

Abstract
Cytoadhesion of Plasmodium falciparum‐infected erythrocytes to the microvascular endothelial lining shares striking similarities to cytoadhesion of leukocytes. In both cases, adhesins are presented in structures that raise them above the cell surface. Another similarity is the enhancement of adhesion under physical force (catch bonding). Here, we review recent advances in our understanding of the molecular and biophysical mechanisms underlying cytoadherence in both cellular systems. We describe how imaging, flow chamber experiments, single‐molecule measurements, and computational modeling have been used to decipher the relevant processes. We conclude that although the parasite seems to induce processes that resemble the cytoadherence of leukocytes, the mechanics of erythrocytes is such that the resulting behavior in shear flow is fundamentally different.
Keywords: catch bond, cytoadhesion, leukocyte, malaria, mesoscopic model, modeling
Abbreviations
AFM, atomic force microscopic
CSA, chondroitin‐4‐sulfate
DPD, dissipative particle dynamics
FENE, finitely extensible nonlinear elastic
GPCR, G‐protein‐coupled receptors
HEV, human endothelial venules
ICAM 1, intercellular adhesion molecule 1
KAHRP, knob‐associated histidine‐rich protein
MPCD, multiparticle collision dynamics
PfEMP1, Plasmodium falciparum erythrocyte membrane protein 1
Pf, Plasmodium falciparum
PSGL‐1, P‐selectin glycoprotein ligand‐1
SEM, scanning electron microscopy
TSP, thrombosporin
A drop of blood (10 μL) contains an estimated 50 million erythrocytes and approximately 5000 leukocytes, among other cells. Both red and white blood cells are easy to spot under the microscope because of their difference in color and shape. Erythrocytes are red and are shaped in the form of a biconcave discoid, whereas leukocytes are colorless and have a spherical appearance. The distinct colors and shapes are a reflection of their different biological functions. Erythrocytes are part of the respiratory and circulatory system, carrying oxygen in a complex with the red chromophore hemoglobin from the lung to the periphery of the body. Leukocytes are cells of the immune system and are involved in controlling infections and removing foreign antigens.
Leukocytes usually move passively with the blood flow. However, in response to an inflammatory stimulus, they can roll along the microvascular endothelium and, eventually, firmly adhere (Fig. 1) 1. Leukocytes then migrate over the endothelium until they extravasate into the surrounding tissue, where they home in on infections 2. In comparison, erythrocytes do not adhere to the endothelial lining under normal physiological conditions. The exceptions are sickle cell erythrocytes that can adhere to postcapillary venule endothelium, a condition that is associated with vaso‐occlusive pain 3. Erythrocytes stay in circulation until they are cleared by the reticuloendothelial system of the spleen and liver after an average lifetime of 120 days.
Figure 1.
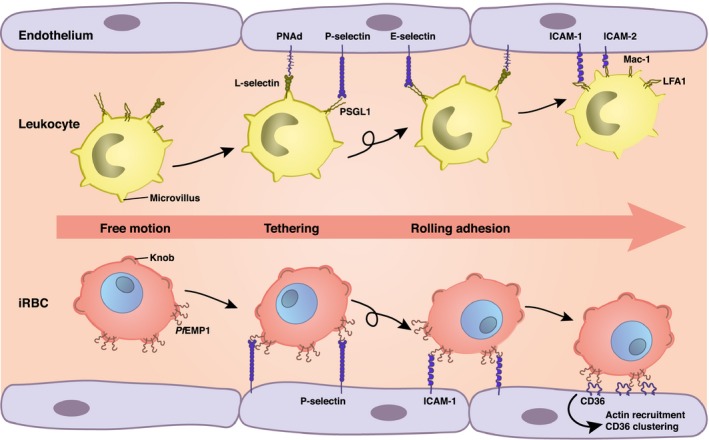
Schematic illustration of the adhesive phenotypes displayed by P. falciparum‐infected erythrocytes and leukocytes. Selected receptor and ligand pairs are shown.
The rheological properties of erythrocytes dramatically change upon infection with the human malaria parasite Plasmodium falciparum. Now erythrocytes acquire adhesive properties mediated by parasite‐encoded adhesins that are presented on the host cell surface in knob‐like protrusions (Fig. 1) 4, 5. Strikingly, the cytoadherence of parasitized erythrocytes mimics the way leukocytes adhere (Table 1) 6. Leukocytes are covered with hundreds of microvilli that carry adhesins such as L‐selectin on their tips 1. These adhesins interact with receptors on the surface of vascular endothelial cells, leading to cytoadhesive events that phenotypically resemble the rolling and firm adhesion displayed by P. falciparum‐infected erythrocytes 1, 6. Thus, both infected erythrocytes and leukocytes use localized and elevated adhesive domains to adhere to vessel walls. Another similarity is the use of tension‐enhanced adhesion systems 7, 8. However, in marked contrast to parasitized erythrocytes, cytoadherence of leukocytes is cooperative with the endothelium and leads to extravasation rather than sustained adhesion as is seen for P. falciparum‐infected erythrocytes.
Table 1.
Comparison of cytoadhesion of leukocytes and P. falciparum‐infected erythrocytes
| Leukocytes | Pf‐infected erythrocytes | |
|---|---|---|
| Adhesive structures | 100s of microvilli, each 300 nm high | 10 000s of knobs, each 20 nm high |
| Mechanics of adhesive structure | Viscoelastic, elongates under shear flow | Compact and stiff |
| Cell size, shape, and mechanics | Diameter 8–30 μm; spherical; flexible envelope | Diameter 6.2–8.2 μm; varies between biconcave and round, but not perfectly spherical; stiff envelope |
| Molecular system | Mainly selectins, very specific ligands on HEV | PfEMP‐1, broad range of receptors specific to distinct microvascular beds |
| On‐set of adhesion | Activated by chemokines, GPCR | Developmentally controlled, commences 16 h post invasion until end of replicative cycle |
| Interplay with endothelium | Inside out signaling to recruit leukocytes | Contact‐dependent intracellular signaling; clustering and activation of host adhesion receptors |
| Movement during cytoadhesion under flow conditions | Rolling on HEV | Sometimes rolling, but often irregular movement with flipping, depending on life cycle |
| Biological purpose | Preface to extravasation | Evasion of splenic clearance |
GPCR, G‐protein‐coupled receptors; HEV, human endothelial venules; Pf, Plasmodium falciparum.
In this review, we discuss the similarities and differences between cytoadhesion in these two medically important cellular systems. In Cytoadhesion: a molecular perspective, we will briefly introduce the molecular players mediating cytoadhesion of P. falciparum‐infected erythrocytes and leukocytes to endothelial cells. For a comprehensive review of the interacting receptor–ligand pairs, we refer the interested reader to specialized literature on this topic 1, 5, 9, 10. In Biophysics of adhesive bonds: slip or catch, we will discuss the phenomenon of tensile force‐enhanced cytoadhesion. Adhesive dynamics of white blood cells and Adhesive dynamics of P. falciparum‐infected erythrocytes will deal with the dynamic of cytoadhesion and how mathematical modeling and simulation can help us understand the mechano‐physical and biophysical principles underpinning cytoadhesion in flow. In particular, we will explain the reason for placing adhesins in structures elevated above the cell surface. We close with a discussion of the similarities and differences between the two systems and an outlook on open issues that should be addressed in the future.
Cytoadhesion: a molecular perspective
Pathology and cytoadhesion during a P. falciparum infection
Malaria remains a leading cause of morbidity and mortality in many developing countries. According to the world malaria report of 2014, there were 198 million cases of malaria in 2013 and 584 000 deaths 11. Of the five Plasmodium species that can cause malaria in humans, P. falciparum is the most virulent and deadly form, being responsible for most of the malaria‐associated deaths. P. falciparum is transmitted to humans by the bite of an infected Anopheles mosquito. Sporozoites released into the skin enter blood vessels and are carried with the blood flow to the liver where they invade hepatocytes. An infected hepatocyte can give rise to more than 10 000 daughter cells, termed merozoites, which are released into the blood stream where they infect erythrocytes. After 48 h the infected erythrocyte bursts, releasing typically 16 or 32 merozoites that then again infect erythrocytes, thereby establishing the intraerythrocytic life cycle. In extreme cases, 40% of the approximately 2.6 × 1013 erythrocytes of an adult malaria patient can be infected by parasites. The entire life cycle is completed when an Anopheles mosquito takes up sexual stages during a blood meal.
The asexual intraerythrocytic stages of P. falciparum are responsible for much of the pathology associated with malaria, whereas the liver stages are asymptomatic 12. The virulence of the asexual blood stages is largely due to the fact that the parasite alters the rheological and morphological properties of the infected erythrocyte. Uninfected erythrocytes move in the middle of the blood vessel where the flow velocity is maximum, leaving a rim around the vessel wall free of cells 13. This phenomenon can be explained by the Magnus effect. As soon as a cell moves out of the central part of the capillary the velocity gradient causes it to spin. The cell will then feel a force perpendicular to the flow direction and the rotational axis, which forces it back to the central region 13. P. falciparum‐infected erythrocytes also move with the blood flow but only for the first hours post invasion. After 16–20 h post invasion, they acquire adhesive properties and sequester in the deep microvascular bed of inner organs, including lung and brain 12. By sequestering in the microvasculature, parasitized erythrocytes avoid passage through the spleen and, hence, elimination by splenic clearance mechanisms that remove senescent and infected erythrocytes from circulation. Cytoadhesion is part of the parasite's strategy to colonize its host. However, it causes severe disease 12. The pathological sequelae that develop in the affected blood vessels include obstruction of microvascular blood flow, disturbed tissue perfusion, hypoxia, and systemic microvascularitis that can progress to life‐threatening complications, as presented by patients suffering from cerebral malaria 12, 14.
