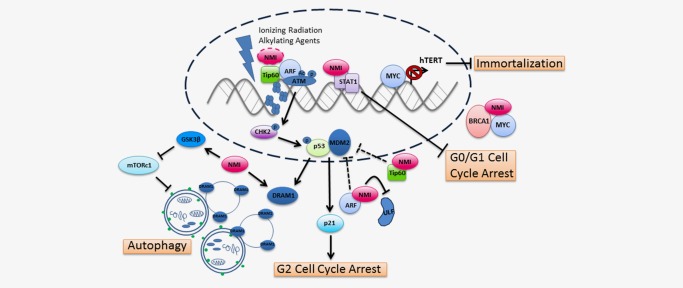
NMI influences cancer initiation. NMI together with BRCA1 sequesters MYC, thereby reducing downstream hTERT gene transcription and inhibiting immortalization of cells during early stages of carcinogenesis. Accumulation of mutations due to DNA damage leads to the transformation of cells. Ionizing radiation and alkylating agents up‐regulate NMI expression and trigger the DNA damage response. Through its interaction with the histone acetyltransferase, Tip60, and tumor suppressor, ARF, NMI could play a role in ATM and p53‐dependent DNA damage responses. The interaction between NMI, ARF and Tip60 may be in a complex to facilitate activation of ATM at sites of DNA damage or independent of each other. Stabilization of ARF by NMI blocks MDM2‐mediated p53 degradation and promotes G2 cell cycle arrest. In addition, NMI may assist Tip60 in its independent inhibition of MDM2 leading to p53 stabilization. However, in contrast, NMI binds STAT1 in glioma cells leading to inhibition of G0/G1 cell cycle arrest. Lastly, NMI facilitates autophagic cell death in response to DNA damage by activating GSK3β and subsequently inhibiting the mTOR pathway—a pathway that blocks autophagy. Expression of DRAM1, a protein essential for the fusing of autophagosomes with lysosomes, positively correlated with NMI levels in breast cancer patients. The dotted connectors indicate possible interactions whereas solid connectors are supported by at least one peer‐reviewed publication.
Immortalization
The first indication that NMI was involved in influencing breast cancer biology came with the discovery of its interaction with c‐Myc, and BRCA1 forming a novel heterotrimeric protein complex. The presence of NMI in this complex led to the sequestration of c‐Myc and a subsequent decrease in the expression of telomerase reverse transcriptase (hTERT).9 Thus, NMI was shown to ameliorate one well‐known cause of cellular immortalization—hTERT expression. This observation is isolated, and many details of this mechanism remain unclear. However, a clearer role of NMI emerges when we consider its involvement in DNA damage repair.
DNA damage
NMI interacts with multiple proteins that are essential for different steps in the DNA damage sensing and repair pathways. BRCA1 is essential for maintaining genomic stability because of its roles in DNA double strand break repair through homologous recombination.10 Naturally occurring DNA damage is identified and repaired through BRCA1 function, thus women with germline mutations in either BRCA1 or BRCA2 have a 50–80% lifetime risk of developing breast cancer.10
Before DNA repair pathways can be activated, areas of damaged DNA need to be detected. NMI may be involved in sensing DNA damage via its interaction with Tip60, a histone acetyltransferase that is activated by radiation‐induced, double‐stranded DNA breaks.11 Interaction of H3K9 tri‐methylated histones and Tip60's chromodomain recruits ATM kinase to the site of double stranded breaks. Acetylation of ATM by Tip60 at lysine 3016 changes the conformation of ATM to allow substrate proteins to access the kinase domain of ATM.12 Ionizing radiation intensifies the interaction between NMI and Tip60 and leads to enhanced expression of both proteins.13 NMI was shown to associate with Tip60 in cytoplasmic speckles. Acetyltransferase activity of Tip60 is necessary to stabilize NMI levels, but the interaction of Tip60 and NMI is not dependent on acyltransferase activity, nor is NMI found to be acetylated by Tip60.13 These studies implicate NMI in this process by association, which again supports the proposition that NMI has a myriad of functions in cancer‐related mechanisms, each dependent on its protein–protein interaction.
In addition to the NMI‐Tip60 interaction, NMI also interacts with another important player in the DNA damage response pathway, ARF.14 ARF responds to DNA damage‐inducing oncogenic stress from hyper‐proliferative genes like c‐Myc.15 Synchronizing DNA damage repair pathways with cellular response requires complex signaling crosstalk converging at the tumor suppressor p53. Both Tip60 and ARF block MDM2‐mediated degradation of p53, and therefore control the p53/MDM2 checkpoint leading to cell cycle arrest or apoptosis.16, 17 Decreased NMI expression, a phenomenon seen in late stage breast tumors, may potentially destabilize ARF as experimental observations showed that NMI inhibits proteasomal degradation of ARF by displacing ULF, the ubiquitin ligase that catalyzes ARF ubiquitination.14 Furthermore, NMI interacts with ARF after cells are stressed with DNA damaging agents such as IFNα, cisplatin or methylmethanosulfate. To summarize these findings, loss of NMI expression in cancer leads to down‐regulation of ARF, destabilization of p53 and subsequent desensitization of cells to DNA damage‐induced cell cycle arrest or cell death. In corroboration with these findings, NMI overexpression also induced CHK2 activation and G2/M cell cycle arrest downstream of p53, particularly after DNA damage.14
Eymin et al. identified a novel p53 independent pathway in response to genotoxic stress. In this pathway, Tip60 and ARF activate ATM to induce G2 cell cycle arrest after DNA damage.18 Moreover, Tip60 and ARF were identified as binding partners that accumulated after treatment with DNA alkylating agents. As NMI binds both Tip60 and ARF, it is plausible that NMI also has a role in this p53‐independent mode of DNA damage sensing and cell cycle arrest, but this idea remains to be tested.
