

Abstract
Patients with hepatitis C virus (HCV) genotype 3 infection, especially those with advanced liver disease, are a challenging population in urgent need of optimally effective therapies. The combination of daclatasvir (DCV; pangenotypic nonstructural protein 5A inhibitor) and sofosbuvir (SOF; nucleotide nonstructural protein 5B inhibitor) for 12 weeks previously showed high efficacy (96%) in noncirrhotic genotype 3 infection. The phase III ALLY‐3+ study (N = 50) evaluated DCV‐SOF with ribavirin (RBV) in treatment‐naïve (n = 13) or treatment‐experienced (n = 37) genotype 3‐infected patients with advanced fibrosis (n = 14) or compensated cirrhosis (n = 36). Patients were randomized 1:1 to receive open‐label DCV‐SOF (60 + 400 mg daily) with weight‐based RBV for 12 or 16 weeks. The primary endpoint was sustained virological response at post‐treatment week 12 (SVR12). SVR12 (intention‐to‐treat) was 90% overall (45 of 50): 88% (21 of 24) in the 12‐week (91% observed) and 92% (24 of 26) in the 16‐week group. All patients with advanced fibrosis achieved SVR12. SVR12 in patients with cirrhosis was 86% overall (31 of 36): 83% (15 of 18) in the 12‐week (88% observed) and 89% (16 of 18) in the 16‐week group; for treatment‐experienced patients with cirrhosis, these values were 87% (26 of 30), 88% (14 of 16; 93% observed), and 86% (12 of 14), respectively. One patient (12‐week group) did not enter post‐treatment follow‐up (death unrelated to treatment). There were 4 relapses (2 per group) and no virological breakthroughs. The most common adverse events (AEs) were insomnia, fatigue, and headache. There were no discontinuations for AEs and no treatment‐related serious AEs. Conclusion: The all‐oral regimen of DCV‐SOF‐RBV was well tolerated and resulted in high and similar SVR12 after 12 or 16 weeks of treatment among genotype 3‐infected patients with advanced liver disease, irrespective of past HCV treatment experience. (Hepatology 2016;63:1430‐1441)
Abbreviations
- AE
adverse event
- AFP
alpha‐fetoprotein
- APRI
aspartate aminotransferase to platelet ratio index
- CI
confidence interval
- DAA
direct‐acting antiviral
- DCV
daclatasvir
- HCC
hepatocellular carcinoma
- HCV
hepatitis C virus
- ITT
intention‐to‐treat
- LLOQ
lower limit of quantitation
- NGS
next‐generation sequencing
- NS5A
nonstructural protein 5A
- NS5B
nonstructural protein 5B
- Peg‐IFN‐α
pegylated interferon alpha
- RAV
resistance‐associated variant
- RBV
ribavirin
- SAE
serious AE
- SOF
sofosbuvir
- SVR
sustained virological response
- SVR4
sustained virological response at post‐treatment week 4
- SVR12
sustained virological response at post‐treatment week 12
Hepatitis C virus (HCV) genotype 3 is globally distributed and most prevalent in South East Asia.1, 2 It is responsible for approximately 12% of chronic HCV infections in the United States,3 up to 30% of infections in parts of Europe,4, 5, 6 and approximately 40% of infections in Australia.7 It is common among infections resulting from injection drug use, tattooing, or piercing.8, 9
Genotype 3 infection is associated with rapid progression of hepatic fibrosis,10 a high rate of steatosis11 that correlates with level of viral replication,12 and a greater risk of developing hepatocellular carcinoma (HCC) than other genotypes.13 Thus, genotype 3‐infected patients urgently require treatment. Although historically genotypes 3 and 2 were considered similarly responsive to pegylated interferon alpha (Peg‐IFN‐α) and ribavirin (RBV) treatment, it is now known that sustained virological response (SVR) rates after Peg‐IFN‐α/RBV treatment are lower for genotype 3 than for genotype 2.14, 15
Achieving sufficient treatment uptake to effectively address the public health burden of HCV‐associated liver disease requires effective, well‐tolerated treatment options for all HCV genotypes. The move away from interferon‐based therapy toward all‐oral combinations of direct‐acting antivirals (DAAs) for HCV has significantly improved the convenience, efficacy, and tolerability of HCV treatment overall; however, treatment of genotype 3 remains a significant challenge. Many of the currently approved DAAs—including ledipasvir,16 simeprevir,17, 18 dasabuvir,19 and asunaprevir20—are genotype specific and have limited activity against genotype 3 in vitro or in vivo. Sofosbuvir (SOF), a pangenotypic nonstructural protein 5B (NS5B) inhibitor,21 is active against genotype 3. The combination of SOF plus RBV (SOF‐RBV) requires a 24‐week treatment duration, and SVR rates are suboptimal among patients with previous treatment experience and/or cirrhosis.22, 23, 24, 25 This response rate can be improved by the addition of Peg‐IFN‐α to SOF‐RBV (SOF‐RBV‐Peg‐IFN‐α),22, 26 though at the expense of introducing a significant burden of interferon‐associated adverse events (AEs)27 that excludes a large proportion of individuals who are unwilling or unable to take interferons.28, 29, 30
Daclatasvir (DCV), a pangenotypic nonstructural protein 5A (NS5A) inhibitor,31 has picomolar activity against wild‐type genotype 3. For patients without cirrhosis, RBV‐free treatment with DCV plus SOF (DCV‐SOF) for 12 weeks is highly effective for treatment of genotype 3 infection. In the phase III ALLY‐3 study, the sustained virological response (SVR) rate at post‐treatment week 12 (SVR12) was 96% in genotype 3‐infected patients without cirrhosis, regardless of past HCV treatment experience, with good tolerability.32 A lower SVR12 rate was observed in ALLY‐3 among genotype 3‐infected patients with cirrhosis treated with DCV‐SOF for 12 weeks. Therefore, there is a need for improved treatment strategies for patients with genotype 3 infection and advanced liver disease.
To this end, we report herein the results of a phase III randomized study (ALLY‐3+) evaluating the efficacy and safety of the combination of DCV‐SOF plus RBV (DCV‐SOF‐RBV) for 12 or 16 weeks in genotype 3‐infected patients with advanced fibrosis or compensated cirrhosis.
Patients and Methods
STUDY DESIGN AND PATIENTS
ALLY‐3+ is an open‐label, randomized phase IIIb study (Study AI444326; ClinicalTrials.gov: NCT02319031) of a 12‐ or 16‐week regimen of DCV‐SOF‐RBV in genotype 3‐infected patients with advanced fibrosis or compensated cirrhosis.