Cytoadhesion is mediated by a family of parasite‐encoded immuno‐variant adhesins collectively called Plasmodium falciparum erythrocyte membrane protein 1 (PfEMP1). PfEMP1 proteins can be classified into different groups based on domain architecture and functional specialization for interaction with particular host receptors 10. While the majority of the 60 PfEMP1 variants encoded in the haploid P. falciparum genome have unique binding properties, a few PfEMP1 variants can interact with several host cell receptors, for example, CD36 and ICAM‐1 10. Collectively, PfEMP1 proteins can interact with a broad range of receptors, including CD36, intercellular adhesion molecule 1 (ICAM 1), and P‐selectin 4, 5. In addition, a certain PfEMP1 variant can mediate attachment of parasitized erythrocytes to chondroitin‐4‐sulfate (CSA), a component of the proteoglycan matrix present in the intervillous space and moderately on the syncytiotrophoblast cell layer of the human placenta 15, 16, 17, 18, 19. Other parasite‐encoded factors implicated in cytoadhesive events include the RIFINS and STEVOR immunovariant antigens that are thought to mediate rosetting of infected erythrocytes with uninfected erythrocytes 20, 21.
For effective cytoadhesion, the major adhesin molecule PfEMP1 needs to be placed in parasite‐induced knob‐like protrusions on the erythrocyte plasma membrane (Figs 1 and 2). Knobs have a typical height of 20 nm and there can be as many as 10 000 knobs on the surface of a mature infected erythrocyte 22. Knobs consist of several parasite and host factors. The major constituent is the knob‐associated histidine‐rich protein (KAHRP) 23. This protein is encoded by the parasite and trafficked into the host erythrocyte cytoplasm. KAHRP expression is essential for cytoadhesion of parasitized erythrocytes, as demonstrated in parasite lines deficient in the kahrp gene 24. These kahrp‐deficient lines do not form knobs and the corresponding infected erythrocytes do not cytoadhere in flow, although they present PfEMP1 on the surface, suggesting that the display of PfEMP1 in knobs is critical for productive cytoadhesive interactions 24. KAHRP contains several protein‐binding domains and is known to interact with both parasite and host factors to form knobs and the cytoadhesion complex (Fig. 2). For instance, KAHRP interacts with several membrane skeletal proteins of the host erythrocyte, including spectrin, actin, ankyrin R, and spectrin–actin band 4.1 complex 25, 26, 27, 28. The ankyrin R and spectrin‐binding domains have been mapped to amino acids 285–362 and 370–441, respectively 25, 26; the actin‐binding domain is contained within residues 253–524 and most likely corresponds to the 5′ repeat unit 28, 29. KAHRP further self‐aggregates possibly via the 5′ repeat unit and it can bind to the C‐terminal cytoplasmic domain of PfEMP1 via one of two domains: the His‐rich domain from residue 60–123 and the domain from residues 359–441 26, 28, 30, 31, 32. PfEMP1 itself is linked to the spectrin network via PHIST proteins, with members of the PHIST family interacting with PfEMP1 proteins in a variant‐specific manner (Fig. 2) 33.
Figure 2.
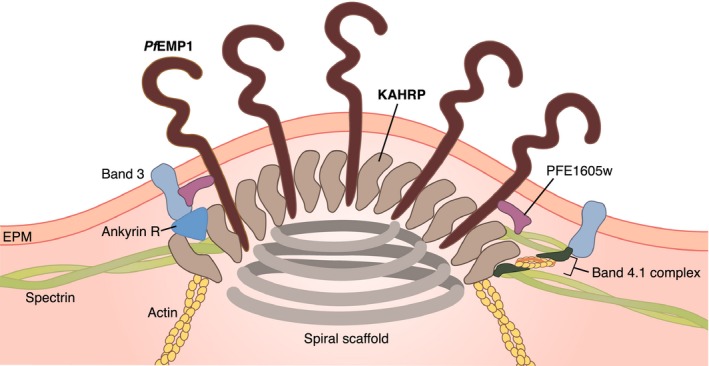
Schematic illustration of the knob structure of P. falciparum‐infected erythrocytes. Proximity between two elements indicates empirically determined interactions. PFE1605w is a member of the PHIST family that link the cytoplasmic domain of PfEMP1 in a variant‐specific manner to spectrin 33.
The interaction of both PfEMP1 and KAHRP with components of the membrane skeleton, but possibly also with parasite‐induced long actin filaments (Fig. 2) 34, may provide the mechanical support for binding of PfEMP1 to endothelial surface receptors under flow conditions. Further mechanical support may be provided by a spiral structure of hitherto unknown nature, underlying the knobs 35. The spiral structure may further contribute to the enhanced rigidity of the erythrocyte plasma membrane 35.
Cytoadhesion of leukocytes
Rolling adhesion of leukocytes can be seen in vivo as the first step toward extravasation during an acute inflammation 1, 36, 37. Initial contact is established through a system of three selectin receptors: L‐selectin is localized at the tips of the leukocyte microvilli and binds to sulfated sialyl‐Lewis X‐like sugars (e.g., PNAd) in high endothelial venules of the endothelium. P‐ and E‐selectin are activated in inflamed endothelium and bind to partners, such as P‐selectin glycoprotein ligand‐1 (PSGL‐1), which are also localized at the tips of the leukocyte microvilli (Fig. 1) 1, 38. Some integrins, such as α4β7 and α4β1, also contribute to rolling adhesion. One essential aspect of rolling is that it exposes the leukocytes to chemoattractant signals, such as the chemokine IL‐8 that then activates G‐protein‐coupled receptors (GPCR) 36, 37. This, in turn, leads to an inside‐out activation of β2 or α4‐integrins that establishes firm adhesion by binding to endothelial immunoglobins. The most prominent integrins in this context are LFA‐1 (αLβ2) or Mac‐1 (αMβ2) that can bind to ICAM‐1 or ICAM‐2 presented on the surface of endothelial cells (Fig. 1). There are hundreds of microvilli on a leukocyte with a typical height of 300 nm. In marked contrast to the knobs on infected erythrocytes, the microvilli of leukocytes can strongly increase their length under force, due to visoelastic flow in the membrane and the cytoskeleton 39. Once firmly attached, the leukocytes start to migrate along the endothelium in order to induce the extravasation process that usually proceeds in cooperativity with the endothelial cells at cell–cell boundaries (paracellular route). However, there is also a second mode of extravasation, in which leukocytes simply cross endothelial cells through their cell body (transcellular route). Both processes require tight regulation of the actin cytoskeleton 40. While rolling adhesion can be reconstituted easily in flow chambers, the study of extravasation requires more complex coculture assays or intravital microscopy.
Biophysics of adhesive bonds: slip or catch
Adhesive bonds are formed by noncovalent interactions between a receptor and a ligand. In a coarse‐grained description, the dissociation of such noncovalent bonds can be understood from Kramers reaction rate theory. The potential minimum of such a receptor–ligand bond corresponds to the bound state, and dissociation occurs by crossing the transition‐state barrier. The rate of dissociation can be written from Kramers theory as , where ΔE represents the energy barrier height and x c is the distance between the potential minimum and the transition point. This means that the dissociation rate increases with decreasing barrier height ΔE (compare the energy landscape sketched in Fig. 3A). In 1978, George Bell formulated a model 41, which was later refined by Evans and Ritchie 42, describing the dissociation rate of molecular bonds in the presence of an external force. The application of an external force f lowers the potential well to a new barrier height ΔE − fx c. Thus, the new rate of dissociation becomes k off ∼ exp(−(ΔE − fx c)/k B T), which leads to . Due to the exponential relation, a small increase in force can strongly increase the dissociation rate. The bonds that become short‐lived under external tensile force are called slip bonds 43 (Fig. 3B). This concept has been confirmed for many receptor–ligand pairs using dynamic force spectroscopy 44. In such experiments, the bonds are loaded by a dynamic ramp of force (which is easier to control than a constant force). From dynamic force spectroscopy measurements, the most probable rupture forces were shown to depend linearly on the logarithm of the loading rate, as expected from the Kramers–Bell–Evans model and as initially validated for biotin–streptavidin and biotin–avidin bonds 44.
Figure 3.
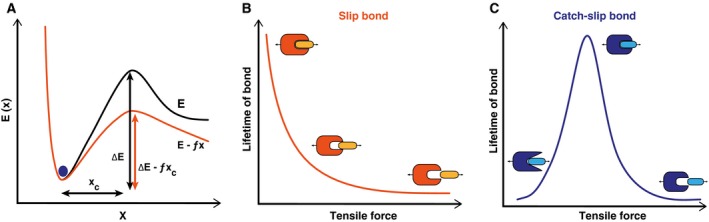
Slip bonds versus catch bonds. (A) Energy (E) landscape of a noncovalent receptor–ligand bond. The bound state corresponds to a metastable minimum separated from the unbound state by a transition state barrier of height ΔE. x c denotes the reactive compliance. Application of force, f, decreases the height of the barrier height to ΔE − fx c and therefore increases the dissociation rate. (B) As a result, the life time of the slip bond decreases when stressed by a tensile force. (C) In contrast, the lifetime of a catch‐slip bond initially increases with tensile force before it decreases at higher forces.