Cell cycle control and growth
Mutations which occur from defects in DNA damage repair pathways can lead to oncogenic insults that alter cell cycle progression. A number of studies have been conducted to assess the role of NMI on tumor cell growth. In vitro, NMI expression in GBM cell lines enhances cell proliferation by inhibiting G0/G1 cell cycle arrest and this effect was thought to be due to NMI binding with STAT1.7 STAT1 blocks the cell cycle by transcriptionally up‐regulating CDK inhibitors or by interacting directly with the Cyclin D1/CDK4 complex.19, 20 In addition, NMI expression increased the growth rate of GBM xenografts.7 NMI expression is significantly higher in GBM when compared to normal controls.7 It is unclear whether NMI is causative in GBM progression or merely a bystander that serves as a prognostic indicator.
NMI was identified through its interaction with Myc family members N‐myc and c‐Myc, as well as their dimerization partner, Max.1 Both members of the Myc family promote cell division and growth in different cancer types. N‐myc is most notable in neuroblastoma, where 20% of tumors exhibit amplification of the N‐myc gene.21 Similarly, c‐Myc expression is high in a wide array of cancers, including breast cancer.22 The interaction between NMI and Myc family members was confirmed in non‐cancerous cell lines and repeatedly observed in breast cancer cells, as mentioned previously. c‐Myc is able to influence all steps of cancer initiation and progression, perhaps due to its wide array of target genes and effects on multiple RNA polymerases.23, 24c‐Myc is a relatively weak transcription factor with the ability to activate or repress transcription in a context‐dependent manner and depending on its transcription co‐factor influence.25 However, a c‐Myc core signature of target genes reveals that transcripts regulated by c‐Myc promote cellular biomass accumulation as well as cell division through up‐regulation of genes involved in ribosome biogenesis and cell cycle advancement, respectively.26 It is not known whether the interaction between NMI and c‐Myc alters transcription or how NMI binding would change the expression of specific c‐Myc transcriptional targets. c‐Myc contributes to multiple aspects of carcinogenesis. Hence, the potential role of NMI regulating c‐Myc‐mediated transcription could provide potential targets in developing novel cancer therapeutics.
Cell death
Carcinogenesis is contingent on cells acquiring resistance to intrinsic cell death mechanisms.27 Resultant tumor growth is the balance between tumor cell growth rates and tumor cell death rates. Early studies of NMI mostly examined its characteristics in hematologic cells. However, a consistent functional role for NMI in hematologic malignancies has not been identified thus far. Nagel et al. discovered that the interaction between STAT5, NMI and N‐myc mediates repression of myocyte enhancing factor 2c leading to increased apoptosis in T cell acute lymphoblastic leukemia.28 Sun et al. identified NMI, from a human leukocyte library, as an interactor of the viral apoptosis‐inducing protein, Apoptin.29 Apoptin is encoded by the chicken anemia virus which infects hematopoietic cells in young chickens, and causes cell death that leads to anemia.30 Interestingly, Apoptin was found to selectively induce apoptosis in tumorigenic cell lines but not in non‐transformed, non‐tumorigenic cells.31 It was observed that Apoptin localized to the nucleus in malignant cells yet it remained cytoplasmic in non‐transformed cells. The nuclear localization sequence (NLS) of Apoptin allows it to interact with the anaphase‐promoting complex/cyclosome, causing G2/M cell cycle arrest and apoptosis.32 The region of Apoptin that interacts with NMI was later confirmed as a nuclear export sequence. Taken together, these data support a potential mechanism by which NMI–Apoptin interaction selectively diminishes nuclear export of Apoptin and subsequently enhances the apoptic activity of Apoptin in transformed cells. This mechanism is speculative, especially since the nature of the NMI–Apoptin interaction is not well understood; yet these data do provide suggestive evidence of NMI being involved in cancer‐cell‐specific apoptosis.
Cytotoxic chemotherapies act by triggering cell death in tumor cells. Expression of NMI sensitized breast cancer cell lines to DNA damaging agents, such as doxorubicin and cisplatin.33 Autophagy, occasionally termed programmed cell death II, was implicated in the enhanced sensitivity imparted by NMI. NMI expression in NMI deficient cell lines elevated basal levels of autophagy in breast cancer cell lines, as indicated by autophagic vacuole formation. This was confirmed molecularly by LC3 processing and expression of Beclin, and p62.33 In certain cellular contexts, autophagy enables cell survival under conditions of stress. However, activation of autophagy beyond a threshold leads to extensive autodigestion and subsequent cell death.34 Numerous novel cancer treatment strategies aim to induce autophagic cell death.35 In the context of clinical specimens, NMI expression significantly correlated with the expression of DNA‐damage regulated autophagy mediator 1(DRAM1) mRNA in breast tumors collected by the Cancer Genome Atlas.33 DRAM1 is essential for the final steps of autophagy and mediates the fusion of autophagosomes with lysosomes.36 Experimentally, decreasing DRAM1 expression in MDA‐MB‐231 breast cancer cells expressing NMI protected the cells against cisplatin treatment.33 Furthermore, Crighton et al. showed that DRAM1 induction of autophagy is essential for p53‐mediated cell death.37 In this study, combinatorial knock down of ATG5 and DRAM1 did not additively affect apoptosis, suggesting that both proteins act through the same cell death pathway—autophagy. Taken together these findings point out that NMI imparts chemosensitivity by enhancing autophagic cell death.
