

Abstract
The synthesis of O‐doped benzorylenes, in which peripheral carbon atoms have been replaced by oxygen atoms, has been achieved for the first time. This includes key high‐yielding ring‐closure steps which, through intramolecular C−O bond formation, allow stepwise planarization of oligonaphthalenes. Single‐crystal X‐ray diffraction showed that the tetraoxa derivative forms remarkable face‐to‐face π–π stacks in the solid state, a favorable solid‐state arrangement for organic electronics.
Keywords: copper, cyclization, macromolecules, nanostructures, supramolecular chemistry
Discrete and extended polycyclic all‐carbon aromatic hydrocarbons (PAHs)1, 2 have polarized great interest3, 4, 5, 6, 7 as ultralight materials for engineering flexible optoelectronic devices. Replacing the carbon atoms with other isostructural atoms at given positions8, 9 is now developing as a versatile functionalization strategy to control the chemical, charge‐carrier, and self‐assembly behaviors of PAHs.10 Specifically, in the last years, we took note of the renaissance of O‐doped aromatics such as peri‐xanthenoxanthene (PXX; Scheme 1). These molecules are in fact characterized by excellent carrier‐transport and injection properties, as well as easy processability, chemical inertness, and high‐thermal stability.11 Because of these properties, PXX has proven exceptional performance when used as an active organic semiconductor (OSC) in transistors for rollable OLEDs.12, 13, 14 However, the expansion of PXX into larger O‐doped frameworks has so far remained unexplored (Figure 1), although understanding and controlling the O‐doping ratio could provide the conceptual basis to engineer a new family of OSCs with tunable optoelectronic properties.
Scheme 1.
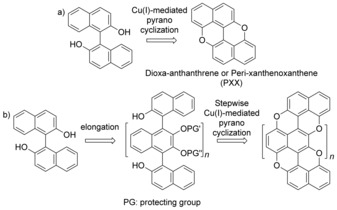
a) Pyranopyran‐fusing approach for preparing a PXX core. b) Synthetic strategy toward the O‐doped benzorylenes.
Figure 1.
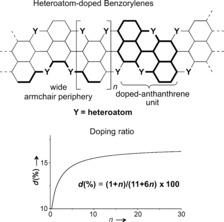
Heteroatom‐doped benzorylenes and its distinctive doping ratio (d) as function of the number (n) of dihydroxynaphthalenyl units.
Herein we describe the synthesis of unprecedented O‐doped benzoryles (Scheme 1), like pentaphenopentaphene and napthotetraphenopyranthrene (n=1 and 2, respectively), featuring a tailored topological periphery and doping ratio, d, with the latter being controlled by the number of the monomeric units. Generally, controlled doping patterns in discrete graphene substructures are obtained through bottom‐up synthesis involving monomeric aromatic heterocyles which are preorganized in a covalent scaffold and successively planarized through oxidative C−C bond formation.10, 15, 16, 17 In our approach, we instead considered the O‐doped benzoryles derived from oligonaphthalenes with 2,3‐dihydroxynaphthalene and 2‐hydroxynaphthalene moieties as the key monomeric and capping units, respectively (Scheme 1 b). At the synthetic planning level, this consideration guided us to contemplate the oxidative metal‐mediated formation of C−O bonds in a pyranopyran motif (Scheme 1 a) as the planarization reaction. As we anticipated potential susceptibility of the 2,3‐dihydroxynaphthalenyl moieties under oxidative conditions, a decision was made to protect the hydroxy groups and to follow a two‐step planarization protocol (Scheme 1 b).
Specifically, two classes of molecules were prepared (Scheme 2): one bearing only 3,5‐di(tert‐butyl)phenyl substituents (13, 17H, and 21H) and another with extra 4‐tert‐butylphenyl side groups (17 tBuPh and 21 tBuPh) to favor solubility. The key 1,4‐linked oligonaphthalene skeletons (12, 14, and 18) were synthesized by sequential oxidative coupling reactions in the presence of racemic phenylethylamine and CuCl2.18 It should be noted that the intermediates 9–12 were prepared and used as racemates. The same applies for the molecules 14–16 and 18–20.
Scheme 2.