Interestingly, not all receptor–ligand pairs behave as slip bonds. Some bonds are enhanced by tensile force and are referred to as catch bonds 45, 46. The concept of catch bonds was first introduced as a theoretical consideration by Dembo et al. 43 and is explained by an increase in the bond's lifetime with increasing loading. Given that every bond slips at very high forces, a pure catch bond is not possible and one rather deals with a catch‐slip bond that is long‐lived at intermediate forces (Fig. 3C). Enhancement of adhesion in shear flow in the case of bacteria 47, 48 and in the case of leukocytes 49 supports the possibility of catch bond behavior at the cellular level. The first direct observation of catch‐slip bonds at the molecular level came from atomic force microscopic (AFM) measurements in which Marshall et al. 7 measured lifetimes of L‐selectin/PSGL‐1 bonds under conditions of external loading. Off‐rates of L‐selectin and PSGL‐1 bonds were also estimated in flow chamber experiments using L‐selectin‐coated microspheres 50. Off‐rates were measured for two sizes of microspheres in the presence and absence of ficoll, a sugar that increases the viscosity of the medium and, therefore, the hydrodynamic force. It was found that the catch bond behavior of L‐selectin and PSGL‐1 bonds is responsible for shear‐enhanced adhesion. Catch bond behavior is also observed in actomyosin bonds, with maximum bond lifetime at around 6 pN below which the bond lifetime decreases 51. Other examples of catch bond behavior include FimH‐mediated adhesion of E. coli bacteria 52 and the cell‐matrix adhesion receptor integrin α5β1 53.
Different biological systems might have evolved different molecular mechanisms for catch bonding. Also different theoretical models have been suggested in this context. For instance, the deformation model suggests that application of an external force optimizes the relative position of the receptor and the ligand to one another, which, in turn, is believed to increase the binding strength 54. Other models evoke allosteric effects or sliding and rebinding of bonds when stressed by a tensile force 55, 56, 57, 58, 59. The two‐pathway model takes kinetic aspects into consideration, by proposing that the molecular bond has two pathways for dissociation, one with a high‐energy barrier and another one with a low‐energy barrier 60, 61, 62, 63.
Adhesive dynamics of white blood cells
Leukocyte rolling has been studied in vitro using flow chamber experiments where rolling of leukocytes was examined on culture dishes and artificial lipid bilayers coated with appropriate ligands. It was found that the dissociation rate of the P‐selectin‐glycoprotein ligand bond increases with tensile force, indicative of a slip bond, making the P‐selectin–glycoprotein ligand interaction suitable for rolling adhesion 64, 65. In comparison, L‐selectin‐mediated adhesion involves a shear threshold, indicative of a catch bond, and only at high tensile force does the bond behave like a slip bond 50, 66.
Catch bonds can be considered as hydrodynamic sensors. Like sites of trauma, sites of inflammation are characterized by vasodilation and increased shear flow. The biological purpose of catch bonds, therefore, seems to be to facilitate the adhesion of leukocytes exactly where it is needed.
Modeling adhesion of white blood cells centers around two key aspects: hydrodynamics and bond kinetics, which together are referred to as adhesive dynamics. The first to address this issue were Hammer and Apte, who took a reductionist approach by assuming that leukocytes are ideal spheres homogenously covered with receptors and that the interacting ligands are homogenously distributed on a planar surface 67, 68, 69. A more refined version of the model was developed by Korn and Schwarz 70 who incorporated explicit positions for receptors and ligands. The adhesion dynamics was simulated using a Langevin equation:
where X(t) is a six‐dimensional vector for both position and orientation. Here, the generalized force, F, includes both the force and the torque. The terms F D and F S are direct and shear forces, respectively. The direct forces include gravitational force and receptor–ligand bond forces. The 6 × 6‐dimensional mobility matrix M converts forces into velocities and u ∞ is the unperturbed flow velocity. The stochastic random force, ξ(t), represents the thermal forces from the environment, which are crucial to bring spatially resolved receptors and ligands into binding range. The spurious drift term with the spatial derivative ∇M ensures that the stochastic dynamics satisfy the requirements of equilibrium physics. In the case of a sphere in unbounded flow, the mobility matrix becomes position independent, eliminating the drift term, ∇M. The ligand–receptor bond forms when the distance between ligand and receptor is less than the capture radius, r 0, with a probability 1 − exp(−κ onΔt), where κ on is the on‐rate. The dissociation of the bond is implemented via the Bell equation and occurs with a probability of 1 − exp(−κ offΔt), where is the unstressed off‐rate and . Here, F d is the detachment force scale that can be estimated directly from flow chamber experiments 71.
Adhesive dynamics is able to reproduce different dynamical states, such as rolling adhesion, firm adhesion, and transient adhesion. When systematically varying the model parameters, the model allows phase diagrams to be constructed, for example, as a function of molecular association and dissociation rates 72 (Fig. 4). The different phases correspond to different characteristics of the individual cell trajectories. During free motion both translational and angular velocities change only in a smooth manner and no interactions occur between the cell and the substrate. Firm adhesion is achieved when the cell is firmly attached to the substrate, thus translational and rotational velocities are almost zero. During both transient and rolling adhesion, the cell moves along the substrate by constantly forming new bonds at the front end and breaking the old bonds at the rear end. This leads to strong variations in the velocities. Translation and rotation are synchronized in the case of rolling adhesion, while they are more variable and more decoupled for transient adhesion. The on–off state diagram depicted in Fig. 4 depends on many other parameters, including the receptor–ligand kinetics, the viscosity of the medium, and receptor and ligand density. For example, increases in viscosity leads to a more stable rolling at the expense of firm adhesion, leaving free motion and transient adhesion unchanged. Particularly, the receptor and ligand density strongly affects the cytoadherence efficiency 38, 70. Simulation further revealed that the binding efficiency is significantly increased if the receptors are elevated above the cell surface 70. The physical reason is that hydrodynamic flow becomes slowed down toward the cell surface. Protrusions are, therefore, exposed to larger flows and can sample more environmental space than the cell surface. This might explain why the adhesion receptors are placed at the tip of microvilli in leukocytes and why the adhesin PfEMP1 is presented in knob‐like protrusions on the surface of P. falciparum‐infected erythrocytes. However, leukocytes with their microvilli of 300 nm in height seem to exploit this principle to a larger extent than do infected erythrocytes where knobs are only a moderate 20 nm in height. However, the large number of ten thousands of knobs as opposed to hundreds of microvilli per cell might compensate for the inferior height.
Figure 4.
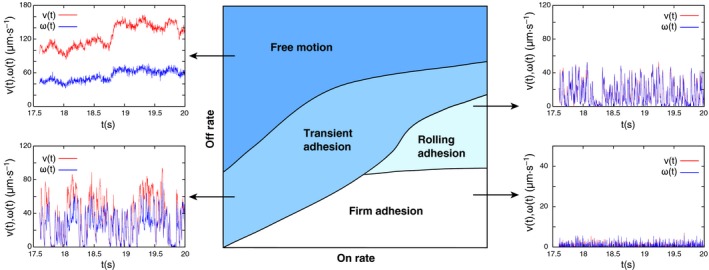
Phase diagram of adhesive dynamics with representative trajectories. Computer simulations can be used to predict how adhesive cells, such as leukocytes or infected red blood cells, move in hydrodynamic shear flow above a wall 69, 70. As a function of molecular association rate (x‐axis) and dissociation rate (y‐axis), one typically finds the four different regimes as shown here. Free motion is characterized by smooth changes in translational and angular velocities. At lower dissociation rates, transient adhesion occurs, with repeated binding and unbinding leading to strongly variable trajectories. Rolling is characterized by synchronization of translation and rotation and occurs for a high association rate and an intermediate dissociation rate, for example, as is seen for selectin‐mediated adhesion of leukocytes. Firm adhesion with low translational and rotational velocities occurs for low dissociation rates, which corresponds to adhesion through activated integrins.
Adhesive dynamics has also been used to investigate the importance of thermal fluctuations at both low and high shear rates on adhesive behaviors 73. Moreover, adhesive dynamics can be applied to other systems, for example, to adhesion of tumor cells 74, 75. The model was also used to study a two receptor system 76 and catch‐slip bond behavior 77, explaining the shear threshold nature of leukocyte cytoadhesion 50.
Adhesive dynamics of P. falciparum‐infected erythrocytes
Cytoadhesion in flow
Plasmodium falciparum‐infected erythrocytes display complex cytoadhesion phenotypes that include tumbling, rolling, and firm adhesion depending on the receptor–ligand interaction (Fig. 1). They firmly attach to CD36 and CSA, but roll over surfaces containing ICAM 1 and P‐selectin at physiological wall shear stresses, as shown in flow chamber assays 78, 79, 80, 81 (Fig. 1). Both rolling and firm adhesion can be observed in flow experiments using endothelial cells that express multiple receptors, such as ICAM1 and CD36 80. Li et al. 82 measured the interaction of parasitized erythrocytes with surface‐coated CD36 and thrombosporin (TSP). They found that TSP initiates the cell adhesion before they firmly adhere to CD36 82.
In a recent development, Rieger et al. 8 have studied adhesion of P. falciparum‐infected erythrocytes to artificial membranes coated with CSA under variable hydrodynamic conditions. They found that the number of adhering cells first increases and then decreases as the wall shear stress increases. Since this behavior is reminiscent of stress‐enhanced adhesion in other systems, Rieger et al. 8 proposed that the interaction between PfEMP1 and CSA behaves like a catch bond when stressed by a tensile force. Shear‐enhanced adhesion is also evident for rolling adhesion of parasitized erythrocytes to ICAM‐1 78, 80, suggesting that it might be a feature common to different PfEMP1/receptor interactions. Shear force‐enhanced bonds might have evolved as sensors for those blood vessels that provide optimal hydrodynamic and nutritional conditions for parasite growth and propagation. Consistent with this conclusion, Rieger et al. reported that the wall shear stress of 0.2 ± 0.1 Pa that affords optimal adhesion of parasitized erythrocytes to CSA in in vitro flow experiments corresponds to the hydrodynamic conditions present in the intervillous space of the placenta where CSA‐binding parasite sequester in high numbers during primigravidae 8, 83. In subsequent pregnancies, mothers and their unborn children are protected from placental malaria by acquired antibody‐mediated immune responses directed against parasite populations expressing the CSA‐binding PfEMP1 variant 83.
