

Abstract
Background and Purpose
Hydrogen sulfide (H2S) modulates many pathophysiological processes, including inflammation and allergic reactions, in which mast cells act as major effector cells. IgE receptor (FcεRI) cross linking leads to an increase in intracellular calcium ([Ca+2]i), a critical step in mast cell degranulation. The aim of this study was to investigate the role of H2S in [Ca+2]i‐dependent mast cell activation.
Experimental Approach
We investigated the effects of H2S, either endogenously produced or released by the slow H2S donor 4‐carboxy‐phenyl isothiocyanate (PhNCS‐COOH), on antigenic‐ and non‐antigenic degranulation of native murine mast cells, and human and rat (RBL‐2H3) mast cell lines. We measured the release of specific mast cell degranulation markers (β‐hexosaminidase and renin), as well as changes in [Ca+2]i and phosphorylation of proteins downstream of FcεRI activation.
Key Results
Endogenously produced H2S inhibited antigen‐induced degranulation in RBL‐2H3. Similarly, H2S released by PhNCS‐COOH (10–300 μM) reduced, in a concentration‐dependent manner, antigenic and non‐antigenic degranulation and renin release in all mast cell types. Notably, PhNCS‐COOH also prevented in a concentration‐dependent mode the increase in [Ca+2]i elicited by Ca+2 ionophore, thapsigargin and FcεRI activation. Moreover, PhNCS‐COOH attenuated the phosphorylation of Syk, cPLA‐2 and PLCγ1 in antigen‐stimulated RBL‐2H3 cells.
Conclusion and Implications
Collectively, our results demonstrate that, by attenuating the phosphorylation of proteins downstream of FcεRI cross‐linking on mast cells, H2S diminishes [Ca+2]i availability and thus mast cell degranulation and renin release. These findings suggest that PhNCS‐COOH could be a strategic therapeutic tool in mast cell‐mediated allergic conditions.
Abbreviations
- Ang I
angiotensin I
- BMMC
bone marrow‐derived murine mast cells
- [Ca2+]i
intracellular Ca2 + level
- CBS
cystathionine β‐synthase
- cPLA2
cytosolic phospholipase A2
- CSE
cystathionine γ‐lyase
- DNP
dinitrophenylated‐human serum albumin
- D‐Pen
D‐penicillamine
- FcεRI
high affinity IgE receptor
- HMC‐1
human mastocytoma cell line
- MEF
mouse embryo fibroblast
- PhNCS‐COOH
4‐carboxy‐phenyl‐isothiocyanate
- PLCγ1
phospholipase Cγ1
- RBL‐2H3
rat basophilic leukaemia cell line
- β‐HEX
β‐hexosaminidase
Tables of Links
| TARGETS | |
|---|---|
| Enzymes | |
| CSE, cystathionine γ‐lyase | |
| JNK | |
| cPLA2, cytosolic phospholipase A2 | |
| PLCγ1, phospholipase C γ1 | |
| Renin | |
| SERCA |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Introduction
Hydrogen sulfide (H2S) is an endogenous gaseous compound widely recognized as a gasotransmitter (Wang, 2002) with important modulatory effects on cardiovascular (Hosoki et al., 1997; Yang et al., 2008), respiratory (Kubo et al., 2007), gastrointestinal (Fiorucci et al., 2006), endocrine (Wu et al., 2009) and neural (Abe and Kimura, 1996) functions. H2S is biosynthesized in mammalian tissues by three pyridoxal‐5′‐phosphate‐dependent enzymes: cystathionine β‐synthase (CBS), cystathionine γ‐lyase (CSE) and cysteine aminotransferase. CBS and CSE are widely expressed (Kamoun, 2004) and use L‐cysteine or other thiols as endogenous substrates (Abe and Kimura, 1996; Singh et al., 2009), while cysteine aminotransferase produces H2S by desulfuration via 3‐mercaptopyruvate sulfurtransferase (Shibuya et al., 2009). CBS and CSE have been recognized as major sources of endogenous H2S in the central nervous and cardiovascular systems respectively. Nevertheless, tissue‐specific expression of H2S‐generating enzymes, their substrate availability and homeostatic disturbances may favour one enzymatic pathway over another (Wallace and Wang, 2015).
As H2S is critical for the maintenance of homeostasis, even a small impairment in its availability can lead to severe disorders (Yang et al., 2008; Martelli et al., 2012). Accordingly, much attention has been devoted to the search for exogenous H2S‐donors capable of slowly generating and releasing H2S, so as to correctly mimic the biological effects of endogenous H2S (Martelli et al., 2012).
H2S is also involved in inflammation, and its role has been debated for many years (Li et al., 2008; Zhang and Bhatia, 2008). However, recent strong evidence indicates that H2S is primarily an anti‐inflammatory substance (Wallace and Wang, 2015). Mast cells are multi‐effector participants in IgE‐mediated hypersensitivity reactions and are ubiquitously distributed in the human body (Reid et al., 2007). They play many pathophysiological roles by synthesizing, storing and releasing a wide variety of pro‐inflammatory and vasoactive mediators, cytokines, chemokines, proteases and renin (Roberts and Brenchley, 2000; Silver et al., 2004; Mackins et al., 2006). Hence, inhibition of mast cell activation is viewed as a rational therapeutic strategy to reduce their degranulation and the consequent release of pro‐inflammatory mediators. In a rat model of isoproterenol‐induced cardiac failure, in which local mast cell number and renin levels increase, administration of NaHS reduced both mast cell density and renin (Liu et al., 2014). Also, exogenous H2S inhibited allergen‐induced airway hyper‐reactivity, which was attributed to a reduction of mast cell degranulation, observed in vitro in rat basophilic leukaemia (RBL‐2H3) cells (Roviezzo et al., 2015). Yet, little is known about a possible role of endogenous H2S and H2S‐releasing compounds in mast cell pathophysiology.
In this study, we report the activity of an H2S‐donor molecule containing an isothiocyanate group, 4‐carboxy‐phenyl isothiocyanate (PhNCS‐COOH), which releases H2S at a slow and controlled rate, only in the presence of endogenous thiols (Martelli et al., 2014). We used PhNCS‐COOH as a tool to investigate the role(s) of H2S in mast cell pathophysiology. We show, for the first time, that endogenous and PhNCS‐COOH‐derived H2S inhibits mast cell degranulation and renin release by reducing intracellular Ca2 + levels ([Ca2 +]i). Our study provides new interesting insights for a better understanding of the role of H2S in inflammation, which has been extensively debated recently and suggests potential pharmacological uses of H2S donors.
Methods
Animals
All animal care and experimental procedures complied with the NIH Guide for Animal Care and Use of Laboratory Animals (NIH Publication No. 85–23, revised 1996) and were approved by the Weill Cornell Medicine Institutional Animal Care and Use Committee or the Laurentian University Animal Care Committee. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). C57BL/6 J mice (male, 10‐ to 12‐week‐old, 20–25 g of weight) were purchased from Jackson Laboratory (Bar Harbor, ME, USA) and kept in the animal care facility under controlled temperature, humidity and light/dark cycle, with food and water ad libitum. CSE knockout mice (CSE‐KO) were generated as described previously (Yang et al., 2008). All animals were maintained on standard rodent chow and water ad libitum. 8‐week‐old CSE‐KO mice and wild‐type (WT) littermates were used for liver tissue isolation in this study,
General procedures
The exact group size (n) for each experimental condition is provided in the Figure legends, where ‘n’ refers to independent values, not replicates. Randomisation of animals was not used for this study since we did not use animal groups for pharmacological experiments. Animals were used to provide liver tissue and to obtain bone marrow and embryonic fibroblasts. Cells were randomly assigned across treatment groups.
Cell cultures
RBL‐2H3 cells were purchased from ATCC (American Type Culture Collection, Manassas, VA, USA) and cultured in Minimum Essential Medium (MEM) supplemented with 10% heat‐inactivated FBS and 1% streptomycin/penicillin at 37°C in a humidified 5% CO2 atmosphere. The human mastocytoma cell line (HMC‐1) was kindly provided to us by Dr. J. H. Butterfield (Mayo Clinic, Rochester, MN, USA). Cells were maintained in suspension culture at high density in Iscove's modified Dulbecco's medium supplemented with 10% heat‐inactivated FBS, 1% penicillin/streptomycin and kept at 37°C in a 5% CO2 atmosphere (Aldi et al., 2015).