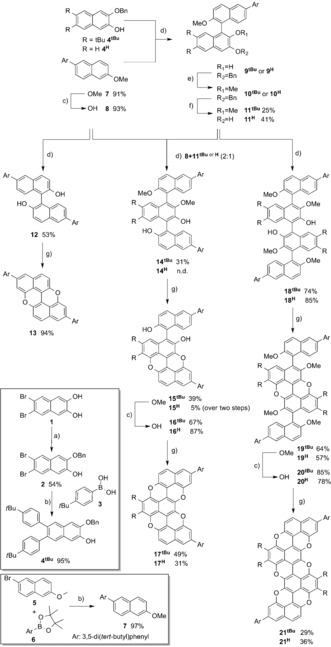
Synthetic path for 13, 17X, and 21X (X=H or tBuPh). In the frames: synthesis of the naphthol building blocks. a) NaHCO3, BnBr, DMF, 100 °C; b) Cs2CO3, [Pd(PPh3)4], toluene/DMF/EtOH, microwave, 100 °C; c) BBr3, CH2Cl2, 0 °C; d) CuCl2, (±)‐1‐phenylethylamine, MeOH, CH2Cl2, 0 °C; e) K2CO3, MeI, acetone, reflux; f) H2, Pd/C, CHCl3, AcOH; g) CuI, PivOH, DMSO, 130–145 °C.
To commence, the naphthol 8 (prepared starting from 6‐bromo methoxynaphthalene 5 by a Suzuki cross‐coupling reaction followed by cleavage of the methoxy group with BBr3) was dimerized by copper(II)‐mediated oxidative coupling into binaphthyl 12, which was used as model substrate. Despite numerous works describing the preparation of dibenzofurans,19 only a few synthetic strategies have been developed to date for the formation of benzopyranes.20 Amongst those, the modified protocol described by Pummerer and co‐workers21 with CuO allowed the transformation of 12 into the PXX derivative 13 in 42 % yield. However, when CuI was used in the presence of O2 and PivOH in DMSO at 140 °C,22 we exceptionally improved the yield to 94 %. In any circumstances, CuOAc gave inferior yields while other transition metals gave either low yields or no conversion.
For preparing the quarternaphthalene derivatives, 8 was cross‐coupled with either the monobenzyl dihydroxynaphthalene 4H or 4 tBuPh (the latter prepared from the 6,7‐dibromo precursor, 1,23 through double Suzuki cross‐coupling and monobenzylation reactions) by copper(II)‐promoted cross‐coupling to yield the monohydroxy binaphthalenes 11H and 11 tBuPh, respectively, after methylation and cleavage of the benzyl ether. Subsequent oxidative dimerization of 11H and 11tBu gave the quarternaphthalenes 18H (85 %) and 18 tBuPh (74 %), respectively, as isomeric mixtures. Intramolecular etherification of 18H and 18 tBuPh afforded the intermediates 19H and 19 tBuPh, respectively, as diastereoisomeric mixtures where the cis‐ (cis‐19H and cis‐19 tBuPh) and trans (trans‐19H and trans‐19 tBuPh) isomers could be easily separated. Small transparent crystals of both isomers of 19 tBuPh were obtained by vapor diffusion. X‐ray analysis confirmed the presence of the central pyranopyran cycle with two naphthalenyl substituents in the cis and trans configurations (Figure 2). Removal of the methyl protecting groups by BBr3 and subsequent the CuI‐mediated ring‐closure reaction led to the formation of the tripyranopyran derivatives 21H and 21 tBuPh in 36 % and 29 % yield, respectively. The fully conjugated tetramers 21H and 21 tBuPh were unambiguously identified by HR‐MALDI through the detection of the peaks corresponding to the molecular ions (M +) at m/z 966.3935 (C68H54O6 +, calc.: 966.3920) and 1494.7526 (C108H102O6 +, calc.: 1494.7556), respectively.
Figure 2.
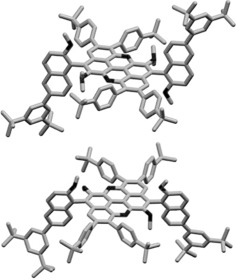
Crystal structures of trans‐19 tBuPh (above) and cis‐19 tBuPh (below).25 Space groups P21/c and , respectively. C gray, O black.