The receptor density not only modulates the efficiency of cytoadherence, it is a defining factor of whether productive cytoadhesive bonds are formed in flow or not. In the case of CSA, P. falciparum‐infected erythrocytes adhered to CSA only when the distance between neighboring CSA molecules is less than 15 nm, corresponding to 4400 CSA molecules per square micrometer 8. This affords cytoadhesion of P. falciparum‐infected erythrocytes in the placental intervillous space, where the CSA density is high, and not elsewhere in the microvasculature where the CSA content on endothelial cells is low—mainly confined to single CSA units attached to thrombomodulin 84.
A recent study has examined rolling of P. falciparum‐infected erythrocytes over ICAM‐1‐coated surfaces as a function of the capillary size, using microfluidic devices 85. Unexpectedly, cytoadherence was reduced in channels the size of an erythrocyte, compared to wider vessels 85. This finding might challenge the long held view that the first capillaries to clog in vivo during a P. falciparum infection are the very fine microvessels.
Some studies have attempted to quantify the adhesion force between parasitized erythrocytes and endothelial cells or receptor‐coated surfaces, by exposing adherent parasitized erythrocytes to gradually increasing wall shear stresses and determining the pressure (τ50) at which 50% of the cells detach 78. For cytoadherence to CSA, this value is approximately 1.3 Pa 8, 81, 86. Interestingly, the wall shear stress at which 50% of the cells detach from CSA‐coated surfaces depends on the receptor density in a manner that suggests cooperative binding 8. Whether binding of PfEMP1 variants to other host receptors is also cooperative remains to be seen.
By attaching infected erythrocyte to an AFM cantilever and then bringing them in contact with endothelial cells, Davis et al. 87 found that the adhesion force from the formation of single or multiple bonds with CD36 is initially in the range of 167 ± 208 pN. Once an initial contact is made, the adhesion force increases by five to sixfold through a signal pathway that leads to CD36 clustering and actin recruitment and polymerization at the focal point of cytoadhesion 87. The newly formed actin filaments are thought to reinforce the interacting CD36 molecules, by anchoring them to the cytoskeleton such that they withstand the shear forces from flow 88. Other studies have pointed out that the adhesion strength increases with parasite maturation and decreases when the temperature is raised from 37 °C to 41 °C 89, 90.
Modeling cytoadhesion of P. falciparum‐infected erythrocytes
Modeling adhesive dynamics of infected erythrocytes requires prior knowledge of cell mechanics and the corresponding physical parameters, such as the elastic Young's modulus. In addition, modeling requires mesoscopic models of the shape of infected erythrocytes and of the fluid flow.
The shape of an infected erythrocyte changes dramatically as the parasite matures. For the first 20 h of parasite development the host cell largely maintains its biconcave discoid shape, although a slight deformation is visible at the periphery of the host cell where the parasite resides. This quiescent phase is followed by a phase of volume increase brought about by the activation of parasite‐encoded solute channels in the host cell plasma membrane 91, 92, 93. These new permeation pathways lead to an influx of Na+, Cl−, and osmotic water. As the host cell swells due to the Na+‐driven fluid gain, it changes its shape form a biconcave discoid to a sphere 91, 94. For characterizing these temporal changes in host cell volume, several microscopic methods have been used 95, 96. For instance, 3D models of surface‐rendered views have been obtained from consecutive serial stacks of confocal fluorescence images 94, providing information regarding surface area and volume. However, only structures larger than 200 nm can be resolved by conventional microscopy. As the size of the knobs is in the range between 90 and 150 nm, with the distance between the knobs varying between 130 and 220 nm 22, standard optical microscopy is not suitable for analyzing these fine structures. Atomic force microscopy (AFM) and scanning electron microscopy (SEM) have recently provided detailed images and quantitative information regarding knob morphology and size, showing that there is a wide range of knob densities depending on the parasite strain and the type of host erythrocyte 8, 22, 97, 98. For instance, parasitized erythrocytes containing the sickle cell hemoglobin S or hemoglobin C (in which glutamic acid at position 6 in β‐globin is substituted by valine and lysine, respectively) display abnormally enlarged and dispersed knobs 99, 100. These aberrations have been associated with the reduced cytoadherence displayed by these infected hemoglobinopathic erythrocytes 99, 100. Moreover, aberrant knob formation and impaired cytoadhesion might underpin the mechanism by which these hemoglobinopathies protect carriers from severe malaria 101, 102.
As a result of the numerous parasite‐induced alterations, the elastic moduli of the host plasma membrane changes during intraerythrocytic development. For instance, the parasite reorganizes the actin of the host cell, mining it from the junctional complexes of the membrane skeleton and using it to generate long actin filaments required for vesicular transport of adhesins and other virulence factors from Maurer's clefts (unilamellar membrane profiles of parasite origin in the cytoplasm of the host cell) to the erythrocyte surface 34, 103, 104. As a result of actin mining and other parasite‐induced modifications of the host cell's membrane skeleton, the original spectrin network is broken down and reorganized in the form of spectrin aggregates around the knobs, as shown by AFM imaging 98. As a consequence of actin and spectrin remodeling and due to the generation of knobs, the erythrocyte plasma membrane becomes stiffer and less deformable 98. Using an optical stretcher, Mauritz et al. 105 detected that the infected erythrocytes have a reduced ability to stretch as compared to uninfected erythrocytes.
Mesoscopic models for red blood cell shape
Study of adhesive dynamics in flow requires mesoscopic modeling of red blood cells. Mesoscopic or coarse‐grained models of any biological system consider lower resolution subunits. One such class of mesoscopic model is a bead‐spring network model where the 3D shape is represented by a triangulated network of beads connected via springs. Using this approach, the red blood cell shape has been modeled at the spectrin level (Fig. 5A,B) 106, 107, 108, 109, 110. The positions of the beads or vertices are given by x j. The total surface area of an erythrocyte is the sum of areas of individual triangles, A i, and the total volume is the sum of volumes contributed by each triangle, . The total free energy of the system is,
Figure 5.

(A) Equilibrium shape of a model red blood cell generated using the mesoscopic model by Li et al. 109 for 5000 vertices. The total volume and surface area was constrained to 80 μm3 and 135 μm2, respectively. (B) Triangulated lattice in which beads are connected by springs. C. Snapshots of rolling uninfected and infected erythrocytes. (i) Snapshots of rolling uninfected erythrocytes are shown. The blue dots are markers on the membrane. (ii) Snapshots of flipping infected erythrocytes (uninfected erythrocytes with high Young's modulus) are shown. (iii) Snapshots of flipping infected erythrocytes with rigid parasite particles inside are shown. Figure reproduced from Ref 120.
where conserves the total surface area of the cell and conserves the total volume of the cell. Both and are implemented as harmonic potentials about A 0 and V 0 with rigidity factors k volume and k area, which set the strengths of the constraining potentials for surface area and volume, respectively, where A 0 and V 0 are total surface area and total volume of the cell, respectively.
The equilibrium length of the edge is ℓ 0. The total bending energy is written as,
where θi is the angle between the normals of adjacent triangles, κ b is the bending constant, and θ0 is the spontaneous curvature angle. The bending constant, κ b, is related to the bending moduli of the lipid membrane, κ c, as 108, 109. The last term in free energy is the elastic energy stored in the membrane. It is written as the sum of the elastic energy stored in spectrin links and the hydrostatic elastic energy stored in each triangle. It is written as
For V s(ℓ i), there exists many nonlinear spring models such as the Worm‐like Chain model (WLC) and the Finitely Extensible Non‐linear Elastic (FENE) polymer model 111. In the second term, C q is a constant that depends on spring parameters 108, 109 and q is an exponent that is chosen to be 1. The elastic moduli, such as Young's modulus and shear modulus, can be deduced in model parameters using a linear analysis. Figure 5A shows the equilibrium shape of a model red blood cell for 5000 vertices, a total volume constrained to V 0 = 80 μm3, and a total surface area of S 0 = 135 μm2. The currently considered mesoscopic model contains a fixed network, meaning that it does not consider the fluid nature of lipid membranes. Noguchi and Gompper took this into account by employing dynamically triangulated lattices 112, 113. Recently, it has been shown that the surface properties of erythrocytes are also strongly determined by active elements (e.g., ion pumps) in the membrane 114.
Modeling adhesion of parasitized erythrocyte under flow
There are several coarse‐grained or mesoscopic models of fluid, such as Multi Particle Collision Dynamics (MPCD) 115 and Dissipative Particle Dynamics (DPD) 116. MPCD and DPD are off‐lattice particle‐based simulation techniques to model the fluid. Noguchi and Gompper 113 used MPCD for simulating the fluid along with a dynamically triangulated network model for red blood cells to study the shape transitions under capillary flow. The dynamics of solvent particles was coupled to the dynamics of membrane vertices, by including membrane vertices in solvent exchange of momentum step 117, 118. They found that model red blood cells transform from nonaxis symmetric discocyte shapes to axis symmetric parachute shapes in capillary flow and that the speed of this transition decreases with increasing elastic moduli, such as the bending modulus and the shear modulus. Fedosov et al. 108 simulated the mechanics and the dynamics of model red blood cells in flow, using the DPD method. The external solvent makes bounce‐back interactions with the membrane, separating the internal and external solvent and preserving no‐slip boundary conditions at the membrane. The membrane and solvent dynamics are linked via viscous force coupling.
