

Abstract
Intra‐tumor heterogeneity is a vivid problem of molecular oncology that could be addressed by imaging mass spectrometry. Here we aimed to assess molecular heterogeneity of oral squamous cell carcinoma and to detect signatures discriminating normal and cancerous epithelium. Tryptic peptides were analyzed by MALDI‐IMS in tissue specimens from five patients with oral cancer. Novel algorithm of IMS data analysis was developed and implemented, which included Gaussian mixture modeling for detection of spectral components and iterative k‐means algorithm for unsupervised spectra clustering performed in domain reduced to a subset of the most dispersed components. About 4% of the detected peptides showed significantly different abundances between normal epithelium and tumor, and could be considered as a molecular signature of oral cancer. Moreover, unsupervised clustering revealed two major sub‐regions within expert‐defined tumor areas. One of them showed molecular similarity with histologically normal epithelium. The other one showed similarity with connective tissue, yet was markedly different from normal epithelium. Pathologist's re‐inspection of tissue specimens confirmed distinct features in both tumor sub‐regions: foci of actual cancer cells or cancer microenvironment‐related cells prevailed in corresponding areas. Hence, molecular differences detected during automated segmentation of IMS data had an apparent reflection in real structures present in tumor.
Keywords: Data clustering, Gaussian mixture model, Head and neck cancer, Imaging mass spectrometry, Technology, Unsupervised analysis
Abbreviations
- GMM
Gaussian mixture model
- HNC
head and neck cancer
- MALDI‐IMS
matrix‐assisted laser desorption/ionization imaging mass spectrometry
1. Introduction
Imaging mass spectrometry (IMS) is a powerful approach allowing unique combination of molecular and morphological information. Mass profiles of different molecular species (proteins, lipids, metabolites, etc.) revealed by IMS can be spatially resolved and annotated with morphological and histological structures which makes this method complementary and superior to classical pathology 1, 2, 3, 4, 5. The idea of IMS based on MALDI (MALDI‐IMS) was introduced about two decades ago and since then this approach has been applied to visualize distribution of proteins and other molecules in different types of tissues. Among many applications of IMS there is molecular characterization of different types of cancer, including lung 6, breast 7, prostate 8, gastric 9, larynx 10 cancers and brain tumors 11. The particular advantage of IMS in cancer research is allocation of molecular profiles to specific cell types, such as cancerous, preneoplastic or inflammatory 12, 13, 14. Moreover, IMS can be used in studies aimed at interfacing tumor and normal tissue (tumor niche) and intra‐tumor heterogeneity 10, 14, 15, 16, 17, 18. It is noteworthy that automated (unsupervised) methods of clustering of IMS data, particularly based on component analysis and spatial segmentation, appeared to be a particularly suitable approach in studies of intra‐tumor heterogeneity and classification of tumor sub‐regions 19. Hence, IMS proved its role as a powerful tool in clinical proteomics, with obvious applicability in biomarker research and molecular tissue classification. This approach revealed its exceptional value in studies of complex heterogeneous systems exemplified by many tumors.
Significance of the study
Due to high dimensionality of IMS data, proper information processing is crucial for knowledge discovery. The majority of existing algorithms focus on image segmentation performed in PCA component domain. The algorithm developed by us transforms a spectrum into a mixture of Gaussian components and performs step‐down k‐means clustering in a domain reduced to the subset of the most dispersed components. The implemented algorithm allowed to detect molecular sub‐regions of oral squamous cell carcinoma which reflected real structures present in a cancerous tissue.
Cancer located in head and neck region (HNC) is the sixth most common cancer worldwide accounting for over 550 000 new cases and about 300 000 deaths per annum. The vast majority of HNC (>95%) are squamous cell carcinomas located in the upper‐aerodigestive tract (including the mouth, pharynx and larynx), and are derived from stratified squamous epithelium lining mucosa of a target organ. HNC has various etiologic factors (including tobacco smoking with alcohol consumption and HPV infection), heterogeneous pathologic and clinical features, and diverse outcome. Despite recent improvements in treatment, HNC prognosis remains rather poor, with less than 40–50% of patients staying alive after 5 years. Therapeutic decisions are solely based on tumor localization and traditional staging, yet HNC is a heterogeneous disease and cases with similar pathologic features can differ in clinical outcome 20, 21, 22. Moreover, since surgery is the primary treatment in most HNC cases, uncompleted resection of a primary tumor can be a reason for local recurrence and treatment failure. Adequacy of surgical resection of a tumor is conventionally determined by classical histopathological examination which can miss out sub‐microscopic and/or pre‐cancerous spots, therefore determination of cancer‐specific molecular factor(s) for proper delineation of tumor area remains a vivid issue in this malignancy 23. Studies on molecular profiling of HNC, mainly based on gene mutations and expression profiles, have been conducted worldwide in recent years 24, 25, 26. However, HNC remains a relatively under‐researched cancer ‐ there is a lack of robust molecular biomarkers to guide HNC patient management and the majority of questions related to potential “molecular subtypes” of this malignancy remain unanswered yet.