Eligible patients were adults (≥18 years old) with chronic HCV genotype 3 infection who were either treatment‐naïve or treatment‐experienced and had HCV‐RNA levels ≥10,000 IU/mL at screening. Treatment‐experienced patients may have received past therapy with any agent or combination of agents, with the exception of NS5A inhibitors. Patients with previous virological failure on SOF‐RBV were permitted, but patients who discontinued SOF‐RBV for intolerance or anemia were excluded. All previous HCV treatment must have been completed or discontinued at least 12 weeks before screening.
Eligible patients required confirmation of the presence of either advanced liver fibrosis or compensated cirrhosis, with advanced fibrosis or cirrhosis determined on the basis of a liver biopsy, a liver stiffness measurement (FibroScan), and/or the results of the serum fibrosis biomarker, FibroTest (scores determined by BioPredictive), plus an aspartate aminotransferase to platelet ratio index (APRI) before randomization. Advanced fibrosis was defined as a METAVIR score of F3 or an Ishak score of 4 on liver biopsy up to 36 months before screening, or a FibroScan ≥9.6 kPa but <14.6 kPa within 1 year of baseline, or a screening FibroTest score of 0.58‐0.74 plus an APRI score above 1 but below 2. Cirrhosis was defined as a METAVIR score of F4 or an Ishak score >4 on liver biopsy within 36 months before screening, a liver stiffness value ≥14.6 kPa within 1 year of baseline, or a screening FibroTest result ≥0.75 plus APRI ≥2. Where different testing methods yielded conflicting results, biopsy data took precedence. If biopsy data were not available, a FibroScan result took precedence over the FibroTest/APRI result.
Key exclusion criteria included chronic liver disease unrelated to HCV infection, infection with HCV genotypes other than 3 or mixed infection, infection with human immunodeficiency virus, previous treatment with an NS5A inhibitor, evidence or documentation of decompensated liver disease (including, but not limited to, radiological criteria, history/presence of ascites, bleeding varices, or hepatic encephalopathy) or HCC, or ineligibility for RBV treatment according to the local product label. Patients with a screening total bilirubin ≥2 mg/dL (unless with a history of Gilbert's disease), albumin <3.5 g/dL, platelets <50,000 cells/mm3, hemoglobin <8.5 g/dL, absolute neutrophil counts <750 cells/mm3, creatinine clearance ≤50 mL/min (Cockcroft‐Gault), or alpha‐fetoprotein (AFP) >100 ng/mL were also excluded. Patients with screening AFP between 50 and 100 ng/mL required liver ultrasound to exclude HCC before being considered eligible.
All patients received open‐label treatment with DCV 60 mg and SOF 400 mg once daily with or without food, plus weight‐based RBV (1,000 mg/day if <75 kg or 1,200 mg/day if ≥75 kg) taken twice daily as a divided dose with food. Dose reduction of RBV was permitted at investigator discretion for patients with low hemoglobin (≤10 g/dL) or creatinine clearance <50 mL/min.
Patients were randomized 1:1 using an interactive voice response system to receive treatment for 12 or 16 weeks, with a subsequent 24‐week follow‐up period. Randomization was stratified by fibrosis stage (advanced fibrosis or cirrhosis, as defined above), with enrollment of advanced fibrosis capped at 40%.
The study was conducted in accord with the ethical principles originating in the Declaration of Helsinki, and the protocol was approved by the institutional review board or independent ethics committee at each clinical site before study initiation. All patients provided written informed consent before study procedures.
STUDY ASSESSMENTS
HCV genotype or subtype was determined using the RealTime HCV Genotype II assay (Abbott Molecular, Abbott Park, IL). Levels of HCV RNA in patient plasma were assessed at the screening and baseline visits, on treatment at weeks 1, 2, 4, 8, 12, and 16 (in the 16‐week treatment group only), and at post‐treatment weeks 4, 12, and 24 using the HCV COBAS TaqMan Test (version 2.0; Roche Molecular Systems, Pleasanton, CA) with a lower limit of quantitation (LLOQ) of 25 IU/mL. On‐treatment virological response was defined as HCV RNA below the LLOQ with no target RNA detected (HCV RNA <LLOQTND). Post‐treatment virological response was defined as HCV RNA below the LLOQ with or without target RNA detected (HCV RNA <LLOQTD/TND). Safety and tolerability were assessed through AE reporting, clinical laboratory tests, vital signs, and physical examinations.
Resistance testing of HCV NS5A and NS5B (sensitivity, 10%‐20%) was performed by direct (population‐based) sequencing of isolated plasma HCV RNA from all patients at baseline and in those with virological failure whose plasma HCV RNA was at least 1,000 IU/mL. In addition, next‐generation sequencing (NGS; sensitivity, ≥1%; DDL Diagnostic Laboratory, Rijswijk, The Netherlands) was performed on NS5A and NS5B regions isolated from baseline and failure samples for patients with virological failure and at baseline for all those with previous SOF‐RBV treatment experience.
Virological failure was defined as virological breakthrough (an on‐treatment increase in HCV RNA of at least 1 log10 IU/mL above nadir or confirmed HCV RNA ≥LLOQ if previously <LLOQTD/TND), relapse (any confirmed HCV‐RNA measurement ≥LLOQ during post‐treatment follow‐up subsequent to an on‐treatment response <LLOQ without target RNA detected [<LLOQTND] at the end‐of‐treatment visit) or any other HCV‐RNA measurement ≥LLOQ that did not meet the criteria for virological breakthrough or relapse.
STATISTICAL ANALYSES
The primary endpoint was the proportion of patients with SVR12, defined as a post‐treatment virological response (HCV RNA <LLOQTD/TND) at week 12 after the treatment period.
The study was not designed to be hypothesis‐testing for establishing a difference between 12 and 16 weeks of treatment; the 16‐week treatment group was exploratory and based on the inclusion of treatment‐experienced patients with cirrhosis for whom data in the literature suggested potentially lower SVR12 rates after 12 weeks of all‐oral treatment. Sample size was based on estimation of SVR12 outcome for DCV‐SOF‐RBV and the confidence with which the estimated outcome could be differentiated from the observed rate of SVR12 among patients with cirrhosis who received DCV‐SOF without RBV in ALLY‐3. Assuming 40% of the enrolled patients were treatment‐naïve and 60% were treatment‐experienced, an overall observed SVR12 rate of 86% after 12 weeks of DCV‐SOF‐RBV treatment was assumed, based on published data for SOF‐RBV in combination with an NS5A inhibitor. For a target sample size of 25 patients in the 12‐week group, an observed SVR12 rate of 86% (22 of 25) or above would yield 95% confidence that the population‐level SVR12 would exceed 68.8% (i.e., that the lower bound of the 95% confidence interval [CI] for the population estimate would exceed 68.8%). A target sample size of 25 patients in the 16‐week treatment group with an observed SVR12 rate of 90% (23 of 25) or above would provide 95% confidence that the population‐level SVR12 rate was above 74.0%.