Fedosov et al. 119, 120 extended the approach to the adhesion of infected erythrocytes. Different dynamical states were recovered, such as firm adhesion, rolling adhesion, and flipping motion. Infected erythrocytes were modeled by increasing the membrane stiffness in relation to uninfected red blood cells and by placing solid particles mimicking the intracellular parasite inside the host cell cytosol. In Fig. 5C, the different adhesive behaviors of uninfected and infected erythrocytes are illustrated. Model uninfected erythrocytes change from rolling motion to flipping behavior when the Young's modulus is increased three times. Irregular flipping dynamics is observed when rigid particles are placed inside the erythrocyte cytosol as models for the parasite. Thus, the transition from rolling to flipping is due to increased stiffness and the presence of an intracellular rigid parasite.
Hosseini et al. modeled infected erythrocytes at the schizont stage based on uninfected red blood cells with an internal spherical rigid parasite to simulate stretching of the cell as observed using optical tweezers 121, 122. They found that the deformability of the cell under external force decreases owing to the increased stiffness and the presence of the internal parasite.
Conclusions and perspectives
Leukocyte rolling adhesion and extravasation is a widely investigated subject in biophysics, cell biology, and immunology. In comparison, the biophysical interactions of infected red blood cells with the endothelium are much less explored in spite of the medical relevance for the pathophysiology of malaria. Table 1 provides an overview of the main differences between the two systems as discussed here. It is evident that, despite the striking similarities that might have evolved due to the physical limitations of cytoadhesion in flow, there are also marked differences that certainly matter for the biological function. Overall the leukocytes seems to be better adapted to cell capture in shear flow, as evidenced by the round shape, the mechanical design of the microvilli and the molecular properties of the different members of the selectin family. Together, these elements lead to a cascade of adhesion events that ensures efficient and specific cell capture. In direct comparision, the adhesive knobs of the infected erythrocyte, which are less elevated and stiffer than the microvilli, seem to be less efficient in this regard, possibly because the biological function does not require high effectivity or specificity. On the other hand, their large number and complex composition and architecture suggest that the situation is more complex, for example, involving interactions with the endothelium and the immune system that we do not yet fully understand.
While there has recently been substantial progress in our understanding of the cytoadherence of P. falciparum‐infected erythrocytes in flow, there are still open questions and technical and methodological challenges to be met before this important pathological behavior of parasitized erythrocytes can be appreciated in its full complexity. From the comparison conducted in this review, it is evident that the mechanics of infected erythrocytes is crucial to a deeper insight into interactions with the endothelium. However, the mesoscopic shape model of parasitized erythrocytes needs to be improved as current models do not yet consider the effect of the knobs or temporal changes in host erythrocyte shape during the course of intracellular parasite development. In a recent development, Zhang et al. 123 have approached this gap in understanding, by calculating the shear modulus of infected erythrocytes, using a coarse‐grained model where the lipid bilayer and the spectrin network were modeled separately and connected with vertical linkage. The knobs were simulated by circular areas with increased membrane stiffness and more connections with the spectrin network 123. Such modeling advances have to be pushed further in the future as empirical data on the relevant parameters become available.
There is further a gap in our knowledge about the number of PfEMP1 molecules on the host cell surface and how many PfEMP1 molecules are present per knob. It is further not fully understood how they are displayed in knobs and with which factors they interact in these protrusions. Single‐molecule counting in combination with super‐resolution microscopy might help address these questions. First experiments using various super‐resolution microscopic techniques [including stimulated emission microscopy (STED), photo‐activated localization microscopy (PALM), stochastic optical reconstruction microscopy (STORM), and structional illumination microscopy (SIM)] are encouraging 124, 125, 126, promising novel insights into host erythrocyte shape, knob formation, and PfEMP1 assembly and presentation in the near future. Other techniques that might help elucidate the mechanical properties of the host cell membrane include flicker microscopy, which has recently been used to study the effect of endotoxins on uninfected erythrocytes 127. A central aspect here will be the role of passive versus active contributions to the flickering spectrum 114. For example, it might well be that the stiffening observed for infected erythrocytes not only results from the remodeling of the spectrin network but also from changes in the cellular metabolism by the parasite. While we have first information regarding the adhesion strength at the whole cell level, the contribution of single‐molecule bonds is unclear. Here, single‐molecule force measurements using atomic force microscopy are anticipated to resolve this issue, following the lead in other systems 128. Once such results are available, they can be used for adhesive dynamics simulations for cell capture from the flow.
Another pending question pertains to cytoadhesion and flow dynamics of parasitized hemoglobinopathic erythrocytes. As mentioned above carriers of the sickle cell hemoglobins S and C are protected from severe malaria (which explains why these hemoglobin polymorphisms are highly prevalent in malaria‐endemic areas). Carriers are not sterile to infection. In fact, they can have parasitemias as high as those seen in patients with normal hemoglobin. However, they are less likely to develop cerebral malaria and to die from severe complications 102. These hemoglobinopathic erythrocytes, and also fetal and α‐thalassemic erythrocytes (which also protect from severe malaria), possess fewer and abnormally large knobs on their surface than wild‐type erythrocytes, when infected with P. falciparum 99, 100, 102, 129, 130, 131, 132. In addition, the amount of PfEMP1 molecules presented is reduced and aberrantly displayed 99, 100, 130. How sickle cell hemoglobins S and C affect knob formation and cytoadherence is largely unclear as are the ramifications of the aberrant knob morphology and density for the interaction of these parasitized hemoglobinopathic erythrocytes with the microvascular endothelial lining in flow. Do they bind to endothelial cells with sufficient strength to activate the endothelium? If the adhesion strength is insufficient to trigger the life‐threatening downstream inflammatory events, as recently proposed 130, then this might explain the malaria‐protective trait of hemoglobinopathic and fetal erythrocytes.
Understanding cytoadhesion dynamics of leukocytes has substantially benefited from computational modeling and the application of biophysical methods and approaches. In the case of parasitized erythrocytes, this research is still at its infancy. However, progress is anticipated to accelerate given the ability of the malaria community to implement novel techniques and to learn from the experience gained in other fields.
Author contributions
USS and ML outlined the manuscript. GH, AKD, USS and ML wrote the article.
Acknowledgements
This work was in part supported by the Deutsche Forschungsgemeinschaft under the Collaborative Research Center SFB 1129. USS and ML are members of the cluster of excellence CellNetworks. We thank Britta Nyboer for the art work.
Edited by Wilhelm Just
References
- 1. Simon SI and Goldsmith HL (2002) Leukocyte adhesion dynamics in shear flow. Ann Biomed Eng 30, 315–332. [DOI] [PubMed] [Google Scholar]
- 2. Carman CV and Springer TA (2008) Trans‐cellular migration: cell‐cell contacts get intimate. Curr Opin Cell Biol 20, 533–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hebbel RP (1991) Beyond hemoglobin polymerization: the red blood cell membrane and sickle disease pathophysiology. Blood 77, 214–237. [PubMed] [Google Scholar]
- 4. Tembo D and Montgomery J (2010) Var gene expression and human Plasmodium pathogenesis. Future Microbiol 5, 801–815. [DOI] [PubMed] [Google Scholar]
- 5. Sherman IW, Eda S and Winograd E (2003) Cytoadherence and sequestration in Plasmodium falciparum: defining the ties that bind. Microbes Infect 5, 897–909. [DOI] [PubMed] [Google Scholar]
- 6. Ho M, Hickey MJ, Murray AG, Andonegui G and Kubes P (2000) Visualization of Plasmodium falciparum‐endothelium interactions in human microvasculature: mimicry of leukocyte recruitment. J Exp Med 192, 1205–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Marshall BT, Long M, Piper JW, Yago T, McEver RP and Zhu C (2003) Direct observation of catch bonds involving cell‐adhesion molecules. Nature 423, 190–193. [DOI] [PubMed] [Google Scholar]
- 8. Rieger H, Yoshikawa HY, Quadt K, Nielsen MA, Sanchez CP, Salanti A, Tanaka M and Lanzer M (2015) Cytoadhesion of Plasmodium falciparum‐infected erythrocytes to chondroitin‐4‐sulfate is cooperative and shear enhanced. Blood 125, 383–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Smith JD (2014) The role of PfEMP1 adhesion domain classification in Plasmodium falciparum pathogenesis research. Mol Biochem Parasitol 195, 82–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Smith JD, Rowe JA, Higgins MK and Lavstsen T (2013) Malaria's deadly grip: cytoadhesion of Plasmodium falciparum‐infected erythrocytes. Cell Microbiol 15, 1976–1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. World Health Organization (2014) World Malaria Report 2014.