Here we implemented IMS approach to characterize intra‐tumor heterogeneity of HNC, and to identify molecular components discriminating normal and cancerous epithelium. Proteins were imaged using MALDI‐IMS in material resected from five patients with oral cancer. A novel method of spectra processing and unsupervised clustering was used to identify regions corresponding to normal oral mucosa and different sub‐regions of cancer, and then components critical for segmentation of tissue regions were detected.
2. Materials and methods
2.1. Clinical material
Tissue material was collected from five patients (36–59 years old; four males) who underwent surgery due to oral cavity squamous cell carcinoma. Tumor was located in tongue (four patients) and in floor of the mouth (one patient): Preparation_1 – cancer stage T4N2M0, Preparation_2 – stage T4N2bM0, Preparation_3 – stage T1N0M0, Preparation_4 – stage T2N0M0, Preparation_5 – stage T2N0M0. Surgery was the primary treatment in all cases (no pre‐surgery chemo‐ or radiotherapy was involved). Tissue specimens containing tumor and surrounding tissues were evaluated by an experienced pathologist in fresh post‐operative material, then immediately frozen and stored at –80°C. Each sample was cut into 10 μm sections using a cryostat, then H&E stained and analyzed by a pathologist; both sections used for IMS and the corresponding fresh serial sections were analyzed. The study was approved by the appropriate Bioethical Committee, and performed in accordance with national and institutional guidelines.
2.2. Preparation of samples for IMS
Frozen 10‐μm thick tissue sections were placed onto indium tin oxide‐coated conductive slides (Bruker Daltonik, Bremen), dried under vacuum for 40 min, then washed twice in 70% ethanol and once in 100% ethanol (1 min each), followed by 1 h drying. Subsequently samples were coated with a solution of trypsin (Promega, 20 μg in 200 μL of 50 mM NH4HCO3) using an automatic spraying device (ImagePrep, Bruker Daltonik), and then incubated in a humid chamber for 18 h at 37°C. Next, methanolic solution of 2,5‐dihydroxybenzoic acid (50% methanol, 30 mg/mL DHB, 0.2% TFA) was deposited onto the surface of tissues with the use of ImagePrep device (using Bruker's standard matrix coating program with doubled phase 5); optical images were registered before matrix deposition.
2.3. MALDI analysis
Tissue sections were subjected to peptide imaging with the use of a MALDI‐TOF ultrafleXtreme mass spectrometer (Bruker Daltonik) equipped with a smartbeam II™ laser operating at 1 kHz repetition rate. Ions were accelerated at 25 kV with PIE time of 100 ns. Spectra were acquired in positive reflectron mode within 800–4000 m/z and externally calibrated with Bruker's Peptide Calibration Standard II. A raster width of 100 μm was applied, 400 spectra were collected from each ablation point. Compass 1.4 for FLEX series (Bruker Daltonik) was employed for spectra acquisition, processing and creation of primary images. After analysis slides were rinsed twice with 100% ethanol to remove the matrix, stained with H&E, and scanned for co‐registration with the MALDI images using flexImaging 4.1 software (Bruker Daltonik). Original spectra were converted into .txt files using flexAnalysis 3.4 software (Bruker Daltonik) for further analyses. The obtained dataset consisted of 45 738 raw spectra with 109 568 mass channels.
2.4. Spectra processing and identification of spectral components
Data processing was performed using MATLAB‐based tools (MathWorks, Natick, USA); a complete library of MATLAB commands together with an exemplary dataset was published at our webpage: http://zaed.aei.polsl.pl/index.php/pl/oprogramowanie‐zaed. Standard preprocessing steps were applied to average spectra: spectrum resampling (to unify mass channels across a dataset), baseline removal (msbackadj() procedure), TIC normalization, and Fast Fourier Transform‐based spectral alignment 27. The Gaussian mixture model (GMM) approach 28 was used for spectra modeling and peak detection. To ensure independence of resuls validation for Preparations_2‐5, the average spectrum for Preparation_1 was used for model construction. Peptide abundance was estimated by pairwise convolution of the GMM components and individual spectra, followed by calculating the area below the obtained curve. Neighboring peaks resulting from right‐skewness of spectral peaks were identified and merged by summing their estimated abundance. Location of the dominant component was set as m/z value of a peptide ion; the resulting dataset featuring 3714 components (45 738 spectra) was used for further analyses.