Preparation and culture of bone marrow‐derived mast cells (BMMC)
Bone marrow was collected from femurs and tibias of WT C57BL/6 J mice (male, 10‐ to 12‐week‐old; Jackson Laboratory, Bar Harbor, ME) killed by cervical dislocation under light CO2 anaesthesia. Bone marrow cells were cultured in RPMI 1640 medium containing 1% penicillin/streptomycin, 10% heat‐inactivated fetal calf serum, 55 μM 2‐mercaptoethanol and recombinant murine IL‐3 and stem cell factor, both at 20 ng·mL−1. Bone marrow cells were counted and placed in culture at a cell density of 5 × 105 cells·mL−1. Cell medium was changed every 3 to 4 days, and non‐adherent cells were transferred to a new flask. Mature BMMC were obtained after 4 weeks of culture and stained positive for Toluidine Blue. Most ( >90% ) of these cells expressed both c‐Kit and the high affinity IgE receptor (FcεRI). All experiments were performed with BMMC cultured for 4–7 weeks (Aldi et al., 2014).
Preparation and culture of mouse embryo fibroblasts (MEFs)
MEFs were obtained from about 13.5‐day WT or CSE‐KO sibling embryos as described previously (Yang et al., 2013). MEFs were cultured in DMEM supplemented with 10% FBS, 1% penicillin/streptomycin (v/v) and 3.7 g/L NaHCO3.
β‐hexosaminidase (β‐HEX) release assay
After reaching 80% confluence, RBL‐2H3 cells were seeded into a 96‐well plate (72 × 103 cell well−1 in 100 μL) at 37°C overnight. Twenty‐four hours later, RBL‐2H3 degranulation was induced by 1 h incubation with Ca+2 ionophore (A23187, 1 μM) or thapsigargin (1 μM). Alternatively, plated cells were sensitized with an overnight anti‐dinitrophenylated‐human serum albumin (DNP)‐IgE treatment (0.50 μg·mL−1), and subsequently, MEM was replaced with phenol‐free DMEM supplemented with 1 mg·mL−1 BSA. In these experiments, L‐cysteine (0.3–10 mM), cromolyn (1 mM), a mast cell stabilizer, the rapidly acting H2S‐donor NaHS (1 mM) and the slow H2S‐donor PhNCS‐COOH (10–1000 μM) were incubated for 5 min at 37°C. When used, the CSE inhibitor D‐penicillamine (D‐Pen, 1 mM) was added 20 min before L‐cysteine. Cells were then treated with DNP (10 ng·mL−1) and, finally, with TRITON‐X‐100 0.1%, used to define the maximal release of β‐HEX. One hour later, 50 μL of supernatants from each well were collected and added of 50 μL to p‐nitrophenyl‐N‐acetyl‐β‐D‐glucosaminide 1.4 mM in citrate buffer 0.2 M, pH 4.2. The enzymatic reaction was terminated after 1 h by adding 100 μL well−1 of Trizma solution 0.3 M pH 9.4. The release of β‐HEX was measured at 405 nM in a multiplate reader (EnSpire, PerkinElmer, Milan, Italy).
For BMMC, after 16 h of sensitization with anti‐DNP IgE (0.125 μg·mL−1), cells were washed and resuspended in Ringer's buffer of the following composition (in mM): 140 NaCl, 5 KCl, 10 HEPES, 1 MgCl2, 2 glucose, 2 CaCl2 and pH 7.4. When the degranulation was induced by Ca2 + ionophore or thapsigargin, the sensitization step was unnecessary. 2 × 105 cells well−1 were seeded in a 96‐well plate with rounded bottom and pre‐incubated for 5 min at 37°C with PhNCS‐COOH (10–300 μM), then challenged with DNP 10 ng·mL−1, A23187 (1 μM) or thapsigargin (300 nM), for 20 min at 37°C. Following the incubation time, cells were placed on ice and the plates were centrifuged at 400 g, for 5 min at 4°C. HMC‐1 were also incubated with A23187 (1 μM) or thapsigargin (1 μM) for 20 min at 37°C, in the presence or absence of PhNCS‐COOH (10–300 μM). At the end of the incubation, samples were placed on ice and centrifuged at 300 g for 5 min. Twenty microliters of BMMC or HMC‐1 supernatants were placed in 96‐well plates, to which 50 μL of p‐nitrophenyl‐N‐acetyl‐β‐D‐glucosaminide (1.3 mg·mL−1 dissolved in 0.1 M citrate buffer at pH 4.5) was added and incubated for 90 min at 37°C. Cell pellets were lysed with 0.5% Triton X‐100, and total lysates were used to determine total β‐HEX content. The reaction was stopped by adding 150 μL of 0.2 M glycine (pH 10.7). Optical density was read at 405 nM on a spectrophotometric plate reader using SoftMax Pro version 4.8 (Molecular Devices, Sunnyvale, CA, USA). β‐HEX release was expressed as % of total β‐HEX (where total β‐HEX denotes the sum of β‐HEX in supernatant and total cell lysate) (Aldi et al., 2014).
Renin assay
Renin activity was measured in supernatants from RBL‐2H3, BMMC and HMC‐1 cells (treated with as described above) using a GammaCoat Plasma Renin Activity 125I Radioimmunoassay kit (DiaSorin, Stillwater, MN, USA) according to the manufacturer's instructions. Briefly, RBL‐2H3 and BMMCs supernatants were diluted 1:200 in Ringer buffer and incubated with porcine angiotensinogen substrate, while 500 μL of each HMC‐1 supernatant was incubated with 0.1 mg·mL−1 human angiotensinogen, maleate buffer and PMSF (part of the DiaSorin kit). The incubation for angiotensin I (Ang I) production was for 1.5 h at 37°C. Total protein concentration was measured using a Bradford protein assay kit. Results were normalized to total protein. The detection limit was approximately 0.01 pmol (Aldi et al., 2015).
Measurement of intracellular calcium ([Ca2 +]i)
[Ca2 +]i was measured using a Fura‐2 No Wash Calcium Assay Kit. For adherent cells such as RBL‐2H3, 72 × 103 cells well−1 100 μL were seeded and in a black‐wall clear‐bottom plate in growth medium at 37°C. For non‐adherent cells, such as BMMC and HMC‐1, 2 × 105 cells well−1 100 μL were pelleted and suspended in HHBS solution (of the following composition in mM: 137 NaCl, 5.4 KCl, 0.5 MgCl2∙6H2O, 0.4 MgSO4∙7H2O, 0.44 KH2PO4, 0.34 Na2HPO4∙7H2O, 1.3 CaCl2, 5.5 glucose, 4.2 NaHCO3 and pH 7.4). Then, the plated cells were centrifuged at 400 × g for 2 min. When needed, RBL‐2H3 and BMMC were sensitized in their own medium overnight with anti‐DNP‐IgE 0.2 μg·mL−1. All cell types, seeded in 96‐well plates, were loaded with 100 μL per well of fura‐2/AM loading solution, prepared according to the manufacturer's instructions and kept from light at 37°C for 45 min. At the end of the incubation, selected wells were exposed to PhNCS‐COOH 300 μM for 5 min, at 37°C. After that, antigen (2 μg·mL−1), Ca2 + ionophore (1 or 10 μM) and thapsigargin (1 μM) were added for 20 min. [Ca2 +]i was monitored during the entire period (25 min total) by detecting, every 30 s, the increase in fluorescence at Ex/Em = 340/510 and 380/510 nM with an HTS fluorescence microplate reader FlexStation using the software SoftMax Pro version 5.0 (Molecular Devices). Ca2 + levels are presented as the ratio of fluorescence at 340 nM to the fluorescence at 380 nM (F340/F380), measured over time.
