

Abstract
Clefting of the lip, with or without palatal involvement (CLP), is associated with a higher incidence of developmental tooth abnormalities, including hypodontia and supernumerary teeth, aberrant crown and root morphologies, and enamel defects, although the underlying mechanistic link is poorly understood. As most CLP genes are expressed throughout the oral epithelium, the authors hypothesized that the expression of CLP genes may persist in the dental epithelium and thus, in addition to their earlier role in labiopalatine development, may play an important functional role in subsequent tooth patterning and amelogenesis. To address this, the authors generated a unique conditional knockout model involving the major CLP gene, Irf6, that overcomes the previously reported perinatal lethality to enable assessment of any posteruption dental phenotypes. A dental epithelium–specific Irf6 conditional knockout (Irf6-cKO) mouse was generated via a Pitx2-Cre driver line. Dental development was analyzed by microcomputed tomography, scanning electron microscopy, histology, immunohistochemistry, and quantitative polymerase chain reaction. Irf6-cKO mice displayed variable hypodontia, occasional supernumerary incisors and molars, as well as crown and root patterning anomalies, including peg-shaped first molars and taurodontic and C-shaped mandibular second molars. Enamel density was reduced in preeruption Irf6-cKO mice, and some shearing of enamel rods was noted in posteruption incisors. There was also rapid attrition of Irf6-cKO molars following eruption. Histologically, Irf6-cKO ameloblasts exhibited disturbances in adhesion and polarity, and delayed enamel formation was confirmed immunohistochemically. Altered structure of Hertwig’s epithelial root sheath was also observed. These data support a role for IRF6 in tooth number, crown and root morphology and amelogenesis that is likely due to a functional role of Irf6 in organization and polarity of epithelial cell types. This data reinforce the notion that various isolated tooth defects could be considered part of the CLP spectrum in relatives of an affected individual.
Keywords: amelogenesis, hypodontia, supernumerary teeth, van der Woude, cleft lip/palate, taurodontism
Introduction
Cleft lip/palate (CLP; i.e., clefting of the lip, with or without palatal involvement) occurs in ~1 in 700 individuals (Dixon et al. 2011) but is a classification that encompasses a diversity of clinical presentations. As a diagnosis, CLP can describe overt clefts of the lip proper, alveolus, secondary palate, or any combination thereof; it also encompasses subclinical findings, such as defects of the orbicularis oris muscle or a Simon’s band (Wang et al. 2014). In addition, an elevated incidence of dental anomalies occurs in CLP patients (Ranta 1986; Aspinall et al. 2014). These resemble the array of dental anomalies seen in the ectodermal dysplasias and include hypodontia and supernumerary teeth, aberrant crown and root morphology, and enamel defects (Ranta 1986; Pegelow et al. 2008; Gomes et al. 2009; Al Jamal et al. 2010; Rullo et al. 2015). These observations support the notion that the primary defect causing CLP is ectodermal in origin.
IRF6, which encodes a transcriptional regulator, is arguably the most significant CLP gene. Common IRF6 sequence variants contribute to ~12% of the genetic contribution to isolated CLP (Zucchero et al. 2004), while more severe mutations cause the allelic conditions of van der Woude syndrome and popliteal pterygium syndrome. Both van der Woude syndrome and popliteal pterygium syndrome exhibit orofacial clefts as well as additional ectodermal abnormalities, including ectopic epithelial adhesions (Kondo et al. 2002). IRF6/Irf6 is expressed throughout the embryonic ectoderm (Knight et al. 2006; Washbourne and Cox 2006). Mice homozygous for either a ubiquitous or broad epithelial-specific Irf6-null allele or a mutation homologous to that causing popliteal pterygium syndrome reproduce many of the features seen in the human syndromes. However, homozygotes die perinatally due to widespread ectopic intraoral adhesions and dehydration as a result of a generalized disruption to epithelial barrier function. Heterozygotes in these studies are grossly normal, with only minor intraoral adhesions reported (Ingraham et al. 2006; Richardson et al. 2006; Peyrard-Janvid et al. 2014). Subsequently, Blackburn et al. (2012) reported that Irf6 is required for proper invagination of incisor epithelium in the early tooth bud stage, and we noted that Irf6 is strongly expressed in secretory-stage ameloblasts in postnatal animals (see Appendix Fig. 1). To investigate its role in postnatal tooth development and amelogenesis, we generated a dental epithelium conditional knockout of Irf6 (Irf6-cKO) using a Pitx2-Cre driver line. Pitx2 is specific to the oral epithelium; it is expressed in the dental lamina, enamel knot, undifferentiated cervical loops, and preameloblasts (Li et al. 2014). This unique knockout has revealed numerous roles for Irf6 in tooth development, providing novel insights into the molecular basis of tooth anomalies in patients with CLP.