- 12. Miller LH, Baruch DI, Marsh K and Doumbo OK (2002) The pathogenic basis of malaria. Nature 415, 673–679. [DOI] [PubMed] [Google Scholar]
- 13. Viallat A and Abkarian M (2014) Red blood cell: from its mechanics to its motion in shear flow. Int J Lab Hematol 36, 237–243. [DOI] [PubMed] [Google Scholar]
- 14. Storm J and Craig AG (2014) Pathogenesis of cerebral malaria–inflammation and cytoadherence. Front Cell Infect Microbiol 4, 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Viebig NK, Gamain B, Scheidig C, Lepolard C, Przyborski J, Lanzer M, Gysin J and Scherf A (2005) A single member of the Plasmodium falciparum var multigene family determines cytoadhesion to the placental receptor chondroitin sulphate A. EMBO Rep 6, 775–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Goel S and Gowda DC (2011) How specific is Plasmodium falciparum adherence to chondroitin 4‐sulfate? Trends Parasitol 27, 375–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fried M and Duffy PE (1996) Adherence of Plasmodium falciparum to chondroitin sulfate A in the human placenta. Science 272, 1502–1504. [DOI] [PubMed] [Google Scholar]
- 18. Clausen TM, Christoffersen S, Dahlback M, Langkilde AE, Jensen KE, Resende M, Agerbaek MO, Andersen D, Berisha B, Ditlev SB et al (2012) Structural and functional insight into how the Plasmodium falciparum VAR2CSA protein mediates binding to chondroitin sulfate A in placental malaria. J Biol Chem 287, 23332–23345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Srivastava A, Gangnard S, Round A, Dechavanne S, Juillerat A, Raynal B, Faure G, Baron B, Ramboarina S, Singh SK et al (2010) Full‐length extracellular region of the var2CSA variant of PfEMP1 is required for specific, high‐affinity binding to CSA. Proc Natl Acad Sci USA 107, 4884–4889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Niang M, Bei AK, Madnani KG, Pelly S, Dankwa S, Kanjee U, Gunalan K, Amaladoss A, Yeo KP, Bob NS et al (2014) STEVOR is a Plasmodium falciparum erythrocyte binding protein that mediates merozoite invasion and rosetting. Cell Host Microbe 16, 81–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Goel S, Palmkvist M, Moll K, Joannin N, Lara P, Akhouri RR, Moradi N, Ojemalm K, Westman M, Angeletti D et al (2015) RIFINs are adhesins implicated in severe Plasmodium falciparum malaria. Nat Med 21, 314–317. [DOI] [PubMed] [Google Scholar]
- 22. Quadt KA, Barfod L, Andersen D, Bruun J, Gyan B, Hassenkam T, Ofori MF and Hviid L (2012) The density of knobs on Plasmodium falciparum‐infected erythrocytes depends on developmental age and varies among isolates. PLoS One 7, e45658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pologe LG, Pavlovec A, Shio H and Ravetch JV (1987) Primary structure and subcellular localization of the knob‐associated histidine‐rich protein of Plasmodium falciparum . Proc Natl Acad Sci USA 84, 7139–7143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Crabb BS, Cooke BM, Reeder JC, Waller RF, Caruana SR, Davern KM, Wickham ME, Brown GV, Coppel RL and Cowman AF (1997) Targeted gene disruption shows that knobs enable malaria‐infected red cells to cytoadhere under physiological shear stress. Cell 89, 287–296. [DOI] [PubMed] [Google Scholar]
- 25. Weng H, Guo X, Papoin J, Wang J, Coppel R, Mohandas N and An X (2014) Interaction of Plasmodium falciparum knob‐associated histidine‐rich protein (KAHRP) with erythrocyte ankyrin R is required for its attachment to the erythrocyte membrane. Biochim Biophys Acta 1838, 185–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pei X, An X, Guo X, Tarnawski M, Coppel R and Mohandas N (2005) Structural and functional studies of interaction between Plasmodium falciparum knob‐associated histidine‐rich protein (KAHRP) and erythrocyte spectrin. J Biol Chem 280, 31166–31171. [DOI] [PubMed] [Google Scholar]
- 27. Oh SS, Voigt S, Fisher D, Yi SJ, LeRoy PJ, Derick LH, Liu S and Chishti AH (2000) Plasmodium falciparum erythrocyte membrane protein 1 is anchored to the actin‐spectrin junction and knob‐associated histidine‐rich protein in the erythrocyte skeleton. Mol Biochem Parasitol 108, 237–247. [DOI] [PubMed] [Google Scholar]
- 28. Kilejian A, Rashid MA, Aikawa M, Aji T and Yang YF (1991) Selective association of a fragment of the knob protein with spectrin, actin and the red cell membrane. Mol Biochem Parasitol 44, 175–181. [DOI] [PubMed] [Google Scholar]
- 29. Rug M, Prescott SW, Fernandez KM, Cooke BM and Cowman AF (2006) The role of KAHRP domains in knob formation and cytoadherence of P. falciparum‐infected human erythrocytes. Blood 108, 370–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Magowan C, Nunomura W, Waller KL, Yeung J, Liang J, Van Dort H, Low PS, Coppel RL and Mohandas N (2000) Plasmodium falciparum histidine‐rich protein 1 associates with the band 3 binding domain of ankyrin in the infected red cell membrane. Biochim Biophys Acta 1502, 461–470. [DOI] [PubMed] [Google Scholar]
- 31. Ganguly AK, Ranjan P, Kumar A and Bhavesh NS (2015) Dynamic association of PfEMP1 and KAHRP in knobs mediates cytoadherence during Plasmodium invasion. Sci Rep 5, 8617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Waller KL, Cooke BM, Nunomura W, Mohandas N and Coppel RL (1999) Mapping the binding domains involved in the interaction between the Plasmodium falciparum knob‐associated histidine‐rich protein (KAHRP) and the cytoadherence ligand P. falciparum erythrocyte membrane protein 1 (PfEMP1). J Biol Chem 274, 23808–23813. [DOI] [PubMed] [Google Scholar]
- 33. Oberli A, Slater LM, Cutts E, Brand F, Mundwiler‐Pachlatko E, Rusch S, Masik MF, Erat MC, Beck HP and Vakonakis I (2014) A Plasmodium falciparum PHIST protein binds the virulence factor PfEMP1 and comigrates to knobs on the host cell surface. FASEB J 28, 4420–4433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cyrklaff M, Sanchez CP, Kilian N, Bisseye C, Simpore J, Frischknecht F and Lanzer M (2011) Hemoglobins S and C interfere with actin remodeling in Plasmodium falciparum‐infected erythrocytes. Science 334, 1283–1286. [DOI] [PubMed] [Google Scholar]
- 35. Watermeyer JM, Hale VL, Hackett F, Clare DK, Cutts EE, Vakonakis I, Fleck RA, Blackman MJ and Saibil HR (2016) A spiral scaffold underlies cytoadherent knobs in Plasmodium falciparum‐infected erythrocytes. Blood, 127, 343–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Springer TA (1994) Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell 76, 301–314. [DOI] [PubMed] [Google Scholar]
- 37. Luster AD, Alon R and von Andrian UH (2005) Immune cell migration in inflammation: present and future therapeutic targets. Nat Immunol 6, 1182–1190. [DOI] [PubMed] [Google Scholar]
- 38. Sundd P, Pospieszalska MK, Cheung LS, Konstantopoulos K and Ley K (2011) Biomechanics of leukocyte rolling. Biorheology 48, 1–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shao JY, Ting‐Beall HP and Hochmuth RM (1998) Static and dynamic lengths of neutrophil microvilli. Proc Natl Acad Sci USA 95, 6797–6802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Heasman SJ and Ridley AJ (2010) Multiple roles for RhoA during T cell transendothelial migration. Small GTPases 1, 174–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bell GI (1978) Models for the specific adhesion of cells to cells. Science 200, 618–627. [DOI] [PubMed] [Google Scholar]
- 42. Evans E and Ritchie K (1997) Dynamic strength of molecular adhesion bonds. Biophys J 72, 1541–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dembo M, Torney DC, Saxman K and Hammer D (1988) The reaction‐limited kinetics of membrane‐to‐surface adhesion and detachment. Proc R Soc Lond B Biol Sci 234, 55–83. [DOI] [PubMed] [Google Scholar]
- 44. Merkel R, Nassoy P, Leung A, Ritchie K and Evans E (1999) Energy landscapes of receptor‐ligand bonds explored with dynamic force spectroscopy. Nature 397, 50–53. [DOI] [PubMed] [Google Scholar]
- 45. Zhu C, Lou J and McEver RP (2005) Catch bonds: physical models, structural bases, biological function and rheological relevance. Biorheology 42, 443–462. [PubMed] [Google Scholar]
- 46. Chakrabarti S, Hinczewski M and Thirumalai D (2016) Phenomenological and microscopic theories for catch bonds. arXiv:1601.02050 [q‐bio.BM]. [DOI] [PMC free article] [PubMed]
- 47. Thomas WE, Nilsson LM, Forero M, Sokurenko EV and Vogel V (2004) Shear‐dependent ‘stick‐and‐roll’ adhesion of type 1 fimbriated Escherichia coli. Mol Microbiol 53, 1545–1557. [DOI] [PubMed] [Google Scholar]
- 48. Nilsson LM, Thomas WE, Trintchina E, Vogel V and Sokurenko EV (2006) Catch bond‐mediated adhesion without a shear threshold: trimannose versus monomannose interactions with the FimH adhesin of Escherichia coli. J Biol Chem 281, 16656–16663. [DOI] [PubMed] [Google Scholar]
- 49. Finger EB, Puri KD, Alon R, Lawrence MB, von Andrian UH and Springer TA (1996) Adhesion through L‐selectin requires a threshold hydrodynamic shear. Nature 379, 266–269. [DOI] [PubMed] [Google Scholar]