Immunoblot analysis
RBL‐2H3 cells were seeded into a 6‐well culture plate (5 × 105 cells well−1 in 2 mL) in MEM with 10% heat‐inactivated FBS at 37°C. On the following day, cells were incubated overnight with anti‐DNP‐IgE (0.5 μM). IgE‐sensitized cells were then treated with PhNCS‐COOH (30–300 μM) for 5 min at 37°C and stimulated with DNP (10 ng·mL−1) for 1 h. β‐HEX release was measured in each well supernatant, and harvested cells were lysed with lysis buffer containing protease and phosphatase inhibitors. Cell lysates were centrifuged at 14 000 × g at 4°C for 10 min, and each supernatant was equalized to the same protein concentration with the Bradford assay. Proteins in a total cell lysate were then separated by SDS‐PAGE and transferred to PVDF membranes. The membrane was blocked with 5% BSA dissolved in Tris‐buffered saline containing 1% Tween 20 (TBST) for 1 h at room temperature. After washing with TBST, the membrane was incubated with a 1:1000 dilution of specific antibodies against phospho‐Lyn, Lyn, phospho‐Syk, Syk, phospho‐SAPK/JNK, SAPK/JNK, phospho‐cytosolic phospholipase A2 (cPLA2), cPLA2, phospho‐phospholipase Cγ1 (PLCγ1), PLCγ1 in 5% BSA‐TBST at 4°C overnight (antibody concentrations were 5, 17, 206, 73, 491, 318, 178, 29, 38, 165 μg·mL−1 respectively). Blots were washed in TBST, and the membranes were incubated in a 1:5000 dilution of HRP‐conjugated IgG secondary antibody (concentration 66 μg·mL−1) in 5% BSA‐TBST at room temperature for 1 h. RBL‐2H3, BMMC and HMC‐1 proteins in the total lysates were then separated by SDS‐PAGE and transferred to PVDF membranes. The membrane was blocked with 5% skim milk dissolved in TBST for 1 h at room temperature. After washing with TBST, the membrane was incubated with a 1:2500 dilution of specific antibody against CSE (final concentration 100 ng·mL−1) in 5% milk‐TBST at 4°C overnight. Blots were washed in TBST, and the membranes were incubated in a 1:5000 dilution of goat anti‐rabbit‐IgG HRP secondary antibody in 5% milk‐TBST at room temperature for 1 h. Total proteins were evaluated by incubating the membranes with a 1:50 000 dilution of β‐actin HRP conjugated (concentration 1 μg·mL−1) in milk 5% at room temperature. After washing with TBST, blots were developed by an ECL Western Blotting Detection Reagent, and the membranes were exposed to Hyperfilm ECL. The intensity of each protein was expressed as a density ratio based on the protein standard size marker. The density of each band was determined with GeneTool software.
To ascertain CSE expression in RBL‐2H3, BMMC and HMC‐1 cells, we initially used an anti‐CSE antibody purchased from Abcam (Cat. No. ab131052), whose specificity was subsequently found to be questionable (see Supporting Information Appendix S1). Thus, to demonstrate the specificity of the CSE antibody, we performed two different experiments: in the first, we blocked the CSE antibody with its immunizing peptide; in the second, we used MEFs and liver tissue from WT and CSE‐KO mice. For the first experiment, we neutralized CSE antibody with an excess of peptide corresponding to the epitope recognized by the antibody (from Proteintech). Accordingly, we incubated 70 ng·mL−1 of CSE antibody in 5% non‐fat dry milk, in the presence (blocked) or in the absence (control) of 1 μg·mL−1 CSE peptide, for 30 min at room temperature under agitation. Then, we performed the experiments on two identical PVDF membranes, using the blocked antibody for one and the control for the other. Blots were washed in PBS, and the membranes were incubated with a 1:5000 dilution of anti‐rabbit‐IgG HRP secondary antibody in 5% non‐fat dry milk‐PBS at room temperature for 1 h. For the second experiment, MEFs, harvested as described previously (Yang et al., 2013), and liver tissue were lysed in ice‐cold RIPA buffer with 0.2 mM PMSF, 1% protease and phosphatase inhibitor cocktail. Proteins were equalized to the same concentration with the Bradford assay, separated by SDS‐PAGE and transferred to nitrocellulose membranes. Membranes were blocked in 5% non‐fat dry milk in PBS containing 0.1% Tween 20 with primary antibody overnight at 4°C. After three times wash with PBS containing 0.1% of Tween 20, the membranes were incubated with specific antibody against CSE (final concentration 70 ng·mL−1) in 5% non‐fat dry milk‐PBS at 4°C overnight. Blots were washed in PBS, and the membranes were incubated with a 1:5000 dilution of anti‐rabbit‐IgG HRP secondary antibody in 5% non‐fat dry milk‐PBS at room temperature for 1 h. Proteins were detected using the ECL chemiluminescence system.
Data and statistical analysis
The data and statistical analysis in this study complies with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). All data are reported as mean ± SEM. Statistical analysis was performed with GraphPad Prism 6.0 software (San Diego, CA, USA). In accordance with Journal policy, statistical analysis was performed only when a minimum of n = 5 independent samples was acquired. We used one‐way ANOVA followed by Dunnett's post hoc test when comparing more than two groups of data and one‐way ANOVA, non‐parametric Kruskal–Wallis test, followed by Dunn's post hoc test when comparing multiple independent groups. When comparing two groups, the unpaired t‐test was used. Differences between group means were considered statistically significant when a value of at least P < 0.05 was achieved.
Materials
DMSO was used as the solvent for stock solutions of PhNCS‐COOH, A23187 and thapsigargin. DMSO was diluted 1/1000–1/300; 1/10 000; 1/1000 for the three compounds respectively. DMSO had no effect of its own at the highest concentration used (i.e. 3%). We found no significant difference (one‐way ANOVA, non‐parametric Kruskal–Wallis post hoc test) between basal levels of β‐HEX in the absence and presence of 1–3% DMSO in RBL‐2H3 (n = 12), BMMC (n = 11) and HMC‐1 (n = 5). Martelli et al., (2014) have shown that 1% DMSO had no effect on cells. PhNCS‐COOH and the specific antibody against goat anti‐rabbit‐IgG HRP (Cat. No. sc‐2030) were purchased from Santa Cruz Biotechnology (Dallas, TX, USA). MEM, DMEM phenol free, DMEM, FBS, trypsin, DNP, BSA, p‐nitrophenyl‐N‐acetyl‐β‐D‐glucosaminide, mouse anti‐DNP‐IgE, thapsigargin, Ca2 + ionophore A23187, NaHS, L‐Cysteine, D‐Pen, renin substrate tetradecapeptide porcine and cromolyn were purchased from Sigma‐Aldrich (St. Louis, MO, USA). Human angiotensinogen was purchased from AnaSpec (Fremont, CA, USA). Medium RPMI 1640 and penicillin/streptomycin were purchased from Life Technologies (Invitrogen Life Technologies, Carlsbad, CA, USA). Recombinant murine IL‐3 and stem cell factor were purchased from PeproTech (Rocky Hill, NJ, USA). Iscove's modified Dulbecco's medium and FBS for MEF cells were purchased from HyClone (South Logan, UT, USA). Specific antibodies against phospho‐Syk (Cat. No. 2710), Syk (Cat. No. 2712), phospho‐Lyn (Cat. No. 2731), Lyn (Cat. No. 2796), phospho‐SAPK/JNK (Cat. No. 9251), SAPK/JNK (Cat. No. 9252), phospho‐cPLA2 (Cat. No. 2831), cPLA2 (Cat. No. 2832), phospho‐PLCγ1 (Cat. No. 2821) and PLCγ1 (Cat. No. 5690) were purchased from Cell Signaling Technology (Beverly, MA, USA), anti β‐actin HRP‐conjugated antibody (Cat. No. ACTB12‐HRP) was bought from Alpha Diagnostic International (San Antonio, TX, USA); specific antibody against CSE (Cat. 12217‐1‐AP) and recombinant human CSE (ag2872, CSE blocking peptide) were purchased from Proteintech Group Inc. (Chicago, IL, USA). For initial experiments, we used anti‐CSE antibody (Cat. No. ab131052) purchased from Abcam (see Supporting Information Appendix S1). PVDF membranes and ECL Western Blotting Detection Reagent were purchased from Millipore (Billerica, MA, USA). Phosphatase inhibitors were purchased from BioVision (Milpitas, CA, USA). The Bradford protein assay kit was purchased from Bio‐Rad Laboratories (Hercules, CA, USA). Fura‐2 No Wash Calcium Assay Kit for the measurements of cytosolic Ca2 + levels was purchased from Abcam (Cambridge, MA, USA).