In parallel, the fused bispyranopyran 17H and 17 tBuPh were also prepared. The naphthol 8 was cross‐coupled with 11H and 11 tBuPh to afford the dihydroxyternaphthalenes 14H and 14 tBuPh, respectively, as isomeric mixtures (dimer 12 and tetramers 18H and 18 tBuPh were also obtained as side‐products). Successive pyranopyran fusion led to the corresponding intermediates 15H and 15 tBuPh. BBr3‐promoted cleavage of the methyl groups followed by the oxidative cyclization yielded 17H and 17 tBuPh in 31 % and 49 % yield, respectively. Again, 17H and 17 tBuPh were clearly identified by HR‐MALDI through detection of the peaks related to the molecular ions (M+) at m/z 812.3848 (C58H52O4 +, calc.: 812.3866) and 1076.5760 (C78H76O4 +, calc.: 1076.5744), respectively. Surprisingly, 1H NMR investigations of the final molecules were inconclusive as only broad peaks were observed after planarization.
However, upon addition of a few drops of NH2NH2 to a [D8]THF solution of some samples of 17 tBuPh, a sharpening of the proton resonances was observed. As shown in Figure 3, the 1H NMR spectrum of the sample containing NH2NH2 features well‐resolved peaks, thus perfectly integrating the 22 aromatic proton resonances expected for 17 tBuPh. This spectrum could suggest that a fraction of the compound is present as a radical cation. Notably, for 17H, 21H and 21 tBuPh the addition of NH2NH2 was fruitless, and only broad resonances in the 1H NMR spectra were observed. To further corroborate the chemical structure of the bis(pyranopyran) derivative, crystals suitable for X‐ray diffraction analysis were obtained by vapor diffusion of iPrOH into a CH2Br2 solution of 17H (Figure 4). The X‐ray structure confirms the nearly flat boomerang‐like shape of the pentaphenopentaphene framework, in which four oxygen atoms have replaced four carbon atoms at the peripheries.
Figure 3.
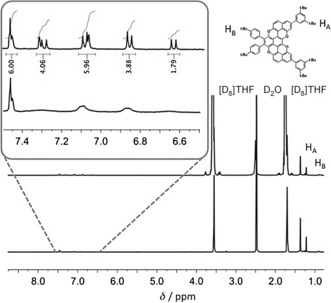
1H‐NMR spectra (400 MHz, [D8]THF) of 17 tBuPh before (below) and after addition of NH2NH2 (above).
Figure 4.
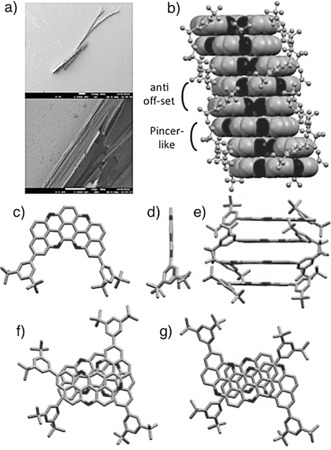
a) SEM images of the crystals of 17H displaying a lamellar‐like texture. b–e) Solid‐state columnar π–π stacks with an interplanar distance of 3.3 Å. c,d) Top‐ and side‐view of the crystal structure. f) Pincerlike and g) anti off‐set π–π stacking arrangements. Space group: . C gray, O black.
Looking at its organization at the solid state (Figures 4 b and e), one can clearly evidence the presence of a columnar arrangement in which the molecules are organized in π–π stacks, with an average interplanar spacing of 3.3 Å. Two face‐to‐face stacking modes are apparent: a pincerlike stack (Figure 4 f), in which two crystallographically independent molecules are facing each other in an antiparallel fashion with a relative angle of about 36°, and an anti offset shift (Figure 4 g), where two molecules stack in an antiparallel arrangement with a lateral offset of about 3.4 Å and 0.8 Å for the other crystallographically independent molecule.
UV‐vis steady‐state absorption spectra of 13, 17X, 21X are shown in Figure 5. While the spectrum of the PXX derivative features the typical electronic transitions at λ=392, 421 and 449 nm, the spectra for conjugates 17X and 21X appear much broader. In particular, the absorption spectra of 17H and 17 tBuPh display unstructured low‐intensity red‐shifted bands at λ=412, 509 and 552 nm, whereas only a long absorption tail reaching λ=650 nm is observed for both 21H and 21 tBuPh. Variable‐temperature measurements did not display any significant sharpening of the electronic transitions even at elevated temperatures (80 °C; see Figure SI5 in the Supporting Information). Together with the 1H NMR results, the intense broadening of the electronic transitions suggests that these O‐doped molecules most likely undergo strong aggregation. This aggregation can possibly occur either by simple π‐stacking interactions between neutral molecules or between a radical‐cation with its neutral counterparts in mixed‐valence complexes,24 or through the formation of covalent oligomers possibly deriving from a radical recombination followed by proton elimination.