- 50. Yago T, Wu J, Wey CD, Klopocki AG, Zhu C and McEver RP (2004) Catch bonds govern adhesion through L‐selectin at threshold shear. J Cell Biol 166, 913–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Guo B and Guilford WH (2006) Mechanics of actomyosin bonds in different nucleotide states are tuned to muscle contraction. Proc Natl Acad Sci USA 103, 9844–9849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Thomas WE, Trintchina E, Forero M, Vogel V and Sokurenko EV (2002) Bacterial adhesion to target cells enhanced by shear force. Cell 109, 913–923. [DOI] [PubMed] [Google Scholar]
- 53. Kong F, Garcia AJ, Mould AP, Humphries MJ and Zhu C (2009) Demonstration of catch bonds between an integrin and its ligand. J Cell Biol 185, 1275–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pereverzev YV and Prezhdo OV (2006) Force‐induced deformations and stability of biological bonds. Phys Rev E Stat Nonlin Soft Matter Phys 73, 050902. [DOI] [PubMed] [Google Scholar]
- 55. Lou J, Yago T, Klopocki AG, Mehta P, Chen W, Zarnitsyna VI, Bovin NV, Zhu C and McEver RP (2006) Flow‐enhanced adhesion regulated by a selectin interdomain hinge. J Cell Biol 174, 1107–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lou J and Zhu C (2007) A structure‐based sliding‐rebinding mechanism for catch bonds. Biophys J 92, 1471–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yago T, Lou J, Wu T, Yang J, Miner JJ, Coburn L, Lopez JA, Cruz MA, Dong JF, McIntire LV et al (2008) Platelet glycoprotein Ibalpha forms catch bonds with human WT vWF but not with type 2B von Willebrand disease vWF. J Clin Invest 118, 3195–3207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Thomas W, Forero M, Yakovenko O, Nilsson L, Vicini P, Sokurenko E and Vogel V (2006) Catch‐bond model derived from allostery explains force‐activated bacterial adhesion. Biophys J 90, 753–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yakovenko O, Sharma S, Forero M, Tchesnokova V, Aprikian P, Kidd B, Mach A, Vogel V, Sokurenko E and Thomas WE (2008) FimH forms catch bonds that are enhanced by mechanical force due to allosteric regulation. J Biol Chem 283, 11596–11605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Pereverzev YV and Prezhdo OV (2007) Universal laws in the force‐induced unraveling of biological bonds. Phys Rev E Stat Nonlin Soft Matter Phys 75, 011905. [DOI] [PubMed] [Google Scholar]
- 61. Pereverzev YV, Prezhdo OV, Forero M, Sokurenko EV and Thomas WE (2005) The two‐pathway model for the catch‐slip transition in biological adhesion. Biophys J 89, 1446–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Pereverzev YV, Prezhdo OV, Thomas WE and Sokurenko EV (2005) Distinctive features of the biological catch bond in the jump‐ramp force regime predicted by the two‐pathway model. Phys Rev E Stat Nonlin Soft Matter Phys 72, 010903. [DOI] [PubMed] [Google Scholar]
- 63. Prezhdo OV and Pereverzev YV (2009) Theoretical aspects of the biological catch bond. Acc Chem Res 42, 693–703. [DOI] [PubMed] [Google Scholar]
- 64. Lawrence MB and Springer TA (1991) Leukocytes roll on a selectin at physiologic flow rates: distinction from and prerequisite for adhesion through integrins. Cell 65, 859–873. [DOI] [PubMed] [Google Scholar]
- 65. Alon R, Hammer DA and Springer TA (1995) Lifetime of the P‐selectin‐carbohydrate bond and its response to tensile force in hydrodynamic flow. Nature 374, 539–542. [DOI] [PubMed] [Google Scholar]
- 66. Schwarz US and Alon R (2004) L‐selectin‐mediated leukocyte tethering in shear flow is controlled by multiple contacts and cytoskeletal anchorage facilitating fast rebinding events. Proc Natl Acad Sci USA 101, 6940–6945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Caputo KE and Hammer DA (2005) Effect of microvillus deformability on leukocyte adhesion explored using adhesive dynamics simulations. Biophys J 89, 187–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Chang KC, Tees DF and Hammer DA (2000) The state diagram for cell adhesion under flow: leukocyte rolling and firm adhesion. Proc Natl Acad Sci USA 97, 11262–11267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hammer DA and Apte SM (1992) Simulation of cell rolling and adhesion on surfaces in shear flow: general results and analysis of selectin‐mediated neutrophil adhesion. Biophys J 63, 35–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Korn C and Schwarz US (2006) Efficiency of initiating cell adhesion in hydrodynamic flow. Phys Rev Lett 97, 138103. [DOI] [PubMed] [Google Scholar]
- 71. Alon R, Chen S, Puri KD, Finger EB and Springer TA (1997) The kinetics of L‐selectin tethers and the mechanics of selectin‐mediated rolling. J Cell Biol 138, 1169–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Korn CB and Schwarz US (2008) Dynamic states of cells adhering in shear flow: from slipping to rolling. Phys Rev E Stat Nonlin Soft Matter Phys 77, 041904. [DOI] [PubMed] [Google Scholar]
- 73. Ramesh KV, Thaokar R, Prakash JR and Prabhakar R (2015) Significance of thermal fluctuations and hydrodynamic interactions in receptor‐ligand‐mediated adhesive dynamics of a spherical particle in wall‐bound shear flow. Phys Rev E Stat Nonlin Soft Matter Phys 91, 022302. [DOI] [PubMed] [Google Scholar]
- 74. Cheung LS‐L, Zheng X, Wang L, Baygents JC, Guzman R, Schroeder JA, Heimark RL and Zohar Y (2011) Adhesion dynamics of circulating tumor cells under shear flow in a bio‐functionalized microchannel. J Micromech Microeng 21, 054033. [Google Scholar]
- 75. Zheng X, Cheung LS, Schroeder JA, Jiang L and Zohar Y (2011) Cell receptor and surface ligand density effects on dynamic states of adhering circulating tumor cells. Lab Chip 11, 3431–3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Bhatia SK, King MR and Hammer DA (2003) The state diagram for cell adhesion mediated by two receptors. Biophys J 84, 2671–2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Caputo KE, Lee D, King MR and Hammer DA (2007) Adhesive dynamics simulations of the shear threshold effect for leukocytes. Biophys J 92, 787–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Nash GB, Cooke BM, Marsh K, Berendt A, Newbold C and Stuart J (1992) Rheological analysis of the adhesive interactions of red blood cells parasitized by Plasmodium falciparum . Blood 79, 798–807. [PubMed] [Google Scholar]
- 79. Cooke BM, Berendt AR, Craig AG, MacGregor J, Newbold CI and Nash GB (1994) Rolling and stationary cytoadhesion of red blood cells parasitized by Plasmodium falciparum: separate roles for ICAM‐1, CD36 and thrombospondin. Br J Haematol 87, 162–170. [DOI] [PubMed] [Google Scholar]
- 80. Yipp BG, Anand S, Schollaardt T, Patel KD, Looareesuwan S and Ho M (2000) Synergism of multiple adhesion molecules in mediating cytoadherence of Plasmodium falciparum‐infected erythrocytes to microvascular endothelial cells under flow. Blood 96, 2292–2298. [PubMed] [Google Scholar]
- 81. Cooke BM, Rogerson SJ, Brown GV and Coppel RL (1996) Adhesion of malaria‐infected red blood cells to chondroitin sulfate A under flow conditions. Blood 88, 4040–4044. [PubMed] [Google Scholar]
- 82. Li A, Lim TS, Shi H, Yin J, Tan SJ, Li Z, Low BC, Tan KS and Lim CT (2011) Molecular mechanistic insights into the endothelial receptor mediated cytoadherence of Plasmodium falciparum‐infected erythrocytes. PLoS One 6, e16929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Duffy PE (2007) Plasmodium in the placenta: parasites, parity, protection, prevention and possibly preeclampsia. Parasitology 134, 1877–1881. [DOI] [PubMed] [Google Scholar]
- 84. Martin FA, Murphy RP and Cummins PM (2013) Thrombomodulin and the vascular endothelium: insights into functional, regulatory, and therapeutic aspects. Am J Physiol Heart Circ Physiol 304, H1585–H1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Antia M, Herricks T and Rathod PK (2007) Microfluidic modeling of cell‐cell interactions in malaria pathogenesis. PLoS Pathog 3, e99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Avril M, Traore B, Costa FT, Lepolard C and Gysin J (2004) Placenta cryosections for study of the adhesion of Plasmodium falciparum‐infected erythrocytes to chondroitin sulfate A in flow conditions. Microbes Infect 6, 249–255. [DOI] [PubMed] [Google Scholar]
- 87. Davis SP, Amrein M, Gillrie MR, Lee K, Muruve DA and Ho M (2012) Plasmodium falciparum‐induced CD36 clustering rapidly strengthens cytoadherence via p130CAS‐mediated actin cytoskeletal rearrangement. FASEB J 26, 1119–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Davis SP, Lee K, Gillrie MR, Roa L, Amrein M and Ho M (2013) CD36 recruits alpha(5)beta(1) integrin to promote cytoadherence of P. falciparum‐infected erythrocytes. PLoS Pathog 9, e1003590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Xu X, Efremov AK, Li A, Lai L, Dao M, Lim CT and Cao J (2013) Probing the cytoadherence of malaria infected red blood cells under flow. PLoS One 8, e64763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Carvalho PA, Diez‐Silva M, Chen H, Dao M and Suresh S (2013) Cytoadherence of erythrocytes invaded by Plasmodium falciparum: quantitative contact‐probing of a human malaria receptor. Acta Biomater 9, 6349–6359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Mauritz JM, Esposito A, Ginsburg H, Kaminski CF, Tiffert T and Lew VL (2009) The homeostasis of Plasmodium falciparum‐infected red blood cells. PLoS Comput Biol 5, e1000339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Desai SA (2012) Ion and nutrient uptake by malaria parasite‐infected erythrocytes. Cell Microbiol 14, 1003–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Lew VL, Tiffert T and Ginsburg H (2003) Excess hemoglobin digestion and the osmotic stability of Plasmodium falciparum‐infected red blood cells. Blood 101, 4189–4194. [DOI] [PubMed] [Google Scholar]