Results
Mast cell degranulation and renin release elicited by Ca2 + ionophore, thapsigargin and antigen
We first ascertained the degranulating and renin‐releasing effect of three different stimuli in three mast cell models. For this purpose, we raised [Ca+2]i by promoting its influx with the Ca2 + ionophore A23187, by preventing Ca2 + storage in the sarcoplasmic reticulum with thapsigargin, or by activating FcεRI. We used two immortalized cell lines, RBL‐2H3 and HMC‐1, as well as native mast cells isolated and differentiated from mouse bone marrow (BMMC). Of these, RBL‐2H3 and BMMC express the FcεRI receptor and contain renin (Shimizu et al., 1988; Aldi et al., 2014), whereas HMC‐1 contain renin but do not express FcεRI (Silver et al., 2004).
Incubation of RBL‐2H3 and HMC‐1 cells with Ca2 + ionophore and thapsigargin caused marked degranulation as shown by β‐HEX release (Figure 1D and F). Ca2 + ionophore and thapsigargin also elicited a release of renin from both RBL‐2H3 and HMC‐1 cells, measured as Ang I formed (Figure 1A and C). Incubation of BMMC cells with A23187 and thapsigargin caused β‐HEX release (Figure 1E). Specific antigen challenge of IgE‐sensitized BMMC and RBL‐2H3 elicited β‐HEX release (Figure 1D and E). Degranulation, by A23187, thapsigargin and antigen, was accompanied by renin release (i.e. Ang I formed) in BMMC (Figure 1B). Renin was also released (i.e. Ang I formed) by antigen‐induced degranulation in RBL‐2H3 cells (Figure 1A).
Figure 1.
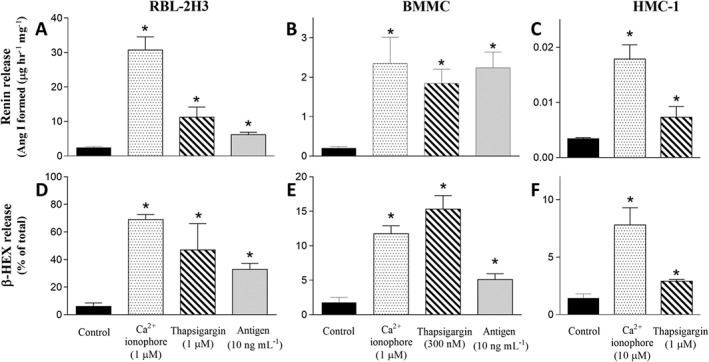
Mast cell degranulation and renin release elicited by Ca2 + ionophore, thapsigargin or antigen. Incubation with Ca2 + ionophore (A23187, 1 μM), thapsigargin (1 μM) or antigen (DNP 10 ng·mL−1), elicits β‐HEX and renin release in RBL‐2H3 and BMMC cells (A, B, D, E). β‐HEX values are expressed as percentage of total β‐HEX present in the corresponding cell lysate. Basal β‐HEX release in RBL‐2H3 and BMMC was 6.08 ± 0.69 (n = 13) and 1.78 ± 0.17 (n = 19) % of total respectively. Basal renin release in RBL‐2H3 and BMMC was 2.42 ± 0.29 (n = 21) and 0.20 ± 0.04 (n = 11) Ang I formed (Ang I, μg·h−1·mg−1 protein) respectively. HMC‐1 cells were incubated with Ca2 + ionophore (10 μM) or thapsigargin (1 μM), and content of both β‐HEX and renin was measured in the supernatants at the end of incubation (C and F). Basal β‐HEX and renin release (Ang I formed) in HMC‐1 was 1.43 ± 0.09% of total (n = 17) and 3.4 ± 0.22 ng·h−1·mg−1 protein (n = 20) respectively. Data shown are means ± SEM. * P < 0.05; significantly different from corresponding basal level; one‐way ANOVA with non‐parametric Kruskal–Wallis, Dunn's post hoc test.
Endogenous H2S inhibits antigen‐induced degranulation in RBL‐2H3 cells
As mentioned above, H2S is mainly produced by three enzymes, whose expressions can vary in different tissues (Wallace and Wang, 2015). Because we found that RBL‐2H3, BMMC and HMC‐1 all express CSE (Figure 2A), we took advantage of this pyridoxal‐5′‐phosphate‐dependent enzyme to assess whether H2S plays a role as an endogenous modulator of antigen‐induced mast cell degranulation. We found that the CSE precursor, L‐cysteine (1–10 mM), elicited a concentration‐dependent inhibition of the degranulation caused by FcεRI activation in RBL‐2H3 cells. The selective CSE inhibitor D‐Pen (1 mM)(Brancaleone et al., 2016) significantly antagonized the effect of 10 mM concentration of L‐cysteine (Figure 2B). The expression of CSE, in the presence of antigen and of increasing concentrations of L‐cysteine, did not differ from control values, indicating that the inhibitory effect of L‐cysteine was not due to a change in CSE expression (Figure 2C).
Figure 2.
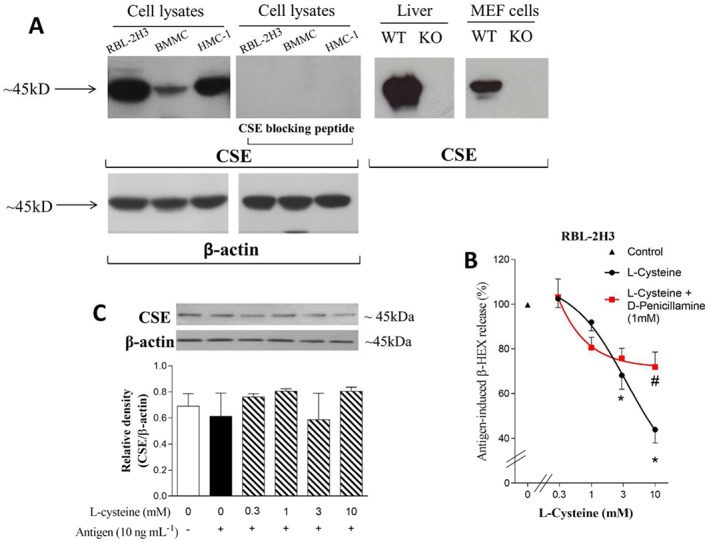
Endogenous H2S inhibits antigen‐induced degranulation in RBL‐2H3 cells. A. Representative Western blot of total lysates obtained from RBL‐2H3 (20 μg per lane), BMMC cells (20 μg per lane), HMC‐1 (20 μg per lane) respectively. Liver and MEFs from WT and CSE‐KO mice were used to verify the specificity of anti‐CSE antibody (15 μg for liver samples and 50 μg for MEF cells lysates). The image shows the presence of CSE in all three cell lines and in liver tissue and MEFs of WT mice. Notably, there was a lack of CSE in all three cell lysates when the antibody was pre‐incubated with its blocking peptide; there was also an absence of CSE in MEFs (Zhao et al., 2014) and liver (Yang et al., 2008) tissue from CSE‐KO mice. β‐actin was used as internal control. B. Concentration‐response curve for L‐cysteine‐induced inhibition of antigen‐elicited (DNP 10 ng·mL−1, 1 h) degranulation of RBL‐2H3 cells pre‐sensitized with anti‐DNP IgE, in the absence and presence of D‐Pen (1 mM). β‐HEX was measured in the supernatants at the end of the incubation. Basal β‐HEX release was 6.08 ± 0.69% of total (n = 13), while antigen‐elicited release was 32.93 ± 4.36% of total (n = 8), which corresponds to 100% in the ordinate scale (▲).Data shown are means ± SEM. * P < 0.05; significantly different from antigen alone; one‐way ANOVA with Dunnett's post hoc test. # P < 0.05; significantly different from L‐cysteine (10 mM); unpaired t‐test. C. Representative Western blot of CSE expression in total lysates (20 μg per lane) of control and antigen‐stimulated RBL‐2H3 in the absence and presence of L‐cysteine (0.3–10 mM).
H2S donors inhibit antigen‐induced degranulation and renin release in RBL‐2H3 and BMMC cells
As the findings presented in Figure 2 suggested that endogenously formed H2S inhibited mast cell degranulation, we next investigated the effect of PhNCS‐COOH, a slow H2S‐releasing donor (Martelli et al., 2014), on the FcεRI‐induced degranulation and renin release in RBL‐2H3 and BMMC cells. We found that PhNCS‐COOH caused a concentration‐dependent (10–300 μM) inhibition of antigen‐induced β‐HEX and renin release (Figure 3A–D). NaHS, a fast H2S donor (Zhao et al., 2001), also inhibited antigen‐induced degranulation of RBL‐2H3 cells (50% inhibition at 1 mM; Figure 3E). NaHS was equipotent with the mast cell stabilizer cromolyn (1 mM), used as positive control. At an equimolar concentration, PhNCS‐COOH (1 mM) abolished antigen‐induced degranulation (Figure 3E).
Figure 3.
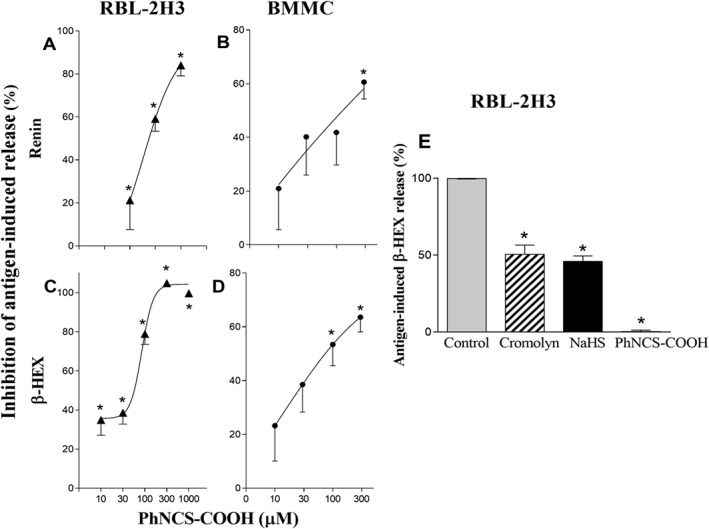
The H2S donor PhNCS‐COOH inhibits antigen‐induced degranulation and renin release in RBL‐2H3 and BMMC cells. A–D. Pre‐incubation with PhNCS‐COOH (10–1000 μM, 5 min) in pre‐sensitized RBL‐2H3 and BMMC cells inhibits antigen‐induced (DNP 10 ng·mL−1) degranulation and renin release in a concentration‐dependent manner. Points represent PhNCS‐COOH‐induced inhibition (%) of β‐HEX and renin released in the supernatants at the end of the antigen incubation. Basal β‐HEX and renin release (Ang I formed) in RBL‐2H3 was 6.08 ± 0.69% (n = 13) of total and 2.42 ± 0.29 μg·h−1·mg−1 protein (n = 21) respectively; basal β‐HEX and renin release (Ang I formed) in BMMC was 1.78 ± 1.17% of total (n = 19) and 0.20 ± 0.04 μg·h−1·mg−1 protein (n = 11) respectively. Points are means ± SEM. * Significantly different (P < 0.05) from DNP by one‐way ANOVA, Dunnett's post hoc test. E. Bars represent DNP‐induced (10 ng·mL−1) release of β‐HEX in the absence (100%, control) and in the presence of equimolar concentrations (1 mM) of cromolyn, a reference mast cell stabilizer, NaHS, a fast H2S‐donor and PhNCS‐COOH. Data shown are means ± SEM of β‐HEX and renin release (n = 15). * P < 0.05; significantly different from antigen alone; one‐way ANOVA with Dunnett's post hoc test.
The H2S donor PhNCS‐COOH inhibits degranulation and renin release elicited by Ca2 + ionophore and thapsigargin in RBL‐2H3, BMMC and HMC‐1 cells
As the H2S‐donor PhNCS‐COOH markedly inhibited the antigen‐induced degranulation of RBL‐2H3 and BMMC (see Figure 3), we next tested the effects of PhNCS‐COOH on degranulation and renin release elicited by agents known to raise [Ca+2]i. As shown in Figure 4, PhNCS‐COOH (10–300 μM) concentration‐dependently inhibited the release of β‐HEX and renin elicited by either A23187 or thapsigargin in RBL‐2H3, BMMC and HMC‐1 cells.
Figure 4.
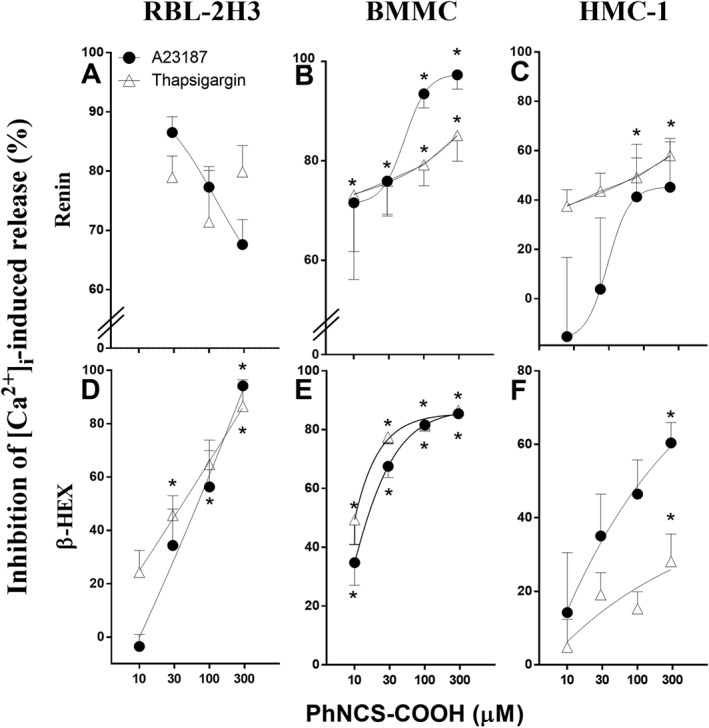
The H2S donor PhNCS‐COOH inhibits degranulation and renin release induced by increased [Ca2 +]i in RBL‐2H3, BMMC and HMC‐1 cells. B, C, E, F. Incubation with PhNCS‐COOH (10–300 μM, 5 min) in BMMC and HMC‐1 significantly inhibits mast cell degranulation (i.e. β‐HEX) and renin release induced by the administration of Ca2 + ionophore (A23187, 1 μM) and thapsigargin (1 μM, incubated for 20 min at 37°C). At the end of the incubation with A23187 and thapsigargin, β‐HEX and renin were measured in the supernatants and in total cell lysates. A, D. Pre‐incubation with PhNCS‐COOH (10–300 μM, 5 min) in RBL‐2H3 cells inhibits A23187‐ and thapsigargin‐induced (1 μM, 1 h) degranulation and renin release in a concentration‐dependent manner. β‐HEX and renin were measured in the supernatants at the end of the antigen incubation. Data shown represent PhNCS‐COOH‐induced inhibition (%) of β‐HEX (n = 9) and renin (n = 5) released, as means ± SEM. * P < 0.05; significantly different from A23187 and thapsigargin in the absence of PhNCS‐COOH; one‐way ANOVA with Dunnett's post hoc test.
PhNCS‐COOH reduces the increase in [Ca+2]i elicited by antigen, Ca2 + ionophore and thapsigargin in RBL‐2H3, BMMC and HMC‐1 cells
We next assessed whether the PhNCS‐COOH‐induced inhibition of degranulation and renin release elicited by antigen, Ca2 + ionophore and thapsigargin was associated with a decreased response of [Ca+2]i. For this, we monitored cytosolic Ca2 + levels fluorimetrically in RBL‐2H3, BMMC and HMC‐1 cells. Fura‐2‐loaded cells were pre‐incubated with PhNCS‐COOH (5 min, 37°C) and then stimulated with Ca2 + ionophore (A23187 1 and 10 μM, 20 min, 37°C), thapsigargin (1 μM and 300 nM, 20 min, 37°C) or antigen (2 μg·mL−1 for RBL‐2H3 and 100 ng·mL−1 for BMMC). Stimulation with antigen, Ca2 + ionophore or thapsigargin each caused a consistent, immediate increase in [Ca+2]i in RBL‐2H3 and BMMC cells. This increase in [Ca+2]i was markedly inhibited in the presence of the H2S‐donor PhNCS‐COOH (300 μM) (Figure 5).
Figure 5.
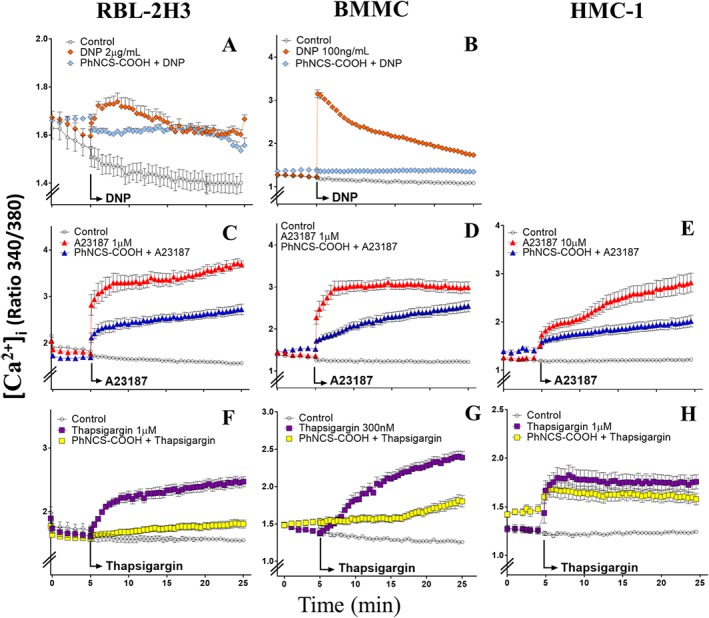
The H2S donor PhNCS‐COOH reduces the increase in [Ca+2]i elicited by antigen, Ca2 + ionophore and thapsigargin in RBL‐2H3, BMMC and HMC‐1 cells. Changes in [Ca2 +]i in RBL‐2H3, BMMC and HMC‐1 cells were monitored using the fluorescent Ca2 +‐indicator dye, fura‐2. Basal [Ca2 +]i levels were recorded throughout the entire experiment in wells containing control cells. In the other wells, antigen (DNP), thapsigargin and Ca2 + ionophore (A23187) were added either alone or in the presence of PhNCS‐COOH (300 μM, pre‐incubated for 5 min). [Ca2 +]i was recorded during the following 20 min in a FlexStation plate reader by measuring the ratio of fura‐2 fluorescence intensity (F340/F380) every 30 s. Data shown are expressed as mean ± SEM of six (A) or five (B–H) independent experiments, each performed in triplicate.
PhNCS‐COOH inhibits antigen‐induced degranulation of RBL‐2H3 cells by targeting proteins downstream of the FcεRI pathway
FcεRI‐receptor activation elicits mast cell degranulation by triggering a tyrosine kinase cascade, which begins with phosphorylation of Syk and Lyn and is followed by activation of several signal transduction pathways including the phosphorylation of PLCγ1, JNK and cPLA2 (Gilfillan and Tkaczyk, 2006). Accordingly, we next investigated whether the inhibitory effects of PhNCS‐COOH involved effects on the tyrosine kinase pathway. When RBL‐2H3 cells were incubated for 5 min with PhNCS‐COOH (30–300 μM) before antigen challenge, the phosphorylation of Syk, but not Lyn, was inhibited in a concentration‐dependent manner. Notably, at 300 μM, PhNCS‐COOH abolished Syk phosphorylation (Figure 6A and B). By Western blot analysis, we also demonstrated that the H2S donor down‐regulates, in a concentration‐dependent manner, the antigen‐induced phosphorylation of PLCγ1 and cPLA2, without inhibiting JNK phosphorylation (Figure 6C, D and E). Further, at the concentrations at which PhNCS‐COOH inhibited protein phosphorylation, this H2S donor failed to modify CSE expression (Figure 6F), indicating that its action did not involve a change in levels of CSE. Collectively, these results suggest that PhNCS‐COOH reduced antigen‐induced degranulation of RBL‐2H3 by targeting proteins downstream of the FcεRI pathway (Figure 7).
Figure 6.
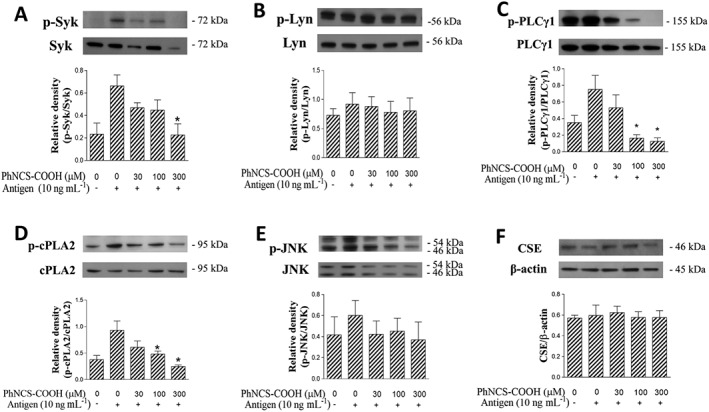
The H2S donor PhNCS‐COOH inhibits antigen‐induced degranulation of RBL‐2H3 cells by targeting proteins downstream of the FcεRI pathway. Sensitized RBL‐2H3 were incubated with PhNCS‐COOH (300 μM, 5 min) and challenged with the antigen (DNP 10 ng·mL−1, 1 h). At the end of the incubation, cells were lysed and total lysates (20 μg per lane) were collected, separated by SDS‐PAGE and transferred to PVDF membranes. Membranes were incubated with anti p‐Syk, Syk, p‐Lyn, Lyn, p‐PLCγ1, PLCγ1, p‐cPLA2, cPLA2, p‐JNK, JNK and CSE. β‐actin was used as internal control. Bars represent means ± SEM of five (A–F) independent experiments. * P < 0.05; significantly different from DNP; unpaired t‐test.
Figure 7.
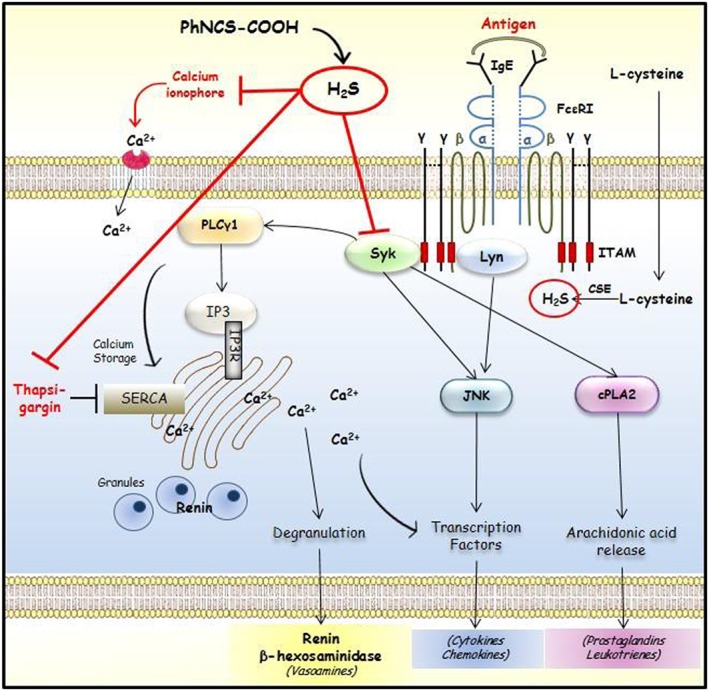
Putative sites of H2S‐induced inhibition of mast cell degranulation and renin release.
Discussion
The aim of our study was to investigate the role of H2S in mast cell degranulation and renin release. Our data clearly show for the first time that H2S, whether endogenously produced or released from H2S donors, targets proteins downstream of the FcεRI pathway, thus inhibiting the elevation of [Ca2 +]i and eventually reducing degranulation and renin release.