Secondary efficacy endpoints included the proportions of patients achieving an on‐treatment virological response (HCV RNA <LLOQTND) at treatment week 4 (rapid virological response), week 12 (complete early virological response), weeks 4 and 12 (extended rapid virological response), at end of treatment, and an off‐treatment virological response (HCV RNA <LLOQTD/TND) at post‐treatment week 4 (SVR4). Exploratory endpoints were SVR12 rates in patients with an IL28B CC or non‐CC genotype and the frequency of genotypic substitutions associated with virological failure.
For all efficacy endpoints, response rates and exact binomial 95% CIs were calculated using an intention‐to‐treat (ITT) approach that included patients who received at least 1 dose of study medication. For the SVR4 and SVR12 endpoints, missing data were derived from the next available HCV‐RNA measurement by next‐observation‐carried‐backward imputation. For other (on‐treatment) ITT analyses, patients with missing data were classed as nonresponders. Where relevant, observed values analyses were also undertaken in which patients with missing data were excluded.
Results
PATIENTS
Fifty‐three patients were screened and 50 randomized at 10 clinical sites in Australia and France, with initial study visits between February 16 and 24, 2015. Three patients did not meet study inclusion criteria for reasons of low platelet count (n = 1), cardiomyopathy (n = 1), or uncontrolled hypertension (n = 1) and were not randomized. All 50 randomized patients received at least 1 dose of study medication, and 49 (98%) completed treatment. Patient disposition is shown in Fig. 1.
Figure 1.
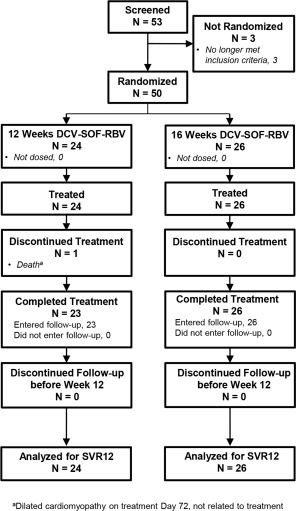
Patient disposition.
Baseline characteristics by treatment group are shown in Table 1 and characteristics by randomization stratum (advanced fibrosis or compensated cirrhosis) in Supporting Table S1. Patients were mostly male (80%) and white (98%), with a median age of 54 years. Fibrosis status was determined by liver biopsy (10 of 50; 20%) or FibroScan data (40 of 50; 80%) with no determinations made by FibroTest/APRI under the testing hierarchy described above. Most patients (72%) had cirrhosis. Baseline plasma HCV‐RNA level was high (median, 6.87 log10 IU/mL), with 76% of patients having a value above 2 million IU/mL and 52% above 6 million. Most patients (74%) were HCV treatment‐experienced; 62% had previously failed treatment with interferon/RBV regimens—mostly attributable to relapse (30%), null response (12%), or intolerance (10%)—and 12% had previously experienced relapse post‐treatment with SOF‐RBV with (1 of 50; 2%) or without (5 of 50; 10%) Peg‐IFN‐α. Baseline characteristics were comparable between treatment groups.
Table 1.
Demographic and Baseline Characteristics
| Parameter | DCV‐SOF‐RBV 12 Weeks (n = 24) | DCV‐SOF‐RBV 16 Weeks (n = 26) | Total (N = 50) |
|---|---|---|---|
| Age, median (range) years | 53.0 (36‐73) | 56.0 (42‐62) | 53.5 (36‐73) |
| Male, n (%) | 18 (75.0) | 22 (84.6) | 40 (80.0) |
| Race, n (%) | |||
| White | 23 (95.8) | 26 (100) | 49 (98.0) |
| Asian | 1 (4.2) | 0 | 1 (2.0) |
| HCV RNA, median (range) log10 IU/mL | 6.70 (4.6‐7.6) | 6.91 (4.7‐7.8) | 6.87 (4.6‐7.8) |
| HCV RNA category, n (%) | |||
| ≥800,000 IU/mL | 20 (83.3) | 21 (80.9) | 41 (82.0) |
| ≥2,000,000 IU/mL | 18 (75.0) | 20 (76.9) | 38 (76.0) |
| ≥6,000,000 IU/mL | 11 (45.8) | 15 (57.7) | 26 (52.0) |
| Fibrosis stratum, n (%)a | |||
| Advanced fibrosis | 6 (25.0) | 8 (30.8) | 14 (28.0) |
| Cirrhosis | 18 (75.0) | 18 (69.2) | 36 (72.0) |
| Albumin, median (range) g/L | 43 (33‐47) | 43 (34‐48) | 43 (33‐48) |
| Platelet count, median (range) × 109 cells/L | 161 (63‐299) | 155 (84‐324) | 161 (63‐324) |
| IL28B (rs12979860) genotype, n (%) | |||
| CC | 11 (45.8) | 11 (42.3) | 22 (44.0) |
| CT | 12 (50.0) | 13 (50.0) | 25 (50.0) |
| TT | 1 (4.2) | 2 (7.7) | 3 (6.0) |
| Past treatment status, n (%) | |||
| Naïve | 6 (25.0) | 7 (26.9) | 13 (26.0) |
| Experienced | 18 (75.0) | 19 (73.1) | 37 (74.0) |
| IFN‐based | 15 (62.5) | 16 (61.5) | 31 (62.0) |
| SOF‐basedb | 3 (12.5) | 3 (11.5) | 6 (12.0) |
| Past treatment outcome | |||
| IFN‐based | |||
| Relapse | 7 (29.2) | 8 (30.8) | 15 (30.0) |
| Null response | 2 (8.3) | 4 (15.4) | 6 (12.0) |
| Partial response | 0 | 1 (3.8) | 1 (2.0) |
| Virological breakthrough | 1 (4.2) | 1 (3.8) | 2 (4.0) |
| Intolerance | 3 (12.5) | 2 (7.7) | 5 (10.0) |
| Indeterminate | 2 (8.3) | 0 | 2 (4.0) |
| SOF‐based | |||
| Relapse | 3 (12.5) | 3 (11.5) | 6 (12.0) |
| DCV‐resistant NS5A polymorphisms, n (%) | |||
| A30K | 6 (25.0) | 0 | 6 (12.0) |
| Y93H | 1 (4.2) | 1 (3.8) | 2 (4.0) |
Stratum was determined by biopsy in 10 patients (20%) and FibroScan in 40 (80%). See Patients and Methods for details.
SOF‐RBV (n = 5); SOF‐RBV‐Peg‐IFN‐α (n = 1; 12‐week group).