The mechanism by which NMI impacts autophagy is through GSK3β activation, which inhibits mechanistic target of rapamycin (mTOR) signaling.33 mTOR signaling functions as a sensor of macromolecular nutrient level within the cell and tightly couples regulation of autophagy to the energy state of the cell.38 The mTOR complex 1 inhibits phosphorylation of autophagy related genes (ATGs) and therefore prevents the formation of the autophagosome.39, 40 Cancer cell lines silenced for NMI, which became more resistant to doxorubicin and cisplatin, were sensitized through the addition of rapamycin, a potent inducer of autophagy and inhibitor of mTOR signaling.33
The interaction of NMI with the above mentioned regulatory molecules suggests it may play an important role in apoptotic and autophagic cell death pathways. These cell death pathways are known to crosstalk extensively, thus it is provocative to consider that NMI may function as a bridge between these parallel pathways. The precise role of NMI in these cell death pathways may be context‐dependent, and additional details need to be elucidated.
Cancer Progression
The distinction between cancer initiation and progression has grown more pronounced with the discovery of signaling pathways specific to each process. Progression of cancer from early to late stage is a critical step after which therapeutic intervention becomes increasingly difficult. At a cellular level, progression often involves gaining resistance to therapeutic agents and assumption of a less differentiated state. These characteristics can result in metastatic disease. Recent studies propose that the loss of NMI enhances the cellular acquisition of these characteristics that ultimately lead to cancer progression (Fig. 3).
Figure 3.
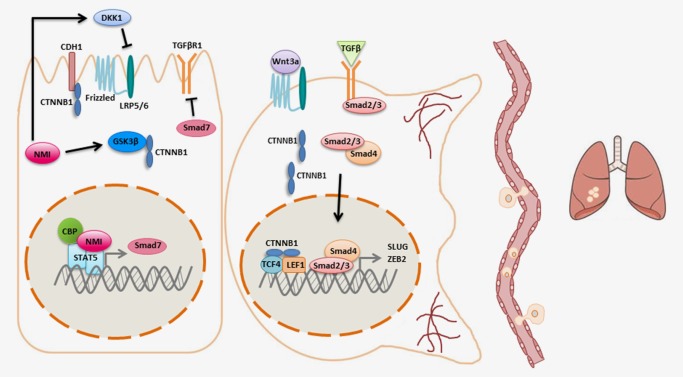
NMI inhibits cancer progression. NMI expression in epithelial cells inhibits the Wnt and TGFβ signaling pathways. NMI also increases the expression of DKK1, the secreted inhibitor of the LRP 5/6 receptor in the Wnt pathway. In addition, NMI increases levels of active GSK3β, thereby targeting β‐catenin for degradation. In parallel, NMI also binds STAT5 to enhance expression of its target gene SMAD7, the inhibitory SMAD, and subsequently dampens TGFβ signaling. In the absence of NMI, β‐catenin accumulates in the cytoplasm and is translocated to the nucleus where it binds TCF/LEF transcription factors to enhance transcription of Wnt pathway target genes. Decreased expression of NMI leads to enhanced transcription of EMT master regulators SLUG and ZEB2 downstream of TGFβ signaling. As a result, cells that have lost NMI undergo morphological changes and become more invasive and metastatic.
Epithelial–mesenchymal transition
Residual tumor cells that survive initial chemotherapy and radiotherapy are often slow growing and may exhibit characteristics of stem cells.41 These cells are phenotypically different from the bulk of the original tumor and contribute to recurrence, dissemination and metastasis. In breast cancer, NMI expression is detected in early stage primary tumors. It is likely that loss of NMI precedes metastatic dissemination of breast tumor cells, as the expression of NMI is significantly lower in primary tumors of patients that have Stage 4 disease.4 Metastasis of epithelial cancers is a complex process that employs epithelial–mesenchymal transition (EMT), entry into the vasculature (intravasation), survival within the circulation and colonization at a secondary site within a foreign microenvironment.42 EMT, the initial step of the metastatic cascade, is a developmental phenomenon whereby epithelial cells undergo a phenotypic change characterized by loss of epithelial markers, such as cytokeratins, followed by gain of mesenchymal markers. These phenotypic changes are accompanied by loss of apical‐basal polarity, reorganization of the actin cytoskeleton and invasion into surrounding tissue.43 EMT is controlled by a variety of signaling pathways that converge at the master regulatory transcription factors SLUG, SNAIL, TWIST and ZEB1/2.44 Silencing NMI in breast cancer cells with inherently epithelial phenotypes caused a morphological change, decreased expression of epithelial markers, as well as an increased expression of master regulators ZEB2 and SLUG.4 Devine and coworkers showed that NMI inhibits the EMT program by antagonizing TGFβ signaling. TGFβ stimulation leads to Smad‐dependent transcription of the master regulators of EMT, and therefore is a potent inducer of EMT.45 NMI expression reversed mesenchymal phenotypes of highly invasive breast cancer cells through enhancing STAT5‐mediated transcription of SMAD7.4 SMAD7, the inhibitory SMAD, provides negative feedback regulation on TGFβ signaling.46, 47
Stemness
Expression of EMT master regulators increases the expression of cell surface markers CD44 and CD24 which are widely accepted as markers of stem cells.48 Also, stem cells often express master regulators of EMT. One of the foremost signaling pathways implicated in EMT and stem cell renewal is the Wnt signaling pathway.49 Wnt signaling, discovered for its role in embryonic development of Drosophila, is essential for stem cell and progenitor cell self‐renewal in multiple tissues, including the intestine and mammary glands.50 The pathway begins at the cell surface, where Wnt ligands bind to a member of the Frizzled family of receptors and either the LRP5 or LRP6 co‐receptor. Tri‐complex formation leads to the stabilization of cytosolic β‐catenin, a transcriptional co‐factor that can enter the nucleus, bind to a member of the TCF/LEF protein family and subsequently activate transcription of Wnt target genes.51 The presence of β‐catenin in cadherin–catenin complexes that comprise epithelial cell tight‐junctions allows for maintenance of epithelial structure and inhibition of EMT.52 Accumulation of β‐catenin in the nucleus of breast carcinoma cells is associated with poor prognosis.53 In breast cancer, deregulation of the Wnt pathway is not due to mutation of the intracellular components, as in colorectal cancers. Instead, many breast cancers are dysregulated at other levels of the pathway, such as overexpression of the LRP6 co‐receptor and the FZD7 receptor, up‐regulation of secreted Wnt ligands and silencing of Wnt inhibitors SFRP‐1 and WIF1.54
In agreement with the findings that NMI inhibits EMT, NMI has also been shown to attenuate Wnt signaling, one of the pathways that can promote EMT. Stably expressing NMI in MDA‐MB‐231 and MDA‐MB‐435 cancer cell lines led to increased levels of DKK1 and concomitantly reduced levels of β‐catenin and c‐Myc, known downstream targets of Wnt signaling.55 DKK1 is a secreted inhibitor of Wnt signaling that has been shown to restrict tumor growth in breast cancer.56 In the same study, stably expressing NMI reduced tumor growth in vitro and in vivo, as well as invasion and migration of tumor cells.55 In addition to its effect on DKK1, NMI has the potential to influence Wnt signaling through its interaction with Casein kinase 2 interacting protein‐1 (CKIP1 or PLEKHO1).57 CKIP1 interacts with the catalytic subunit of casein kinase 2 (CK2α) through its pleckstrin homology domain.58 CK2α phosphorylates and inhibits Dishevelled, a negative regulator of Wnt signaling, and also phosphorylates and stabilizes β‐catenin.59 This ultimately enhances the transcription of Wnt target genes. How CKIP1 affects CK2α, and whether its interaction with NMI directly influences Wnt signaling, are questions that remain unanswered.