2.5. Unsupervised clustering
Looking at complex composition of a tissue specimen, one can imagine that only a small subset of hundreds of measured molecular species might be specific for the observed sub‐regions. The signal obtained from these species is overpowered by the remaining less informative ones and standard clustering approaches may not give satisfactory results. Furthermore, heterogeneity of tissue sub‐regions can be hidden behind predominant main tissue structure. Hence, we have developed a novel iterative k‐means algorithm, with feature domain optimization at every step of clustering. A flowchart of the proposed algorithm of spectra processing and clustering is presented schematically in Fig. 1. The elements of the procedure are: (i) step‐down recursive sub‐region splitting; (ii) independent unsupervised feature selection during every sub‐region splitting; (iii) k‐means initial condition setting based on the maximum distance criterion.
Figure 1.
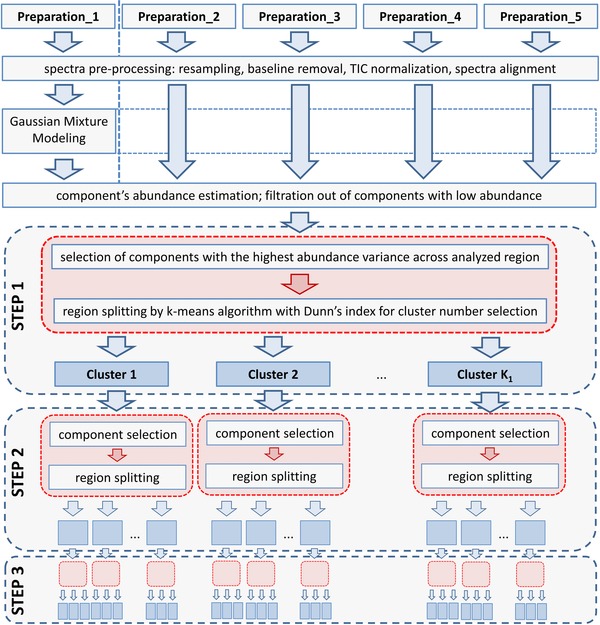
Flowchart of the proposed algorithm of IMS data analysis.
The recursive nature of the developed algorithm allows sub‐region detection in spite of the driving character of the main tissue structures. After the first sample split, the k‐means algorithm is applied independently to each sub‐region obtained in the antecedent split. The splitting is then continued until a specified number of recursions is reached. After having tested several distance metrics, Pearson's correlation coefficient was chosen due to its best performance in capturing spectral similarity. The number of clusters at each splitting was not predefined, k‐means clustering was performed for two to ten clusters and Dunn index was used for selection of the optimal number of clusters 29, 30.
At the beginning of the segmentation process, components with relatively low abundance were filtered out; the data‐driven abundance threshold was found through modeling abundance distribution as a sum of Gaussian‐shaped functions, with the smallest mean component treated as noise‐related 31. This reduced the number of components to 3671. During each sub‐region splitting step, independent of the recursion step, the most informative features (i.e. the ones with the highest variance within an individual sub‐region of interest) were selected out of the set of 3671. In uninformative peak filtering procedure, the Gaussian mixture component with the highest mean value (top right) was chosen from the model of signal variance distribution and variance threshold was calculated.
Since the final result of k‐means partitioning strongly depends on the initial configuration, we developed a novel procedure for setting highly effective initial partitions. Rigorous numerical evaluation (data not included) demonstrated its predominance over standard approaches. The procedure does not require repetitions to protect against hitting local optima. First, a linear regression model is built using the most locally informative features for a given subset of spectra from a sub‐region. The most distant data point, defined as a spectrum with the highest residuum, is chosen as the initial center of the first cluster. The remaining K‐1 initial centers are chosen sequentially in such a way that the minimal distance from the new center to all of the centers found so far has to be maximal.
2.6. Statistical analyses
The permutation ANOVA‐type test with the Games‐Howell post hoc testing was applied to identify molecular signatures of specific sub‐regions. The effect size was estimated by Cohen's d statistics. For comparison of expert‐defined regions, henceforth referred to as a supervised analysis, the testing was performed for every preparation independently. In order to be a part of this analysis the components had to be classified as differentially expressed among at least four out of five preparations whilst maintaining a clear regulatory trend (i.e., always significantly upregulated or always significantly downregulated). For comparison of superclusters detected with the use of our novel algorithm, the statistical analysis was performed for all preparations together. Components were assigned as differentially expressed if: (i) the ANOVA p‐value was less than the significance threshold, Bonferroni corrected for multiple testing, (ii) the p‐value from the Games‐Howell test was less than the significance level with Bonferroni correction, and (iii) the effect size was bigger than 0.5 (for supervised analysis) or 0.8 (for unsupervised analysis).