Materials and Methods
Animals
Irf6-cKO was generated with floxed Irf6 mice (Biggs et al. 2012; Schutte et al. unpublished data) and a Pitx2-Cre driver line (Liu et al. 2003; Li et al. 2014). For all analyses, Irf6-cKO (Pitx2Cre/+;Irf6fl/fl) animals were compared with 4 control genotypes: Pitx2+/+;Irf6+/+, Pitx2+/+;Irf6fl/+, Pitx2Cre/+;Irf6fl/+, and Pitx2+/+;Irf6fl/fl. Genotyping for Irf6fl and Pitx2Cre alleles was performed as described (Liu et al. 2003; Biggs et al. 2012). All animal work was approved by the Animal Care and Use Committee of the Seattle Children’s Research Institute.
Microcomputed Tomography
Fixed heads were scanned in a SkyScan 1076 microcomputed tomography (microCT) system at 9-µm resolution with the following settings: 55 kV, 180 µA, 1,450-ms exposure, 1-mm Al filter, 0.7° rotation step, and 3-frame averaging. Raw scan data were reconstructed with NRecon 1.6.9, then resliced in DataViewer (Bruker) for segmentation. Total tooth volume, enamel volume, dentin volume, enamel volume fraction, dentin volume fraction, enamel mineral density, and dentin mineral density were analyzed following threshold-based segmentation in CTan software (Bruker). Hydroxyapatite phantoms were used to determine definitive enamel mineral density. Data were statistically analyzed through 1-way analysis of variance with Tukey’s post hoc test (P < 0.05; GraphPad Prism 6.01), with Bonferroni correction for pairwise comparisons of groups. Differences were considered significant for comparisons of data sets yielding P < 0.01. Three-dimensional renderings were generated with Drishti 2.4 (Limaye 2012) or CTVox (Bruker).
Histology
Heads from mice euthanized at postnatal (P) days 4, 7, 14, 28, and 84 were fixed in 4% paraformaldehyde or Bouin’s fixative (Sigma Aldrich). Tissues were demineralized in 14% ethylenediaminetetraacetic acid (EDTA) solution (pH 7.1) or acid-fast stain (AFS) (10% acetic acid, 4% formalin, and 0.86% sodium chloride) and embedded in paraffin. Serial 5-µm sections were collected and deparaffinized for standard histologic procedures.
In Situ Hybridization
In situ hybridization was performed with an RNAscope 2.0 Assay kit and mouse amelogenin probe, per the kit recommendations (Advanced Cell Diagnostics; Wang et al. 2012).
Immunohistochemistry
Immunohistochemistry was performed as previously described (Foster et al. 2013) with biotinylated secondary antibodies and AEC (3-amino 9-ethylcarbazole) peroxidase substrate for detection (Vectastain Elite ABC Kit; Vector Labs). Primary antibodies, used at the manufacturer’s recommended dilutions, included anti-NME2, anti-IRF6, and anti-KLK4 (LS-B9880, LS-B3231, and LS-B1736, respectively; Lifespan Biosciences), as well as anti-amelogenin (sc-32892; Santa Cruz Biotechnology) and anti-Wnt10b (ab150608; Abcam).
RNA Extraction and Quantitative Polymerase Chain Reaction
P4 and P13 mandibular molars were dissected and placed in RNALater (Life Technologies). Samples were homogenized (MP FastPrep; MP Biomedicals) and RNA extracted with TriPure Isolation Reagent (Roche Life Science), then column purified (Qiagen). cDNA was generated for quantitative polymerase chain reaction (qPCR) with a Lightcycler 480 (Roche Life Science) and various primer pairs (see Appendix Table 1).
Scanning Electron Microscopy
Samples for scanning electron microscopy (SEM) were fixed in 4% paraformaldehyde and stored in 70% ethanol. Samples were air-dried, mounted in room temperature cure epoxy (Allied High Tech Products) and then polished sequentially with 600-grit SiC paper, followed by diamond lapping films of 6 µm and 1 µm (Allied High Tech Products) to a mirror finish. After polishing, samples were lightly etched with 0.5% aqueous HCl for 30 s, mounted on SEM stubs, and sputter coated with ~5 nm of platinum (SPI Supplies, Inc.), then imaged in a JSM 6010 SEM system (JEOL USE) at 10 kV in secondary electron imaging mode.