We had previously shown that the immortalized cell line, HMC‐1(Silver et al., 2004) and BMMC (Aldi et al., 2014) contain enzymatically active renin, whose release is exocytotic and can be considered a marker of mast cell degranulation along with β‐HEX. Here, we report the novel finding that the mast cell‐like RBL‐2H3 cells also contain and release active renin as a consequence of degranulation.
In mammalian cells, H2S is predominantly produced by the enzymatic action of CSE on L‐cysteine (Zhao et al., 2001; Wang, 2002). We hypothesized that endogenous H2S might play a regulatory role in mast cell degranulation and renin release. We found that by activating CSE with its endogenous substrate, L‐cysteine, IgE‐mediated degranulation in RBL‐2H3 cells progressively decreased with increasing concentrations of L‐cysteine. In fact, the selective CSE inhibitor D‐Pen markedly antagonized the effect of the highest L‐cysteine concentration. Notably, CSE expression remained stable in antigen‐challenged cells at increasing L‐cysteine concentrations. These findings suggest that endogenously generated H2S is likely to modulate allergic reactions. It is conceivable that other enzymes, yet to be discovered in mast cells, might also be involved in the endogenous production of H2S.
We next tested the hypothesis that, as observed with endogenous H2S, H2S donors would also diminish mast cell degranulation and renin release. For this, we used a compound containing an isothiocyanate group, PhNCS‐COOH, which we had shown to release H2S at a slow and controlled rate, only in the presence of endogenous thiols (Martelli et al., 2014). Notably, when measuring H2S by amperometry and gas/mass chromatography (Martelli et al., 2014), a strong correlation was found between the amounts of H2S released from PhNCS‐COOH or other donors and their effects on mast cells (Marino, 2015). Indeed, PhNCS‐COOH caused a significant concentration‐dependent inhibition of antigen‐mediated degranulation and renin release in RBL‐2H3 and BMMC cells. HMC‐1 degranulation could not be triggered by antigen since these cells do not express the FcεRI receptor (Nilsson et al., 1994). Interestingly, the rapidly releasing H2S donor NaHS (Li et al., 2009) also inhibited the antigen‐induced degranulation of RBL‐2H3 cells and was equipotent with cromolyn, a reference mast cell stabilizer (Norris and Alton, 1996).
Although antigen, Ca2 + ionophore and thapsigargin act differently, they share the capacity to increase [Ca2 +]i, which ultimately promotes the fusion of mast cell granules with the plasma membrane and the release of pro‐inflammatory mediators (Metcalfe et al., 1997). Accordingly, we evaluated whether H2S‐donors also modulate non‐antigenic‐induced mast cell degranulation. For this, we activated mast cells by increasing [Ca2 +]i levels with the Ca2 + ionophore A23187 (Liu and Hermann, 1978) or the blocker of the SERCA (sarco/endoplasmic reticulum Ca2+ ATP‐ase) pump thapsigargin (Lytton et al., 1991). PhNCS‐COOH clearly inhibited, as a function of its concentration, the degranulation and renin release associated with this increase in [Ca2 +]i in RBL‐2H3, BMMC and HMC‐1 cells. Collectively, these findings suggested that the anti‐degranulating action of the H2S donor is associated with a decrease in [Ca2 +]i. Indeed, by measuring [Ca2 +]i in RBL‐2H3, HMC‐1 and BMMC, we found that whether [Ca2 +]i was increased by antigen/IgE reaction, Ca2 + ionophore or thapsigargin, PhNCS‐COOH always markedly reduced this Ca2 +response.
We next investigated the mechanism(s) of this H2S‐induced reduction in [Ca2 +]i. Indeed, activation of the FcεRI on the mast cell surface leads to an increase in [Ca2 +]i. Antigen‐induced mast cell degranulation is initiated by FcεRI cross‐linking, which ultimately elicits the release of many inflammatory mediators via phosphorylation of the Lyn/Syk pathway (Roth et al., 2008). Accordingly, we hypothesized that H2S released by the isothiocyanate might act at a site on the FcεRI signalling cascade. FcεRI is a tetrameric structure formed by αβγ2 chains. Aggregation of the receptor results in phosphorylation of Lyn and Syk, which leads to downstream propagation of signalling (Figure 7). We found that PhNCS‐COOH caused a marked reduction of phosphorylated Syk and inhibition of its downstream effectors, suggesting that Syk might be a primary target of the H2S‐donor. Interestingly, only Syk phosphorylation, but not that of Lyn, was reduced by PhNCS‐COOH in a concentration‐dependent fashion. This suggests a potential interaction only with the two γ‐subunits of FcεRI, which are associated with Syk kinase, rather than the β‐subunit, which is associated with Lyn kinase. In fact, the H2S‐donor caused a concentration‐dependent reduction of cPLA2 and PLCγ1 phosphorylation. PLCγ1 ultimately generates inositol‐1,4,5‐triphosphate, which releases Ca2 + from intracellular stores. Hence, PhNCS‐COOH is likely to reduce the antigen‐induced increase in [Ca2 +]i via PLCγ1 inhibition. The finding that PhNCS‐COOH did not affect JNK phosphorylation suggests that other Syk‐independent pathways might be involved in JNK phosphorylation (Kawakami et al., 1998).
In conclusion, our study demonstrated that H2S plays an important modulatory role in mast cell degranulation and renin release. We propose that the inhibitory effects of H2S in IgE‐mediated mast cell activation result from an action at different levels of the tyrosine kinase cascade initiated by FcεRI cross linking. The physiological role of H2S remains largely unknown, as it is difficult to distinguish between intracellular and extracellular H2S, with a possible feedback action. However, our findings suggest that the use of novel and suitable H2S‐donors, such as PhNCS‐COOH, could represent a new therapeutic strategy for those inflammatory disorders associated with a marked mast cell component. Moreover, given the pathophysiological relevance of local mast cell‐based renin–angiotensin systems in the heart and lungs (Veerappan et al., 2008; Reid et al., 2011), it is conceivable that H2S‐donors will prove useful in the clinical treatment of cardiovascular and pulmonary disorders in which mast cell‐derived renin may play a primary contributory role.
Author contributions
A.M. designed and performed the research, analysed the data and wrote the manuscript. A.M. and V.C. contributed to preliminary studies. M.F. and R.W. performed the immunoblotting experiment shown in Figure 2A. V.C. designed the research, analysed the data and contributed to the writing of the manuscript. R.L. designed the research, analysed the data and wrote the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Appendix S1 Validation of anti‐CSE‐antibody.
Supporting info item
Acknowledgements
Supported in part by NIH grant HL034215 and by an American Heart Association Grant‐in‐Aid and in part by a Discovery Grant from Natural Sciences and Engineering Research Council of Canada to RW. We thank our colleague Dr. Steven S. Gross for allowing us to use his FlexStation for [Ca2 +]i measurements.
Marino, A. , Martelli, A. , Citi, V. , Fu, M. , Wang, R. , Calderone, V. , and Levi, R. (2016) The novel H2S donor 4‐carboxy‐phenyl isothiocyanate inhibits mast cell degranulation and renin release by decreasing intracellular calcium. British Journal of Pharmacology, 173: 3222–3234. doi: 10.1111/bph.13583.