In addition to Wnt signaling, NMI also influences other factors known to promote stemness. The SOX10 protein was identified as yet another NMI binding partner.3 SOX10 is a member of the Sox family of transcription factors that are evolutionarily conserved and classified by their HMG box, which contains a DNA binding and bending domain. SOX proteins are essential for embryonic development and control many cell fate decisions, including programming of embryonic stem cells.60 SOX10 is well‐known for its role in neural crest development during embryogenesis.61 The SOX10 protein contains a nuclear localization signal and a nuclear export sequence, allowing it to shuttle in and out of the nucleus.62 NMI and SOX10 co‐localized in the nucleus of C6 glioma cells.3 This may explain how NMI, repeatedly observed in the cytoplasm, physically interacts with other transcription factors without a nuclear localization signal of its own. Shlierf et al. showed that NMI differentially influenced SOX10‐mediated transcriptional activity in a promoter‐dependent manner. NMI increased SOX10‐mediated transcription of one target gene, myelin protein zero, a major structural component of peripheral myelin. However, in the same study, the activity of a second SOX10 target gene, dopachrome tautomerase promoter, was unchanged by NMI.3 This suggests that NMI does not act as a global co‐factor for SOX10‐mediated transcription, and that genomic context and other factors may play a determinative role.
While SOX10 is one potential mechanism by which NMI may travel to the nucleus, it is also possible that cytoplasmic transcription factors bind NMI and shuttle it into the nucleus to facilitate transcription. As mentioned previously, NMI expression has not been reported in fetal or adult brain tissue, and SOX10 is most notable for its role in the development of the neural crest, the peripheral nervous system and melanocytes.61, 63 However, the amount of NMI protein in various adult and fetal tissues remains to be investigated. The interaction between SOX10 and NMI may have a broader functional impact, as emerging evidence shows that SOX10 is also expressed in myoepithelial cells in the breast, salivary and bronchial glands.63 In addition, SOX10 expression was detected by immunohistochemistry in most molecular subtypes of breast cancer patient samples.64
Immunosurveillance
The most effective, intrinsic, protective force against disseminated tumor cells is the immune system. Immune cells rely heavily on JAK/STAT signaling to direct cellular responses upon cytokine stimulation. Compared to other adult tissues, hematopoietic cells have significantly higher endogenous levels of NMI mRNA indicating that NMI may play an integral role in the function of these cells.1 The first study that identified NMI's ability to enhance or alter activity of transcription factors described NMI's effect on signal transducer and activator (STAT) proteins. NMI interacts with all STATs except STAT2 in activated peripheral blood lymphocytes.65 The interaction between STAT1 or STAT5 and NMI significantly increased the activity of their respective target gene luciferase reporters.65 STAT1 activity promotes an inflammatory anti‐tumor immune response in innate immune cells, such as dendritic cells or macrophages.66 Furthermore, STAT1 signaling stimulates the T helper 1 phenotype in CD4+ T cells of the adaptive immune system, creating an inflammatory response through the production of IFNγ.66 STAT5, on the other hand, impacts differentiation of CD8+ T cells, CD4+ Th1 versus Th2 differentiation and development of regulatory T cells.67 It is feasible that modulation of STAT1 or STAT5 activity by NMI impacts the maturation of innate and adaptive immune cells. Interestingly, the activity of STAT1 and STAT5 reporters was enhanced further when the histone acetyltransferases, CBP and p300, were independently co‐transfected; and it was proposed that NMI enhances the interaction between STATs and CBP/p300.65 This study suggests that NMI facilitates binding between epigenetic modifiers and transcription factors to enhance transcription of downstream target genes. The specific contribution of NMI to the anti‐tumor immune response is an area that has not been studied.
The importance of viral infection in cancer biology was solidified fifty years ago with the discovery of the Rous sarcoma virus and the first proto‐oncogene, Src.68, 69 Since then, this connection has only been strengthened with the discoveries of human viruses initiating carcinogenesis such as HPV in cervical cancer and EBV in Burkitt's lymphoma. In addition, the pivotal role of the host immune system in eliminating cancer cells has been garnered for treatment. Based on its ability to influence JAK/STAT signaling, it is feasible that NMI plays a role in the host's viral response. Indeed, NMI has been independently identified by groups studying viral infection as a mediator of host–viral response.70 NMI was proposed to facilitate apoptosis through interaction with FMDV 2C, a key protein from a virus that causes foot and mouth disease in swine.71 The FMDV 2C–NMI complexes were speculated to associate with the endoplasmic reticulum. In an independent study, microarray data from cells overexpressing HIV viral proteins showed a twofold decrease of NMI mRNA, suggesting it may have a mechanism in host cell viral defense during HIV infection.72 Most recently however, NMI was identified as a binding partner of the severe acute respiratory syndrome (SARS) coronavirus non‐structural protein 6.70, 73 Protein 6 is a transmembrane protein that localizes to the cytosolic side of the endoplasmic reticulum and is assembled into a replication complex that facilitates viral replication. Interestingly, it inhibits the host's innate immune response by binding and preventing nuclear import of STAT1.74 The NMI protein 6 interaction led to the destabilization of NMI.70 Although these studies were conducted independently it is intriguing to speculate that degradation or sequestration of NMI by SARS coronavirus protein 6 could contribute to the inhibitory effect on STAT1.
NMI Regulation
Restoring or inhibiting NMI expression based on the specific cancer type may be an attractive therapeutic option in the future. Therefore, it is essential to know what pathways directly regulate the transcription, translation, and stability of NMI. Early studies of NMI induction indicate that its subcellular localization may be coupled to its regulation and function. Seminal studies identified that NMI expression is transcriptionally induced downstream of inflammatory cytokines, such as IFNγ, IFNα and IL‐2.65 Downstream mediators such as STAT1 and STAT5 are most likely responsible for binding the NMI promoter. NMI, in turn, facilitates the activity of STATs, as previously described, allowing for a robust positive feedback response after cytokine stimulation. Interferon treatment resulted in the formation of high molecular mass complexes (HMMCs) of 300–400 kDa. These complexes were visualized as NMI/IFP35 speckles (NIS) in previous publications and they formed following dephosphorylation of IFP35.75, 76 The function of the NIS complex warrants further exploration, as it may play a role in the human viral response. Other interactors of NMI such as c‐Myc, N‐myc and c‐fos are not thought to be contained in the HMMC. Molecular analysis confirmed that the fractionation properties of NMI change after treatment with IFNγ, indicating a shift in subcellular location.77 Multiple groups have shown that induction of NMI leads to its translocation into the nucleus.77, 78 This may allow NMI to interact with nuclear transcription factors. It has also been reported that cytokine‐induced NMI expression leads to reorganization of the NMI protein into puncta within the cytoplasm.76 Thus, NMI seems to have a distinct cytoplasmic role(s).
Another potential transcriptional regulator of NMI is Ets‐1. The Ets family of transcription factors is highly conserved among species and has documented roles in physiologic and pathologic processes.79 Ets‐1 is an upstream regulator of NMI. When expressed in MCF‐7 breast cancer cells, Ets‐1 increased NMI mRNA and protein expression, as well as STAT1 expression.80 Although the study suggests NMI has a role in Ets‐1 related breast carcinogenesis, it remains unclear whether that role is permissive or inhibitory.
No permanent genetic alterations of NMI expression such as mutation or deletion have been observed in cancer and many studies have identified “plastic” regulation of the NMI gene. In particular, loss of NMI in late stage breast cancer suggests that NMI is transiently down‐regulated. Rostas et al. found that NMI is subject to post‐transcriptional regulation by the micro‐RNA 29 family in breast cancer cell lines, as well as in clinical samples.81 Interestingly, miR‐29 targeting of the NMI transcript led to a decrease in epithelial morphology and markers, as well as an enhancement of invasive properties and these data further support the previously published observations by Devine and coworkers.81 Moreover, it suggests that NMI regulation by miR‐29 is a key event in the progression of breast cancer. Additionally, miR‐29 down‐regulation of NMI created a feed‐forward regulatory loop promoting unrestricted Wnt signaling.
Regulation of NMI by post‐translational modifications remains to be studied. NMI is a relatively labile protein with a half‐life of approximately 8 hr, and this half‐life is extended through interaction with other cellular proteins.14 As discussed previously, Zhang et al. reported that interaction between Tip60 and NMI in the cytoplasm led to stabilization of the NMI protein. Interaction of the two proteins was not dependent on acetyltransferase activity, thus the mechanism of stabilization remains unknown.13 Of note, NMI and its paralog, IFP35, are both labile proteins targeted for degradation by the proteasome; yet, heterodimerization stabilizes both proteins. To date the only protein that has been shown to destabilize NMI is the previously mentioned SARS coronavirus protein 6. The interaction between NMI and Protein 6 causes ubiquitination and subsequent degradation of NMI.70
Conclusion
Additional studies investigating the function and clinical relevance of NMI are needed. However, at this stage of NMI research, experimental data support the notion that NMI affects numerous mechanisms essential to the initiation and progression of tumors. NMI has clearly emerged as a protein that binds to both transcription factors as well as epigenetic modifiers and modulates their activities. Thus far, the only chromatin modifiers shown to bind NMI are histone acetyltransferases (HATs). Luciferase reporter studies indicate that these interactions influence promoter activity of downstream target genes. The functional consequences of the molecular interaction between NMI, transcription factors and HATs remain unclear. Clinical studies suggest that NMI plays a tumor suppressive role in epithelial cancers such as breast cancer while it indicates a poor prognosis in mesenchymal‐like tumors such as gliomas. It is likely that the stage in which NMI is most critical depends on the specific histological tumor type and how far it has progressed. It is conceivable that NMI may contribute to intra‐tumor heterogeneity, a concept that needs experimental validation.
Non‐transformed cells in the tumor microenvironment directly influence tumor progression. Therefore, the role of NMI in the tumor microenvironment and host cells may impact the course of disease progression. Studies using genetic model systems will provide critical insights into these roles of NMI. Lastly, studies of NMI regulation will allow a unique opportunity for the development of therapeutic strategies that target NMI which may differ depending on the cancer type.
References
- 1. Bao J, Zervos AS. Isolation and characterization of Nmi, a novel partner of Myc proteins. Oncogene 1996;12:2171–6. [PubMed] [Google Scholar]
- 2. Lee ND, Chen J, Shpall RL, et al. Subcellular localization of interferon‐inducible Myc/Stat‐interacting protein Nmi is regulated by a novel IFP 35 homologous domain. J Interferon Cytokine Res 1999;19:1245–52. [DOI] [PubMed] [Google Scholar]
- 3. Schlierf B, Lang S, Kosian T, et al. The high‐mobility group transcription factor Sox10 interacts with the N‐myc‐interacting protein Nmi. J Mol Biol 2005;353:1033–42. [DOI] [PubMed] [Google Scholar]
- 4. Devine DJ, Rostas JW, Metge BJ, et al. Loss of N‐Myc interactor promotes epithelial‐mesenchymal transition by activation of TGF‐beta/SMAD signaling. Oncogene 2014;33:2620–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Quaye L, Song H, Ramus SJ, et al. Tagging single‐nucleotide polymorphisms in candidate oncogenes and susceptibility to ovarian cancer. Br J Cancer 2009;100:993–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vega A, Salas A, Milne RL, et al. Evaluating new candidate SNPs as low penetrance risk factors in sporadic breast cancer: a two‐stage Spanish case‐control study. Gynecol Oncol 2009;112:210–14. [DOI] [PubMed] [Google Scholar]
- 7. Meng D, Chen Y, Yun D, et al. High expression of N‐myc (and STAT) interactor predicts poor prognosis and promotes tumor growth in human glioblastoma. Oncotarget 2015;6:4901–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Meng D, Li X, Zhang S, et al. Genetic variants in N‐myc (and STAT) interactor and susceptibility to glioma in a Chinese Han population. Tumour Biol 2015;36:1579–88. [DOI] [PubMed] [Google Scholar]
- 9. Li H, Lee TH, Avraham H. A novel tricomplex of BRCA1, Nmi, and c‐Myc inhibits c‐Myc‐induced human telomerase reverse transcriptase gene (hTERT) promoter activity in breast cancer. J Biol Chem 2002;277:20965–73. [DOI] [PubMed] [Google Scholar]
- 10. Roy R, Chun J, Powell SN. BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat Rev Cancer 2012;12:68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sun Y, Jiang X, Chen S, et al. A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc Natl Acad Sci USA 2005;102:13182–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sun Y, Jiang X, Price BD. Tip60: connecting chromatin to DNA damage signaling. Cell Cycle 2012;9:930–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang K, Zheng G, Yang YC. Stability of Nmi protein is controlled by its association with Tip60. Mol Cell Biochem 2007;303:1–8. [DOI] [PubMed] [Google Scholar]
- 14. Li Z, Hou J, Sun L, et al. NMI mediates transcription‐independent ARF regulation in response to cellular stresses. Mol Biol Cell 2012;23:4635–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zindy F, Eischen CM, Randle DH, et al. Myc signaling via the ARF tumor suppressor regulates p53‐dependent apoptosis and immortalization. Genes Dev 1998;12:2424–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Legube G, Linares LK, Tyteca S, et al. Role of the histone acetyl transferase Tip60 in the p53 pathway. J Biol Chem 2004;279:44825–33. [DOI] [PubMed] [Google Scholar]
- 17. Ozenne P, Eymin B, Brambilla E, et al. The ARF tumor suppressor: structure, functions and status in cancer. Int J Cancer 2010;127:2239–47. [DOI] [PubMed] [Google Scholar]
- 18. Eymin B, Claverie P, Salon C, et al. p14ARF activates a Tip60‐dependent and p53‐independent ATM/ATR/CHK pathway in response to genotoxic stress. Mol Cell Biol 2006;26:4339–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chin YE, Kitagawa M, Su WC, et al. Cell growth arrest and induction of cyclin‐dependent kinase inhibitor p21 WAF1/CIP1 mediated by STAT1. Science 1996;272:719–22. [DOI] [PubMed] [Google Scholar]
- 20. Dimco G, Knight RA, Latchman DS, et al. STAT1 interacts directly with cyclin D1/Cdk4 and mediates cell cycle arrest. Cell Cycle 2010;9:4638–49. [DOI] [PubMed] [Google Scholar]
- 21. Beltran H. The N‐myc oncogene: maximizing its targets, regulation, and therapeutic potential. Mol Cancer Res 2014;12:815–22. [DOI] [PubMed] [Google Scholar]
- 22. Singhi AD, Cimino‐Mathews A, Jenkins RB, et al. MYC gene amplification is often acquired in lethal distant breast cancer metastases of unamplified primary tumors. Mod Pathol 2012;25:378–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Oskarsson T, Trumpp A. The Myc trilogy: lord of RNA polymerases. Nat Cell Biol 2005;7:215–17: [DOI] [PubMed] [Google Scholar]
- 24. Gomez‐Roman N, Felton‐Edkins ZA, Kenneth NS, et al. Activation by m‐Myc of transcription by RNA polymerases I, II and III. Biochem Soc Symp 2006;73:141–54. [DOI] [PubMed] [Google Scholar]
- 25. Cowling VH, Cole MD. Mechanism of transcriptional activation by the Myc oncoproteins. Semin Cancer Biol 2006;16:242–52. [DOI] [PubMed] [Google Scholar]
- 26. Ji H, Wu G, Zhan X, et al. Cell‐type independent MYC target genes reveal a primordial signature involved in biomass accumulation. PLoS One 2011;6:e26057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646–74. [DOI] [PubMed] [Google Scholar]
- 28. Nagel S, Venturini L, Meyer C, et al. Transcriptional deregulation of oncogenic myocyte enhancer factor 2C in T‐cell acute lymphoblastic leukemia. Leuk Lymphoma 2011;52:290–7. [DOI] [PubMed] [Google Scholar]
- 29. Sun GJ, Tong X, Dong Y, et al. Identification of a protein interacting with apoptin from human leucocyte cDNA library by using yeast two‐hybrid screening. Sheng Wu Hua Xue Yu Sheng Wu Wu Li Xue Bao (Shanghai) 2002;34:369–72. [PubMed] [Google Scholar]
- 30. Noteborn MH, Todd D, Verschueren CA, et al. A single chicken anemia virus protein induces apoptosis. J Virol 1994;68:346–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dan‐Van Ooeschot AAAM, Fischer DF, Grimbergen JM, et al. Apoptin induces apoptosis in human transformed and malignant cells but not in normal cells. Proc Natl Acad Sci USA 1997;94:5843–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Piao Z, Lee KS, Kim H, et al. Identification of novel deletion regions on chromosome arms 2q and 6p in breast carcinomas by amplotype analysis. Genes Chromosomes Cancer 2001;30:113–22. [PubMed] [Google Scholar]
- 33. Metge BJ, Mitra A, Chen D, et al. N‐Myc and STAT Interactor regulates autophagy and chemosensitivity in breast cancer cells. Sci Rep 2015;5:11995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Munoz‐Pinedo C, Martin SJ. Autosis: a new addition to the cell death Tower of Babel. Cell Death Dis 2014;5:e1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fulda S, Kogel D. Cell death by autophagy: emerging molecular mechanisms and implications for cancer therapy. Oncogene 2015;34:5105–5113. [DOI] [PubMed] [Google Scholar]
- 36. Zhang XD, Qi L, Wu JC, et al. DRAM1 regulates autophagy flux through lysosomes. PLoS One 2013;8:e63245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Crighton D, Wilkinson S, O'Prey J, et al. DRAM, a p53‐induced modulator of autophagy, is critical for apoptosis. Cell 2006;126:121–34. [DOI] [PubMed] [Google Scholar]
- 38. Laplante M, Sabatini DM. mTOR signaling at a glance. J Cell Sci 2009;122:3589–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hosokawa N, Hara T, Kaizuka T, et al. Nutrient‐dependent mTORC1 association with the ULK1‐Atg13‐FIP200 complex required for autophagy. Mol Biol Cell 2009;20:1981–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jung CH, Jun CB, Ro SH, et al. ULK‐Atg13‐FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell 2009;20:1992–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wicha MS, Liu S, Dontu G. Cancer stem cells: an old idea—a paradigm shift. Cancer Res 2006;66:1883–90. discussion 95–6. [DOI] [PubMed] [Google Scholar]
- 42. Yang J, Weinberg RA. Epithelial‐mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell 2008;14:818–29. [DOI] [PubMed] [Google Scholar]
- 43. Kalluri R, Weinberg RA. The basics of epithelial‐mesenchymal transition. J Clin Invest 2009;119:1420–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zheng H, Kang Y. Multilayer control of the EMT master regulators. Oncogene 2014;33:1755–63. [DOI] [PubMed] [Google Scholar]
- 45. Xu J, Lamouille S, Derynck R. TGF‐beta‐induced epithelial to mesenchymal transition. Cell Res 2009;19:156–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hayashi H, Abdollah S, Qiu Y, et al. The MAD‐related protein Smad7 associates with the TGFbeta receptor and functions as an antagonist of TGFbeta signaling. Cell 1997;89:1165–73. [DOI] [PubMed] [Google Scholar]
- 47. Samant RS, Shevde LA. NMI and EMT. Oncoscience 2014;1:476–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mani SA, Guo W, Liao MJ, et al. The epithelial‐mesenchymal transition generates cells with properties of stem cells. Cell 2008;133:704–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chang YW, Su YJ, Hsiao M, et al. Diverse targets of beta‐catenin during the epithelial‐mesenchymal transition define cancer stem cells and predict disease relapse. Cancer Res 2015;75:3398–410. [DOI] [PubMed] [Google Scholar]
- 50. Klaus A, Birchmeier W. Wnt signalling and its impact on development and cancer. Nat Rev Cancer 2008;8:387–98. [DOI] [PubMed] [Google Scholar]
- 51. Rudnicki MA, Williams BO. Wnt signaling in bone and muscle. Bone 2015;80:60–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Heuberger J, Birchmeier W. Interplay of cadherin‐mediated cell adhesion and canonical Wnt signaling. Cold Spring Harb Perspect Biol 2010;2:a002915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lin SY, Xia W, Wang JC, et al. Beta‐catenin, a novel prognostic marker for breast cancer: its roles in cyclin D1 expression and cancer progression. Proc Natl Acad Sci USA 2000;97:4262–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mukherjee N, Bhattacharya N, Alam N, et al. Subtype‐specific alterations of the Wnt signaling pathway in breast cancer: clinical and prognostic significance. Cancer Sci 2012;103:210–20. [DOI] [PubMed] [Google Scholar]
- 55. Fillmore RA, Mitra A, Xi Y, et al. Nmi (N‐Myc interactor) inhibits Wnt/beta‐catenin signaling and retards tumor growth. Int J Cancer 2009;125:556–64. [DOI] [PubMed] [Google Scholar]