3. Results and discussion
In each of the analyzed samples, specific regions corresponding to different types of tissue were identified by an experienced pathologist based on histological features of H&E stained sections (Fig. 2A). Each specimen included tumor, normal epithelium, muscles and connective tissue (moreover, Preparation_1 contained fragments of a salivary gland); expert‐defined areas corresponding to tumor and normal epithelium (delineated with red and blue lines in Fig. 2A, respectively) were used as a reference in further analyses. During the supervised analysis peptide components with significantly different abundance between the regions of normal epithelium and tumor were detected (Supporting Information Table S1). There were 108 peptides significantly upregulated and 26 peptides significantly downregulated in tumor area compared to histologically normal epithelium. It is noteworthy that when all peptide components detected during IMS were hypothetically annotated as tryptic peptides identified by LC‐MS/MS in the same tissue preparations, GO terms related to negative regulation of apoptosis, cell motility and protein folding were associated with proteins whose fragments were putatively upregulated in tumor area, while GO terms related to canonical glucose metabolism were associated with the downregulated ones (data not shown in this work). Peptides upregulated in tumor are exemplified by the component 2172.08 m/z (putatively a fragment of pyruvate kinase, an enzyme involved in the Warburg effect), whose distribution is shown in Fig. 2B.
Figure 2.
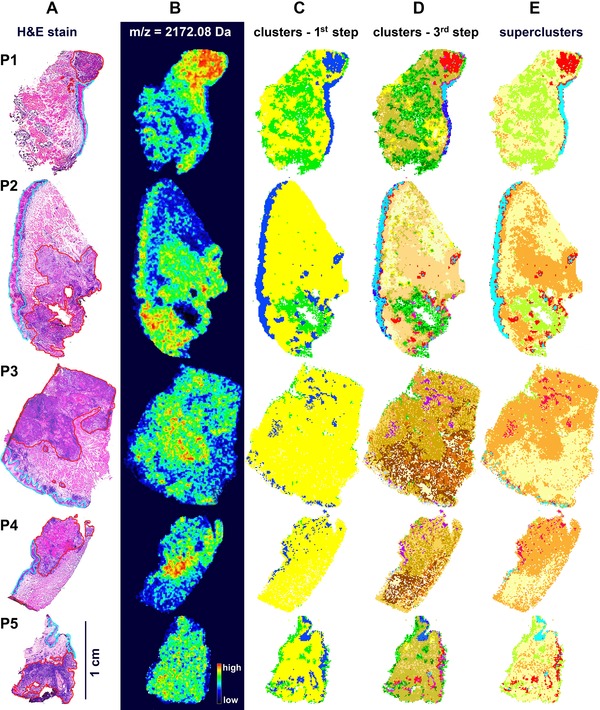
MALDI‐IMS analysis of oral squamous cell cancer. A – H&E stained tissue preparations; areas corresponding to normal epithelium and tumor were marked with blue and red lines, respectively. B – Distribution of an exemplary component (2172.08 m/z) relatively upregulated in tumor. C – Representation of regions corresponding to clusters A, B and C (navy blue, yellow and green, respectively) detected in the first step of segmentation. D – Representation of regions corresponding to clusters detected in the third step of segmentation. E – Illustration of “superclusters” corresponding to normal epithelium (Normal A, blue) and tumor (Tumor A, red, and Tumor B, orange) regions.
Our original algorithm of unsupervised clustering was implemented to define segmentation maps of tissue regions basing on similarity of their molecular profiles. In contrast to many other unsupervised approaches, the clustering procedure proposed in this work took into consideration only selected components that showed the highest variance in the analyzed area (therefore, the results of clustering were not confounded by components with low discrimination potential). Moreover, pixels corresponding to all five tissue preparations (i.e., more than 45 000 spectra) were subjected to segmentation simultaneously, hence the identified clusters could be used directly to describe similarities among analyzed specimens. As a result, only a minority of the identified components (usually 20–30%) were used for cluster discrimination; 1697 components (46%) showed generally low variance and were not used in any step of clustering, while 492 components (13%) showed high variance and were used in all steps of clustering (for details see Supporting Information Table S2). At the first step of segmentation four major clusters were detected. All analyzed tissues consisted of three clusters, namely A, B and C, while the fourth one (cluster 0) corresponded to “empty” areas adjacent to the actual tissue and was excluded from further analysis; tissue regions corresponding to Cluster A, B and C are depicted in Fig. 2C (importantly, these three major clusters were detected in all five tissue preparations). We performed two additional steps of segmentation to discover heterogeneous structure of the primarily detected clusters (Fig. 3). Clusters generated during the third step are depicted in Fig. 2D; further steps of segmentation were