- 94. Esposito A, Choimet JB, Skepper JN, Mauritz JM, Lew VL, Kaminski CF and Tiffert T (2010) Quantitative imaging of human red blood cells infected with Plasmodium falciparum . Biophys J 99, 953–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Cho S, Kim S, Kim Y and Park Y (2012) Optical imaging techniques for the study of malaria. Trends Biotechnol 30, 71–79. [DOI] [PubMed] [Google Scholar]
- 96. Mauritz JM, Esposito A, Tiffert T, Skepper JN, Warley A, Yoon YZ, Cicuta P, Lew VL, Guck JR and Kaminski CF (2010) Biophotonic techniques for the study of malaria‐infected red blood cells. Med Biol Eng Comput 48, 1055–1063. [DOI] [PubMed] [Google Scholar]
- 97. Subramani R, Quadt K, Jeppesen AE, Hempel C, Petersen JE, Hassenkam T, Hviid L and Barfod L (2015) Plasmodium falciparum‐infected erythrocyte knob density is linked to the PfEMP1 variant expressed. MBio 6, e01456–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Shi H, Liu Z, Li A, Yin J, Chong AG, Tan KS, Zhang Y and Lim CT (2013) Life cycle‐dependent cytoskeletal modifications in Plasmodium falciparum infected erythrocytes. PLoS One 8, e61170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Cholera R, Brittain NJ, Gillrie MR, Lopera‐Mesa TM, Diakite SA, Arie T, Krause MA, Guindo A, Tubman A, Fujioka H et al (2008) Impaired cytoadherence of Plasmodium falciparum‐infected erythrocytes containing sickle hemoglobin. Proc Natl Acad Sci USA 105, 991–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Fairhurst RM, Baruch DI, Brittain NJ, Ostera GR, Wallach JS, Hoang HL, Hayton K, Guindo A, Makobongo MO, Schwartz OM et al (2005) Abnormal display of PfEMP‐1 on erythrocytes carrying haemoglobin C may protect against malaria. Nature 435, 1117–1121. [DOI] [PubMed] [Google Scholar]
- 101. Taylor SM, Cerami C and Fairhurst RM (2013) Hemoglobinopathies: slicing the Gordian knot of Plasmodium falciparum malaria pathogenesis. PLoS Pathog 9, e1003327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Taylor SM, Parobek CM and Fairhurst RM (2012) Haemoglobinopathies and the clinical epidemiology of malaria: a systematic review and meta‐analysis. Lancet Infect Dis 12, 457–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Cyrklaff M, Sanchez CP, Frischknecht F and Lanzer M (2012) Host actin remodeling and protection from malaria by hemoglobinopathies. Trends Parasitol 28, 479–485. [DOI] [PubMed] [Google Scholar]
- 104. Kilian N, Dittmer M, Cyrklaff M, Ouermi D, Bisseye C, Simpore J, Frischknecht F, Sanchez CP and Lanzer M (2013) Haemoglobin S and C affect the motion of Maurer's clefts inPlasmodium falciparum‐infected erythrocytes. Cell Microbiol 15, 1111–1126. [DOI] [PubMed] [Google Scholar]
- 105. Mauritz JM, Tiffert T, Seear R, Lautenschlager F, Esposito A, Lew VL, Guck J and Kaminski CF (2010) Detection of Plasmodium falciparum‐infected red blood cells by optical stretching. J Biomed Opt 15, 030517. [DOI] [PubMed] [Google Scholar]
- 106. Boey SK, Boal DH and Discher DE (1998) Simulations of the erythrocyte cytoskeleton at large deformation. I. Microscopic models. Biophys J 75, 1573–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Discher DE, Boal DH and Boey SK (1998) Simulations of the erythrocyte cytoskeleton at large deformation. II. Micropipette aspiration. Biophys J 75, 1584–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Fedosov DA, Caswell B and Karniadakis GE (2010) A multiscale red blood cell model with accurate mechanics, rheology, and dynamics. Biophys J 98, 2215–2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Li J, Dao M, Lim CT and Suresh S (2005) Spectrin‐level modeling of the cytoskeleton and optical tweezers stretching of the erythrocyte. Biophys J 88, 3707–3719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Pivkin IV and Karniadakis GE (2008) Accurate coarse‐grained modeling of red blood cells. Phys Rev Lett 101, 118105. [DOI] [PubMed] [Google Scholar]
- 111. Marko JF and Siggia ED (1995) Stretching DNA. Macromolecules 28, 8759–8770. [Google Scholar]
- 112. Gompper G and Kroll DM (1997) Network models of fluid, hexatic and polymerized membranes. J Phys Condens Matter 9, 8795–8834. [Google Scholar]
- 113. Noguchi H and Gompper G (2005) Shape transitions of fluid vesicles and red blood cells in capillary flows. Proc Natl Acad Sci USA 102, 14159–14164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Turlier H, Fedosov DA, Audoly B, Auth T, Gov NS, Sykes C, Joanny J‐F, Gompper G and Betz T (2016) Equilibrium physics breakdown reveals the active nature of red blood cell flickering. Nat Phys, doi:10.1038/nphys3621. [Google Scholar]
- 115. Malevanets A and Kapral R (1999) Mesoscopic model for solvent dynamics. J Chem Phys 110, 8605–8613. [Google Scholar]
- 116. Hoogerbrugge PJ and Koelman JMVA (1992) Simulating microscopic hydrodynamic phenomena with dissipative particle dynamics. Europhys Lett 19, 155–190. [Google Scholar]
- 117. Kikuchi N, Pooley CM, Ryder JF and Yeomans JM (2003) Transport coefficients of a mesoscopic fluid dynamics model. J Chem Phys 119, 6388–6395. [Google Scholar]
- 118. Ripoll M, Mussawisade K, Winkler RG and Gompper G (2004) Low‐Reynolds‐number hydrodynamics of complex fluids by multi‐particle‐collision dynamics. Europhys Lett 68, 106–112. [Google Scholar]
- 119. Fedosov DA, Caswell B, Suresh S and Karniadakis GE (2011) Quantifying the biophysical characteristics of Plasmodium falciparum‐parasitized red blood cells in microcirculation. Proc Natl Acad Sci USA 108, 35–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Fedosov DA, Caswell B and Karniadakis GE (2011) Wall shear stress‐based model for adhesive dynamics of red blood cells in malaria. Biophys J 100, 2084–2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Dao M, Lim CT and Suresh S (2003) Mechanics of the human red blood cell deformed by optical tweezers. J Mech Phys Solids 51, 2259–2280. [Google Scholar]
- 122. Hosseini SM and Feng JJ (2012) How malaria parasites reduce the deformability of infected red blood cells. Biophys J 103, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Zhang Y, Huang C, Kim S, Golkaram M, Dixon MW, Tilley L, Li J, Zhang S and Suresh S (2015) Multiple stiffening effects of nanoscale knobs on human red blood cells infected with Plasmodium falciparum malaria parasite. Proc Natl Acad Sci USA 112, 6068–6073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Hans N, Relan U, Dubey N, Gaur D and Chauhan VS (2015) Identification and localization of a Novel Invasin of Plasmodium falciparum . Mol Biochem Parasitol 202, 38–43. [DOI] [PubMed] [Google Scholar]
- 125. Hliscs M, Millet C, Dixon MW, Siden‐Kiamos I, McMillan P and Tilley L (2015) Organization and function of an actin cytoskeleton in Plasmodium falciparum gametocytes. Cell Microbiol 17, 207–225. [DOI] [PubMed] [Google Scholar]
- 126. Riglar DT, Richard D, Wilson DW, Boyle MJ, Dekiwadia C, Turnbull L, Angrisano F, Marapana DS, Rogers KL, Whitchurch CB et al (2011) Super‐resolution dissection of coordinated events during malaria parasite invasion of the human erythrocyte. Cell Host Microbe 9, 9–20. [DOI] [PubMed] [Google Scholar]
- 127. Ito H, Kuss N, Rapp BE, Ichikawa M, Gutsmann T, Brandenburg K, Poschl JM and Tanaka M (2015) Quantification of the iInfluence of endotoxins on the mechanics of adult and neonatal red blood cells. J Phys Chem B 119, 7837–7845. [DOI] [PubMed] [Google Scholar]
- 128. Wijeratne SS, Martinez JR, Grindel BJ, Frey EW, Li J, Wang L, Farach‐Carson MC and Kiang CH (2016) Single molecule force measurements of perlecan/HSPG2: a key component of the osteocyte pericellular matrix. Matrix Biol 50, 27–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Amaratunga C, Lopera‐Mesa TM, Brittain NJ, Cholera R, Arie T, Fujioka H, Keefer JR and Fairhurst RM (2011) A role for fetal hemoglobin and maternal immune IgG in infant resistance to Plasmodium falciparum malaria. PLoS One 6, e14798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Fairhurst RM, Bess CD and Krause MA (2012) Abnormal PfEMP1/knob display on Plasmodium falciparum‐infected erythrocytes containing hemoglobin variants: fresh insights into malaria pathogenesis and protection. Microbes Infect 14, 851–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Fairhurst RM, Fujioka H, Hayton K, Collins KF and Wellems TE (2003) Aberrant development of Plasmodium falciparum in hemoglobin CC red cells: implications for the malaria protective effect of the homozygous state. Blood 101, 3309–3315. [DOI] [PubMed] [Google Scholar]
- 132. Krause MA, Diakite SA, Lopera‐Mesa TM, Amaratunga C, Arie T, Traore K, Doumbia S, Konate D, Keefer JR, Diakite M et al (2012) alpha‐Thalassemia Impairs the cytoadherence of Plasmodium falciparum‐infected erythrocytes. PLoS One 7, e37214. [DOI] [PMC free article] [PubMed] [Google Scholar]