VIROLOGICAL RESPONSE
Key virological responses on‐ and post‐treatment are summarized in Table 2. SVR12 rates were similar for both 12 and 16 weeks of treatment with DCV‐SOF‐RBV. By ITT analysis, SVR12 was 88% (21 of 24) in the 12‐week treatment group and 92% (24 of 26) in the 16‐week group, giving an overall rate in all treated patients of 90% (45 of 50). All patients with advanced fibrosis achieved SVR12 (14 of 14; 100%). Among patients with cirrhosis, 83% (15 of 18) achieved SVR12 in the 12‐week group and 89% (16 of 18) in the 16‐week group, for an overall rate of 86% (31 of 36). In the subgroup of patients with cirrhosis and previous HCV treatment experience, SVR12 was 88% (14 of 16) in the 12‐week group and 86% (12 of 14) in the 16‐week group, giving an overall SVR rate of 87% (26 of 30).
Table 2.
Virological Response
| Parameter | DCV‐SOF‐RBV 12 Weeks (n = 24) | DCV‐SOF‐RBV 16 Weeks (n = 26) |
|---|---|---|
| Post‐treatment response, n (%) [95% CI]a | ||
| SVR12 (primary endpoint) | 21 (87.5) [67.6, 97.3] | 24 (92.3) [74.9, 99.1] |
| SVR4 | 21 (87.5) [67.6, 97.3] | 25 (96.2) [80.4, 99.9] |
| On‐treatment response, n (%) [95% CI]b | ||
| Week 4 (RVR) | 20 (83.3) [62.6, 95.3] | 23 (88.5) [69.8, 97.6] |
| Weeks 4 and 12 (eRVR) | 19 (79.2) [57.8, 92.9] | 23 (88.5) [69.8, 97.6] |
| Week 12 (cEVR) | 23 (95.8) [78.9, 99.9] | 26 (100) [86.8, 100] |
| End of treatment | 24 (100) [85.8, 100] | 26 (100) [86.8, 100] |
| Patients without SVR12, n | ||
| Virological breakthrough | 0 | 0 |
| Relapse | 2 | 2 |
| Other on‐treatment failure (death) | 1c | 0 |
HCV RNA <LLOQTD/TND.
HCV RNA <LLOQTND.
Dilated cardiomyopathy at treatment day 72. See text for details.
Abbreviations: cEVR, complete early virological response; eRVR, extended rapid virological response; RVR, rapid virological response.
Using observed data, which excluded a single patient from the 12‐week group who did not enter post‐treatment follow‐up because of death from causes unrelated to treatment, SVR12 was 91% (21 of 23) in the 12‐week group and 92% (24 of 26) in the 16‐week group, for an overall SVR12 of 92% (45 of 49). Both ITT and observed results for key groups are shown in Fig. 2. Overall SVR12 rates were also comparable among other subgroups (Supporting Fig. S1; Supporting Table S2). There was no decline in SVR12 at high baseline HCV RNA; overall SVR12 was 83% (20 of 24) in those patients with HCV RNA <6 million IU/mL versus 96% (25 of 26) in those with HCV RNA ≥6 million IU/mL.
Figure 2.
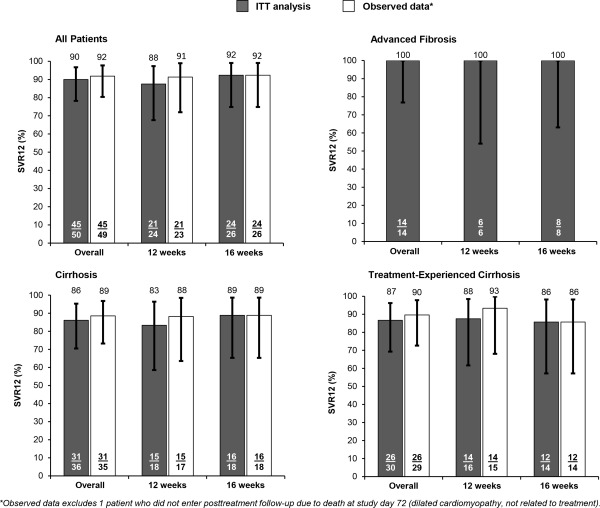
SVR12 and 95% CIs for all patients, by fibrosis stage, and for treatment‐experienced patients with cirrhosis.
Reductions in HCV RNA on treatment were rapid in both treatment groups (mean, ‐5.2 to ‐5.3 log10 IU/mL at week 2), and all patients had undetectable HCV RNA at their end‐of‐treatment visit. Because of the small number of patients with virological failure and the rapidity of the on‐treatment response, it was not possible to assess any correlation between on‐ and post‐treatment response rates.
VIROLOGICAL FAILURE AND RESISTANCE
No virological breakthroughs occurred in the study; post‐treatment relapse occurred in 4 patients overall, 2 in each treatment group. Characteristics of these 4 patients are shown in Table 3. All had compensated cirrhosis, and 3 were treatment‐experienced, including 2 who had previously relapsed on SOF‐RBV. The only treatment‐naïve patient who relapsed had several markers of very advanced liver disease (screening FibroScan 66.5 kPa, baseline albumin 33 g/L, and baseline platelets 83 × 109 cells/L) and harbored the NS5A‐Y93H variant associated with DCV resistance (see below). Three relapses were identified at week 4 post‐treatment, and 1 occurred between weeks 4 and 12 post‐treatment.
Table 3.
Baseline Characteristics of Patients Who Experienced Relapse
| Patient (age/gender) | Treatment Group | Past HCV treatment (outcome) | IL28B GT | HCV RNA (log10 IU/mL) | FibroScan score (kPa) | Albumin (g/L) | Platelets (× 109 cells/L) | NS5A RAVs |
|---|---|---|---|---|---|---|---|---|
| 1 (51/M) | 12 weeks | None | CC | 6.7 | 66.5 | 33 | 83 | Y93Y/H |
| 2 (53/M) | 12 weeks | IFN‐based (VBT) | CT | 7.0 | 19.0 | 43 | 157 | None |
| 3 (61/M) | 16 weeks | SOF‐RBV (relapse) | CT | 5.3 | NA (biopsy) | 41 | 188 | None |
| 4 (57/M) | 16 weeks | SOF‐RBV (relapse) | CT | 6.8 | 14.6 | 46 | 201 | None |
All patients had NS5A‐Y93H at relapse. No patient had NS5B RAVs at positions 282, 159, 320 or 321 detected at baseline or at relapse by direct sequencing or by NGS.
Abbreviations: GT, genotype; M, male; NA, not applicable; RAV, resistance‐associated variant; VBT, virological breakthrough.