Figure 5.
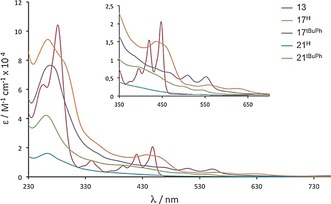
Steady‐state UV‐vis absorption spectra of 13, 17X, and 21X in CH2Cl2.
While the formation of mixed‐valence species can be realistically excluded because of the absence of electronic transitions fingerprinting a charge transfer in the NIR spectral region (see Figure SI2),24 HRMS‐MALDI and tandem mass (MSMS) measurements unambiguously displayed the presence of oligomeric species, and are supportive of the presence of aggregates even in the gas phase. In particular, peaks at m/z 1624 and 3247 could be discerned, thus suggesting the presence of dimeric and tetrameric species for 17H (Figure SI8 a). MSMS analysis at m/z 3247 and 1624 suggest that the dimers are formed by a combination of covalent [(17H)2−2 H] and noncovalent [(17H)2] complexes (Figure SI8 c,d,e,f,h). In contrast, the tetrameric species (Figure SI8 b,g,h) are constituted by noncovalent complexes of covalent dimers, [(17H)2−2 H]2.
In conclusion, we have described the first methodology to prepare unprecedented O‐doped benzorylenes by using a stepwise planarization strategy. This approach involves the simultaneous formation of C−O bonds through an intramolecular copper(I)‐mediated oxidative reaction originating pyranopyran rings. First X‐ray diffraction showed that the tetraoxa derivative undergoes strong π‐stacking in the solid state to form lamellar‐like microstructures. The remarkable propensity of this class of molecules to undergo self‐aggregation is intriguing in view of the design of organic materials to be used in optoelectronic devices. Detailed electron paramagnetic resonance, electrochemical, and conductivity studies are under investigation to fully understand the chemical behavior of this class of O‐doped π‐conjugated framework, as well as their potential for engineering transistors.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
D.B. gratefully acknowledges the EU through the ERC Starting Grant “COLORLANDS” project, the FRS‐FNRS (FRFC contract n° 2.4.550.09), the Science Policy Office of the Belgian Federal Government (BELSPO‐IAP 7/05 project), and the “TINTIN” ARC project (09/14‐023). D.S. thanks the FNRS for her doctoral fellowship. We thank B. Norberg and Prof. J. Wouters (UNamur) for the crystal structure n° 1415364.
D. Stassen, N. Demitri, D. Bonifazi, Angew. Chem. Int. Ed. 2016, 55, 5947.
References
- 1. Chen L., Hernandez Y., Feng X., Müllen K., Angew. Chem. Int. Ed. 2012, 51, 7640–7654; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 7758–7773. [Google Scholar]
- 2. Dössel L., Gherghel L., Feng X., Müllen K., Angew. Chem. Int. Ed. 2011, 50, 2540–2543; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 2588–2591. [Google Scholar]
- 3. Schwierz F., Nat. Nanotechnol. 2010, 5, 487–496. [DOI] [PubMed] [Google Scholar]
- 4. Hoheisel T. N., Schrettl S., Szilluweit R., Frauenrath H., Angew. Chem. Int. Ed. 2010, 49, 6496–6515; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 6644–6664. [Google Scholar]
- 5. Chen F., Tao N. J., Acc. Chem. Res. 2009, 42, 429–438. [DOI] [PubMed] [Google Scholar]
- 6. Cai J., Ruffieux P., Jaafar R., Bieri M., Braun T., Blankenburg S., Muoth M., Seitsonen A. P., Saleh M., Feng X., Müllen K., Fasel R., Nature 2010, 466, 470–473. [DOI] [PubMed] [Google Scholar]
- 7. Grill L., Dyer M., Lafferentz L., Persson M., Peters M. V., Hecht S., Nat. Nanotechnol. 2007, 2, 687–691. [DOI] [PubMed] [Google Scholar]
- 8. Maiti U. N., Lee W. J., Lee J. M., Oh Y., Kim J. Y., Kim J. E., Shim J., Han T. H., Kim S. O., Adv. Mater. 2014, 26, 40–67. [DOI] [PubMed] [Google Scholar]