not presented and analyzed because the resulting clusters consisted of rather few spectra in most cases. Contribution of each cluster to expert‐defined region (i.e., what percentage of a region was filled by a cluster) and the overlap (coverage) between clusters and regions (i.e., what percentage of a cluster was enclosed in a region) was assessed to allow unbiased detection of clusters corresponding to normal epithelium and cancerous tissue (Supporting Information Table S3). The majority of tissue areas defined as normal epithelium were found primarily in Cluster A: almost 50% of Cluster A overlapped with expert‐defined epithelium, and almost 70% of this region consisted of Cluster A. In marked contrast, expert‐defined tumor region was much more heterogeneous and substantially overlapped with all three major clusters. Twenty‐three clusters were identified in the third step of segmentation (Fig. 3): 01–10, 11–19 and 20–23 within Cluster A, B and C, respectively (cluster 23 corresponded to minor “gaps” inside tissue detected mostly in Preparation_2). We found five clusters (namely 01, 02, 03, 04 and 05) revealing high overlap with normal epithelium (more than 75%), all of them within Cluster A. These five clusters contributed to about 55% of normal epithelium (in the complete dataset) and were merged as “supercluster” Normal A. Moreover, we found eight clusters showing high overlap with tumor (more than 50% coverage). Four clusters (namely 07, 08, 09 and 10) within Cluster A, and four clusters (namely 12, 13, 14 and 15) within Cluster B were merged as superclusters Tumor A and Tumor B, respectively (Fig. 3). These superclusters contributed to about 10% and 60% of tumor area, respectively (in the complete dataset). Hence, when the selected clusters were merged basing on their high overlap with expert‐defined tumor and normal epithelium, five areas (superclusters) were established: Normal A (corresponding to normal epithelium), Tumor A, Tumor B, Normal B (the remaining clusters within Cluster B showing lower coverage with tumor) and Cluster C (without cluster 23); these five superclusters were represented in each tissue preparation (Fig. 2E). It is noteworthy that the results of primary segmentation indicated molecular similarity between areas corresponding to histologically normal epithelium and specific sub‐region of tumor (Tumor A), and apparently discriminated between two major tumor sub‐regions (A and B). We also found differences between tumor samples when relative contribution of superclusters Tumor A and Tumor B was analyzed: significantly higher proportion of Tumor A was detected in Preparations 1 and 2 representing an advanced disease (T4N2), yet this interesting observation would need further validation. We concluded that unsupervised segmentation of tissue helped identify distinct sub‐regions of tumor characterized by different molecular profiles. Pathologist's re‐inspection of the tissue corresponding to supercluster Tumor A revealed substantial presence of foci of squamous cell carcinoma, i.e., transformed cells derived from normal epithelium. The other tumor sub‐region, corresponding to Tumor B, showed molecular similarity with connective tissues present in Cluster B, yet was markedly different from epithelial cells. Re‐analysis of the corresponding tissue by a pathologist revealed substantial contribution of inflammation‐related cells and other features putatively related to cancer microenvironment. Hence, molecular differences detected during automated segmentation of IMS data had an apparent reflection in functional structures present in cancer area.
Figure 3.
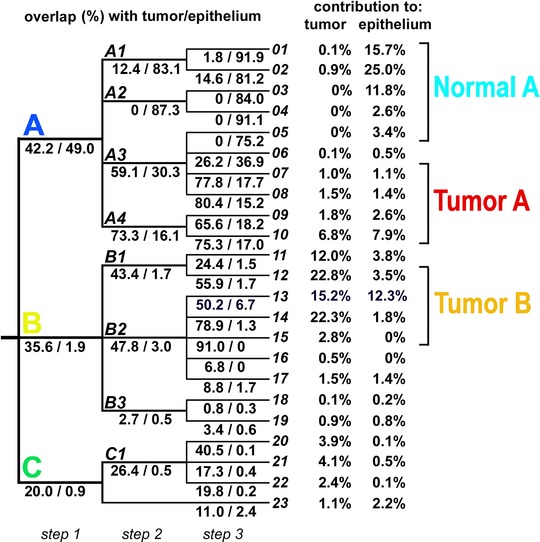
Description of clusters detected in three steps of concomitant unsupervised clustering of all tissue preparations. Shown is an overlap between each cluster and regions corresponding to tumor and normal epithelium. Contribution of each cluster detected in the third step of segmentation (clusters 01 to 23) to expert‐defined areas is presented on the right.
