

Abstract
The present study investigated the influence of PGE2, E prostanoid (EP) receptors, and their signaling pathways on matrix metalloproteinase (MMP)-1 and IL-6 expression in synovial fibroblasts (SFs) from rheumatoid arthritis (RA) patients. RASFs expressed all four EP receptors, with selective induction of EP2 by TNF-α. TNF-α time-dependently increased intracellular cAMP/protein kinase A signaling (maximum, 6–12 h) and PGE2 secretion (maximum, 24 h). PGE2 and the EP2 agonists butaprost or ONO-AE1-259 ((16)-9-deoxy-9β-chloro-15-deoxy-16-hydroxy-17,17-trimethylene-19,20-didehydro PGE1), in turn, induced a rapid, time-dependent (maximum, 15–30 min) increase of cAMP. Additionally, cyclooxygenase-2 inhibition by NS-398 (N-(2-cyclohexyloxy-4-nitrophenyl)-methanesulfonamide) reduced the TNF-α-induced increase in IL-6 mRNA/protein, which was restored by stimulation with PGE2 or EP2, EP3, and EP4 agonists. In contrast, TNF-α-induced MMP-1 secretion was not influenced by NS-398 and diminished by PGE2 via EP2. Finally, 3-isobutyl-1-methylxanthine enhanced the effects of PGE2 on MMP-1, but not on IL-6 mRNA. In conclusion, PGE2 differentially affects TNF-α-induced mRNA expression of proinflammatory IL-6 and prodestructive MMP-1 regarding the usage of EP receptors and the dependency on cAMP. Although specific blockade of EP2 receptors is considered a promising therapeutic strategy in RA, opposite regulation of proinflammatory IL-6 and prodestructive MMP-1 by PGE2 via EP2 may require more complex approaches to successfully inhibit the cyclooxygenase-1/2 cAMP axis.
In rheumatoid arthritis (RA),4 activated synovial fibroblasts (RASFs) contribute to the inflammatory/destructive potential of the aggressive synovial tissue by producing proinflammatory mediators and matrix-degrading enzymes including matrix metalloproteinases (MMPs) (1, 2, 3, 4, 5, 6). Under the influence of proinflammatory cytokines, for example, TNF-α or IL-1β, RASFs secrete PGE2 (7, 8, 9). PGE2 can raise cAMP levels in RASFs (10, 11, 12, 13, 14), and cAMP is involved in the IL-1β-induced expression of several target genes, e.g., IL-6, M-CSF, and vascular endothelial growth factor (11). PGE2 belongs to the family of prostanoid, autocrine, and paracrine lipid mediators released by cells following mechanical injury or stimulation with cytokines or growth factors. The synthesis of the prostanoid is catalyzed by the cyclooxygenase (COX) pathway (15).
PGE2 mediates its biological functions via binding to four types of membrane-bound, G protein-coupled receptors termed E prostanoid (EP)1 to EP4 (15, 16). Following ligand binding, the EP receptors activate different signal transduction pathways. EP1 raises intracellular Ca2+, whereas EP3 reduces or increases cAMP by activating inhibitory G (Gi) or stimulatory G (Gs) proteins depending on the particular splice variant expressed by the cell (17). The EP2 and EP4 receptors increase intracellular cAMP by activating adenylate cyclase via Gs proteins. However, differences in the strength of Gs coupling, activation of other signal transduction pathways, agonist-induced desensitization, and agonist-induced internalization result in a differential response of the target cell to a ligand-induced activation of the EP2 or EP4 receptors (18).
Human RASFs have consistent mRNA expression for the PGE2 receptors EP2 and EP4, while there have been inconsistent reports of EP1 and EP3 mRNA expression (10, 19, 20, 21). Surprisingly, although EP2 and EP4 receptors are regarded as attractive pharmacological targets for RA treatment, the exact role of cAMP or other signals issued by PGE2-challenged EP receptor subtypes, as well as their influence on the effects stimulation with TNF-α or IL-1β, remains largely unknown. The present study sought to analyze the involvement of PGE2-dependent cAMP signaling in TNF-α-induced proinflammatory IL-6 and/or prodestructive MMP-1 effector functions of RASFs.
Materials and Methods
Materials
Rabbit anti-human polyclonal Abs against the EP1, EP2, EP3, and EP4 receptors, PGE2, NS-398 (N-(2-Cyclohexyloxy-4-nitrophenyl)-methanesulfonamide), butaprost, sulprostone, and an ELISA kit for cAMP were purchased from Cayman Chemical. A competition assay for the detection of PGE2 was obtained from Biotrak (Amersham Pharmacia Biotech). Research grade 3-isobutyl-1-methylxanthine (IBMX) was purchased from Calbiochem. Selective EP1 (ONO-DI-004, (17S)-2,5-ethano-6-oxo-17, 20-dimethyl PGE1), EP2 (ONO-AE1-259, (16)-9-deoxy-9β-chloro-15-deoxy-16-hydroxy-17,17-trimethylene-19,20-didehydro PGE1), EP3 (ONO-AE-248, 11,15-O-dimethyl PGE2), and EP4 (ONO-AE1-329, 16-(3-methoxymethyl)phenyl-ω-tetranor-3,7-dithia PGE1) receptor agonists were provided by Ono Pharmaceutical. Recombinant human TNF-α was purchased from R&D Systems, DMEM from Invitrogen, FCS from Cambrex Bio Science, TriPure reagent from Roche Applied Science, and Hot-StarTaq polymerase from Qiagen.
Patients, tissue digestion, and cell culture
Synovial tissue from RA patients was obtained during open joint replacement/arthroscopic synovectomy from the Clinic of Orthopedics, Eisenberg, Germany. All patients fulfilled the respective American Rheumatism Association criteria (22). The study was approved by the Ethics Committee of the University Hospital Jena (Jena, Germany), and patient informed consent was obtained. Immediately after synovectomy, tissue was placed in culture medium at ambient temperature and subjected to digestion within 2 h.
RA synovial samples were digested, subsequently cultured for 7 days, and RASFs were negatively isolated as previously described (23, 24). RASFs were cultured in the virtual absence of contaminating nonadherent cells and macrophages. Third-passage cells were used for all experiments. Stimulation of the cells was performed in DMEM/0.2% lactalbumin hydrolysate. Mycoplasma contamination of the cells was excluded by 4′-6-diamidino-2-phenylindole (DAPI) staining.
Cell stimulation
For kinetic analysis of the TNF-α-induced expression of EP receptors, RASFs (2.5 × 105 cells/well of a 12-well plate) were allowed to adhere for 24 h in DMEM/10% FCS at 37°C and 5% CO2. Thereafter, cells were stimulated with 10 ng/ml TNF-α (R&D Systems). After stimulation for 0, 1, 2, 4, 8, 10, and 24 h, the cells were lysed in buffer for RNA isolation.
For analysis of intracellular cAMP and protein kinase A (PKA) substrate phosphorylation, as well as mRNA and protein expression of proinflammatory/prodestructive IL-6 and MMP-1, RASFs (4 × 105 cells/well of 6-well plates or 1.5–2.0 × 105 cells/well of a 12-well plate) were allowed to adhere for 24 h in DMEM/10% FCS at 37°C. Thereafter, cells were pretreated with 1 μM NS-398 for 30–45 min followed by treatment with TNF-α (10 ng/ml), PGE2 (1 μM), the EP2 receptor agonist butaprost or the EP3/EP1 agonist sulprostone (each 1 μM), or selective EP agonists (EP1, EP2, EP3, and EP4; ONO; 10 μM). In selected experiments (see Figs. 2, A and B, and 4, as well as supplemental Fig. 2),5 500 μM IBMX was added to each well 4 h before the end of the experiment to increase the signal for intracellular cAMP production and PKA substrate phosphorylation; in other experiments, the results were compared for cultivation with and without prior coincubation with IBMX (see Figs. 2E and 6, 100 μM IBMX; supplemental Fig. 1, A and B, 500 μM; and supplemental Fig. 1C, 100 μM IBMX). Supernatants of the cells were collected for analysis of protein secretion.
FIGURE 2.
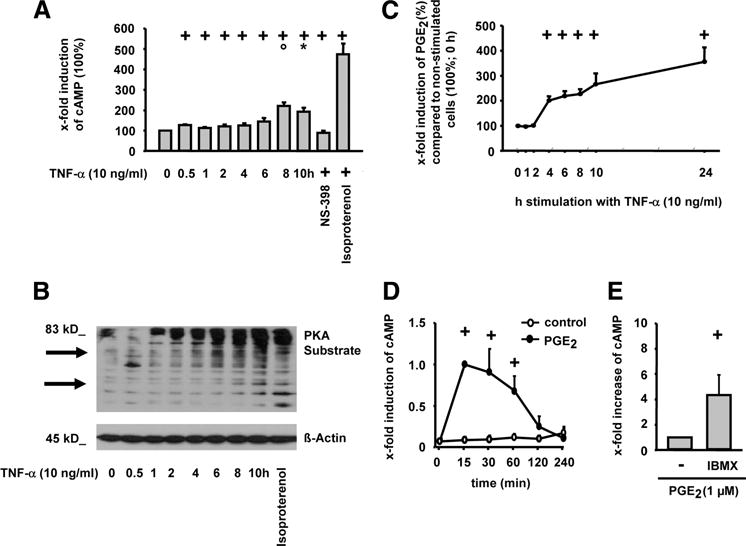
Influence of TNF-α on intracellular cAMP levels, PKA substrate phosphorylation, and PGE2 secretion in RASFs. RASFs were stimulated with TNF-α (10 ng/ml; different time points), TNF-α/NS-398 (1 μM; 10 h), or isoproterenol (100 μM; 10 min) in the presence of IBMX (500 μM; A and B) or the absence of IBMX (C). Alternatively, cells were stimulated with PGE2 for different time points (D) or for 30 min with/without IBMX (100 μM) (E). Intracellular cAMP was determined by RIA (A) or ELISA (D), PKA substrate phosphorylation by Western blot (B), and PGE2 secretion by ELISA (C); means ± SEM for three patients with RA; +, p ≤ 0.05 Mann-Whitney U test vs 0 h (A, C, and D) or vs without IBMX (E); ○, p ≤ 0.05 Mann-Whitney U test vs ≤4 h (A);*, p ≤ 0.05 Mann-Whitney U test vs 6 h (A).
FIGURE 4.
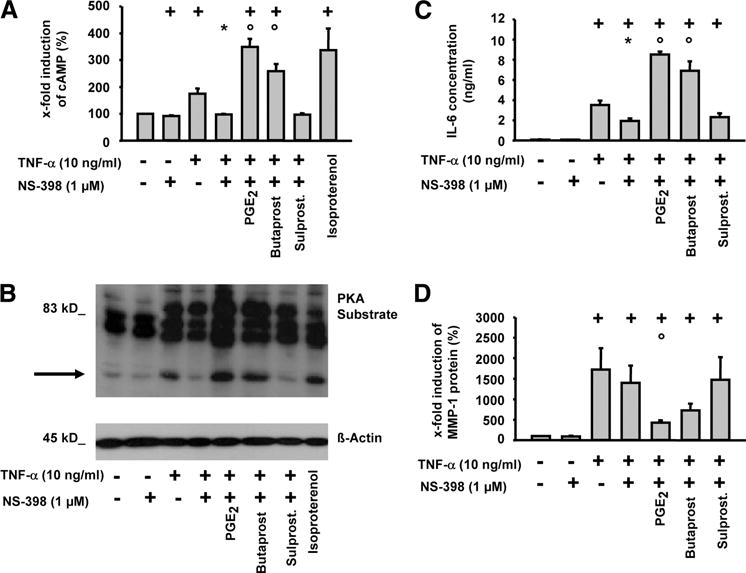
Role of EP2 and EP3 receptors in TNF-α-induced cAMP production, PKA substrate phosphorylation, and IL-6 or MMP-1 secretion in RASFs. Cells were stimulated with TNF-α (10 ng/ml) in the absence or presence of NS-398, PGE2, butaprost, or sulprostone (Sulprost.; 1 μM each) with IBMX for 10 h. Intracellular cAMP was determined by RIA (A) and PKA substrate phosphorylation by Western blot (B); IL-6 secretion was analyzed by ELISA (C) and MMP-1 secretion by Western blot (D); means ± SEM for three patients with RA; +, p ≤ 0.05 Mann-Whitney U test vs control; *, p ≤ 0.05 Mann-Whitney U test vs TNF-α; ○, p ≤ 0.05 Mann-Whitney U test vs TNF-α/NS-398.
FIGURE 6.
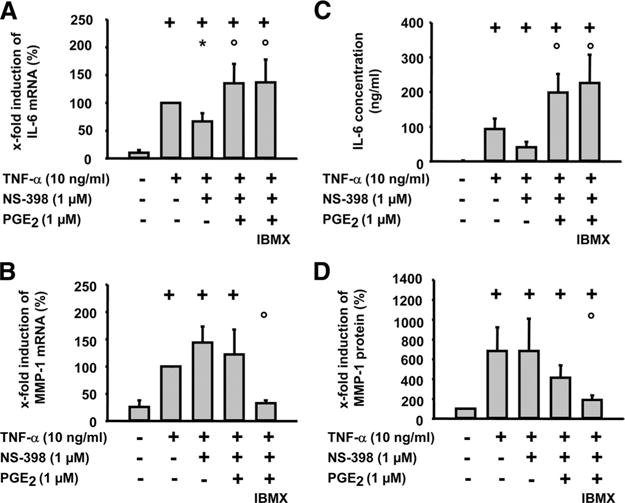
Effect of the phosphodiesterase inhibitor IBMX on the regulation of MMP-1 and IL-6 by PGE2 (RT-PCR). RASFs were harvested at 24 h after TNF-α (10 ng/ml) stimulation with or without NS-398 (1 μM), PGE2 (1 μM), and/or IBMX (100 μM). mRNA expression of IL-6 and MMP-1 was detected by real-time RT-PCR (A and B). IL-6 secretion was analyzed by ELISA (C), and MMP-1 secretion was analyzed by Western blot (D). For each experiment, a value of 100% was assigned to TNF-α. Results are expressed as means ± SEM for 3 patients with RA; +, p ≤ 0.05 Mann-Whitney U test vs control; *, p ≤ 0.05 Mann-Whitney U test vs TNF-α; ○, p ≤ 0.05 Mann-Whitney U test vs TNF-α/NS-398.
Analysis of EP receptor, MMP-1, and IL-6 expression by real-time RT-PCR
Total RNA was isolated from RASFs using a commercially available RNA isolation kit (Macherey & Nagel) and 1 μg was reverse-transcribed using SuperScript II reagents (Invitrogen). EP1, EP2, EP3, EP4, MMP-1, and IL-6 mRNA expression was analyzed by real-time PCR in a RealPlex PCR machine (Eppendorf). PCR reactions were performed in a total volume of 20 μl in 96-well plates containing a reaction mix of HotMaster DNA polymerase (0.05 U; Eppendorf), 10 × Taq buffer with 15 mM magnesium (Eppendorf), MgCl2 (final concentration, 3.5 mM; Invitrogen), dNTPs (0.4 mM; Roche), BSA (40 ng/ml), SYBR Green (1/1250; SYBR Green I, 10,000 concentrate; Molecular Probes), sense and antisense primers (each 0.3 μM), and cDNA. To normalize the amount of cDNA in each sample, the housekeeping gene aldolase was also amplified. The sequences of the PCR primers used in this study and the real-time PCR conditions are described in Tables I and II. The fluorescence emitted by dsDNA-bound SYBR Green was measured once at the end of each additional heating step and continuously during the melting curve program. The concentration of EP1, EP2, EP3, EP4, MMP-1, and IL-6 mRNA in each sample was calculated by the RealPlex software using an external standard curve. Product specificity of the real-time PCRs was confirmed by 1) melting curve analysis (see Table II), 2) agarose gel electrophoresis, and 3) cycle sequencing of the PCR products.
Table I.
Sequences of PCR primers used in this study
| Gene | Primer |
kb | |
|---|---|---|---|
| Real-Time PCR | Conventional PCR | ||
| Aldolase | |||
| Sense | 5′-tcatcctcttccatgagacactct-3′ | 313 | |
| Anti-sense | 5′-attctgctggcagatactggcataa-3′ | ||
| GAPDH | |||
| Sense | 5′-cca ccc atg gca aat tcc atg gca-3′ | 606 | |
| Anti-sense | 5′-tct aga cgg gag gtc agg tcc acc-3′ | ||
| Sense | 5′-tcagcaatgcctcctgcac-3′ | 250 | |
| Anti-sense | 5′-ccagtgagcttcccgttcag-3′ | ||
| IL-6 | |||
| Sense | 5′-atgaactccttctccacaagcg-3′ | 5′-atgaactccttctccacaagcgc-3′ | 199/627 |
| Anti-sense | 5′-ctcctttctcagggctgag-3′ | 5′-gaagagccctcaggctggactg-3′ | |
| MMP-1 | |||
| Sense | 5′-gacctggaggaaatcttgc-3′ | 5′-aactctggagtaatgtcacac-3′ | 321/584 |
| Anti-sense | 5′-gttagcttactgtcacacgc-3′ | 5′-attcgtaagcagcttcaagcc-3′ | |
| EP1 | |||
| Sense | 5′-cttgtcggtatcatggtggtgtc-3′ | 317 | |
| Anti-sense | 5′-ggttgtgcttagaagtggctgagg-3′ | ||
| EP2 | |||
| Sense | 5′-ccacctcattctcctggcta-3′ | 216 | |
| Anti-sense | 5′-cgacaacagaggactgaacg-3′ | ||
| EP3 | |||
| Sense | 5′-cttcgcataactggggcaac-3′ | 300 | |
| Anti-sense | 5′-tctccgtgtgtgtcttgcag-3′ | ||
| EP4 | |||
| Sense | 5′-tggtatgtgggctggctg-3′ | 429 | |
| Anti-sense | 5′-gaggacggtggcgagaat-3′ | ||
Table II.
Real-time and conventional PCR conditions
| Gene | Real-Time PCR |
Conventional RT-PCR
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Initial Denaturation (°C; min) | Denaturation (°C; s) | Annealing (°C; s) | Elongation (°C; s) | Melting (°C; s) | Initial Denaturation (°C; min) | Denaturation (°C; s) | Annealing (°C; s) | Elongation (°C; s) | Terminal Elongation (°C; min) | No Cycles | |
| Aldolase | 95; 10 | 95; 20 | 58; 20 | 68; 20 | 82; 8 | ||||||
| GAPDH | 94; 3 | 94; 60 | 59; 60 | 72; 60 | 72; 7 | 25 | |||||
| 95; 15 | 95; 30 | 56; 30 | 72; 45 | 72; 5 | 26 | ||||||
| IL-6 | 95; 15 | 95; 10 | 62; 10 | 68; 20 | 81; 8 | 95; 15 | 95; 30 | 56; 30 | 72; 45 | 72; 5 | 25 |
| MMP-1 | 95; 10 | 95; 15 | 58; 15 | 68; 20 | 81; 8 | 95; 15 | 95; 30 | 56; 30 | 72; 45 | 72; 5 | 26 |
| EP1 | 95; 10 | 95; 20 | 60; 10 | 68; 20 | 83; 8 | 95; 2 | 95; 45 | 59; 45 | 72; 60 | 72; 5 | 35 |
| EP2 | 95; 10 | 95; 15 | 60; 15 | 68; 20 | Not performed | 95; 2 | 95; 45 | 61; 45 | 72; 60 | 72; 5 | 35 |
| EP3 | 95; 10 | 95; 20 | 60; 15 | 68; 20 | 83; 8 | 95; 2 | 95; 45 | 59; 45 | 72; 60 | 72; 5 | 40 |
| EP4 | 95; 15 | 95; 15 | 60; 15 | 68; 20 | 81; 8 | 94; 3 | 95; 60 | 60; 45 | 72; 60 | 72; 7 | 35 |
For conventional RT-PCR of EP receptor expression, PCR reactions were performed in a total volume of 50 μl containing a reaction mix of Taq-polymerase (50 mU/μl; Jena Bioscience), 10× PCR buffer, 4% DMSO, dNTP (50 μM), as well as sense and antisense primers (each 0.5 pmol; Jena Bioscience). For EP1 and EP3 PCR, 5% 5× Q-Solution (Qiagen) was added to the mix. The sequences of the PCR primers used in this study and the PCR conditions are stated in Tables I and II. Verification of the PCR products was performed by cycle sequencing.
RT-PCR for MMP-1 and IL-6
For conventional RT-PCR of MMP-1 and IL-6, RNA from the cells was extracted with TriPure reagent according to the manufacturer’s instructions. Reverse transcription was performed according to the manufacturer’s instructions using a SuperScript preamplification system with 1 μg of total RNA as a template. Subsequent amplifications of the cDNA fragments by PCR with HotStarTaq polymerase were performed using 0.5 μl of the reverse-transcribed mixture as a template with specific primers and PCR conditions as mentioned in Tables I and II. The amplified cDNA fragments were resolved electrophoretically on 2% agarose gels and then visualized under UV illumination using a Bio-Rad ChemiDoc apparatus after staining with ethidium bromide.
Analysis of EP receptor and MMP-1 protein in RASFs by Western blot
For the analysis of EP receptor protein, 35 μg of protein from nonstimulated or TNF-α-stimulated RASFs (30 h) was separated by denaturing SDS-PAGE (12%) and transferred onto blotting membranes (Hybond-C Extra; Amersham Life Sciences). In the case of MMP-1, cell culture supernatant was used. After blocking with 2.5% skim milk in Tris-buffered saline-Tween 20 (10 mM Tris, 150 mM NaCl, 0.1% Tween 20 (pH 7.4)), membranes were probed overnight at 4°C with specific primary Abs against the EP1, EP2, EP3, or EP4 receptors (Cayman Chemical) or against MMP-1 (clone 50647; R&D Systems), washed, and incubated with HRP-conjugated goat anti-rabbit IgG as a secondary Ab. Proteins were visualized by chemiluminescence (Supersignal West chemiluminescent substrate; Pierce). The intensity of each band was quantified using an integration image software (Scion Corporation).
cAMP measurements
Intracellular cAMP was determined in RASFs using either the cAMP enzyme immunoassay kit (Cayman Chemical) or the cAMP [3H] assay system TRK 432 (Amersham Bioscience). Samples were prepared exactly as described by the manufacturer.
Phosphorylation of PKA substrates
Phosphorylation of PKA substrates was determined by Western blot analysis of RASFs with an anti phospho-PKA substrate Ab (clone 100G7; Cell Signaling Technology). The blots were subsequently reprobed with β-actin to ascertain equal protein loading.
IL-6 measurements
Human IL-6 was measured in diluted cell culture supernatants using a quantitative sandwich enzyme immunoassay (OptEIA; BD Biosciences). A wavelength of 450 nm with a wavelength correction at 570 nm was used. Sample concentrations of IL-6 were determined by comparison with a standard curve (range, 2.34–300 pg/ml).
PGE2 measurements
PGE2 concentrations in the supernatants of TNF-α-stimulated cells were determined using a competition assay (sensitivity, 40 pg/ml PGE2, Biotrak; Amersham Pharmacia Biotech). The OD in each well was determined at 450 nm. The concentration of PGE2 was determined by comparison with a standard curve (range, 50–6400 pg/ml).
Statistical analysis
The data were expressed as means ± SEM. Significance was tested using the nonparametric Mann-Whitney U test. Differences were considered statistically significant for p ≤ 0.05. Analyses were performed using the SPSS 13.0 program.
Results
Role of PGs in TNF-α-induced IL-6 and MMP-1 mRNA expression and secretion in RASFs
Exposure of RASFs to TNF-α led to a marked induction of IL-6 mRNA expression and secretion (Fig. 1, A and C). To test the involvement of PGs in this process, a pharmacological approach was used to inhibit COXs. The COX-2-selective inhibitor NS-398 significantly reduced TNF-α-induced IL-6 mRNA expression and protein secretion, pointing to an enhancing role of COX-2-derived PGs in this process. In line with this notion, NS-398-blocked expression and secretion of IL-6 was restored by simultaneous administration of exogenous PGE2 (Fig. 1, A and C). Taken together, these data point to a critical role of PGs, possibly PGE2, as modulators of the proinflammatory actions of TNF-α.
FIGURE 1.
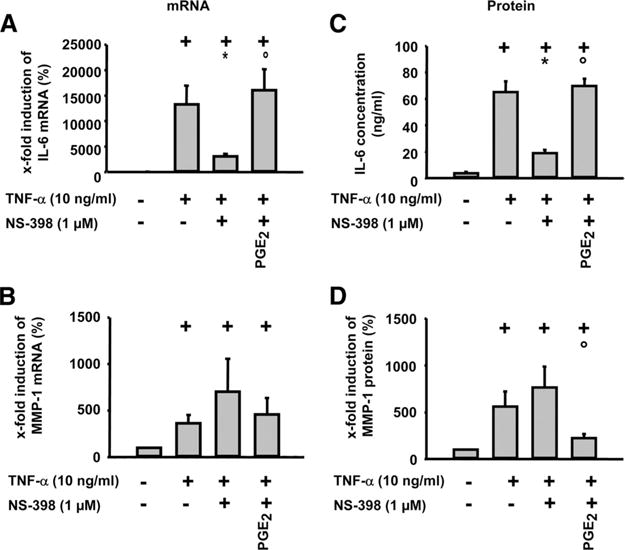
Influence of PGE2 on TNF-α-induced IL-6 and MMP-1 mRNA expression and protein secretion in RASFs. Cells were stimulated with TNF-α (10 ng/ml) in the absence or presence of NS-398 and PGE2 (1 μM each) for 24 h. IL-6 and MMP-1 mRNA expression was analyzed by real-time PCR (A and B). IL-6 secretion was analyzed by ELISA (C), and MMP-1 secretion was analyzed by Western blot (D); means ± SEM for six patients with RA; +, p ≤ 0.05 Mann-Whitney U test vs control; *, p ≤ 0.05 Mann-Whitney U test vs TNF-α; ○, p ≤ 0.05 Mann-Whitney U test vs TNF-α/NS-398.
As observed for IL-6, TNF-α also significantly induced the mRNA expression and secretion of MMP-1 in RASFs (Fig. 1, B and D). However, NS-398 did not significantly reduce, but even numerically enhanced, MMP-1 mRNA expression and secretion. Concordantly, addition of exogenous PGE2 significantly reduced the NS-398-enhanced mRNA expression and secretion of MMP-1 upon TNF-α stimulation (Fig. 1, B and D). Therefore, PGE2 and possibly other PG species appear to have critical and partially opposite effects on proinflammatory and prodestructive signaling by TNF-α in RASFs.
TNF-α activates the cAMP/PKA signaling pathway in RASFs
TNF-α induced a gradual, time-dependent increase in cAMP levels that reached a maximum after 8–10 h of stimulation (Fig. 2A). The β-adrenoreceptor agonist isoproterenol, a well-known cAMP-elevating agent, was used as a positive control in this and forthcoming experiments. To confirm with an independent approach that TNF-α addressed the cAMP/PKA signaling cassette, the phosphorylation status of PKA target proteins was assessed using a phosphorylation-specific Ab that selectively detects the minimum RRXS/T consensus target sequence for PKA in its phosphorylated state. The corresponding experiment (Fig. 2B) illustrated a time-dependent increase in the phosphorylation of multiple PKA targets, in full agreement with the cAMP measurements, and thus confirming that TNF-α activates the cAMP/PKA pathway.
Since TNF-α induced both cAMP/PKA signaling and PGE2 release with similar kinetics (Fig. 2A–C) and PGE2 increased cAMP in RASFs (Fig. 2D; enhanced by IBMX, Fig. 2E), PGs may mediate the activation of the cAMP/PKA pathway by TNF-α.
TNF-α induces the expression of EP2 receptors in RASFs
PCR of total RNA preparations from RASFs was performed and mRNA for all four EP receptors was detected in these cells (Fig. 3, A and B). Intriguingly, TNF-α induced a time-dependent increase in EP2 mRNA (maximum, 10 h), as assessed by both real-time (Fig. 3A) or conventional PCR (Fig. 3B). In contrast, stimulation with TNF-α reduced EP4 mRNA levels and left EP1 and EP3 mRNA unchanged.
FIGURE 3.
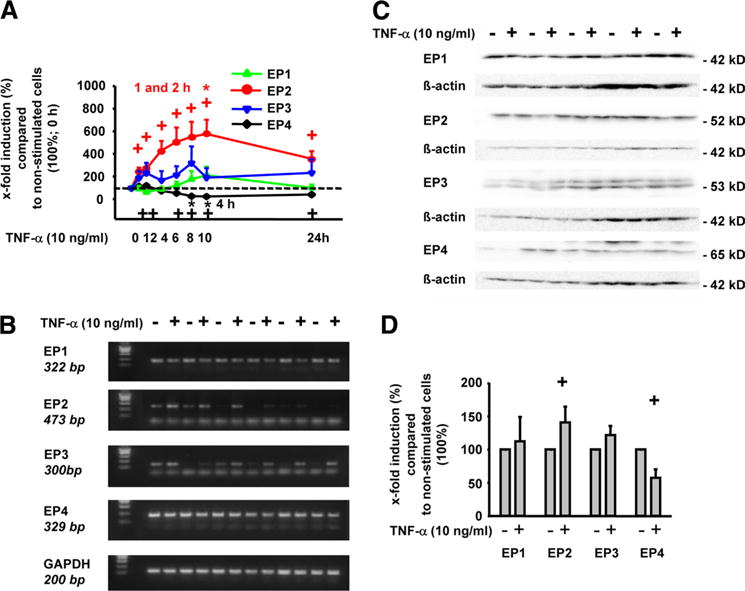
Influence of TNF-α on the EP receptor expression in RASFs. RASFs were stimulated with 10 ng/ml TNF-α for different time points (A), 8 h (B), or 30 h (C and D). EP receptor expression was analyzed by quantitative real-time RT-PCR (A) or conventional RT-PCR (B). To analyze the influence of TNF-α on EP receptor protein levels, protein extracts were subjected to Western blot analysis using specific Abs against the EP1, EP2, EP3, and EP4 receptors (C; quantification in D); means ± SEM for five patients with RA; +, p ≤ 0.05 Mann-Whitney U test vs the 0 h time point (A) or vs culture without TNF-α (D); *, p ≤ 0.05 Mann-Whitney U test vs indicated time points (A).
To confirm these data, Western blots of cell lysates were performed. As shown in Fig. 3C, RASF extracts contained all four EP receptor proteins (i.e., EP1 (42 kDa), EP2 (52 kDa), EP3 (53 kDa), and EP4 (65 kDa)). In agreement with the PCR data, TNF-α significantly up-regulated EP2 protein expression following stimulation for 30 h (1.4-fold; see quantification in Fig. 3D). In contrast, EP4 protein was significantly down-regulated (43% reduction), whereas EP1 or EP3 protein levels were not altered by TNF-α stimulation.
PGE2 and selective EP receptor agonists modulate cAMP/PKA pathway activation by TNF-α and TNF-α-induced secretion of IL-6 and MMP-1 in RASFs
COX-2 inhibition with NS-398 completely prevented the increase in cAMP levels induced by TNF-α (Fig. 4A), further underlining that PG release by TNF-α was involved in the up-regulation of cAMP levels (see also Fig. 2A). Accordingly, addition of PGE2 or the EP2 selective agonist butaprost restored the increase in cAMP levels to a level above that in TNF-α/NS-398-treated cells. In contrast, the EP1/3-specific agonist sulprostone did not revert the blockade exerted by NS-398. All cAMP measurement data were also confirmed by phospho-(PKA-substrate) Western blots (Fig. 4B). The effects of PGE2 were dose-dependent, both in the presence and the absence of NS-398 (supplemental Fig. 2).
To test whether the TNF-α/PGE2/cAMP axis was physiologically relevant in RASFs, the IL-6 and MMP-1 secretion was examined under the same experimental conditions (Fig. 4, C and D). Except for the induction of IL-6 by TNF-α in the presence of NS-398, IL-6 production showed a pattern identical to that of cAMP levels and PKA activity (Fig. 4, A–C). EP3 signaling induced a marginal, nonsignificant raise of the mean IL-6 concentration compared with that of the TNF-α/NS-398 treatment group, however, with a consistent increase in the paired comparison for all three individual patients (supplemental Fig. 3).
In contrast, PGE2-elicited signaling and, more specifically, EP2-dependent signals, diminished the MMP-1 secretion induced by TNF-α (Fig. 4D), demonstrating that PGE2/EP2 signals have directly opposite effects on IL-6 and MMP-1 release by RASFs (for PGE2 effects, see also Fig. 1).
Effects of PGE2 and selective EP receptor agonists on TNF-α-induced expression of mRNAs for IL-6 and MMP-1 in RASFs
In the case of IL-6, the mRNA expression in comparison with TNF-α-stimulated RASFs in the presence of NS-398 was significantly increased by PGE2 and selective receptor agonists for EP2 (3.2-fold and 2.7-fold, respectively), but also for EP3 and EP4 (1.7-fold and 1.9-fold, respectively; Fig. 5, A and B).
FIGURE 5.
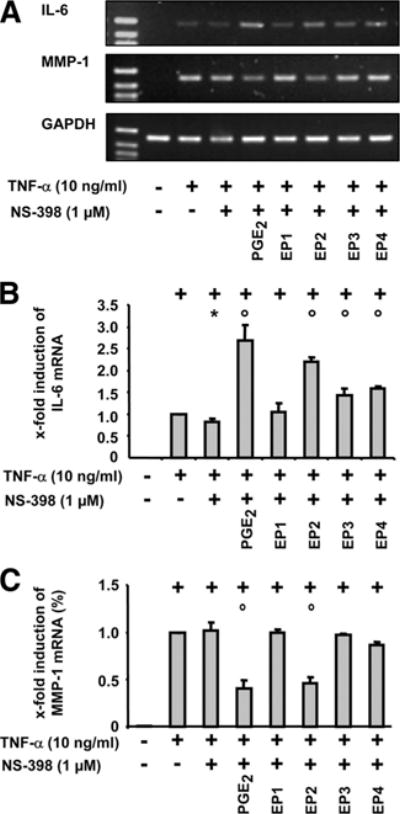
Effect of selective EP receptor agonists on the regulation of MMP-1 and IL-6 (RT-PCR). A, RASFs were harvested at 24 h after TNF-α (10 ng/ml) stimulation with or without NS-398 (1 μM), PGE2 (1 μM), and/or selective agonists of the EP receptors 1–4 (10 μM each). mRNA expression of (B) IL-6 and (C) MMP-1 was detected by conventional RT-PCR. For each experiment, a value of 1 was assigned to the stimulation with TNF-α. Results are expressed as means ± SEM for three patients with RA; +, p ≤ 0.05 Mann-Whitney U test vs control; *, p ≤ 0.05 Mann-Whitney U test vs TNF-α; ○, p ≤ 0.05 Mann-Whitney U test vs TNF-α/NS-398.
The above results for MMP-1 were confirmed with selective EP receptor agonists; that is, in the presence of COX-2 inhibitors only PGE2 and the EP2 receptor agonist significantly suppressed the mRNA expression in TNF-α-stimulated RASFs (Fig. 5, A and C).
Effect of the phosphodiesterase inhibitor IBMX on TNF-α-induced expression of mRNAs for IL-6 and MMP-1 in RASFs
To directly assess the relevance of cAMP for the effects of PGE2 on the TNF-α-induced gene expression, the degradation of cAMP was inhibited by addition of IBMX. Strikingly, the mRNA expression and secretion of IL-6 remained unaffected (Fig. 6, A and C), whereas the mRNA expression and secretion of MMP-1 was further suppressed in the presence of IBMX (Fig. 6, B and D).
Discussion
This study demonstrates for the first time that PGE2 has opposite effects on MMP-1 and IL-6 synthesis, uses different PGE2 receptors for these effects, and differentially applies the postreceptor signaling molecule cAMP. Thus, PGE2 is a differential key mediator of inflammatory/destructive functions in TNF-α-stimulated RASFs and may exhibit both proinflammatory (10, 11, 12, 25) and antidestructive capacities (Ref. 18 and the present study). These bipolar effects of PGE2 in RASFs may also be the reason for the inefficacy of COX-2 inhibitors to arrest joint destruction and should be considered in future studies focused on the therapeutic inhibition of COX-1/2 in RA.
PGE2 has opposite effects on the TNF-α-induced protein expression of MMP-1 and IL-6
In RASFs, TNF-α induces the secretion of proinflammatory and prodestructive mediators, for example, IL-6, PGE2, and MMP-1 (present study and Refs. 9, 26). The stimulatory effect of PGE2 on the TNF-α-induced IL-6 secretion in RASFs is in agreement with previously published data in IL-1β-stimulated RASFs (11, 14). However, the molecular mechanisms involved in the interplay between PGE2 and TNF-α for the control of IL-6 secretion in RASFs have so far remained largely undeciphered.
In marked contrast to the effects on IL-6, TNF-α-induced MMP-1 secretion was significantly reduced by PGE2, a finding also reported in IL-1β-stimulated RASFs (13, 27, 28). This clearly implicates PGE2 as a negative feedback molecule in the signaling pathway linking TNF-α to MMP-1 production. Whether this involves phosphorylation of p53 or expression of NURR1, or else inhibition of Erk and NF-κB activation by PGE2, as previously reported in the context of IL-1β/TNF-α signaling, remains to be investigated (13, 29, 31).
Interestingly, in TNF-α-stimulated periodontal ligament fibroblasts PGE2 down-regulates MMP-13 (but not MMP-1 or MMP-3) (32, 33). This emphasizes the antidestructive properties of PGE2, but also shows clear differences between fibroblasts of different origin. Contrasting results for IL-6 in periodontal ligament fibroblasts may also indicate cell-specific differences (34).
TNF-α induces an increase of intracellular cAMP and activation of PKA
The present study shows the novel finding that stimulation of RASFs with TNF-α induces an increased production of intracellular cAMP and PKA activity. Similar findings have been previously reported for IL-1β (35), suggesting that the cAMP system represents a critical regulatory pathway in RASFs.
With regard to the increased levels of intracellular cAMP induced by TNF-α, the participation of PGs appears plausible because previous data show an increase of intracellular cAMP in RASFs following PGE2 stimulation (35, 36). In contrast to the slow effects of TNF-α, the increase of cAMP induced by PGE2 occurred as early as 15 min after the start of stimulation (see Fig. 2D). This difference can be explained by the delayed synthesis of PGE2 following TNF-α stimulation (Fig. 2C) (35). In turn, cAMP may directly contribute to a further increase of PGE2 synthesis in RASFs (37). Concurrent with the increase of intracellular cAMP, TNF-α induced a phosphorylation of PKA substrates in RASFs in a strictly COX-2-dependent manner (see Fig. 4B), showing that the increase in cAMP translated into downstream PKA signaling (supplemental Fig. 4). This establishes PKA as a target of TNF-α (and PGE2) in RASFs, a notion previously only inferred from the use of pharmacological inhibitors (H89) or activators (Rp-cAMP) (27, 28, 38).
Only the effects of PGE2 on TNF-α-induced MMP-1 production are cAMP-dependent (but not those on IL-6 production)
Down-regulation of TNF-α-induced MMP-1 expression by PGE2 was mediated via cAMP. Therefore, the cAMP increase may have partial specificity for the antidestructive properties of PGE2, because phosphodiesterase IV inhibitors reduce joint damage in arthritis models (39, 40) or RA (41, 42) by further increasing cAMP levels. Also, cAMP-dependent regulation of MMP-1 has been shown after stimulation of RASFs with IL-1β (27), indicating partially common mechanisms for postreceptor signaling of these two pivotal proinflammatory cytokines.
The insensitivity of the augmentation of TNF-α-induced IL-6 expression by PGE2 to an increase of cAMP suggests a relevance of other pathways. Indeed, cAMP-independent pathways (PI3K/ERK) are involved in the signaling of the EP4 receptor (43). Alternatively, IL-6 expression may only depend on cAMP at very high intracellular concentrations (44).
In contrast to our findings with TNF-α, Inoue et al. have reported that the regulation of IL-1β-induced IL-6 expression involves cAMP-dependent pathways (11) This may indicate specific and differential regulation of IL-6 expression by different proinflammatory cytokines well below the receptor level.
TNF-α differentially regulates the expression of EP receptors
The biological function of PGE2 is mediated by four membrane-bound receptors (15, 16), all of which are expressed in RASFs (present study and Refs. 10, 11, 12, 45). In agreement with previously published data following IL-1β stimulation, TNF-α up-regulates the expression of the EP2 (and to some degree the EP3) (11, 45). Thus, up-regulation of EP2 and/or EP3 may be a widespread response to proinflammatory signals in RASFs. In contrast to previous reports (11), TNF-α down-regulated EP4. This difference may be explained by the different cytokines used for stimulation or by different culture conditions. The induction of the enzymes involved in the synthesis of PGE2 (8, 46, 47), in conjunction with the up-regulation of certain EP receptors by proinflammatory cytokines (present study and Refs. 11, 45), suggests that the PGE2 signaling cascade is tightly controlled by proinflammatory cytokines, not only at the level of PGE2 synthesis, but also at the level of expression of particular prostanoid receptor subclasses.
Individual PGE2 receptors differentially modulate the functional effects of TNF-α
Up-regulation of the EP2 receptor by TNF-α points to a prominent role of this receptor for the TNF-α/PGE2-elicited signal in RASFs. This was confirmed by the PGE2/TNF-α-induced increase of intracellular cAMP and phosphorylation of PKA substrates via EP2 (butaprost), but not via EP3 (Fig. 4, A and B), and, to a minor degree, by EP4 (under IBMX; supplemental Fig. 1). The unresponsiveness of intracellular cAMP levels to sulprostone/EP3 stimulation has been reported before for other cell types (48) and likely reflects the fact that EP3 receptors are mostly coupled to Gi proteins. The weaker ability of the EP4 receptor to stimulate cAMP formation compared with EP2 has also been described (45, 50). This may be due to rapid desensitization of the EP4 receptor via internalization (50, 51).
Analysis of the secretion of proinflammatory IL-6 and prodestructive MMP-1 further underlined the dominant role of the EP2 receptor. Although the IL-6 mRNA expression was significantly increased by stimulation of EP2, EP3, and EP4, the magnitude of IL-6 induction via EP2 (>2.5-fold) was larger than via EP3/EP4 (<2.0-fold). This is somewhat in contrast to the results of Inoue et al. (11), who reported that the IL-6 secretion in IL-1α-stimulated RASFs was only induced by agonists for the EP receptors 2 and 4. Explanations include the usage of different proinflammatory cytokines (IL-1β vs TNF-α), different agonist concentrations (20 nM vs 10 μM), and variable EP3 mRNA/protein expression (11). The selective regulation of MMP-1 via the EP2 receptor is a novel observation.
The dominant role of EP2 in the regulation of TNF-α-induced functions of RASFs suggests that the EP2 receptor is a potential therapeutic target in RA. However, the findings presented herein indicate that this point needs to be regarded with caution. In particular, the radically opposite consequences of PGE2/EP2 signaling on TNF-α-induced IL-6 and MMP-1 secretion suggest that a blockade of EP2 activity, while being beneficial in reducing inflammatory parameters, may on the other hand exacerbate tissue destruction. Also, proinflammatory IL-6 is induced by PGE2 predominantly via EP2, but to some degree also by EP3 and EP4, making difficult the exclusive targeting of just one EP receptor.
In agreement with the present data, previous reports have also shown an influence of both EP2 and EP4 on the secretion of IL-6 (11). However, the down-regulation of EP4 by TNF-α suggests a minor role of EP4 in RASFs. On the other hand, a possible involvement of the EP4 receptor in the pathogenesis of RA is supported by results in chondrocytes (52) and in animal models (20, 53, 54, 55). Taken together, the present data indicate that the concerted therapeutic manipulation of both the EP2 and EP4 receptors may represent a promising approach for the treatment of RA.
Supplementary Material
Acknowledgments
Bärbel Ukena, Bianca Lanick (Experimental Rheumatology Unit, University Hospital Jena, Jena, Germany) and Lihua Yang (Kentucky Clinic, University of Kentucky, Lexington, KY) are gratefully acknowledged for technical assistance, and Dr. Ernesta Palombo-Kinne for critical revision of the manuscript. We also thank Ono Pharmaceutical for providing us with PGE2 receptor subtype agonists (ONO-DI-004, ONO-AE1-259, ONO-AE-248, and ONO-AE1-329).
Footnotes
The study was supported by the German Federal Ministry of Education and Research (BMBF; Grants FKZ 01ZZ9602, 01ZZ0105, and 010405 to R.W.K., Interdisciplinary Center for Clinical Research (IZKF) Jena, including a grant for junior researchers to E.K.; Grants FKZ 0312704B and 0313652B to R.W.K., Jena Center for Bioinformatics; Grant 01GS0413, NGFN-2 to R.W.K.), the German Research Foundation (DFG; Grants KI 439/7-1 and KI 439/6-1 to R.W.K.), National Institutes of Health/National Institute of Arthritis and Musculoskeletal and Skin Diseases Grant 1 R01 AR049010 to L.J.C., and a grant for the advancement of female scientists to Elke Kunisch (LUBOM Thuringia). Anne Jansen was supported by a stipend from the Friends of the Friedrich Schiller University Jena, as well as by travel allowances from Jenapharm and the Boehringer Ingelheim Foundation.
Abbreviations used in this paper: RA, rheumatoid arthritis; RASF, rheumatoid arthritis synovial fibroblast; MMP, matrix metalloproteinase; COX, cyclooxygenase; EP, E prostanoid; NS-398, N-(2-cyclohexyloxy-4-nitrophenyl)-methanesulfonamide; IBMX, 3-isobutyl-1-methylxanthine; PKA, protein kinase A.
The online version of this article contains supplemental material.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Firestein GS. Evolving concepts of rheumatoid arthritis. Nature. 2003;423:356–361. doi: 10.1038/nature01661. [DOI] [PubMed] [Google Scholar]
- 2.Kinne RW, Palombo-Kinne E, Emmrich F. Activation of synovial fibroblasts in rheumatoid arthritis. Ann Rheum Dis. 1995;54:501–504. doi: 10.1136/ard.54.6.501-b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yamanishi Y, Firestein GS. Pathogenesis of rheumatoid arthritis: the role of synoviocytes. Rheum Dis Clin N Am. 2001;27:355–371. doi: 10.1016/s0889-857x(05)70206-4. [DOI] [PubMed] [Google Scholar]
- 4.Chomarat P, Rissoan MC, Pin JJ, Banchereau J, Miossec P. Contribution of IL-1, CD14, and CD13 in the increased IL-6 production induced by in vitro monocyte-synoviocyte interactions. J Immunol. 1995;155:3645–3652. [PubMed] [Google Scholar]
- 5.Gay S. Rheumatoid arthritis. Curr Opin Rheumatol. 2001;13:191–192. doi: 10.1097/00002281-200105000-00007. [DOI] [PubMed] [Google Scholar]
- 6.Smolen JS, Steiner G. Therapeutic strategies for rheumatoid arthritis. Nat Rev Drug Discov. 2003;2:473–488. doi: 10.1038/nrd1109. [DOI] [PubMed] [Google Scholar]
- 7.Dayer JM, Krane SM, Russell RG, Robinson DR. Production of collagenase and prostaglandins by isolated adherent rheumatoid synovial cells. Proc Natl Acad Sci USA. 1976;73:945–949. doi: 10.1073/pnas.73.3.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alaaeddine N, Di Battista JA, Pelletier JP, Kiansa K, Cloutier JM, Martel-Pelletier J. Inhibition of tumor necrosis factor α-induced prostaglandin E2 production by the antiinflammatory cytokines interleukin-4, interleukin-10, and interleukin-13 in osteoarthritic synovial fibroblasts: distinct targeting in the signaling pathways. Arthritis Rheum. 1999;42:710–718. doi: 10.1002/1529-0131(199904)42:4<710::AID-ANR14>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 9.Alsalameh S, Amin RJ, Kunisch E, Jasin HE, Kinne RW. Preferential induction of prodestructive matrix metalloproteinase-1 and proinflammatory interleukin 6 and prostaglandin E2 in rheumatoid arthritis synovial fibroblasts via tumor necrosis factor receptor-55. J Rheumatol. 2003;30:1680–1690. [PubMed] [Google Scholar]
- 10.Kojima F, Naraba H, Sasaki Y, Beppu M, Aoki H, Kawai S. Prostaglandin E2 is an enhancer of interleukin-1β-induced expression of membrane-associated prostaglandin E synthase in rheumatoid synovial fibroblasts. Arthritis Rheum. 2003;48:2819–2828. doi: 10.1002/art.11261. [DOI] [PubMed] [Google Scholar]
- 11.Inoue H, Takamori M, Shimoyama Y, Ishibashi H, Yamamoto S, Koshihara Y. Regulation by PGE2 of the production of interleukin-6, macrophage colony stimulating factor, and vascular endothelial growth factor in human synovial fibroblasts. Br J Pharmacol. 2002;136:287–295. doi: 10.1038/sj.bjp.0704705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yoshida T, Sakamoto H, Horiuchi T, Yamamoto S, Suematsu A, Oda H, Koshihara Y. Involvement of prostaglandin E2 in interleukin-1 α-induced parathyroid hormone-related peptide production in synovial fibroblasts of patients with rheumatoid arthritis. J Clin Endocrinol Metab. 2001;86:3272–3278. doi: 10.1210/jcem.86.7.7687. [DOI] [PubMed] [Google Scholar]
- 13.Pillinger MH, Rosenthal PB, Tolani SN, Apsel B, Dinsell V, Greenberg J, Chan ESL, Gomez PF, Abramson SB. Cyclooxygenase-2-derived E prostaglandins down-regulate matrix metalloproteinase-1 expression in fibroblast-like synoviocytes via inhibition of extracellular signal-regulated kinase activation. J Immunol. 2003;171:6080–6089. doi: 10.4049/jimmunol.171.11.6080. [DOI] [PubMed] [Google Scholar]
- 14.Largo R, Diez-Ortego I, Sanchez-Pernaute O, Lopez-Armada MJ, Alvarez-Soria MA, Egido J, Herrero-Beaumont G. EP2/EP4 signalling inhibits monocyte chemoattractant protein-1 production induced by interleukin 1β in synovial fibroblasts. Ann Rheum Dis. 2004;63:1197–1204. doi: 10.1136/ard.2003.011163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Narumiya S, Sugimoto Y, Ushikubi F. Prostanoid receptors: structures, properties, and functions. Physiol Rev. 1999;79:1193–1226. doi: 10.1152/physrev.1999.79.4.1193. [DOI] [PubMed] [Google Scholar]
- 16.Breyer MD, Breyer RM. Prostaglandin E receptors and the kidney. Am J Physiol. 2000;279:F12–F23. doi: 10.1152/ajprenal.2000.279.1.F12. [DOI] [PubMed] [Google Scholar]
- 17.Kotani M, Tanaka I, Ogawa Y, Usui T, Mori K, Ichikawa A, Narumiya S, Yoshimi T, Nakao K. Molecular cloning and expression of multiple isoforms of human prostaglandin E receptor EP3 subtype generated by alternative messenger RNA splicing: multiple second messenger systems and tissue-specific distributions. Mol Pharmacol. 1995;48:869–879. [PubMed] [Google Scholar]
- 18.Akaogi J, Nozaki T, Satoh M, Yamada H. Role of PGE2 and EP receptors in the pathogenesis of rheumatoid arthritis and as a novel therapeutic strategy. Endocr Metab Immune Disord Drug Targets. 2006;6:383–394. doi: 10.2174/187153006779025711. [DOI] [PubMed] [Google Scholar]
- 19.Kapoor M, Kojima F, Qian M, Yang L, Crofford LJ. Microsomal prostaglandin E synthase-1 deficiency is associated with elevated peroxisome proliferator-activated receptor γ: regulation by prostaglandin E2 via the phosphatidylinositol 3-kinase and AKT pathway. J Biol Chem. 2007;282:5356–5366. doi: 10.1074/jbc.M610153200. [DOI] [PubMed] [Google Scholar]
- 20.Kurihara Y, Endo H, Akahoshi T, Kondo H. Up-regulation of prostaglandin E receptor EP2 and EP4 subtypes in rat synovial tissues with adjuvant arthritis. Clin Exp Immunol. 2001;123:323–330. doi: 10.1046/j.1365-2249.2001.01442.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ben Av P, Crofford LJ, Wilder RL, Hla T. Induction of vascular endothelial growth factor expression in synovial fibroblasts by prostaglandin E and interleukin-1: a potential mechanism for inflammatory angiogenesis. FEBS Lett. 1995;372:83–87. doi: 10.1016/0014-5793(95)00956-a. [DOI] [PubMed] [Google Scholar]
- 22.Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, Healey LA, Kaplan SR, Liang MH, Luthra HS. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31:315–324. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- 23.Zimmermann T, Kunisch E, Pfeiffer R, Hirth A, Stahl HD, Sack U, Laube A, Liesaus E, Roth A, Palombo-Kinne E, et al. Isolation and characterization of rheumatoid arthritis synovial fibroblasts from primary culture: primary culture cells markedly differ from fourth-passage cells. Arthritis Res. 2001;3:72–76. doi: 10.1186/ar142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hirth A, Skapenko A, Kinne R, Emmrich F, Schulze-Koops H, Sack U. Cytokine mRNA and protein expression in primary-culture and repeated-passage synovial fibroblasts from patients with rheumatoid arthritis. Arthritis Res. 2002;4:117–125. doi: 10.1186/ar391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Isomaki P, Punnonen J. Pro- and anti-inflammatory cytokines in rheumatoid arthritis. Ann Med. 1997;29:499–507. doi: 10.3109/07853899709007474. [DOI] [PubMed] [Google Scholar]
- 26.Westra J, Limburg PC, De Boer P, Van Rijswijk MH. Effects of RWJ 67657, a p38 mitogen activated protein kinase (MAPK) inhibitor, on the production of inflammatory mediators by rheumatoid synovial fibroblasts. Ann Rheum Dis. 2004;63:1453–1459. doi: 10.1136/ard.2003.013011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.DiBattista JA, Pelletier JP, Zafarullah M, Fujimoto N, Obata K, Martel-Pelletier J. Coordinate regulation of matrix metalloproteases and tissue inhibitor of metalloproteinase expression in human synovial fibroblasts. J Rheumatol. 1995;43(Suppl):123–128. [PubMed] [Google Scholar]
- 28.DiBattista JA, Martel-Pelletier J, Fujimoto N, Obata K, Zafarullah M, Pelletier JP. Prostaglandins E2 and E1 inhibit cytokine-induced metalloprotease expression in human synovial fibroblasts: mediation by cyclic-AMP signalling pathway. Lab Invest. 1994;71:270–278. [PubMed] [Google Scholar]
- 29.Faour WH, He Q, Mancini A, Jovanovic D, Antoniou J, Di Battista JA. Prostaglandin E2 stimulates p53 transactivational activity through specific serine 15 phosphorylation in human synovial fibroblasts: role in suppression of c/EBP/NF-κB-mediated MEKK1-induced MMP-1 expression. J Biol Chem. 2006;281:19849–19860. doi: 10.1074/jbc.M601293200. [DOI] [PubMed] [Google Scholar]
- 30.Mix KS, Attur MG, Al Mussawir H, Abramson SB, Brinckerhoff CE, Murphy EP. Transcriptional repression of matrix metalloproteinase gene expression by the orphan nuclear receptor NURR1 in cartilage. J Biol Chem. 2007;282:9492–9504. doi: 10.1074/jbc.M608327200. [DOI] [PubMed] [Google Scholar]
- 31.Gomez PF, Pillinger MH, Attur M, Marjanovic N, Dave M, Park J, Bingham CO, III, Al Mussawir H, Abramson SB. Resolution of inflammation: prostaglandin E2 dissociates nuclear trafficking of individual NF-κB subunits (p65, p50) in stimulated rheumatoid synovial fibroblasts. J Immunol. 2005;175:6924–6930. doi: 10.4049/jimmunol.175.10.6924. [DOI] [PubMed] [Google Scholar]
- 32.Ruwanpura SMPM, Noguchi K, Ishikawa I. Prostaglandin E2 regulates interleukin-1β-induced matrix metalloproteinase-3 production in human gingival fibroblasts. J Dent Res. 2004;83:260–265. doi: 10.1177/154405910408300315. [DOI] [PubMed] [Google Scholar]
- 33.Noguchi K, Ruwanpura SM, Yan M, Yoshida N, Ishikawa I. Down-regulation of interleukin-1α-induced matrix metalloproteinase-13 expression via EP1 receptors by prostaglandin E2 in human periodontal ligament cells. Oral Microbiol Immunol. 2005;20:56–59. doi: 10.1111/j.1399-302X.2004.00187.x. [DOI] [PubMed] [Google Scholar]
- 34.Noguchi K, Maeda M, Ruwanpura SMPM, Ishikawa I. Prostaglandin E2 (PGE2) downregulates interleukin (IL)-1-induced IL-6 production via EP2/EP4 subtypes of PGE2 receptors in human periodontal ligament cells. Oral Dis. 2005;11:157–162. doi: 10.1111/j.1601-0825.2005.01059.x. [DOI] [PubMed] [Google Scholar]
- 35.Case JP, Lafyatis R, Kumkumian GK, Remmers EF, Wilder RL. IL-1 regulation of transin/stromelysin transcription in rheumatoid synovial fibroblasts appears to involve two antagonistic transduction pathways, an inhibitory, prostaglandin-dependent pathway mediated by cAMP, and a stimulatory, protein kinase C-dependent pathway. J Immunol. 1990;145:3755–3761. [PubMed] [Google Scholar]
- 36.Newcombe DS, Ciosek CP, Jr, Ishikawa Y, Fahey JV. Human synoviocytes: activation and desensitization by prostaglandins and 1-epinephrine. Proc Natl Acad Sci USA. 1975;72:3124–3128. doi: 10.1073/pnas.72.8.3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baker DG, Baumgarten DF, Bomalaski JS, Zurier RB. Cyclic adenosine 3′5′ monophosphate stimulates prostaglandin E production by human adherent synovial cells. Prostaglandins. 1985;30:669–682. doi: 10.1016/0090-6980(85)90028-0. [DOI] [PubMed] [Google Scholar]
- 38.Takeba Y, Suzuki N, Wakisaka S, Takeno M, Kaneko A, Asai T, Sakane T. Involvement of cAMP responsive element binding protein (CREB) in the synovial cell hyperfunction in patients with rheumatoid arthritis. Clin Exp Rheumatol. 2000;18:47–55. [PubMed] [Google Scholar]
- 39.Ross SE, Williams RO, Mason LJ, Mauri C, Marinova-Mutafchieva L, Malfait AM, Maini RN, Feldmann M. Suppression of TNF-α expression, inhibition of Th1 activity, and amelioration of collagen-induced arthritis by rolipram. J Immunol. 1997;159:6253–6259. [PubMed] [Google Scholar]
- 40.Sekut L, Yarnall D, Stimpson SA, Noel LS, Bateman-Fite R, Clark RL, Brackeen MF, Menius JA, Jr, Connolly KM. Anti-inflammatory activity of phosphodiesterase (PDE)-IV inhibitors in acute and chronic models of inflammation. Clin Exp Immunol. 1995;100:126–132. doi: 10.1111/j.1365-2249.1995.tb03613.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Anaya JM, Espinoza LR. Phosphodiesterase inhibitor pentoxifylline: an antiinflammatory/immunomodulatory drug potentially useful in some rheumatic diseases. J Rheumatol. 1995;22:595–599. [PubMed] [Google Scholar]
- 42.Maksymowych WP, Avina-Zubieta A, Luong MH, Russell AS. An open study of pentoxifylline in the treatment of severe refractory rheumatoid arthritis. J Rheumatol. 1995;22:625–629. [PubMed] [Google Scholar]
- 43.Fujino H, West KA, Regan JW. Phosphorylation of glycogen synthase kinase-3 and stimulation of T-cell factor signaling following activation of EP2 and EP4 prostanoid receptors by prostaglandin E2. J Biol Chem. 2002;277:2614–2619. doi: 10.1074/jbc.M109440200. [DOI] [PubMed] [Google Scholar]
- 44.Dendorfer U, Oettgen P, Libermann TA. Multiple regulatory elements in the interleukin-6 gene mediate induction by prostaglandins, cyclic AMP, and lipopolysaccharide. Mol Cell Biol. 1994;14:4443–4454. doi: 10.1128/mcb.14.7.4443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mathieu MC, Lord-Dufour S, Bernier V, Boie Y, Burch JD, Clark P, Denis D, Han Y, Mortimer JR, Therien AG. Mutual antagonistic relationship between prostaglandin E2 and IFN-γ: implications for rheumatoid arthritis. Eur J Immunol. 2008;38:1900–1912. doi: 10.1002/eji.200838170. [DOI] [PubMed] [Google Scholar]
- 46.Crofford LJ. COX-2 in synovial tissues. Osteoarthritis Cartilage. 1999;7:406–408. doi: 10.1053/joca.1999.0226. [DOI] [PubMed] [Google Scholar]
- 47.Kojima F, Naraba H, Miyamoto S, Beppu M, Aoki H, Kawai S. Membrane-associated prostaglandin E synthase-1 is upregulated by proinflammatory cytokines in chondrocytes from patients with osteoarthritis. Arthritis Res Ther. 2004;6:R355–R365. doi: 10.1186/ar1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Carbonne B, Jannet D, Dallot E, Pannier E, Ferre F, Cabrol D. Synthesis of glycosaminoglycans by human cervical fibroblasts in culture: effects of prostaglandin E2 and cyclic AMP. Eur J Obstet Gynecol Reprod Biol. 1996;70:101–105. doi: 10.1016/s0301-2115(96)02536-5. [DOI] [PubMed] [Google Scholar]
- 49.Fujino H, Xu W, Regan JW. Prostaglandin E2 induced functional expression of early growth response factor-1 by EP4, but not EP2, prostanoid receptors via the phosphatidylinositol 3-kinase and extracellular signal-regulated kinases. J Biol Chem. 2003;278:12151–12156. doi: 10.1074/jbc.M212665200. [DOI] [PubMed] [Google Scholar]
- 50.Desai S, April H, Nwaneshiudu C, Ashby B. Comparison of agonist-induced internalization of the human EP2 and EP4 prostaglandin receptors: role of the carboxyl terminus in EP4 receptor sequestration. Mol Pharmacol. 2000;58:1279–1286. doi: 10.1124/mol.58.6.1279. [DOI] [PubMed] [Google Scholar]
- 51.Wilson RJ, Rhodes SA, Wood RL, Shield VJ, Noel LS, Gray DW, Giles H. Functional pharmacology of human prostanoid EP2 and EP4 receptors. Eur J Pharmacol. 2004;501:49–58. doi: 10.1016/j.ejphar.2004.08.025. [DOI] [PubMed] [Google Scholar]
- 52.Fushimi K, Nakashima S, You F, Takigawa M, Shimizu K. Prostaglandin E2 downregulates TNF-α-induced production of matrix metalloproteinase-1 in HCS-2/8 chondrocytes by inhibiting Raf-1/MEK/ERK cascade through EP4 prostanoid receptor activation. J Cell Biochem. 2007;100:783–793. doi: 10.1002/jcb.21099. [DOI] [PubMed] [Google Scholar]
- 53.Akaogi J, Yamada H, Kuroda Y, Nacionales DC, Reeves WH, Satoh M. Prostaglandin E2 receptors EP2 and EP4 are up-regulated in peritoneal macrophages and joints of pristane-treated mice and modulate TNF-α and IL-6 production. J Leukocyte Biol. 2004;76:227–236. doi: 10.1189/jlb.1203627. [DOI] [PubMed] [Google Scholar]
- 54.McCoy JM, Wicks JR, Audoly LP. The role of prostaglandin E2 receptors in the pathogenesis of rheumatoid arthritis. J Clin Invest. 2002;110:651–658. doi: 10.1172/JCI15528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Honda T, Segi-Nishida E, Miyachi Y, Narumiya S. Prostacyclin-IP signaling and prostaglandin E2-EP2/EP4 signaling both mediate joint inflammation in mouse collagen-induced arthritis. J Exp Med. 2006;203:325–335. doi: 10.1084/jem.20051310. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.