- 9.See some examples for N:
- 9a. Takase M., Narita T., Fujita W., Asano M. S., Nishinaga T., Benten H., Yoza K., Müllen K., J. Am. Chem. Soc. 2013, 135, 8031–8040; [DOI] [PubMed] [Google Scholar]
- 9b. Gońka E., Chmielewski P. J., Lis T., Stępień M., J. Am. Chem. Soc. 2014, 136, 16399–16410; [DOI] [PubMed] [Google Scholar]
- 9c. Kivala M., Pisula W., Wang S., Mavrinskiy A., Gisselbrecht J.-P., Feng X., Müllen K., Chem. Eur. J. 2013, 19, 8117–8128; for S: [DOI] [PubMed] [Google Scholar]
- 9d. Cinar M. E., Ozturk T., Chem. Rev. 2015, 115, 3036–3140; [DOI] [PubMed] [Google Scholar]
- 9e. Takimiya K., Osaka I., Mori T., Nakano M., Acc. Chem. Res. 2014, 47, 1493–1502; for B: [DOI] [PubMed] [Google Scholar]
- 9f. Dou C., Saito S., Matsuo K., Hisaki I., Yamaguchi S., Angew. Chem. Int. Ed. 2012, 51, 12206–12210; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 12372–12376; [Google Scholar]
- 9g. Matsuo K., Saito S., Yamaguchi S., J. Am. Chem. Soc. 2014, 136, 12580–12583; [DOI] [PubMed] [Google Scholar]
- 9h. Hertz V. M., Bolte M., Lerner H.-W., Wagner M., Angew. Chem. Int. Ed. 2015, 54, 8800–8804; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 8924–8928; for P: [Google Scholar]
- 9i. Matano Y., Imahori H., Org. Biomol. Chem. 2009, 7, 1258–1271; [DOI] [PubMed] [Google Scholar]
- 9j. Baumgartner T., Réau R., Chem. Rev. 2006, 106, 4681–4727; [DOI] [PubMed] [Google Scholar]
- 9k. Baumgartner T., Acc. Chem. Res. 2014, 47, 1613–1622; for BN: [DOI] [PubMed] [Google Scholar]
- 9l. Wang X.-Y., Wang J.-Y., Pei J., Chem. Eur. J. 2015, 21, 3528–3539; [DOI] [PubMed] [Google Scholar]
- 9m. Bonifazi D., Fasano F., Lorenzo-Garcia M. M., Marinelli D., Oubaha H., Tasseroul J., Chem. Commun. 2015, 51, 15222–15236. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Narita A., Wang X.-Y., Feng X., Müllen K., Chem. Soc. Rev. 2015, 44, 6616–6643; [DOI] [PubMed] [Google Scholar]
- 10b. Wang X., Sun G., Routh P., Kim D.-H., Huang W., Chen P., Chem. Soc. Rev. 2014, 43, 7067–7098. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Lv N., Xie M., Gu W., Ruan H., Qiu S., Zhou C., Cui Z., Org. Lett. 2013, 15, 2382–2385; [DOI] [PubMed] [Google Scholar]
- 11b. Wang L., Duan G., Ji Y., Zhang H., J. Phys. Chem. C 2012, 116, 22679–22686; [Google Scholar]
- 11c. Kobayashi N., Sasaki M., Nomoto K., Chem. Mater. 2009, 21, 552–556. [Google Scholar]
- 12.“Semiconductor Device, Method of Manufacturing the Same, and Method of Forming Multilayer Semiconductor Thin Film”: Kobayashi N., Sasaki M., Ohe T., U.S. Patent 8,399,288 B2, 2013.
- 13.“Novel Materials for Organic Electroluminescent Devices”: Stoessel P., Buesing A., Heil H., U.S. Patent 2010/0013381 Al, 2010.
- 14. Noda M., Kobayashi N., Katsuhara M., Yumoto A., Ushikura S.-I., Yasuda R.-I., Hirai N., Yukawa G., Yagi I., Nomoto K., Urabe T., Dig. Tech. Pap. Soc. Inf. Disp. Int. Symp. 2010, 41, 710–713. [Google Scholar]
- 15. Bronner C., Stremlau S., Gille M., Brauße F., Haase A., Hecht S., Tegeder P., Angew. Chem. Int. Ed. 2013, 52, 4422–4425; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 4518–4521. [Google Scholar]
- 16. Cai J., Pignedoli C. A., Talirz L., Ruffieux P., Söde H., Liang L., Meunier V., Berger R., Li R., Feng X., Müllen K., Fasel R., Nat. Nanotechnol. 2014, 9, 896–900. [DOI] [PubMed] [Google Scholar]
- 17. Kawai S., Saito S., Osumi S., Yamaguchi S., Foster A. S., Spijker P., Meyer E., Nat. Commun. 2015, 6, 8098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.