In the next step we searched for molecular components with significantly different abundances between superclusters identified above (Table 1 and Supporting Information Table S4). We found that differences between superclusters Normal A and Tumor B were the most frequent (993 differentiating components), while relative similarity between superclusters Normal A and Tumor A was observed (240 differentiating components). Moreover, only 149 components (4%) showed significantly different abundance between Normal A and both Tumor A and Tumor B. On the other hand, there were 198 components differentiating Tumor B from Tumor A, but only one third of them were specific for Tumor A (the others similarly differentiated Tumor B from both Tumor A and Normal A). Furthermore, high degree of similarity was observed between Tumor B and Normal B, which possibly reflected some overlap of clusters forming Normal B and tumor area. We observed that unsupervised segmentation of tissue apparently facilitated detection of components characteristic for cancer, since several species differentiating Normal A from Tumor A and/or Tumor B (Supporting Information Table S4) were not revealed in primary supervised comparison between normal epithelium and tumor (Supporting Information Table S1). Moreover, clustering of spectra helped reveal quantitative features of specific cancer‐related species, which could be exemplified by component 2172.08 m/z (putatively fragment of pyruvate kinase). Upregulation of this peptide in tumor region of each tissue preparation, as well as in corresponding superclusters is shown in Fig. 4A and B, respectively. Abundance of this component was similar among major clusters A, B and C, yet segmentation of Cluster A revealed differences between clusters corresponding to normal epithelium (clusters A1 and A2) and “epithelium‐like” tumor (cluster A4). Moreover, further segmentation of ambiguous cluster A3 allowed detection of differences between areas considered as tumor (cluster 07 and 08) and normal epithelium (cluster 05) (Fig. 4C). We concluded that unsupervised segmentation of sample could facilitate detection of “cancer markers” differentiating between tumor and normal epithelium, which might be complementary to supervised comparison between expert‐defined tissue regions.
Table 1.
Number of components with abundances significantly different between superclusters corresponding to expert‐defined tissue regions
| 1st | 2nd | All changes | Upregulated (1st) | Downregulated (1st) |
|---|---|---|---|---|
| Normal A | Tumor A | 240 | 135 | 105 |
| Normal A | Tumor B | 993 | 350 | 643 |
| Tumor A | Tumor B | 198 | 198 | 0 |
| Normal A | Both Tumor A and Tumor B | 149 | 122 | 27 |
| Tumor B | Both Tumor A and Normal A | 136 | 0 | 136 |
| Normal A | Normal B | 323 | 296 | 27 |
| Tumor A | Normal B | 198 | 198 | 0 |
| Tumor B | Normal B | 1 | 1 | 0 |
Figure 4.
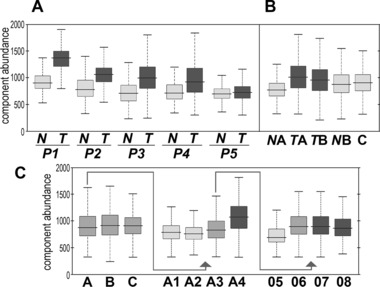
Abundance of a spectral component 2172.08 m/z characteristic for cancer. A – Comparison of areas corresponding to normal epithelium (N) and tumor (T) regions defined in each tissue preparation. B – Comparison of superclusters Normal A (NA), Tumor A (TA), Tumor B (TB), Normal B (NB) and cluster C (C). C – Comparison of the major cluster detected in the first step of segmentation (A, B and C), and selected clusters detected in the second (A1, A2, A3 and A4) and the third (05, 06, 07 and 08) step of segmentation.
It was already documented in several works that unsupervised segmentation (or clusterization) of IMS data enabled classification of complex human tissues and opened new ways for in situ identification of cancer‐related biomarkers. Combination of PCA and hierarchical clustering allowed separation of gastric cancer foci from non‐malignant gastric mucosa 9. Six different methods of unsupervised analysis were tested in dataset generated by MALDI‐IMS for myxofibrosarcoma samples and all of them allowed identification of intra‐tumor heterogeneity showing relatively good concordance 32. Moreover, semi‐supervised segmentation of MALDI‐IMS data based on spatial k‐means clustering on PCA component heat maps allowed to reveal distinct sub‐regions of laryngeal cancer that could be annotated to different stages of tissue dysplasia and neoplasia 10. More recently, unsupervised segmentation was performed for HNC lipidome imaging by MALDI FT‐ICR IMS 33. The authors used the SCiLS Lab pipeline for OMP based peak picking and bisecting k‐means segmentation of spectra processed by spatial denoising. However, in contrast to their previous works, peak detection was done on the mean spectrum instead of each spectrum individually. Our approach uses information preserving GMM‐based dimension reduction technique, where a full model of a mean spectrum is constructed and data‐driven amplitude threshold is estimated for noise level detection. Fully automated filtration of uninformative components done individually per every cluster allows for identification of hidden structure in tissue samples. The obtained significant reduction of the data dimension enables simultaneous analysis of many samples. Complete interpretation of several tissue samples does not require comparison of various PCA component or mass channel images. Thanks to the developed step‐down spectra clusterization, uniquely performed in a variable data‐driven component domain, done together with unsupervised choice of the number of clusters, molecular complexity of the analyzed tissue specimens could be revealed.