There was no apparent difference in the time to undetectable HCV RNA (<LLOQTND) on treatment between patients who did or did not experience relapse. Three of the four patients who relapsed first achieved stable, undetectable on‐treatment HCV RNA at week 4 and the fourth at week 8. Of the 45 patients who entered the post‐treatment period without undergoing relapse, 12 (27%) first achieved stable, undetectable on‐treatment HCV RNA at week 1 or 2, 27 (60%) at week 4, and 6 (13%) at week 8.
Eight patients (16%; 2 advanced fibrosis and 6 cirrhosis) had a single NS5A resistance‐associated NS5A variant (RAV) to DCV at baseline: Y93H (2 patients) or A30K (6 patients); Y93H and A30K were detected as mixed populations with wild‐type sequence in 1 patient each. No patient had NS5A‐L31 variants at baseline. No patient had NS5B RAVs to SOF detected at baseline (NS5B‐S282T or NS5B amino acid substitutions at L159, L320, or V321) by direct sequencing, as well as by NGS in the case of the 4 relapsers and 4 patients who had previously received SOF‐RBV regimens and achieved SVR12.
Of the 6 patients with baseline NS5A‐A30K, of whom 4 had cirrhosis and 2 advanced fibrosis, all achieved SVR12. Of the 2 patients with Y93H, both with cirrhosis, 1 achieved SVR12 and 1 (Y93Y/H mixed population) relapsed. All 4 relapsing patients had NS5A‐Y93H at failure by both direct sequencing and NGS; Y93H was enriched in the patient with baseline Y93Y/H and emergent in the 3 without baseline RAVs. No NS5B RAVs related to SOF were detected at relapse by direct sequencing (n = 4) or NGS (n = 3). An NS5A‐M28M/I polymorphism was noted at baseline by direct sequencing (though not by NGS) in 1 relapsed patient without baseline Y93H or A30K, but M28I—which does not affect HCV susceptibility to DCV in vitro (data not shown)—was not detected at relapse by either sequencing method.
SAFETY AND TOLERABILITY
DCV‐SOF‐RBV was well tolerated. Overall, 94% of patients reported at least 1 on‐treatment AE; the most common AEs occurring in at least 10% of patients were insomnia, fatigue, headache, irritability, asthenia, diarrhea, and dyspnea. There were no AEs leading to discontinuation of treatment. A summary of AEs is shown in Table 4 and a full list in Supporting Table S3.
Table 4.
Safety and Tolerability on Treatment
| Parameter | DCV‐SOF‐RBV 12 Weeks (n = 24) | DCV‐SOF‐RBV 16 Weeks (n = 26) | Total (N = 50) |
|---|---|---|---|
| Any AE | 23 (95.8) | 24 (92.3) | 47 (94.0) |
| Deatha | 1 (4.2) | 0 | 1 (2.0) |
| SAEsb | 2 (8.3) | 3 (11.5) | 5 (10.0) |
| Congestive cardiomyopathy | 1 (4.2) | 0 | 1 (2.0) |
| Somnolence | 1 (4.2) | 0 | 1 (2.0) |
| Pneumonia | 0 | 1 (3.8) | 1 (2.0) |
| Arteriosclerosis | 0 | 1 (3.8) | 1 (2.0) |
| Basal cell carcinoma | 0 | 1 (3.8) | 1 (2.0) |
| AE leading to discontinuation | 0 | 0 | 0 |
| Grade 3‐4 AEsc | 2 (8.3) | 2 (7.7) | 4 (8.0) |
| RBV dose reductions | 2 (8.3) | 4 (15.4) | 6 (12.0) |
| AEs in ≥10% of patients overall (all grades) | |||
| Insomnia | 8 (33.3) | 7 (26.9) | 15 (30.0) |
| Fatigue | 6 (25.0) | 7 (26.9) | 13 (26.0) |
| Headache | 7 (29.2) | 5 (19.2) | 12 (24.0) |
| Irritability | 5 (20.8) | 2 (7.7) | 7 (14.0) |
| Asthenia | 2 (8.3) | 5 (19.2) | 7 (14.0) |
| Diarrhea | 1 (4.2) | 4 (15.4) | 5 (10.0) |
| Dyspnea | 2 (8.3) | 3 (11.5) | 5 (10.0) |
| Grade 3‐4 laboratory abnormalitiesd | |||
| Hemoglobin | 0 | 1 (3.8) | 1 (2.0) |
| Total bilirubin | 1 (4.2) | 1 (3.8) | 2 (4.0) |
Dilated cardiomyopathy on treatment day 72, considered unrelated to study treatment by the investigator. This single cardiac event is reported here as an SAE and a grade 3‐4 AE under the preferred term of “congestive cardiomyopathy”.
None were considered related to study treatment by the investigator.
Congestive cardiomyopathy (grade 4) plus gastrointestinal infection (grade 3; n = 1); somnolence (grade 3; n = 1); and pneumonia (grade 3; n = 1)—all unrelated to treatment. Treatment‐related anemia (grade 3; n = 1).
All listed events were of grade 3 intensity.
Five serious AEs in 5 patients were reported on treatment—somnolence, pneumonia, basal cell carcinoma, and arteriosclerosis in 1 patient each and 1 death from dilated cardiomyopathy on study day 72. No serious AE (SAE), including patient death, was considered to be related to treatment by investigators. The patient who died was a 56‐year‐old Caucasian male with biopsy‐proven cirrhosis and a history of alcohol abuse, who had previously relapsed post‐treatment with SOF‐RBV‐Peg‐IFN‐α and was assigned to the 12‐week group. There was no known history of cardiac disease. Death occurred shortly after the week 8 visit, at which time the patient had undetectable HCV RNA and symptoms consistent with infectious gastroenteritis (reported as a grade 3 AE), which improved spontaneously over the next few days and for which he was receiving symptomatic treatment. The patient was not on amiodarone or beta‐blockers.
Three grade 3 laboratory abnormalities were reported on treatment (hemoglobin decrease [n = 1] and total bilirubin elevation [n = 2]); there were no grade 4 abnormalities. Six patients reduced their dose of RBV for AEs: 3 from 1,200 mg to 400, 600, or 800 mg daily for 1‐20 days; 2 from 1,000 mg to 400 or 600 mg daily for 1 and 27 days, respectively; and 1 from 1,000 mg to 800 mg for 55 days, then 600 mg for 23 days. None of these patients relapsed. There were no RBV discontinuations or dose interruptions.
Discussion
This study demonstrated a high level of efficacy and safety with DCV‐SOF‐RBV administered for 12 or 16 weeks to a challenging group of genotype 3‐infected patients, most of whom had compensated cirrhosis and the rest advanced fibrosis, were treatment‐experienced, and had high baseline HCV RNA levels. In this difficult‐to‐treat patient cohort, the overall SVR12 rate was 90%, and observed SVR12 did not differ with 12 versus 16 weeks of treatment. The SVR12 rate in patients with advanced fibrosis was 100%. The SVR12 rate in patients with cirrhosis was 86% overall and was not lower in those patients with past treatment experience (87% overall). SVR12 was broadly comparable across subgroups and did not decline with high baseline viral load. Furthermore, 7 of the 8 patients with baseline NS5A RAVs achieved SVR12.
ALLY‐3+ is the first randomized study to formally explore strategies to optimize interferon‐free treatment response in genotype 3‐infected patients with cirrhosis. The high SVR12 rate among patients with cirrhosis, irrespective of previous treatment experience, compares favorably with the 63% SVR12 rate achieved in patients with cirrhosis in the earlier ALLY‐3 study and strongly suggests a benefit to adding RBV to DCV‐SOF in this patient group. However, extending treatment duration with DCV‐SOF‐RBV beyond 12 weeks to 16 weeks did not appear to have an effect on response rate, given that 2 patients experienced relapse in each of the 12‐ and 16‐week arms and the SVR12 difference observed between arms (83% vs. 89%, respectively) was driven entirely by a single patient with an undetectable final HCV‐RNA measurement who died of causes deemed unrelated to treatment before entering follow‐up. The benefit of prolonging treatment duration beyond 16 weeks was not evaluated.
Subject to the usual caveats around cross‐study comparisons, observed SVR in patients with cirrhosis after either 12 or 16 weeks of treatment with DCV‐SOF‐RBV in ALLY‐3+ was numerically higher than that generally observed with up to 24 weeks of SOF‐RBV in both randomized studies and observational cohorts. SVR to SOF‐RBV in clinical studies is typically only ≈20% after 12 weeks of treatment in genotype 3‐infected patients with cirrhosis,24 ≈50%‐60% after 16 weeks,22, 24 and ≈70%‐80% after 24 weeks (≈60%‐75% in treatment‐experienced patients with cirrhosis).22, 25 Limited observational data suggest that SVR12 to SOF‐RBV among genotype 3‐infected patients with cirrhosis may be lower in the routine clinic than in clinical studies: reported SVR12 rates were 53% (39 of 73) from the HCV‐TARGET cohort (74% [17 of 23] in treatment‐naïve and 44% [22 of 50] in treatment‐experienced)33 and 57% among a small sample of 14 patients from the TRIO health network after 24 weeks of SOF‐RBV.34
Addition of RBV to DCV‐SOF in ALLY‐3+ improved SVR12 response in patients with cirrhosis relative to that of DCV‐SOF without RBV in ALLY‐3, but did not appear to significantly alter the safety and tolerability profile. Neither ALLY‐3 nor ALLY‐3+ had any discontinuations for AEs nor any SAEs that were considered treatment‐related; common AEs were broadly similar and general. There was no increase in overall grade 3‐4 laboratory abnormalities in ALLY‐3+ (3 events in 50 patients) compared with ALLY‐3 (8 events in 152 patients), and the addition of RBV to DCV‐SOF resulted in only a single case of grade 3 treatment‐related anemia in ALLY‐3+. By comparison, whereas the addition of Peg‐IFN‐α to SOF‐RBV for 12 weeks in the BOSON study22 similarly improved SVR12 in genotype 3‐infected patients with cirrhosis (88%) relative to SOF‐RBV alone for 16 weeks (51%) or 24 weeks (79%), consistent with the established safety profile of Peg‐IFN‐α/RBV treatment, the addition of Peg‐IFN‐α resulted in a higher incidence of constitutional symptoms (myalgia, pyrexia, chills, and influenza‐like illness), laboratory cytopenias, low hemoglobin, and study drug dose modifications or interruptions.
DCV‐SOF, with or without RBV, is currently the only regimen option recommended by both U.S. treatment guidelines (American Association for the Study of Liver Diseases/Infectious Diseases Society of America/International Antiviral Society USA recommendations; see www.hcvguidelines.org) and European guidelines (European Association for the Study of the Liver recommendations35) for use in all genotype 3‐infected patients irrespective of HCV treatment experience or cirrhosis status. Both guidelines recommend 12 weeks of DCV‐SOF without RBV for patients without cirrhosis, and this recommendation is supported by the similarly high (≥96%) SVR12 rates noted in this patient group with and without RBV in ALLY‐3 and ALLY‐3+. Recommendations for RBV use and treatment duration in genotype 3‐infected patients with cirrhosis differ between U.S. and EU guidelines and are based on limited empirical data. The results of ALLY‐3+ suggest that 12 or 16 weeks of DCV‐SOF‐RBV is an effective therapeutic option for both HCV treatment‐naïve and treatment‐experienced patients with compensated cirrhosis. The SVR12 rates observed are similar to those noted recently for genotype 3‐infected patients with cirrhosis treated with SOF and the investigational agent velpatasvir,36 suggesting that a 100% response rate may be hard to achieve in this difficult‐to‐treat patient group.
The optimal duration of treatment for some genotype 3 patient groups—such as those with decompensated cirrhosis or patients with cirrhosis for whom RBV may be contraindicated—remains an open question. There are currently no randomized clinical data assessing DCV‐SOF treatment of genotype 3 beyond 12 weeks or DCV‐SOF‐RBV beyond 16 weeks. Unrandomized, real‐world observations for 24 weeks of DCV‐SOF with and without RBV have recently been reported from interim analyses of two European early access programs that provided DCV ahead of its marketing authorization to patients with advanced liver disease and no other HCV treatment options. The French “Autorisations Temporaires d'Utilisation” (ATU) program observed an SVR12 rate of 86% for 24 weeks of DCV‐SOF without RBV in 135 genotype 3‐infected patients with cirrhosis (mostly Child‐Pugh A [85%] or B [13%]), with no incremental benefit observed in a similar group of 53 patients with cirrhosis treated for 24 weeks with DCV‐SOF‐RBV (SVR12 of 81%).37 Similar results were observed in the multicenter European Compassionate Use Program for a group of 71 genotype 3‐infected patients with cirrhosis, most of whom were treated for 24 weeks, where SVR12 rates were 88% on DCV‐SOF and 86% on DCV‐SOF‐RBV.38
In conclusion, the all‐oral combination of DCV, SOF, and RBV given for either 12 or 16 weeks at their standard doses and dosing schedules demonstrated high and similar efficacy and good tolerability in HCV genotype 3‐infected patients with compensated cirrhosis or advanced fibrosis, irrespective of past treatment experience or high baseline HCV RNA levels. DCV‐SOF‐RBV represents an important option for HCV genotype 3‐infected patients with advanced liver disease in urgent need of effective treatment.
Supporting information
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep.28473/suppinfo.
Supporting Information
Acknowledgment
We thank Patricia Mendez and Melissa Harris for their contributions to the study conception, Kimberly Brown for contributing to the study execution, and Eric Y. Wong for contributing to the development of the manuscript. Editorial support was provided by Nick Fitch, Ph.D., of Articulate Science and was funded by Bristol‐Myers Squibb.
Potential conflict of interest: Dr. Pol consults and received grants from Bristol‐Myers Squibb, Gilead, Roche, and MSD. He consults Boehringer‐Ingelheim, Janssen, Vertex, Novartis, AbbVie, Sanofi, and GlaxoSmithKline. Dr. Leroy consults and is on the speakers' bureau of Janssen and Merck. He advises and is on the speakers' bureau of AbbVie, Gilead, and Bristol‐Myers Squibb. Dr. Bronowicki consults, advises, is on the speakers' bureau of, and received grants from Bristol‐Myers Squibb and Gilead. Dr. Dore consults, advises, is on the speakers' bureau of, and received grants from AbbVie and Merck. He advises, is on the speakers' bureau of, and received grants from Gilead. He advises and received grants from Bristol‐Myers Squibb. He advises Janssen. Dr. Pianko consults, advises, and is on the speakers' bureau of Gilead. He is on the speakers' bureau of Bristol‐Myers Squibb. Dr. Thompson advises or has served on review panels for, has acted as a speaker or teacher for, and has received grants from Gilead, AbbVie and Bristol‐Myers Squibb. He advises or has served on review panels for, and has acted as a speaker or teacher for Merck. He advises or has served on review panels for Spring Bank Pharmaceuticals and Arrowhead. Dr. Stuart is on the speakers' bureau of AbbVie and Bristol‐Myers Squibb. She received grants from Gilead and Bayer. Dr. McPhee is employed by and owns stock in Bristol‐Myers Squibb. Dr. Jimenez‐Exposito is employed by and owns stock in Bristol‐Myers Squibb. Dr. Bhore is employed by and owns stock in Bristol‐Myers Squibb.
This study was funded by Bristol‐Myers Squibb.
REFERENCES
- 1. Shepard CW, Finelli L, Alter MJ. Global epidemiology of hepatitis C virus infection. Lancet Infect Dis 2005;5:558‐567. [DOI] [PubMed] [Google Scholar]
- 2. Idrees M, Riazuddin S. Frequency distribution of hepatitis C virus genotypes in different geographical regions of Pakistan and their possible routes of transmission. BMC Infect Dis 2008;8:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Germer JJ, Mandrekar JN, Bendel JL, Mitchell PS, Yao JD. Hepatitis C virus genotypes in clinical specimens tested at a national reference testing laboratory in the United States. J Clin Microbiol 2011;49:3040‐3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Stamouli M, Panagiotou I, Kairis D, Michopoulou A, Skliris A, Totos G. Genotype distribution in chronic hepatitis C patients in Greece. Clin Lab 2012;58:173‐176. [PubMed] [Google Scholar]
- 5. Chlabicz S, Flisiak R, Kowalczuk O, Grzeszczuk A, Pytel‐Krolczuk B, Prokopowicz D, et al. Changing HCV genotypes distribution in Poland‐relation to source and time of infection. J Clin Virol 2008;42:156‐159. [DOI] [PubMed] [Google Scholar]
- 6. de Vries MJ, te Rijdt B, van Nieuwkerk CM. Genotype distribution amongst hepatitis C patients in the Netherlands. Neth J Med 2006;64:109‐113. [PubMed] [Google Scholar]
- 7. McCaw R, Moaven L, Locarnini SA, Bowden DS. Hepatitis C virus genotypes in Australia. J Viral Hepat 1997;4:351‐357. [DOI] [PubMed] [Google Scholar]
- 8. Roman F, Hawotte K, Struck D, Ternes AM, Servais JY, Arendt V, et al. Hepatitis C virus genotypes distribution and transmission risk factors in Luxembourg from 1991 to 2006. World J Gastroenterol 2008;14:1237‐1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bourliere M, Barberin JM, Rotily M, Guagliardo V, Portal I, Lecomte L, et al. Epidemiological changes in hepatitis C virus genotypes in France: evidence in intravenous drug users. J Viral Hepat 2002;9:62‐70. [DOI] [PubMed] [Google Scholar]
- 10. Bochud PY, Cai T, Overbeck K, Bochud M, Dufour JF, Mullhaupt B, et al. Genotype 3 is associated with accelerated fibrosis progression in chronic hepatitis C. J Hepatol 2009;51:655‐666. [DOI] [PubMed] [Google Scholar]
- 11. Adinolfi LE, Gambardella M, Andreana A, Tripodi MF, Utili R, Ruggiero G. Steatosis accelerates the progression of liver damage of chronic hepatitis C patients and correlates with specific HCV genotype and visceral obesity. Hepatology 2001;33:1358‐1364. [DOI] [PubMed] [Google Scholar]
- 12. Rubbia‐Brandt L, Quadri R, Abid K, Giostra E, Male PJ, Mentha G, et al. Hepatocyte steatosis is a cytopathic effect of hepatitis C virus genotype 3. J Hepatol 2000;33:106‐115. [DOI] [PubMed] [Google Scholar]
- 13. Nkontchou G, Ziol M, Aout M, Lhabadie M, Baazia Y, Mahmoudi A, et al. HCV genotype 3 is associated with a higher hepatocellular carcinoma incidence in patients with ongoing viral C cirrhosis. J Viral Hepat 2011;18:e516‐e522. [DOI] [PubMed] [Google Scholar]
- 14. Zeuzem S, Hultcrantz R, Bourliere M, Goeser T, Marcellin P, Sanchez‐Tapias J, et al. Peginterferon alfa‐2b plus ribavirin for treatment of chronic hepatitis C in previously untreated patients infected with HCV genotypes 2 or 3. J Hepatol 2004;40:993‐999. [DOI] [PubMed] [Google Scholar]
- 15. Andriulli A, Mangia A, Iacobellis A, Ippolito A, Leandro G, Zeuzem S. Meta‐analysis: the outcome of anti‐viral therapy in HCV genotype 2 and genotype 3 infected patients with chronic hepatitis. Aliment Pharmacol Ther 2008;28:397‐404. [DOI] [PubMed] [Google Scholar]
- 16. Harvoni (ledipasvir and sofosbuvir) [summary of product characteristics]. Cambridge, UK: Gilead Sciences International Ltd; 2014. [Google Scholar]