- 56. Menezes ME, Devine DJ, Shevde LA, et al. Dickkopf1: a tumor suppressor or metastasis promoter? Int J Cancer 2012;130:1477–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhang L, Tang Y, Tie Y, et al. The PH domain containing protein CKIP‐1 binds to IFP35 and Nmi and is involved in cytokine signaling. Cell Signal 2007;19:932–44. [DOI] [PubMed] [Google Scholar]
- 58. Bosc DG, Graham KC, Saulnier RB, et al. Identification and characterization of CKIP‐1, a novel pleckstrin homology domain‐containing protein that interacts with protein kinase CK2. J Biol Chem 2000;275:14295–306. [DOI] [PubMed] [Google Scholar]
- 59. Song DH, Sussman DJ, Seldin DC. Endogenous protein kinase CK2 participates in Wnt signaling in mammary epithelial cells. J Biol Chem 2000;275:23790–7. [DOI] [PubMed] [Google Scholar]
- 60. Sarkar A, Hochedlinger K. The sox family of transcription factors: versatile regulators of stem and progenitor cell fate. Cell Stem Cell 2013;12:15–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Paratore C, Goerich DE, Suter U, et al. Survival and glial fate acquisition of neural crest cells are regulated by an interplay between the transcription factor Sox10 and extrinsic combinatorial signaling. Development 2001;128:3949–61. [DOI] [PubMed] [Google Scholar]
- 62. Rehberg S, Lischka P, Glaser G, et al. Sox10 is an active nucleocytoplasmic shuttle protein, and shuttling is crucial for Sox10‐mediated transactivation. Mol Cell Biol 2002;22:5826–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Nonaka D, Chiriboga L, Rubin BP. Sox10: a pan‐schwannian and melanocytic marker. Am J Surg Pathol 2008;32:1291–8. [DOI] [PubMed] [Google Scholar]
- 64. Cimino‐Mathews A, Subhawong AP, Elwood H, et al. Neural crest transcription factor Sox10 is preferentially expressed in triple‐negative and metaplastic breast carcinomas. Hum Pathol 2013;44:959–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zhu M, John S, Berg M, et al. Functional association of Nmi with Stat5 and Stat1 in IL‐2 and IFNgamma‐mediated signaling. Cell 1999;96:121–30. [DOI] [PubMed] [Google Scholar]
- 66. Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer 2009;9:798–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Heltemes‐Harris LM, Willette MJ, Vang KB, et al. The role of STAT5 in the development, function, and transformation of B and T lymphocytes. Ann N Y Acad Sci 2011;1217:18–31. [DOI] [PubMed] [Google Scholar]
- 68. Rous P. A sarcoma of the fowl transmissible by an agent separable from the tumor cells. J Exp Med 1911;13:397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Brugge JS, Erikson RL. Identification of a transformation‐specific antigen induced by an avian sarcoma virus. Nature 1977;269:346–8. [DOI] [PubMed] [Google Scholar]
- 70. Cheng W, Chen S, Li R, et al. Severe acute respiratory syndrome coronavirus protein 6 mediates ubiquitin‐dependent proteosomal degradation of N‐Myc (and STAT) interactor. Virol Sin 2015;30:153–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Wang J, Wang Y, Liu J, et al. A critical role of N‐myc and STAT interactor (Nmi) in foot‐and‐mouth disease virus (FMDV) 2C‐induced apoptosis. Virus Res 2012;170:59–65. [DOI] [PubMed] [Google Scholar]
- 72. Park S‐E, Lee MJ, Yang MH, et al. Expression profiles and pathway analysis in HEK 293 T cells overexpressing HIV‐1 Tat and nucleocapsid using cDNA microarray. J Microbiol Biotechnol 2007;17:154–61. [PubMed] [Google Scholar]
- 73. Zhao J, Falcon A, Zhou H, et al. Severe acute respiratory syndrome coronavirus protein 6 is required for optimal replication. J Virol 2009;83:2368–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Frieman M, Yount B, Heise M, et al. Severe acute respiratory syndrome coronavirus ORF6 antagonizes STAT1 function by sequestering nuclear import factors on the rough endoplasmic reticulum/Golgi membrane. J Virol 2007;81:9812–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zhou X, Liao J, Meyerdierks A, et al. Interferon‐alpha induces nmi‐IFP35 heterodimeric complex formation that is affected by the phosphorylation of IFP35. J Biol Chem 2000;275:21364–71. [DOI] [PubMed] [Google Scholar]
- 76. Chen J, Shpall RL, Meyerdierks A, et al. Interferon‐inducible Myc/STAT‐interacting protein Nmi associates with IFP 35 into a high molecular mass complex and inhibits proteasome‐mediated degradation of IFP 35. J Biol Chem 2000;275:36278–84. [DOI] [PubMed] [Google Scholar]
- 77. Lebrun S, Shpall R, Naumovski L. Interferon‐induced upregulation and cytoplasmic localization of Myc‐interacting protein Nmi. J Interferon Cytokine Res 1998;18:767–71. [DOI] [PubMed] [Google Scholar]
- 78. Bannasch D, Weis I, Schwab M. Nmi protein interacts with regions that differ between MycN and Myc and is localized in the cytoplasm of neuroblastoma cells in contrast to nuclear MycN. Oncogene 1999;18:6810–17. [DOI] [PubMed] [Google Scholar]
- 79. Sharrocks AD. The ETS‐domain transcription factor family. Nat Rev Mol Cell Biol 2001;2:827–37. [DOI] [PubMed] [Google Scholar]
- 80. Jung HH, Lee J, Kim JH, et al. STAT1 and Nmi are downstream targets of Ets‐1 transcription factor in MCF‐7 human breast cancer cell. FEBS Lett 2005;579:3941–6. [DOI] [PubMed] [Google Scholar]
- 81. Rostas JW, III , Pruitt HC, Metge BJ, et al. microRNA‐29 negatively regulates EMT regulator N‐myc interactor in breast cancer. Mol Cancer 2014;13:200. [DOI] [PMC free article] [PubMed] [Google Scholar]