4. Concluding remarks
The novel idea of performing iterative k‐means clustering in the Gaussian mixture model‐transformed mass spectrum domain, combined with adaptive feature selection and data‐driven tuning of the total cluster count, allowed for stepwise discovery of tissue segments exhibiting molecular similarity across specimens. Two sub‐regions of cancerous tissues demonstrating different molecular signatures were discovered within expert‐defined tumor areas across five independent specimens. Our study proved that application of advanced data mining algorithms and artificial intelligence techniques was crucial for discovery of knowledge based on IMS data.
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Table S1. Difference in abundance of spectral components between expert‐defined tissue regions corresponding to normal epithelium and tumor
Acknowledgments
This work was supported by the NCN Grant 2011/03/D/NZ4/03507. Calculations were carried out using the GeCONiI infrastructure funded by POIG.02.03.01‐24‐099/13 grant.
Colour Online: See the article online to view Figs. 1 and 3 in colour.
5 References
- 1. Caldwell, R. L. , Caprioli, R. M. , Tissue profiling by mass spectrometry: a review of methodology and applications. Mol. Cell. Proteomics 2005, 4, 394–401. [DOI] [PubMed] [Google Scholar]
- 2. Cornett, D. S. , Reyzer, M. L. , Chaurand, P. , Caprioli, R. M. , MALDI imaging mass spectrometry: molecular snapshots of biochemical systems. Nat. Methods 2007, 4, 828–833. [DOI] [PubMed] [Google Scholar]
- 3. McDonnell, L. A. , Heeren, R. M. A. , Imaging mass spectrometry. Mass Spectrom. Rev. 2007, 26, 606–643. [DOI] [PubMed] [Google Scholar]
- 4. Seeley, E. H. , Caprioli, R. M. , MALDI imaging mass spectrometry of human tissue: method challenges and clinical perspectives. Trends Biotechnol. 2011, 29, 136–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Aichler, M. , Walch, A. , MALDI Imaging mass spectrometry: current frontiers and perspectives in pathology research and practice. Lab. Invest. 2015, 95, 422–431. [DOI] [PubMed] [Google Scholar]
- 6. Yanagisawa, K. , Shyr, Y. , Xu, B. J. , Massion, P. P. et al., Proteomic patterns of tumour subsets in non‐small‐cell lung cancer. Lancet 2003, 362, 433–439. [DOI] [PubMed] [Google Scholar]
- 7. Cornett, D. S. , Mobley, J. A. , Dias, E. C. , Andersson, M. et al., A novel histology‐directed strategy for MALDI‐MS tissue profiling that improves throughput and cellular specificity in human breast cancer. Mol. Cell. Proteomics 2006, 5, 1975–1983. [DOI] [PubMed] [Google Scholar]
- 8. Schwamborn, K. , Krieg, R. C. , Reska, M. , Jakse, G. et al., Identifying prostate carcinoma by MALDI‐Imaging. Int. J. Mol. Med. 2007, 20, 155–159. [PubMed] [Google Scholar]
- 9. Deininger, S. O. , Ebert, M. P. , Futterer, A. , Gerhard, M. et al., MALDI imaging combined with hierarchical clustering as a new tool for the interpretation of complex human cancers. J. Proteome Res. 2008, 7, 5230–5236. [DOI] [PubMed] [Google Scholar]
- 10. Alexandrov, T. , Becker, M. , Guntinas‐Lichius, O. , Ernst, G. et al., MALDI‐imaging segmentation is a powerful tool for spatial functional proteomic analysis of human larynx carcinoma. J. Cancer Res. Clin. Oncol. 2013, 139, 85–95. [DOI] [PubMed] [Google Scholar]
- 11. Schwartz, S. A. , Weil, R. J. , Thompson, R. C. , Shyr, Y. et al., Proteomic‐based prognosis of brain tumor patients using direct‐tissue matrix‐assisted laser desorption ionization mass spectrometry. Cancer Res. 2005, 65, 7674–7681. [DOI] [PubMed] [Google Scholar]
- 12. Schwamborn, K. , Caprioli, R. M. , Molecular imaging by mass spectrometry ‐ looking beyond classical histology. Nat. Rev. Cancer 2010, 10, 639–646. [DOI] [PubMed] [Google Scholar]
- 13. Schöne, C. , Höfler, H. , Walch, A. , MALDI imaging mass spectrometry in cancer research: Combining proteomic profiling and histological evaluation. Clin. Biochem. 2013, 46, 539–545. [DOI] [PubMed] [Google Scholar]
- 14. Balluff, B. , Frese, C. K. , Maier, S. K. , Schöne, C. et al., De novo discovery of phenotypic intratumour heterogeneity using imaging mass spectrometry. J. Pathol. 2015, 235, 3–13. [DOI] [PubMed] [Google Scholar]