References
- Abe K, Kimura H (1996). The possible role of hydrogen sulfide as an endogenous neuromodulator. J Neurosci 16: 1066–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldi S, Robador PA, Tomita K, Di Lorenzo A, Levi R (2014). IgE receptor‐mediated mast‐cell renin release. Am J Pathol 184: 376–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldi S, Marino A, Tomita K, Corti F, Anand R, Olson KE et al. (2015). E‐NTPDase1/CD39 modulates renin release from heart mast cells during ischemia/reperfusion: a novel cardioprotective role. FASEB J 29: 61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brancaleone V, Esposito I, Gargiulo A, Vellecco V, Asimakopoulou A, Citi V et al. (2016). D‐penicillamine modulates hydrogen sulfide (H S) pathway through selective inhibition of cystathionine‐gamma‐lyase (CSE). Br J Pharmacol 173: 1556–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorucci S, Distrutti E, Cirino G, Wallace JL (2006). The emerging roles of hydrogen sulfide in the gastrointestinal tract and liver. Gastroenterology 131: 259–271. [DOI] [PubMed] [Google Scholar]
- Gilfillan AM, Tkaczyk C (2006). Integrated signalling pathways for mast‐cell activation. Nat Rev Immunol 6: 218–230. [DOI] [PubMed] [Google Scholar]
- Hosoki R, Matsuki N, Kimura H (1997). The possible role of hydrogen sulfide as an endogenous smooth muscle relaxant in synergy with nitric oxide. Biochem Biophys Res Commun 237: 527–531. [DOI] [PubMed] [Google Scholar]
- Kamoun P (2004). Endogenous production of hydrogen sulfide in mammals. Amino Acids 26: 243–254. [DOI] [PubMed] [Google Scholar]
- Kawakami Y, Hartman SE, Holland PM, Cooper JA, Kawakami T (1998). Multiple signaling pathways for the activation of JNK in mast cells: involvement of Bruton's tyrosine kinase, protein kinase C, and JNK kinases, SEK1 and MKK7. J Immunol 161: 1795–1802. [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo S, Doe I, Kurokawa Y, Kawabata A (2007). Hydrogen sulfide causes relaxation in mouse bronchial smooth muscle. J Pharmacol Sci 104: 392–396. [DOI] [PubMed] [Google Scholar]
- Li T, Zhao B, Wang C, Wang H, Liu Z, Li W et al. (2008). Regulatory effects of hydrogen sulfide on IL‐6, IL‐8 and IL‐10 levels in the plasma and pulmonary tissue of rats with acute lung injury. Exp Biol Med (Maywood) 233: 1081–1087. [DOI] [PubMed] [Google Scholar]
- Li YF, Xiao CS, Hui RT (2009). Calcium sulfide (CaS), a donor of hydrogen sulfide (H(2)S): a new antihypertensive drug? Med Hypotheses 73: 445–447. [DOI] [PubMed] [Google Scholar]
- Liu C, Hermann TE (1978). Characterization of ionomycin as a calcium ionophore. J Biol Chem 253: 5892–5894. [PubMed] [Google Scholar]
- Liu YH, Lu M, Xie ZZ, Hua F, Xie L, Gao JH et al. (2014). Hydrogen sulfide prevents heart failure development via inhibition of renin release from mast cells in the isoproterenol treated rats. Antioxid Redox Signal 20: 759–769. [DOI] [PubMed] [Google Scholar]
- Lytton J, Westlin M, Hanley MR (1991). Thapsigargin inhibits the sarcoplasmic or endoplasmic reticulum Ca‐ATPase family of calcium pumps. J Biol Chem 266: 17067–17071. [PubMed] [Google Scholar]
- Mackins CJ, Kano S, Seyedi N, Schafer U, Reid AC, Machida T et al. (2006). Cardiac mast cell‐derived renin promotes local angiotensin formation, norepinephrine release, and arrhythmias in ischemia/reperfusion. J Clin Invest 116: 1063–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martelli A, Testai L, Breschi MC, Blandizzi C, Virdis A, Taddei S et al. (2012). Hydrogen sulphide: novel opportunity for drug discovery. Med Res Rev 32: 1093–1130. [DOI] [PubMed] [Google Scholar]
- Martelli A, Testai L, Citi V, Marino A, Bellagambi FG, Ghimenti S et al. (2014). Pharmacological characterization of the vascular effects of aryl isothiocyanates: is hydrogen sulfide the real player? Vascul Pharmacol 60: 32–41. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metcalfe DD, Baram D, Mekori YA (1997). Mast cells. Physiol Rev 77: 1033–1079. [DOI] [PubMed] [Google Scholar]
- Nilsson G, Blom T, Kusche‐Gullberg M, Kjellen L, Butterfield JH, Sundstrom C et al. (1994). Phenotypic characterization of the human mast‐cell line HMC‐1. Scand J Immunol 39: 489–498. [DOI] [PubMed] [Google Scholar]
- Norris AA, Alton EW (1996). Chloride transport and the action of sodium cromoglycate and nedocromil sodium in asthma. Clin Exp Allergy 26: 250–253. [PubMed] [Google Scholar]
- Reid AC, Silver RB, Levi R (2007). Renin: at the heart of the mast cell. Immunol Rev 217: 123–140. [DOI] [PubMed] [Google Scholar]
- Reid AC, Brazin JA, Morrey C, Silver RB, Levi R (2011). Targeting cardiac mast cells: pharmacological modulation of the local renin‐angiotensin system. Curr Pharm Des 17: 3744–3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts IS, Brenchley PE (2000). Mast cells: the forgotten cells of renal fibrosis. J Clin Pathol 53: 858–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth K, Chen WM, Lin TJ (2008). Positive and negative regulatory mechanisms in high‐affinity IgE receptor‐mediated mast cell activation. Arch Immunol Ther Exp (Warsz) 56: 385–399. [DOI] [PubMed] [Google Scholar]
- Roviezzo F, Bertolino A, Sorrentino R, Terlizzi M, Matteis M, Calderone V et al. (2015). Hydrogen sulfide inhalation ameliorates allergen induced airway hypereactivity by modulating mast cell activation. Pharmacol Res 100: 85–92. [DOI] [PubMed] [Google Scholar]
- Shibuya N, Mikami Y, Kimura Y, Nagahara N, Kimura H (2009). Vascular endothelium expresses 3‐mercaptopyruvate sulfurtransferase and produces hydrogen sulfide. J Biochem 146: 623–626. [DOI] [PubMed] [Google Scholar]
- Shimizu A, Tepler I, Benfey PN, Berenstein EH, Siraganian RP, Leder P (1988). Human and rat mast cell high‐affinity immunoglobulin E receptors: characterization of putative alpha‐chain gene products. Proc Natl Acad Sci U S A 85: 1907–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver RB, Reid AC, Mackins CJ, Askwith T, Schaefer U, Herzlinger D et al. (2004). Mast cells: a unique source of renin. Proc Natl Acad Sci U S A 101: 13607–13612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S, Padovani D, Leslie RA, Chiku T, Banerjee R (2009). Relative contributions of cystathionine beta‐synthase and gamma‐cystathionase to H2S biogenesis via alternative trans‐sulfuration reactions. J Biol Chem 284: 22457–22466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veerappan A, Reid AC, Estephan R, O'Connor N, Thadani‐Mulero M, Salazar‐Rodriguez M et al. (2008). Mast cell renin and a local renin‐angiotensin system in the airway: role in bronchoconstriction. Proc Natl Acad Sci U S A 105: 1315–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace JL, Wang R (2015). Hydrogen sulfide‐based therapeutics: exploiting a unique but ubiquitous gasotransmitter. Nat Rev Drug Discov 14: 329–345. [DOI] [PubMed] [Google Scholar]
- Wang R (2002). Two's company, three's a crowd: can H2S be the third endogenous gaseous transmitter? FASEB J 16: 1792–1798. [DOI] [PubMed] [Google Scholar]
- Wu L, Yang W, Jia X, Yang G, Duridanova D, Cao K et al. (2009). Pancreatic islet overproduction of H2S and suppressed insulin release in Zucker diabetic rats. Lab Invest 89: 59–67. [DOI] [PubMed] [Google Scholar]
- Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K et al. (2008). H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma‐lyase. Science 322: 587–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Zhao K, Ju Y, Mani S, Cao Q, Puukila S et al. (2013). Hydrogen sulfide protects against cellular senescence via S‐sulfhydration of Keap1 and activation of Nrf2. Antioxid Redox Signal 18: 1906–1919. [DOI] [PubMed] [Google Scholar]
- Zhang H, Bhatia M (2008). Hydrogen sulfide: a novel mediator of leukocyte activation. Immunopharmacol Immunotoxicol 30: 631–645. [DOI] [PubMed] [Google Scholar]
- Zhao W, Zhang J, Lu Y, Wang R (2001). The vasorelaxant effect of H(2)S as a novel endogenous gaseous K(ATP) channel opener. EMBO J 20: 6008–6016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao K, Ju Y, Li S, Altaany Z, Wang R, Yang G (2014). S‐sulfhydration of MEK1 leads to PARP‐1 activation and DNA damage repair. EMBO Rep 15: 792–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 Validation of anti‐CSE‐antibody.
Supporting info item