- 17. Moreno C, Berg T, Tanwandee T, Thongsawat S, Van Vlierberghe H, Zeuzem S, et al. Antiviral activity of TMC435 monotherapy in patients infected with HCV genotypes 2‐6: TMC435‐C202, a phase IIa, open‐label study. J Hepatol 2012;56:1247‐1253. [DOI] [PubMed] [Google Scholar]
- 18. Lenz O, Vijgen L, Berke JM, Cummings MD, Fevery B, Peeters M, et al. Virologic response and characterisation of HCV genotype 2‐6 in patients receiving TMC435 monotherapy (study TMC435‐C202). J Hepatol 2013;58:445‐451. [DOI] [PubMed] [Google Scholar]
- 19. Kati W, Koev G, Irvin M, Beyer J, Liu Y, Krishnan P, et al. In vitro activity and resistance profile of dasabuvir, a nonnucleoside hepatitis C virus polymerase inhibitor. Antimicrob Agents Chemother 2015;59:1505‐1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. McPhee F, Sheaffer AK, Friborg J, Hernandez D, Falk P, Zhai G, et al. Preclinical profile and characterization of the hepatitis C virus NS3 protease inhibitor asunaprevir (BMS‐650032). Antimicrob Agents Chemother 2012;56:5387‐5396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sofia MJ, Bao D, Chang W, Du J, Nagarathnam D, Rachakonda S, et al. Discovery of a β‐d‐2'‐deoxy‐2'‐α‐fluoro‐2'‐β‐C‐methyluridine nucleotide prodrug (PSI‐7977) for the treatment of hepatitis C virus. J Med Chem 2010;53:7202‐7218. [DOI] [PubMed] [Google Scholar]
- 22. Foster GR, Pianko S, Brown A, Forton D, Nahass RG, George J, et al. Efficacy of sofosbuvir plus ribavirin with or without peginterferon‐alfa in patients with HCV genotype 3 infection and treatment‐experienced patients with cirrhosis and HCV genotype 2 infection. Gastroenterology 2015;149:1462‐1470. [DOI] [PubMed] [Google Scholar]
- 23. Lawitz E, Mangia A, Wyles D, Rodriguez‐Torres M, Hassanein T, Gordon SC, et al. Sofosbuvir for previously untreated chronic hepatitis C infection. N Engl J Med 2013;368:1878‐1887. [DOI] [PubMed] [Google Scholar]
- 24. Jacobson IM, Gordon SC, Kowdley KV, Yoshida EM, Rodriguez‐Torres M, Sulkowski MS, et al. Sofosbuvir for hepatitis C genotype 2 or 3 in patients without treatment options. N Engl J Med 2013;368:1867‐1877. [DOI] [PubMed] [Google Scholar]
- 25. Zeuzem S, Dusheiko GM, Salupere R, Mangia A, Flisiak R, Hyland RH, et al. Sofosbuvir and ribavirin in HCV genotypes 2 and 3. N Engl J Med 2014;370:1993‐2001. [DOI] [PubMed] [Google Scholar]
- 26. Lawitz E, Poordad F, Brainard DM, Hyland RH, An D, Dvory‐Sobol H, et al. Sofosbuvir with peginterferon‐ribavirin for 12 weeks in previously treated patients with hepatitis C genotype 2 or 3 and cirrhosis. Hepatology 2015;61:769‐775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sulkowski MS, Cooper C, Hunyady B, Jia J, Ogurtsov P, Peck‐Radosavljevic M, et al. Management of adverse effects of peg‐IFN and ribavirin therapy for hepatitis C. Nat Rev Gastroenterol Hepatol 2011;8:212‐223. [DOI] [PubMed] [Google Scholar]
- 28. Talal AH, Lafleur J, Hoop R, Pandya P, Martin P, Jacobson I, et al. Absolute and relative contraindications to pegylated‐interferon or ribavirin in the US general patient population with chronic hepatitis C: results from a US database of over 45 000 HCV‐infected, evaluated patients. Aliment Pharmacol Ther 2013;37:473‐481. [DOI] [PubMed] [Google Scholar]
- 29. Vukotic R, Gamal N, Andreone P. Prospective, observational real‐life study on eligibility for and outcomes of antiviral treatment with peginterferon alpha plus ribavirin in chronic hepatitis C. Dig Liver Dis 2015;47:151‐156. [DOI] [PubMed] [Google Scholar]
- 30. Gatti F, Nasta P, Matti A, Manno D, Mendeni M, Puoti M, et al. Treating hepatitis C virus in HIV patients: are side effects a real obstacle? AIDS Rev 2007;9:16‐24. [PubMed] [Google Scholar]
- 31. Gao M, Nettles RE, Belema M, Snyder LB, Nguyen VN, Fridell RA, et al. Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature 2010;465:96‐100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nelson DR, Cooper JN, Lalezari JP, Lawitz E, Pockros PJ, Gitlin N, et al. All‐oral 12‐week treatment with daclatasvir plus sofosbuvir in patients with hepatitis C virus genotype 3 infection: ALLY‐3 phase 3 study. Hepatology 2015;61:1127‐1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Alqahtani S, Zeuzem S, Manns M, Kuo A, Di Bisceglie AM, Reddy R, et al. Safety and effectiveness of sofosbuvir‐based regimens for the treatment of hepatitis C genotype 3 and 4 infections: interim analysis of a prospective observational study. J Hepatol 2015;62(Suppl):S652‐S653. [Google Scholar]
- 34. Kowdley K, Bacon B, Dieterich D, Lawitz E, Milligan S, Tsai N, et al. Efficacy evaluation of 24 week SOF + RBV in a heterogeneous, real‐world population of genotype 3 HCV patients: data from the TRIO network. J Hepatol 2015;62(Suppl):S665‐S666. [Google Scholar]
- 35. European Association for the Study of the Liver . EASL recommendations on treatment of hepatitis C 2015. J Hepatol 2015;63:199‐236. [DOI] [PubMed] [Google Scholar]
- 36. Foster GR, Afdhal N, Roberts SK, Brau N, Gane EJ, Pianko S, et al.; ASTRAL‐2 Investigators ; ASTRAL‐3 Investigators . Sofosbuvir and velpatasvir for HCV genotype 2 and 3 infection. N Engl J Med 2015;373:2608‐2617. [DOI] [PubMed] [Google Scholar]
- 37. Hezode C, de Ledinghen V, Fontaine H, Zoulim F, Lebray P, Boyer N, et al. Daclatasvir plus sofosbuvir with or without ribavirin in patients with HCV genotype 3 infection: interim analysis of a French multicenter compassionate use program [abstract]. Hepatology 2015;62(Suppl 1):314A. 25914200 [Google Scholar]
- 38. Welzel TM, Petersen J, Ferenci P, Gschwantler M, Herzer K, Cornberg M, et al. Safety and efficacy of daclatasvir plus sofosbuvir with or without ribavirin for the treatment of chronic HCV genotype 3 infection: interim results of a multicenter European compassionate use program. Hepatology 2015;62(Suppl):225A‐226A. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep.28473/suppinfo.
Supporting Information