- 15. Caldwell, R. L , Gonzalez, A. , Oppenheimer, S. R. , Schwartz, H. S. et al., Molecular assessment of the tumor protein microenvironment using imaging mass spectrometry. Cancer Genomics Proteomics 2006, 3, 279–288. [Google Scholar]
- 16. Oppenheimer, R. S. , Mi, D. , Sanders, M. E. , Caprioli, R. M. Molecular analysis of tumor margins by MALDI mass spectrometry in renal carcinoma. J. Proteome Res. 2010, 9, 2182–2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kang, S. , Shim, H. S. , Lee, J. S. , Kim, D. S. et al., Molecular proteomics imaging of tumor interfaces by mass spectrometry. J. Proteome Res. 2010, 9, 1157–1164. [DOI] [PubMed] [Google Scholar]
- 18. Jones, E. A. , Schmitz, N. , Waaijer, C. J. , Frese, C. K. et al., Imaging mass spectrometry‐based molecular histology differentiates microscopically identical and heterogeneous tumors. J. Proteome Res. 2013, 12, 1847–1855. [DOI] [PubMed] [Google Scholar]
- 19. Alexandrov, T. , MALDI imaging mass spectrometry: statistical data analysis and current computational challenges. BMC Bioinformatics 2012, 13 (Suppl 16), S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sturgis, E. M. , Cinciripini, P. M. , Trends in head and neck cancer incidence in relation to smoking prevalence: an emerging epidemic of human papillomavirus‐associated cancers? Cancer 2007, 110, 1429–1435. [DOI] [PubMed] [Google Scholar]
- 21. Corvò, R. , Evidence‐based radiation oncology in head and neck squamous cell carcinoma. Radiother. Oncol. 2007, 85, 156–170. [DOI] [PubMed] [Google Scholar]
- 22. Bose, P. , Brockton, N. T. , Dort, J. C. , Head and neck cancer: from anatomy to biology. Int. J. Cancer 2013, 133, 2013–2023. [DOI] [PubMed] [Google Scholar]
- 23. Carvalho, A. C. , Kowalski, L. P. , Campos, A. H. , Soares, F. A. et al., Clinical significance of molecular alterations in histologically negative surgical margins of head and neck cancer patients. Oral Oncol. 2012, 48, 240–248. [DOI] [PubMed] [Google Scholar]
- 24. Leemans, C. R. , Braakhuis, B. J. , Brakenhoff, R. H. , The molecular biology of head and neck cancer. Nat. Rev. Cancer 2011, 11, 9–22. [DOI] [PubMed] [Google Scholar]
- 25. Stransky, N. , Egloff, A. M. , Tward, A. D. , Kostic, A. D. et al., The mutational landscape of head and neck squamous cell carcinoma. Science 2011, 333, 1157–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rothenberg, S. M. , Ellisen, L. W. , The molecular pathogenesis of head and neck squamous cell carcinoma. J. Clin. Invest. 2012, 122, 1951–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wong, J. W. , Durante, C. , Cartwright, H. M. , Application of fast Fourier transform cross‐correlation for the alignment of large chromatographic and spectral datasets. Anal. Chem. 2005, 77, 5655–5661. [DOI] [PubMed] [Google Scholar]
- 28. Polanski, A. , Marczyk, M. , Pietrowska, M. , Widlak, P. , Polanska, J. , Signal partitioning algorithm for highly efficient Gaussian mixture modeling in mass spectrometry. Plos One 2015, 10, e0134256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bolshakova, N. , Azuaje, F. , Machaon, C. V. E , Cluster validation for gene expression data. Bioinformatics 2003, 19, 2494–2495. [DOI] [PubMed] [Google Scholar]
- 30. Celebi, M. E. , Kingravi, H. A. , in: Celebi, M. E. (Ed.), Partitional Clustering Algorithms, Springer, Cham Heidelberg, New York, 2015, pp. 79–98. [Google Scholar]
- 31. Marczyk, M. , Jaksik, R. , Polanski, A. , Polanska, J. , Adaptive filtering of microarray expression data based on Gaussian mixture decomposition. BMC Bioinformatics 2013, 14, 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jones, E. A. , van Remoortere, A. , van Zeijl, R. J. M. , Hogendoorn, P. C. W. et al., Multiple statistical analysis techniques corroborate intratumor heterogeneity in Imaging Mass Spectrometry datasets of Myxofibrosarcoma. PLoS One 2011, 6, e24913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Krasny, L. , Hoffmann, F. , Ernst G., Trede, D. et al., Spatial segmentation of MALDI FT‐ICR MSI data: a powerful tool to explore the head and neck tumor in situ lipidome. J. Am. Soc. Mass Spectrom. 2015, 26, 36–43. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Table S1. Difference in abundance of spectral components between expert‐defined tissue regions corresponding to normal epithelium and tumor