- 18a. Hovorka M., Scigel R., Gunterova J., Tichy M., Zavada J., Tetrahedron 1994, 50, 9503–9516; [Google Scholar]
- 18b. Hovorka M., Zavada J., Tetrahedron 1994, 50, 9517–9530; [Google Scholar]
- 18c. Feringa B., Wynberg H., Bioorg. Chem. 1978, 7, 397–408; [Google Scholar]
- 18d. Noji K. M., Nakajima M., Koga K., Tetrahedron Lett. 1994, 35, 7983–7984; [Google Scholar]
- 18e. Tanaka K., Furuta T., Fuji K., Miwa Y., Taga T., Tetrahedron: Asymmetry 1996, 7, 2199–2202; [Google Scholar]
- 18f. Furuta T., Tanaka K., Tsubaki K., Fuji K., Tetrahedron 2004, 60, 4431–4441; [Google Scholar]
- 18g. Tsubaki K., Tanaka H., Takaishi K., Miura M., Morikawa H., Furuta T., Tanaka K., Fuji K., Sasamori T., Tokitoh N., Kawabata T., J. Org. Chem. 2006, 71, 6579–6587; [DOI] [PubMed] [Google Scholar]
- 18h. Egami H., Matsumoto K., Oguma T., Kunisu T., Katsuki T., J. Am. Chem. Soc. 2010, 132, 13633–13635. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Nakanishi K., Fukatsu D., Takaishi K., Tsuji T., Uenaka K., Kuramochi K., Kawabata T., Tsubaki K., J. Am. Chem. Soc. 2014, 7101–7109; [DOI] [PubMed] [Google Scholar]
- 19b. Nakanishi K., Sasamori T., Kuramochi K., Tokitoh N., Kawabata T., Tsubaki K., J. Org. Chem. 2014, 79, 2625–2631; [DOI] [PubMed] [Google Scholar]
- 19c. Eskildsen J., Reenberg T., Christensen J. B., Eur. J. Org. Chem. 2000, 1637–1640; [Google Scholar]
- 19d. Nielsen C. B., Brock-Nannestad T., Reenberg T. K., Hammershoj P., Christensen J. B., Stouwdam J. W., Pittelkow M., Chem. Eur. J. 2010, 16, 13030–13034. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. Lv N., Xie M., Gu W., Ruan H., Qiu S., Zhou C., Cui Z., Org. Lett. 2013, 15, 2382–2385; [DOI] [PubMed] [Google Scholar]
- 20b. Asari T., Kobayashi N., Naito T., Inabe T., Bull. Chem. Soc. Jpn. 2001, 74, 53–58. [Google Scholar]
- 21.
- 21a. Bollweg B., Zeh L., Kramer E., Patent No. 545212, 1932;
- 21b. Pummerer R., Prell E., Rieche A., Ber. Dtsch. Chem. Ges. 1926, 59, 2159–2161; [Google Scholar]
- 21c. Pummerer R., Rieche A., Ber. Dtsch. Chem. Ges. 1926, 59, 2161–2175. [Google Scholar]
- 22. Zhao J., Zhang Q., Liu L., He Y., Li J., Li J., Zhu Q., Org. Lett. 2012, 14, 5362–5365. [DOI] [PubMed] [Google Scholar]
- 23. Cammidge A. N., Chambrier I., Cook M. J., Garland A. D., Heeney M. J., Welford K., J. Porphyrins Phthalocyanines 1997, 1, 77–86. [Google Scholar]
- 24.
- 24a. Kaim W., Angew. Chem. Int. Ed. Engl. 1980, 19, 911–912; [Google Scholar]; Angew. Chem. 1980, 92, 940–941; [Google Scholar]
- 24b. Kochi J. K., Rathore R., Le Maguères P., J. Org. Chem. 2000, 65, 6826–6836. [DOI] [PubMed] [Google Scholar]
- 25. CCDC 1415364 (trans-19 tBuPh), 1416668 (cis-19 tBuPh), 1416669 (17H) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary