

ABSTRACT
CRISPR (clustered regularly interspaced short palindromic repeat)-Cas (CRISPR-associated protein) systems are diverse and found in many archaea and bacteria. These systems have mainly been characterized as adaptive immune systems able to protect against invading mobile genetic elements, including viruses. The first step in this protection is acquisition of spacer sequences from the invader DNA and incorporation of those sequences into the CRISPR array, termed CRISPR adaptation. Progress in understanding the mechanisms and requirements of CRISPR adaptation has largely been accomplished using overexpression of cas genes or plasmid loss assays; little work has focused on endogenous CRISPR-acquired immunity from viral predation. Here, we developed a new biofilm-based assay system to enrich for Pseudomonas aeruginosa strains with new spacer acquisition. We used this assay to demonstrate that P. aeruginosa rapidly acquires spacers protective against DMS3vir, an engineered lytic variant of the Mu-like bacteriophage DMS3, through primed CRISPR adaptation from spacers present in the native CRISPR2 array. We found that for the P. aeruginosa type I-F system, the cas1 gene is required for CRISPR adaptation, recG contributes to (but is not required for) primed CRISPR adaptation, recD is dispensable for primed CRISPR adaptation, and finally, the ability of a putative priming spacer to prime can vary considerably depending on the specific sequences of the spacer.
IMPORTANCE Our understanding of CRISPR adaptation has expanded largely through experiments in type I CRISPR systems using plasmid loss assays, mutants of Escherichia coli, or cas1-cas2 overexpression systems, but there has been little focus on studying the adaptation of endogenous systems protecting against a lytic bacteriophage. Here we describe a biofilm system that allows P. aeruginosa to rapidly gain spacers protective against a lytic bacteriophage. This approach has allowed us to probe the requirements for CRISPR adaptation in the endogenous type I-F system of P. aeruginosa. Our data suggest that CRISPR-acquired immunity in a biofilm may be one reason that many P. aeruginosa strains maintain a CRISPR-Cas system.
INTRODUCTION
CRISPR (clustered regularly interspaced short palindromic repeat)-Cas (CRISPR-associated protein) systems are diverse, widespread, and currently classified into six different types (1, 2) and are found in over 84% and 45% of sequenced archaeal and bacterial genomes, respectively (3). The general function of a CRISPR-Cas system is to provide adaptive immunity against mobile genetic elements (MGEs), including viruses and plasmids (4–6). This CRISPR-mediated immunity is generated through three stages. In the first stage, termed CRISPR adaptation, a small section of invading MGE nucleic acid (usually 30 to 40 bp, termed the protospacer) is incorporated into the CRISPR array as a spacer along with an additional CRISPR repeat. In the second step, CRISPR RNA (crRNA) generation, the CRISPR array is transcribed into long, noncoding RNA and subsequently processed into individual spacers which associate with Cas proteins, forming the crRNA ribonucleoprotein complex. The final step is CRISPR interference, in which the spacer region of the crRNA binds invading MGEs at a complementary site through Watson-Crick base pairing and recruits a Cas nuclease to degrade the bound target (for a recent review, see reference 7).
Our understanding of CRISPR adaptation has greatly increased in the last several years but still lags behind advances in understanding crRNA generation and CRISPR interference. Most CRISPR adaptation research in the type I system focuses on the Escherichia coli type I-E system, but since this CRISPR-Cas system is naturally H-NS silenced, CRISPR adaptation studies have relied on either overexpression of Cas1 and Cas2 in the absence of CRISPR interference (8) or the use of a Δhns strain of E. coli (9). E. coli has also been used to investigate adaptation in the type I-F system through expression of a heterologous type I-F system from Pseudomonas aeruginosa in E. coli (10). These studies have been instrumental in advancing our understanding of the mechanisms and genetic requirements for adaptation, but the approaches used limit our understanding of the role of the CRISPR-Cas system in its native context. The endogenous CRISPR-Cas system of P. aeruginosa and its importance during challenge with DMS3vir have been studied previously (11, 12), and we aim to expand upon these types of experiments by studying the role of the P. aeruginosa CRISPR-Cas system in biofilm growth.
Pseudomonas aeruginosa strain UCBPP-PA14 (here abbreviated P. aeruginosa PA14) contains a type I-F CRISPR-Cas system organized such that the canonical type I-F cas genes are flanked by two CRISPR arrays, termed CRISPR1 and CRISPR2, containing 13 and 21 spacers, respectively (Fig. 1) (13). Our lab has previously shown that this CRISPR-Cas system is active under normal laboratory growth conditions (14) and capable of acquiring protective spacers during challenge with the bacteriophage DMS3vir, a lytic-only variant of the bacteriophage DMS3 (15). However, even though the native CRISPR2 locus contains 3 putative DMS3-targeting priming spacers (spacers whose sequences partially match the phage target sequence and increase the likelihood of incorporation of new spacers against the phage), after phage infection and isolation of resistant strains, less than 1% of these resistant isolates had acquired new spacers, while the vast majority (>99%) had gained resistance through mutation of the type IV pilus (T4P), the known receptor of DMS3vir (16) as well as many other Pseudomonas phages (17). Thus, while coinoculation of P. aeruginosa with DMS3vir in a culture tube results in spacer acquisition, this event is too inefficient to be used as a means of investigating natural CRISPR adaptation in P. aeruginosa, necessitating a strategy for increasing the frequency of native spacer acquisition.
FIG 1.

Cartoon of the type I-F CRISPR-Cas system in Pseudomonas aeruginosa strain UCBPP-PA14. Gene names and spacers numbers are indicated and described in the text.
Given that the T4P is known to play an important role in biofilm formation in P. aeruginosa (18), we hypothesized that cells grown in a biofilm during phage challenge would be less likely to gain resistance to DMS3vir through mutations of T4P. We show here that a biofilm-based enrichment system greatly increases the ability to detect CRISPR adaptation events, allowing us to investigate the efficiency of different priming spacers, protospacer sequences, and the genetic requirements of the P. aeruginosa type I-F system in the context of viral immunity. Furthermore, CRISPR-Cas systems are widespread in P. aeruginosa strains (19, 20); thus, a better understanding of the role of the P. aeruginosa type I-F CRISPR-Cas system in the context of biofilm growth may help explain the prevalence of these systems.
MATERIALS AND METHODS
Strains and media.
The strains, plasmids, and primers used in this study are listed in Table S1 in the supplemental material. P. aeruginosa strain PA14 was used in this study. P. aeruginosa and E. coli strains were routinely cultured in lysogeny broth (LB) at 37°C. The growth media were supplemented with gentamicin (Gm) at the following concentrations: 10 μg ml−1 for E. coli and 50 μg ml−1 for P. aeruginosa. The ΔrecD, ΔrecG, ΔCRISPR1ΔCRISPR2sp1-19, ΔCRISPR1ΔCRISPR2sp1-19 + sp20-5MM, and CRISPR2-minimum strains and deletion plasmids were generated using allelic exchange and Saccharomyces cerevisiae recombineering techniques described previously (21). The recG::TnM strain used here is from the P. aeruginosa PA14 nonredundant library (22), and the transposon insertion was confirmed via PCR.
Biofilm enrichment assay.
The indicated P. aeruginosa strains were grown overnight and standardized to an optical density at 600 nm of 3.0, and then 100 μl of the culture (∼2.5 × 108 CFU) was added to 5 ml of M63 minimal medium supplemented with 0.4% arginine and 1 mM magnesium sulfate (a biofilm-inducing medium) per well in a 6-well tissue culture dish. The P. aeruginosa strains indicated below were coinoculated with 2.5 × 106 PFU of the DMS3vir phage in each well (multiplicity of infection, 0.01) and grown overnight at 37°C. After incubation, the medium was aspirated and the wells were washed once with 5 ml of phosphate-buffered saline (PBS), removing any planktonic bacteria and leaving behind only the biofilm population. The biofilm population was isolated by using a 25-cm Sarstedt cell scraper to remove any attached bacterial cells at the air-liquid interface, and the cells were subsequently collected in 1 ml of PBS. These cells were then washed with 1 ml PBS and resuspended in 50 μl of PBS, and the entire 50 μl was inoculated into 5 ml LB and grown for either 10 h (for the slow-growing ΔrecD mutant) or 6 h (for all other strains) at 37°C. After the biofilm was allowed to form, the cells were exposed to fresh DMS3vir at concentrations and under conditions exactly the same as those described above. After coincubation of the biofilm cells and phage, the remaining biofilms cells were collected as described above. Additionally, an aliquot of the planktonic population from the wild-type (WT) P. aeruginosa plus DMS3vir condition was collected prior to aspiration. Both planktonic and biofilm cells were serially diluted, plated on LB agar, and incubated at 37°C for 16 h to yield single colonies. For each condition, at least 200 single colonies were repatched onto LB agar using a sterile pipette tip and grown for 16 h at 37°C to assess their twitching phenotype, as describe in the next section.
Twitch assay.
The twitch assay was performed as described previously (23). Briefly, a sample from repatched isolates not displaying any bacteriophage-mediated lysis (as judged by a lack of obvious plaques) was collected using a sterile pipette tip, punctured through LB agar, and deposited onto the hard plastic of the petri plate. The samples were then allowed to grow for 36 h at 37°C, at which point the LB agar was carefully peeled away. The bacteria were visualized with the addition of 0.1% crystal violet. Twitch-positive strains move along the plastic away from the inoculation point, while twitch-negative cells remain at the inoculation point.
Detecting newly acquired spacers.
Any repatched isolates from the biofilm-based spacer enrichment assay not showing signs of bacteriophage-mediated lysis and displaying a twitch-positive phenotype were used as a template for PCR, using primers amplifying the leader end of either the CRISPR1 or CRISPR2 array. For the isolates indicated below, one of the same primers was used for Sanger sequencing to characterize the newly acquired spacers.
Statistical analysis.
The data presented in Fig. 3 were analyzed using GraphPad Prism (version 5) software. The data represent the means ± standard deviations from three independent experiments with multiple replicates. All data were treated as normally distributed, and comparisons were tested with Student's t test.
FIG 3.

Number of viable P. aeruginosa cells in the planktonic and biofilm populations after the biofilm enrichment assay. After both the first (A) and second (B) 24-h challenge in biofilm-inducing medium, 1 ml of the planktonic culture was collected, serially diluted, and plated to measure the number of CFU. Additionally, after both the first (C) and second (D) 24-h challenge in biofilm-inducing medium, the total biofilm population at the air-liquid interface in each well was isolated using a cell scraper, washed, resuspended in PBS, serially diluted, and plated to measure the number of CFU. Error bars represent standard deviations from three replicates. *, significant difference (P < 0.05, Student's t test) of the specified condition from the equivalent condition without DMS3vir; ns, no significant difference.
RESULTS
P. aeruginosa rapidly gains resistance to the lytic bacteriophage DMS3vir under biofilm-inducing conditions.
The biofilm enrichment system involves two consecutive incubations of P. aeruginosa and DMS3vir under biofilm-inducing conditions. All the bacterial cells used to inoculate the medium for the second biofilm incubation were isolated from the biofilm formed at the end of the first incubation. Because the biofilm cells are exposed to phage DMS3vir, this approach strongly enriches for isolates that efficiently form biofilms and that are resistant to DMS3vir infection. The enrichment procedure is outlined in Fig. 2.
FIG 2.

Schematic of biofilm enrichment assay. Pseudomonas aeruginosa and DMS3vir were coinoculated at a multiplicity of infection of 0.01 using 2.5 × 108 CFU and 2.5 × 106 PFU of P. aeruginosa and DMS3vir, respectively, during each of the two biofilm incubations.
To assess the impact of phage infection on the WT strain and a strain lacking the CRISPR-Cas region, 2.5 × 108 CFU of either P. aeruginosa or P. aeruginosa in which the entire CRISPR-Cas region was deleted (ΔCR) was added either alone or with 2.5 × 106 PFU of DMS3vir to 5 ml of biofilm-inducing minimal medium in a 6-well plate. After 24 h of static growth at 37°C, both the biofilm and planktonic populations were isolated and aliquots from each population were serially diluted and plated to measure the amount of viable bacteria (the number of CFU). As illustrated in Fig. 3, DMS3vir infection significantly reduced the levels of both WT and ΔCR P. aeruginosa cells in the planktonic population (Fig. 3A) as well as the biofilm population (Fig. 3C) after 24 h of incubation. The planktonic population of WT and ΔCR cells grown with DMS3vir had, on average, 91% and 85% fewer cells, respectively, than the planktonic population of WT and ΔCR cells grown without DMS3vir (Fig. 3A). Similarly, the biofilm population of WT and ΔCR cells grown with DMS3vir had, on average, 90% and 97% fewer cells, respectively, than the biofilm population grown without DMS3vir (Fig. 3C).
The biofilm cells isolated after the first incubation step were subsequently grown planktonically for 6 h in LB at 37°C to generate enough cells to inoculate the second biofilm assay plates required for our enrichment protocol. After the 6 h of planktonic growth, the WT and ΔCR cells were added to 5 ml of a biofilm-inducing minimal medium either with or without fresh DMS3vir at the same counts used in the assay described above. After 24 h of growth at 37°C, both the biofilm and planktonic populations were again isolated, and aliquots were serially diluted and plated to measure the number of CFU. In contrast to the results from the first round of incubation (Fig. 3A and C), there was no significant decrease in the number of either WT or ΔCR cells incubated with DMS3vir compared to the number of cells grown in the absence of the bacteriophage in the planktonic population (Fig. 3B), indicating that both the WT and ΔCR planktonic cells had become resistant to DMS3vir-mediated lysis. Interestingly, while the biofilm population of WT cells grown with DMS3vir was not significantly different from the biofilm population of WT cells grown alone, the biofilm population of ΔCR cells grown with DMS3vir had, on average, 79% fewer cells than the population of ΔCR cells grown alone, a significant (P < 0.05) decrease (Fig. 3D). It should be noted that while the difference between the WT biofilm and WT plus DMS3vir biofilm population was not significant, there were still, on average, 40% fewer cells in the WT plus DMS3vir population, and the variability of the assay may partially contribute to the lack of significance measured by Student's t test. Nevertheless, these data indicate that both WT and ΔCR P. aeruginosa can rapidly gain resistance to DMS3vir during biofilm growth and that the P. aeruginosa cells with a functional CRISPR-Cas system had a slight but reproducible advantage over CRISPR-deficient P. aeruginosa cells when growing in a biofilm in the presence of a lytic phage.
Pseudomonas aeruginosa incorporates spacers protective against lytic bacteriophage DMS3vir under biofilm-inducing conditions.
The biofilm-specific increase of WT versus ΔCR P. aeruginosa cells when cells were grown in the presence of DMS3vir suggested that the WT cells gained resistance to DMS3vir through spacer acquisition, in addition to or perhaps in preference to through T4P mutations, especially given that T4P mutations negatively impact the ability of P. aeruginosa to form a biofilm. To test this idea, after isolation and serial dilution plating of the biofilm population from the second biofilm incubation step (Fig. 2), at least 200 colonies for each condition were repatched onto LB agar in three separate assays. Not all cells in the biofilm gained resistance to DMS3vir, and to eliminate nonresistant cells, any isolate that showed signs of lysis from DMS3vir particles that had been retained from the infection step were eliminated from further analysis (Fig. 2; compare nonresistant [NR] versus resistant [R] patches). All the remaining isolates were then assayed for twitching motility, as described previously (24) and as shown in Fig. 2. Isolates that had gained DMS3vir resistance through a T4P mutation were easily distinguishable due to their lack of twitching, and any isolate that was twitch positive was presumed to have acquired a new spacer that blocked infection and thus was screened for spacer acquisition using primers that amplify the leader-proximal ends of both CRISPR1 and CRISPR2.
Unlike the low rate (>1%) of spacer acquisition previously observed during P. aeruginosa and DMS3vir coincubation (15), on average, 37.7% of the DMS3vir-resistant isolates of the WT strain had acquired new spacers, while the remaining 62.3% had gained resistance through the T4P mutation (Fig. 4A). The biofilm-resistant population of the ΔCR mutant strain was 100% twitch negative in these experiments, suggesting that no other means of resistance besides a mutation in the T4P apparatus was acquired.
FIG 4.

Resistance to DMS3vir in the biofilm enrichment assay is gained through either type IV pilus loss of function or Cas1-dependent spacer acquisition. After the second biofilm incubation in the biofilm enrichment assay, both the biofilm population and the planktonic population of P. aeruginosa were plated for single colonies. At least 200 colonies under each specified condition from at least two replicates were repatched on LB agar. (A) Isolates that were resistant to DMS3vir and twitch negative were scored as a type IV pilus (T4P) mutant, while isolates that were resistant to DMS3vir, that were twitch positive, and that demonstrated spacer acquisition, as determined by PCR of the CRISPR arrays, were scored as spacer acquisition positive. Type IV pilus mutant and spacer acquisition-positive isolates are displayed under each condition as a percentage of the total DMS3vir resistant population under that condition. (B) After 24 h of coincubation of either WT or CRISPR-deficient (ΔCR) P. aeruginosa isolates with DMS3vir at a multiplicity of infection of 0.01 at 37°C in 5 ml of LB, 200 single colonies from two replicates under each condition were isolated, scored, and displayed as described in the legend to panel A. (C and D) One hundred randomly selected spacer acquisition-positive isolates from the WT and DMS3vir coincubation conditions in the biofilm enrichment assay were scored for insertion of either a single spacer or multiple spacers (C) and insertion of a new spacer into the CRISPR1, CRISPR2, or both CRISPR1 and CRISPR2 arrays (D), as determined by PCR.
Cas1 is required for new spacer acquisition.
Cas1 is universally conserved across CRISPR-Cas systems and has been shown to be required for CRISPR adaptation (8, 25), including for a heterologous P. aeruginosa type I-F system overexpressed in E. coli (10) and for the endogenous type I-F system of Pectobacterium atrosepticum during CRISPR adaptation against plasmids (26). To test the requirement for endogenous Cas1 in CRISPR adaptation by P. aeruginosa versus a lytic phage, a Δcas1 mutant was assayed for spacer acquisition using the biofilm enrichment system. Similar to the CRISPR-deficient ΔCR mutant, all of the resistant isolates after the second biofilm incubation had lost T4P functionality (Fig. 4A), confirming that the cas1 gene is required for CRISPR adaptation by P. aeruginosa challenged with lytic bacteriophage.
Biofilm growth enhances spacer acquisition.
The underlying premise of our assay is that growth in a biofilm should enhance the frequency of spacer acquisition by selection for the subpopulation of cells with functional T4P, a cell appendage required for robust biofilm formation (18). It has been shown previously that P. aeruginosa more likely gains resistance to DMS3vir through CRISPR adaptation rather than through mutations in genes encoding T4P during coincubation in a minimal medium (11). Therefore, to determine if the CRISPR adaptation frequency of ∼38% observed in WT P. aeruginosa is specifically the result of biofilm growth and not simply growth in a minimal medium (previous assays were performed in rich LB medium), the planktonic population of WT P. aeruginosa incubated with DMS3vir isolated after the second incubation step was assayed for CRISPR adaptation in the same manner as the biofilm population in three separate experiments. Of the resistant planktonic cells, on average, 20.3% had gained resistance through CRISPR adaptation, with the remaining 79.7% gaining resistance through T4P mutations (Fig. 4A), significantly less (P = 0.01) than the CRISPR adaptation frequency (37.7%) observed in the resistant biofilm population (see Table S2 in the supplemental material for the raw data used in the statistical analysis).
Since the level of spacer acquisition in the planktonic population was higher than what was previously observed after coinoculating P. aeruginosa with DMS3vir in LB medium overnight at 37°C with shaking (15), the culture tube-based P. aeruginosa and DMS3vir coincubation was repeated using WT and ΔCR P. aeruginosa isolates in LB medium to confirm the low adaptation frequency observed previously. Of the >300 resistant isolates, 2.3% of the WT P. aeruginosa isolates and 0% of the ΔCR isolates had gained resistance through CRISPR adaptation, with the remaining isolates gaining resistance through T4P mutation (Fig. 4B), confirming the low frequency of spacer acquisition in rich medium for planktonically growing bacteria.
Taken together, these data suggest that P. aeruginosa isolates grown planktonically and challenged with DMS3vir acquire resistance to DMS3vir via CRISPR adaptation at a much higher rate (20% versus 2%) when grown in a minimal medium with static growth than when grown in a rich medium with shaking. Furthermore, the highest rate of CRISPR adaptation to DMS3vir (38%) was observed in the biofilm population of cells grown in a minimal medium, supporting our strategy for increasing the rate of spacer acquisition in P. aeruginosa by enriching for cells with a functional T4P during challenge with DMS3vir.
Multiple spacers are preferentially incorporated with a biased incorporation into the CRISPR2 array.
Of the P. aeruginosa isolates that incorporated new spacers providing resistance to DMS3vir, 100 were randomly selected to assay the specific number of spacers acquired and to determine into which CRISPR array the spacers were inserted. A strong bias for multiple CRISPR insertions was observed in our system, with only 27 out of 100 isolates inserting a single spacer (Fig. 4C). These data are similar to observations in the type I-F system of P. atrosepticum; during CRISPR adaptation against a transformed plasmid, only 24% (9/37) of plasmid-insensitive strains inserted a single spacer (27), suggesting similarities in CRISPR adaptation against a plasmid and versus a lytic bacteriophage in the type I-F system. Multiple incorporated spacers are likely important since it has been shown in P. aeruginosa that spacer diversity against DMS3vir is beneficial to the survival of the bacteria (12).
Additionally, of the 100 resistant isolates, 76 inserted new spacers into only CRISPR2, 5 inserted new spacers into only CRISPR1, and 19 inserted new spacers into both CRISPR1 and CRISPR2 (Fig. 4D), demonstrating that CRISPR2 and CRISPR1 are both functional in CRISPR interference, although there appears to be a bias for inserting new spacers into CRISPR2 in these experiments, similar to what had previously been observed in P. aeruginosa (11). These data are also similar to observations in the type I-F system of P. atrosepticum; of 105 new spacers inserted into three CRISPR arrays in P. atrosepticum, 65%, 32%, and <3% were found in CRISPR1, CRISPR2, and CRISPR3, respectively (27), further suggesting common themes of the type I-F system regardless of target (plasmid versus lytic bacteriophage) or host (P. atrosepticum versus P. aeruginosa).
Spacers incorporated using the biofilm enrichment system are the result of primed CRISPR adaptation.
Recent studies of the type I-E CRISPR-Cas system of E. coli separate type I CRISPR adaptation into two distinct types. In the first type, termed naive adaptation, new spacers are incorporated into a CRISPR array in the absence of CRISPR interference components. This naive adaptation was demonstrated via the overexpression of the cas1 and cas2 genes (8, 28). In the second type of adaptation, termed primed adaptation, a nucleic acid target is bound by a crRNA ribonucleoprotein complex that cannot engage in CRISPR interference due to mismatches between the protospacer and the spacer or a nonconsensus protospacer adjacent motif (PAM), but the presence of this mismatched spacer results in rapid and efficient spacer acquisition from regions proximal to the priming protospacer (25, 29).
P. aeruginosa contains three spacers in the native CRISPR2 array that can potentially participate in primed adaptation against bacteriophage DMS3, as illustrated in Fig. 5. The first, CRISPR2 spacer 20 (CR2_sp20), is 100% complementary to DMS3 but cannot interfere due to a nonconsensus PAM (AG instead of GG). The second, CR2_sp17, is partially complementary to DMS3 with 5 mismatches, including 4 in the seed region, and, in addition, targets a protospacer with a nonconsensus PAM (GA instead of GG). It should be noted that the mismatches between CR2_sp17 and DMS3 likely exist because of a single nucleotide deletion on the phage genome; if a thymine is added where the identity between the spacer and target ends, CR2_sp17 would have a 100% match with the correct PAM. The third, CR2_sp1, is partially complementary to DMS3 with 5 mismatches, but the target has a consensus PAM. Notably, binding of CR2_sp1 to the DMS3 protospacer inserted on the P. aeruginosa chromosome is responsible for the CRISPR-dependent modification of biofilm formation reported previously by our group (14).
FIG 5.
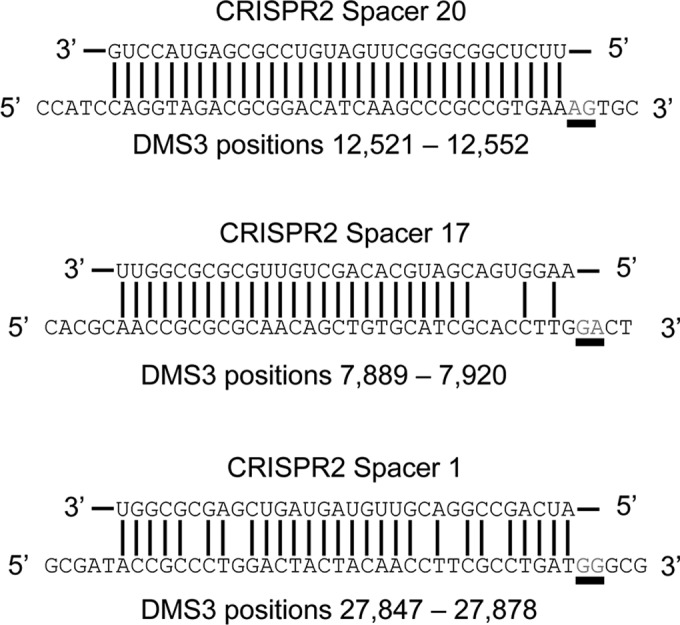
The three putative priming spacers located in the native CRISPR2 locus of P. aeruginosa strain UCBPP-PA14 (top sequences) along with their cognate DMS3 target (bottom sequences). The PAM position is underlined, and the DMS3 location listed indicates the position of the 32-bp protospacer in the DMS3 genome.
To assay if the CRISPR adaptation observed in the biofilm enrichment system is the result of primed adaptation or naive adaptation, a mutant was generated in which the CRISPR1 array was deleted and all spacers in the CRISPR2 array were deleted, leaving only a single repeat and the leader sequence (see Fig. S1 in the supplemental material), since a single repeat and leader sequence has been shown to be sufficient for naive adaptation in the E. coli type I-E system (8). Therefore, any newly acquired spacer in this mutant background would be the result of naive adaptation. This construct, termed CRISPR-minimum, was then analyzed via the biofilm enrichment assay. To increase the likelihood of detection of an acquired spacer, the assay was modified slightly in that rather than repatching the colonies (step 6 in Fig. 2), the bacteria were plated on freshly poured LB agar (therefore, the plates had a higher moisture level than typical plates) so that any twitching-positive colony developed a distinct rough appearance, as previously reported (15), and could therefore be easily identified. While this modification of the assay prevents the determination of the rates of T4P mutations in resistant isolates, since DMS3vir-resistant isolates are indistinguishable from non-DMS3vir-resistant isolates and not every twitch-positive colony displays the rough appearance, it allows the rapid screening of a large number of isolates for putative spacer acquisition events. Over 10,000 colonies were screened, and 768 rough colonies were assayed for spacer acquisition by PCR, with no new spacers being detected. Therefore, since no naive adaptation was detected, our data suggest that primed adaptation is the predominant mode of spacer acquisition in the P. aeruginosa type I-F CRISPR-Cas system.
To determine which of the three putative priming spacers illustrated in Fig. 5 contribute to primed adaptation, the CRISPR arrays of 35 of the CRISPR-positive resistant bacteria isolated from the biofilm enrichment system were sequenced, resulting in the characterization of 87 new spacers (see Table S3 in the supplemental material). Of the 87 spacers examined, the vast majority (84/87) were likely primed by CR2_sp1, on the basis of their distribution around the CR2_sp1 DMS3 target (see Table S3). This finding is similar to that of previous work, in which the vast majority of newly acquired spacers in resistant P. aeruginosa isolates after coincubation with DMS3vir were likely primed by CR2_sp1 (11). Previously, when the P. aeruginosa type I-F CRISPR-Cas system was overexpressed in E. coli, newly acquired primed spacers targeting regions up to 5,000 bp from the location of the priming protospacer were detected (10). In our system, putative primed spacers targeting regions up to 6,000 bp away from the CR2_sp1 target were detected, suggesting that the long range of type I-F CRISPR priming observed previously in E. coli is not due to an artifact of heterologous expression. Additionally, of these 84 CR2_sp1-primed spacers, only 4 targeted a region of DMS3 with a nonconsensus PAM (GC, AG, TG, and GC in spacer numbers 11, 27, 33, and 69, respectively), with three of these spacers (spacers 11, 27, and 33) targeting a region of DMS3 with a consensus PAM shifted 1 nucleotide away (i.e., GG in the +1/−1 or −2/−3 position instead of the canonical −1/−2 position), an occurrence observed in other type I CRISPR systems and termed “PAM slippage” (27, 30).
The remaining 3 spacers that we identified from this collection of 87 spacers were potentially primed by spacer CR2_sp20. The first two of these spacers target DMS3 within 250 bp of the CR2_sp20 target, and the third spacer targets DMS3 within 3,000 bp (see Table S3 in the supplemental material). All three of these putative CR2_sp20-primed spacers were found in the same isolate, strongly suggesting that spacer incorporation likely occurred only once in these resistant isolates, with the number of insertions being dependent on how many new spacers were acquired during a single priming event, in line with the model proposed in the type I-F system of P. atrosepticum (27).
Spacer acquisition occurs in a biased manner in the biofilm enrichment assay.
A key difference between type I-E- and type I-F-primed spacer acquisition is the biases observed during protospacer selection. It was shown in the type I-E CRISPR-Cas system of E. coli that during CRISPR priming, new spacers that target the same strand targeted by the priming spacer are incorporated (9, 25). Conversely, in type I-F CRISPR priming, it was demonstrated that acquired spacers show no bias in respect to the targeted versus nontargeted strand but, instead, that new spacers preferentially target sites 5′ of the priming spacer target on either the targeted or the nontargeted strand (10, 11, 27); the same bias has also been reported in the type I-B system (31).
Of the 83 CR2_sp1 putatively primed spacers that target DMS3 within 6,000 bp of the CR2_sp1 target, protospacer selection was similar to that in other type I-F systems. There was minimal bias between the target strand and the nontarget strand, with 53% of new spacers targeting the same strand as CR2_sp1 and 46% targeting the opposite strand (Fig. 6). However, there was an observed bias between protospacers selected in the 3′ or 5′ direction relative to the CR2_sp1 target, with 73% of new spacers targeting a protospacer present in the 5′ direction relative to the CR2_sp1 target and only 27% targeting a protospacer in the 3′ direction of the CR2_sp1 target (Fig. 6). These data are similar to what has previously been shown with endogenous type I-F CRISPR adaptation against a lytic bacteriophage (11) and confirm that the protospacer selection biases previously observed reflect a general property of the type I-F system.
FIG 6.
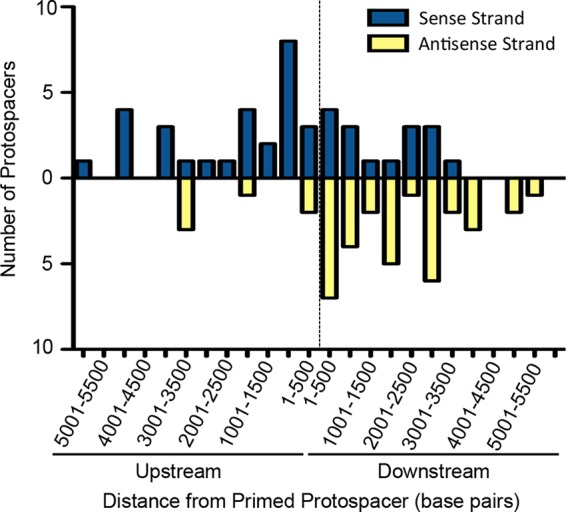
DMS3 targets of newly acquired spacers demonstrate a bias. The 83 sequenced spacers acquired by WT P. aeruginosa, when incubated with DMS3vir in the biofilm enrichment assay, within 6,000 bp of the CRISPR2 spacer 1 (CR2_sp1) target are displayed as a function of both the distance from the CR2_sp1 target and which DMS3 strand (sense or antisense) that each targets. The dashed line represents the CR2_sp1 target location, with the x axis representing the distance from the CR2_sp1 target, divided into segments of 500 bp either upstream or downstream of the CR2_sp1 target. Each bar represents the number of spacers targeting within that particular 500-bp segment of DMS3, with blue bars representing targets on the positive-sense DMS3 strand and with the yellow bars representing targets on the negative-sense DMS3 strand.
RecD is dispensable for primed acquisition in the P. aeruginosa type I-F CRISPR-Cas system, while RecG contributes to efficient priming.
Recent work has demonstrated the involvement of both the RecBCD complex and the RecG protein in naive and primed CRISPR adaptation, respectively (28, 32). The RecBCD complex is required for naive adaptation in the type I-E system of E. coli and likely contributes to adaptation through single-stranded DNA generation at double-stranded breaks, such as those encountered during replication, thereby providing a substrate for Cas1 (28). Regarding primed adaptation, in the E. coli type I-E system, RecG and PriA were shown to be required for primed adaptation, presumably through R-loop removal after crRNA binding, allowing the Cas1-Cas2 complex access to the single-stranded DNA generated by Cas3 (32). We tested if these proteins were playing a similar role in the type I-F system of P. aeruginosa using the biofilm enrichment system and hypothesized that since all spacer acquisition in our system is likely primed, deletion of the recD gene would not impact spacer acquisition, while deletion of the recG gene would impact new spacer insertion.
A deletion of the recD gene was generated in P. aeruginosa, and this mutant strain was incubated with DMS3vir in the biofilm enrichment scheme outlined in Fig. 2. Of note, during the LB growth step between the two biofilm incubations, the cells carrying the recD mutation were incubated for 10 h instead of 6 h due to a slight growth defect caused by the deletion of the recD gene; otherwise, the biofilm enrichment was performed as described above. At least 200 colonies were repatched after isolation of the biofilm population from the second incubation, and of the isolates that had gained resistance to DMS3vir, 30.1% had gained resistance through spacer acquisition, while the remaining 69.9% were T4P mutants (Fig. 7), similar to the spacer acquisition frequency observed for the WT population described above (Fig. 6). To test if the acquired spacers were primed, the CRISPR arrays of 7 ΔrecD isolates with newly acquired spacers were sequenced, resulting in the characterization of 14 newly acquired spacers. As listed in Table S4 in the supplemental material, CR2_sp1 likely primed all 14 newly acquired spacers, supporting the hypothesis that RecD and, presumably, the RecBCD complex are dispensable for primed adaptation in the type I-F CRISPR-Cas system of P. aeruginosa.
FIG 7.
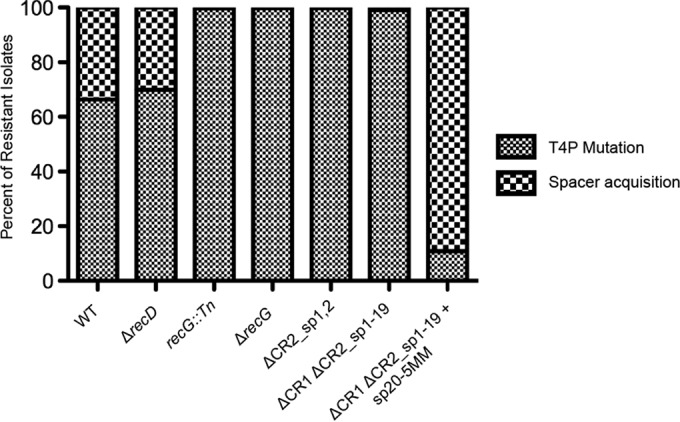
Contributions to new spacer acquisition. RecD is dispensable for primed spacer acquisition, RecG and CRISPR2 spacer 1 are necessary for efficient primed spacer acquisition, and CRISPR2 spacer 20 is sufficient for primed spacer acquisition when the mismatches are in positions similar to those of CRISPR2 spacer 1 in the biofilm enrichment assay. After the second challenge in the biofilm enrichment assay, the biofilm population of P. aeruginosa (either the WT or the specified mutant) coincubated with either DMS3vir (first six columns) or DMS3vir-PC (last column on the right) was plated for single colonies. At least 200 colonies under each specified condition from at least two replicates were repatched on LB agar. Isolates that were resistant to DMS3vir and twitch negative were scored as type IV pilus (T4P) mutants, while isolates that were resistant to DMS3vir, twitch positive, and demonstrated spacer acquisition, as determined by PCR of the CRISPR arrays, were scored as spacer acquisition positive. The type IV pilus mutant and spacer acquisition-positive isolates from each condition are displayed as a percentage of the total DMS3vir-resistant population under that condition.
To test the requirement for RecG in primed CRISPR adaptation, a P. aeruginosa strain with a transposon stably inserted into the recG gene was assayed in the biofilm enrichment assay at the same counts of bacteria and bacteriophages described above (Fig. 2). Of the resistant mutants isolated from the biofilm population after the second incubation, 99.6% were T4P mutants, while 0.4% (two isolates) had gained resistance through CRISPR adaptation (Fig. 7). The CRISPR arrays of these two isolates were sequenced, and the 4 newly acquired spacers were likely primed by CR2_sp1 (see Table S4 in the supplemental material). To confirm that these data were not due to any polar effect of the transposon in recG, an in-frame deletion of recG was generated and assayed in the biofilm enrichment assay at the same counts of bacteria and bacteriophages described above. Of 479 resistant isolates, 99.6% were T4P mutants, with only 2 isolates (0.4%) gaining resistance through spacer acquisition, confirming that while RecG is necessary for efficient CRISPR adaptation, priming can still occur in a recG mutant strain, albeit at a greatly reduced rate.
Spacers incorporated using biofilm enrichment are biased toward CR2_sp1 priming over CR2_sp20 priming due to the specific location of mismatches between the spacer and the target.
One observation made using the biofilm enrichment assay was that almost all of the newly incorporated sequenced spacers were apparently primed by CR2_sp1. The CRISPR2 array of isolate 33 listed in Table S3 in the supplemental material contains three newly acquired spacers which were likely primed by CR2_sp20 due to the proximity of the spacer targets on DMS3 to the target of CR2_sp20. This is the only example obtained using the biofilm enrichment assay of a spacer other than CR2_sp1 likely mediating priming, and we hypothesized that deletion of CR2_sp1 would enrich for alternatively primed spacer acquisitions. A strain in which the first two spacers of CRISPR2 were deleted (the ΔCR2_sp1,2 strain) was coinoculated with DMS3vir in the biofilm enrichment assay under the same counts of bacteria and bacteriophage described above. Interestingly, CRISPR adaptation was greatly reduced without CR2_sp1, with only a single isolate (0.3%) of the resistant biofilm population gaining resistance through spacer acquisition (Fig. 7). This isolate was sequenced, and the resulting single acquired spacer appeared to be primed by CR2_sp20 since the target of the new spacer was only 1.2 kb upstream of the CR2_sp20 DMS3 target. These data suggest that a large difference in priming efficiency can exist between two priming spacers in an endogenous type I-F CRISPR-Cas system.
One possibility for the reduced rate of spacer acquisition observed with the ΔCR2_sp1,2 strain is the distance between CR2_sp20 and the leader sequence of CRISPR2, since it has been observed in Pyrococcus furiosus that crRNA from the spacers closest to the leader accumulates to a higher level than crRNA from spacers farther from the leader sequence (33). To test if the lower ability of CR2_sp20 to prime is related to the distance of CR2_sp20 to the CRISPR2 leader sequence, a strain was generated in which the entire CRISPR1 array and CRISPR2 spacers 1 to 19 were deleted (the ΔCR1 ΔCR2_sp1-19 strain; see Fig. S1 in the supplemental material), resulting in CR2_sp20 being the spacer closest to the CRISPR2 leader sequence. To confirm that CR2_sp20 was functional in this strain, a mutant of DMS3vir was generated in which the CR2_sp20 target contained the correct PAM (GG) instead of the nonfunctional PAM (AG) normally present in wild-type DMS3. This A-to-G mutation is synonymous and therefore unlikely to affect the ability of the phage to infect P. aeruginosa. This mutant phage, referred to as DMS3vir-PC (in reference to PAM corrected), should therefore not be able to infect the ΔCR1 ΔCR2_sp1-19 strain (as measured by plaque assay) if CR2_sp20 is functional due to CRISPR interference. As illustrated in Fig. S2, DMS3vir-PC could infect CRISPR-deficient P. aeruginosa (the ΔCR strain) but not the ΔCR1 ΔCR2_sp1-19 strain, confirming that CR2_sp20 was still functional.
The ΔCR1 ΔCR2_sp1-19 strain was assayed in the biofilm enrichment assay at the same counts of bacteria and bacteriophages described above. Interestingly, even with CR2_sp20 being proximal to the leader, the rate of spacer acquisition was still low, with 99% (513 out of 518) of the resistant isolates gaining resistance to DMS3vir through T4P mutations and only 1% (5 out of 518) of the isolates gaining resistance to DMS3vir through spacer acquisition (Fig. 7, bar second from the right).
The CRISPR array of isolates of the ΔCR1 ΔCR2_sp1-19 mutant that gained resistance through spacer acquisition was sequenced to determine the location on the DMS3 genome from which the newly acquired spacers were derived. As expected, three of the isolates had spacers derived from target regions close to the target of CR2_sp20 (see Table S4 in the supplemental material); however, all of the spacers in isolate 4 target regions of DMS3 that were at least 14.5 kb from the target region of CR2_sp20. Interestingly, the region of DMS3 targeted by these new spacers, while far from the target region of CR2_sp20, was very close (about 1,000 bp) to the target region of CR2_sp1, even though CR2_sp1 was deleted in this strain, suggesting that the region near the CR2_sp1 target is potentially more amenable to spacer acquisition.
An alternate explanation as to why CR2_sp1 is more efficient at priming than CR2_sp20 is the difference in sequences that prevent CRISPR interference. While CR2_sp1 has 5 mismatches between the spacer and the protospacer, CR2_sp20 instead has only a single mutation in the PAM. Therefore, to test the hypothesis that CR2_sp20 would be more efficient at priming if it had sequence changes at a position similar to that with sequence changes in CR2_sp1, a version of the ΔCR1 ΔCR2_sp1-19 strain in which CR2_sp20 had mismatches at positions identical to the locations of mismatches in CR2_sp1, referred to as ΔCR1 ΔCR2_sp1-20 + sp20-5MM (for 5 mismatches), was constructed and is illustrated in Fig. S3 in the supplemental material. This strain was assayed in the biofilm enrichment assay at the same counts of bacteria and bacteriophages described above, with a key difference being that DMS3vir-PC was used instead of DMS3vir. We used DMS3vir-PC here because, to be similar to CR2_sp1, CR2_sp20-5MM would need to target a region of DMS3 with the correct PAM. For the biofilm enrichment assay, three biological replicates were performed with at least 200 isolates each. Strikingly, unlike the low rate of CRISPR adaptation observed with the ΔCR1 ΔCR2_sp1-19 strain (5 out of 518 resistant isolates), with the ΔCR1 ΔCR2_sp1-20 + sp20-5MM strain, 424 out of 476 resistant isolates gained resistance through spacer acquisition (89%) (Fig. 7). This was the highest rate of CRISPR adaptation in resistant isolates observed among isolates of any strain, including the wild-type P. aeruginosa strain.
Overall, these data suggest that certain spacers are much better at priming than others and that one reason for the variability is the specific sequences causing mismatches between the spacer and the protospacer, rather than the location of the target or the overall sequence of the spacer. Furthermore, primed adaptation is the predominant (and perhaps only) mode of spacer acquisition in the P. aeruginosa type I-F CRISPR-Cas system, as we were unable to detect naive adaptation using the biofilm enrichment assay.
DISCUSSION
By developing a strategy of enriching for biofilm-grown cells during challenge with the lytic bacteriophage DMS3vir, we have reduced the frequency of phage receptor mutations dominating the resistant population. We also developed an assay system that allows robust spacer acquisition through endogenous expression of the P. aeruginosa type I-F CRISPR system. This assay system allowed us to characterize the genetic requirements of primed CRISPR adaptation against a lytic bacteriophage and find that the cas1 gene is required for CRISPR adaptation, the recG gene contributes to efficient primed CRISPR adaptation, and the recD gene is dispensable for primed CRISPR adaptation. Furthermore, due to the presence of 3 putative priming spacers against DMS3vir in the native CRISPR2 array in P. aeruginosa, the contribution of specific spacers to CRISPR-acquired immunity against DMS3vir was investigated, and we found a strong bias wherein CR2_sp1 is efficient in conferring CRISPR-acquired immunity through primed adaptation. This high efficiency of priming by CR2_sp1 is not simply due to the proximity of CR2_sp1 to the CRISPR2 leader sequence and is likely the result of the specific sequences that prevent functional CRISPR interference.
Preventing bacteriophage adsorption is a common strategy employed by bacteria to gain resistance to lytic bacteriophages and is analogous to plasmid-based adsorption interference (34), but mutations that directly affect the function of surface receptors can come with a fitness cost (35). Accordingly, the goal of the analysis of the biofilm enrichment system was not simply to investigate mechanisms of CRISPR adaptation in P. aeruginosa but also to develop an assay that may provide insight into why the type I-F CRISPR-Cas system is typically found in a broad range of P. aeruginosa strains. The advantage to P. aeruginosa cells provided by CRISPR adaptation when they were grown in a biofilm in the presence of bacteriophage over CRISPR-deficient P. aeruginosa cells was significant and reproducible, perhaps suggesting one reason why this system is conserved in microbes.
Similar to what was observed in other studies involving the native type I-F system of P. aeruginosa, there were many similarities between the spacers incorporated using the biofilm enrichment assay described here and those incorporated using a plasmid loss-based system with the type I-F system of P. atrosepticum published previously (27), despite the differences between the two assay systems. In both systems, during a primed spacer acquisition event, multiple spacers are incorporated at a higher rate than single spacers (∼75% versus 25%), spacer incorporation is not equal among CRISPR arrays, and a nearly identical strand bias exists regarding selected protospacers. Therefore, these observations likely reflect general characteristics of the type I-F system and are not associated with the type of target (i.e., plasmid versus lytic bacteriophage) or the bacterium that encodes the CRISPR system.
The observation that multiple spacers were typically inserted during an acquisition event suggests the ability of P. aeruginosa to rapidly modify its CRISPR loci. This finding is at odds with the findings of our previous work (19) showing surprisingly conserved spacer arrays across isolates of this microbe from India and two sites in the United States (Hanover, NH, and Pittsburgh, PA). It is possible that the rate of acquisition observed in the lab does not reflect the rates actually occurring in natural settings, or alternatively, perhaps such newly acquired spacers are easily lost.
The biofilm selection assay was used to test the requirements for genome stability proteins previously implicated in CRISPR adaptation. We found that RecD was dispensable for CRISPR adaptation in P. aeruginosa, which was not surprising, since the hypothesized role of the RecBCD complex in P. aeruginosa adaptation is to generate single-stranded DNA for Cas1 during naive adaptation and only primed adaptation was observed in the biofilm selection assay. Intriguingly, we found that priming could still occur in the absence of RecG, albeit at a greatly reduced rate. The putative role of RecG in CRISPR adaptation is to relieve the R loops formed at sites where the crRNA ribonucleoprotein complex has partially bound its target, thereby allowing Cas1 to access the single-stranded DNA generated by Cas3. Our data support this model but demonstrate that RecG is not completely required for Cas1 to access the single-stranded target and incorporate new spacers.
One of the most striking results obtained with the biofilm enrichment assay was the difference in priming efficiency between the CR2_sp20 and CR2_sp1 spacers. It has been shown in the E. coli type I-E CRISPR-Cas system that spacers that match their protospacer with 100% complementarity but lack a consensus PAM (such as CR2_sp20) are efficient priming spacers (29), yet when either CR2_sp1 or all spacers except CR2_sp20 and CR2_sp21 were deleted, the spacer acquisition frequency was greatly reduced to levels similar to those observed in the recG mutant, indicating that CR2_sp20 is a very poor priming spacer, despite 100% complementarity with a nonconsensus PAM. The inefficiency of CR2_sp20 at priming is not due to the location of CR2_sp20 relative to the CRISPR2 leader, since both a leader-distant CR2_sp20 and a leader-proximal CR2_sp20 had a poor priming efficiency. Instead, the inefficiency of CR2_sp20 at priming can be resolved by engineering CR2_sp20 to have the same mismatch sites as CR2_sp1 between the spacer and the bacteriophage target, which results in rates of priming more efficient than the rate for CR2_sp1. Further research is needed to determine which of the 5 mismatches are the most important for this phenomenon and why.
Interestingly, none of the newly acquired spacers sequenced were likely primed by CR2_sp17, suggesting that when escaping CRISPR interference, a single nucleotide deletion can be more beneficial to the bacteriophage than a single nucleotide substitution, since our data indicate that the substitution can be more easily overcome through CRISPR priming. Avoidance of priming for a bacteriophage infecting P. aeruginosa is especially important since no naive adaptation was detected using the biofilm enrichment assay, suggesting that primed adaptation is the dominant form of CRISPR adaptation in P. aeruginosa type I-F.
Overall, the biofilm enrichment assay described here provides a system to study endogenous CRISPR adaptation in the P. aeruginosa type I-F system against a lytic bacteriophage and provides a complementary data set generated by CRISPR studies in which artificial expression constructs or mutants are used to investigate the mechanisms of CRISPR adaptation.
Supplementary Material
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00458-16.
REFERENCES
- 1.Makarova KS, Wolf YI, Alkhnbashi OS, Costa F, Shah SA, Saunders SJ, Barrangou R, Brouns SJ, Charpentier E, Haft DH, Horvath P, Moineau S, Mojica FJ, Terns RM, Terns MP, White MF, Yakunin AF, Garrett RA, van der Oost J, Backofen R, Koonin EV. 2015. An updated evolutionary classification of CRISPR-Cas systems. Nat Rev Microbiol 13:722–736. doi: 10.1038/nrmicro3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shmakov S, Abudayyeh OO, Makarova KS, Wolf YI, Gootenberg JS, Semenova E, Minakhin L, Joung J, Konermann S, Severinov K, Zhang F, Koonin EV. 2015. Discovery and functional characterization of diverse class 2 CRISPR-Cas systems. Mol Cell 60:385–397. doi: 10.1016/j.molcel.2015.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grissa I, Vergnaud G, Pourcel C. 2007. The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinformatics 8:172. doi: 10.1186/1471-2105-8-172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, Horvath P. 2007. CRISPR provides acquired resistance against viruses in prokaryotes. Science 315:1709–1712. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- 5.Brouns SJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJ, Snijders AP, Dickman MJ, Makarova KS, Koonin EV, van der Oost J. 2008. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 321:960–964. doi: 10.1126/science.1159689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marraffini LA, Sontheimer EJ. 2008. CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science 322:1843–1845. doi: 10.1126/science.1165771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barrangou R, Marraffini LA. 2014. CRISPR-Cas systems: prokaryotes upgrade to adaptive immunity. Mol Cell 54:234–244. doi: 10.1016/j.molcel.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yosef I, Goren MG, Qimron U. 2012. Proteins and DNA elements essential for the CRISPR adaptation process in Escherichia coli. Nucleic Acids Res 40:5569–5576. doi: 10.1093/nar/gks216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Swarts DC, Mosterd C, van Passel MW, Brouns SJ. 2012. CRISPR interference directs strand specific spacer acquisition. PLoS One 7:e35888. doi: 10.1371/journal.pone.0035888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vorontsova D, Datsenko KA, Medvedeva S, Bondy-Denomy J, Savitskaya EE, Pougach K, Logacheva M, Wiedenheft B, Davidson AR, Severinov K, Semenova E. 2015. Foreign DNA acquisition by the I-F CRISPR-Cas system requires all components of the interference machinery. Nucleic Acids Res 43:10848–10860. doi: 10.1093/nar/gkv1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Westra ER, van Houte S, Oyesiku-Blakemore S, Makin B, Broniewski JM, Best A, Bondy-Denomy J, Davidson A, Boots M, Buckling A. 2015. Parasite exposure drives selective evolution of constitutive versus inducible defense. Curr Biol 25:1043–1049. doi: 10.1016/j.cub.2015.01.065. [DOI] [PubMed] [Google Scholar]
- 12.van Houte S, Ekroth AK, Broniewski JM, Chabas H, Ashby B, Bondy-Denomy J, Gandon S, Boots M, Paterson S, Buckling A, Westra ER. 2016. The diversity-generating benefits of a prokaryotic adaptive immune system. Nature 532:385–388. doi: 10.1038/nature17436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zegans ME, Wagner JC, Cady KC, Murphy DM, Hammond JH, O'Toole GA. 2009. Interaction between bacteriophage DMS3 and host CRISPR region inhibits group behaviors of Pseudomonas aeruginosa. J Bacteriol 191:210–219. doi: 10.1128/JB.00797-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cady KC, O'Toole GA. 2011. Non-identity-mediated CRISPR-bacteriophage interaction mediated via the Csy and Cas3 proteins. J Bacteriol 193:3433–3445. doi: 10.1128/JB.01411-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cady KC, Bondy-Denomy J, Heussler GE, Davidson AR, O'Toole GA. 2012. The CRISPR/Cas adaptive immune system of Pseudomonas aeruginosa mediates resistance to naturally occurring and engineered phages. J Bacteriol 194:5728–5738. doi: 10.1128/JB.01184-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Budzik JM, Rosche WA, Rietsch A, O'Toole GA. 2004. Isolation and characterization of a generalized transducing phage for Pseudomonas aeruginosa strains PAO1 and PA14. J Bacteriol 186:3270–3273. doi: 10.1128/JB.186.10.3270-3273.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ceyssens PJ, Lavigne R. 2010. Bacteriophages of Pseudomonas. Future Microbiol 5:1041–1055. doi: 10.2217/fmb.10.66. [DOI] [PubMed] [Google Scholar]
- 18.O'Toole GA, Kolter R. 1998. Flagellar and twitching motility are necessary for Pseudomonas aeruginosa biofilm development. Mol Microbiol 30:295–304. doi: 10.1046/j.1365-2958.1998.01062.x. [DOI] [PubMed] [Google Scholar]
- 19.Cady KC, White AS, Hammond JH, Abendroth MD, Karthikeyan RS, Lalitha P, Zegans ME, O'Toole GA. 2011. Prevalence, conservation and functional analysis of Yersinia and Escherichia CRISPR regions in clinical Pseudomonas aeruginosa isolates. Microbiology 157:430–437. doi: 10.1099/mic.0.045732-0. [DOI] [PubMed] [Google Scholar]
- 20.van Belkum A, Soriaga LB, LaFave MC, Akella S, Veyrieras JB, Barbu EM, Shortridge D, Blanc B, Hannum G, Zambardi G, Miller K, Enright MC, Mugnier N, Brami D, Schicklin S, Felderman M, Schwartz AS, Richardson TH, Peterson TC, Hubby B, Cady KC. 2015. Phylogenetic distribution of CRISPR-Cas systems in antibiotic-resistant Pseudomonas aeruginosa. mBio 6:e01796-15. doi: 10.1128/mBio.01796-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shanks RM, Caiazza NC, Hinsa SM, Toutain CM, O'Toole GA. 2006. Saccharomyces cerevisiae-based molecular tool kit for manipulation of genes from gram-negative bacteria. Appl Environ Microbiol 72:5027–5036. doi: 10.1128/AEM.00682-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liberati NT, Urbach JM, Miyata S, Lee DG, Drenkard E, Wu G, Villanueva J, Wei T, Ausubel FM. 2006. An ordered, nonredundant library of Pseudomonas aeruginosa strain PA14 transposon insertion mutants. Proc Natl Acad Sci U S A 103:2833–2838. doi: 10.1073/pnas.0511100103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ha DG, Richman ME, O'Toole GA. 2014. Deletion mutant library for investigation of functional outputs of cyclic diguanylate metabolism in Pseudomonas aeruginosa PA14. Appl Environ Microbiol 80:3384–3393. doi: 10.1128/AEM.00299-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuchma SL, Ballok AE, Merritt JH, Hammond JH, Lu W, Rabinowitz JD, O'Toole GA. 2010. Cyclic-di-GMP-mediated repression of swarming motility by Pseudomonas aeruginosa: the pilY1 gene and its impact on surface-associated behaviors. J Bacteriol 192:2950–2964. doi: 10.1128/JB.01642-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Datsenko KA, Pougach K, Tikhonov A, Wanner BL, Severinov K, Semenova E. 2012. Molecular memory of prior infections activates the CRISPR/Cas adaptive bacterial immunity system. Nat Commun 3:945. doi: 10.1038/ncomms1937. [DOI] [PubMed] [Google Scholar]
- 26.Wilkinson ME, Nakatani Y, Staals RH, Kieper SN, Opel-Reading HK, McKenzie RE, Fineran PC, Krause KL. 2016. Structural plasticity and in vivo activity of Cas1 from the type I-F CRISPR-Cas system. Biochem J 8:1063–1072. doi: 10.1042/BCJ20160078. [DOI] [PubMed] [Google Scholar]
- 27.Richter C, Dy RL, McKenzie RE, Watson BN, Taylor C, Chang JT, McNeil MB, Staals RH, Fineran PC. 2014. Priming in the type I-F CRISPR-Cas system triggers strand-independent spacer acquisition, bi-directionally from the primed protospacer. Nucleic Acids Res 42:8516–8526. doi: 10.1093/nar/gku527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Levy A, Goren MG, Yosef I, Auster O, Manor M, Amitai G, Edgar R, Qimron U, Sorek R. 2015. CRISPR adaptation biases explain preference for acquisition of foreign DNA. Nature 520:505–510. doi: 10.1038/nature14302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fineran PC, Gerritzen MJ, Suarez-Diez M, Kunne T, Boekhorst J, van Hijum SA, Staals RH, Brouns SJ. 2014. Degenerate target sites mediate rapid primed CRISPR adaptation. Proc Natl Acad Sci U S A 111:E1629–E1638. doi: 10.1073/pnas.1400071111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shmakov S, Savitskaya E, Semenova E, Logacheva MD, Datsenko KA, Severinov K. 2014. Pervasive generation of oppositely oriented spacers during CRISPR adaptation. Nucleic Acids Res 42:5907–5916. doi: 10.1093/nar/gku226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li M, Wang R, Zhao D, Xiang H. 2014. Adaptation of the Haloarcula hispanica CRISPR-Cas system to a purified virus strictly requires a priming process. Nucleic Acids Res 42:2483–2492. doi: 10.1093/nar/gkt1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ivancic-Bace I, Cass SD, Wearne SJ, Bolt EL. 2015. Different genome stability proteins underpin primed and naive adaptation in E. coli CRISPR-Cas immunity. Nucleic Acids Res 43:10821–10830. doi: 10.1093/nar/gkv1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hale C, Kleppe K, Terns RM, Terns MP. 2008. Prokaryotic silencing (psi)RNAs in Pyrococcus furiosus. RNA 14:2572–2579. doi: 10.1261/rna.1246808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Forde A, Daly C, Fitzgerald GF. 1999. Identification of four phage resistance plasmids from Lactococcus lactis subsp. cremoris HO2. Appl Environ Microbiol 65:1540–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Avrani S, Wurtzel O, Sharon I, Sorek R, Lindell D. 2011. Genomic island variability facilitates Prochlorococcus-virus coexistence. Nature 474:604–608. doi: 10.1038/nature10172. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.