Results
Loss of Irf6 Causes Defects in Tooth Crown and Root Patterning and Initiation
At birth, mice homozygous for a dental epithelium–restricted Irf6-null allele (i.e., Irf6-cKO) were grossly indistinguishable from control littermates. However, at P14, Irf6-cKO incisors were variably hypoplastic with pitted labial surfaces. By P28, enamel was noticeably whiter and more translucent than controls (Fig. 1A). Three-dimensional renderings of microCT data revealed that Irf6-cKO mice also exhibited defects in molar crown morphology that were not seen in any control group. At preeruption ages (P7 and P14), Irf6-cKO molar cusps were already noticeably shallower in appearance than in controls (arrow, top row, Fig. 1B). Posteruption (P28 and P84), crown anomalies were even more evident (middle row, Fig. 1B). In 25% of Irf6-cKO mice examined (total n = 20; see Appendix Table 2), complete loss of cusp patterning was observed (arrow, bottom row, Fig. 1B). Three Irf6-cKO mice were found to be missing mandibular third molars unilaterally (Fig. 1B, C): 1 was missing a maxillary third molar, and 2 had supernumerary teeth—1 with an extra mandibular incisor and 1 with a diastema tooth (Fig. 1C).
Figure 1.

Loss of Irf6 causes alterations in crown and root morphology. (A) At postnatal day 14 (P14), incisors of Irf6-cKO (conditional knockout of Irf6) animals were variably hypoplastic with pitted labial surfaces. By P28, incisors of Irf6-cKO mice were noticeably whiter and more translucent than controls. (B) Altered crown morphology (note shallower cusps) is already apparent at preeruptive stages (P7, data not shown; P14, yellow arrow, top row) and more evident in P28 (posteruption) Irf6-cKO mice (middle and lower rows). First molars were also frequently reduced in size (yellow arrow, lower row) and third molars occasionally missing unilaterally (yellow arrowhead, lower row). (C) Ten percent of Irf6-cKO mice examined also exhibited supernumerary teeth, including a diastema tooth (black arrowhead) and additional incisor (yellow arrow). (D) Three-dimensional rendering of segmented molar teeth from microcomputed tomography–scanned P84 mice showed various root morphology defects in addition to worn cusps in Irf6-cKO animals. The root anomalies in Irf6-cKO mice included severely taurodontic C-shaped mandibular second molars and peg-shaped mandibular first molars (yellow arrows and arrowheads, upper and lower rows). The C-shaped second molars and the loss of separate mesial and distal first molar canals in Irf6-cKO mice were readily apparent in transverse midroot sections of the mandibular molars. Taurodontism was also seen in Pitx2Cre/+; Irf6fl/+ mice (yellow arrow, second column), although this phenotype was of lower penetrance and severity when compared with Irf6-cKO mice (see Appendix Table 2).
Root patterning defects were also present in all Irf6-cKO mice. These defects included severely taurodontic mandibular second molars and peg-shaped mandibular first molar roots (Fig. 1D). Teeth were characterized as taurodontic on the basis of enlarged pulp chambers, apical displacement of the pulpal floor, reduced separation or fusion of mesial and distal roots, and reduced constriction at the cementum-enamel junction. Midroot transverse microCT sections (apical to the bifurcation) revealed C-shaped roots in Irf6-cKO mandibular second molars (arrow, lower row, Fig. 1D) and a single canal in single-rooted mandibular first molars (arrowhead, lower row, Fig. 1D). Notably, taurodontic second molars were also seen in Pitx2Cre/+;Irf6+/+ and Pitx2Cre/+;Irf6fl/+ mice, albeit at reduced frequency and severity (Fig. 1D). No other genotype exhibited peg-shaped mandibular first molars.
Delayed Enamel Maturation in Irf6-cKO Mice
Irf6-cKO molars exhibited increased attrition following eruption. In some animals, by 7 d posteruption (P28), crown wear reached the dentin in first molars. By P84, Irf6-cKO molar cusps were worn almost completely flat, whereas all mice of control genotypes had normal cusp morphology (Fig. 1D). Cross-sectional microCT data from P28 mice revealed a more radiolucent enamel layer in immature stages of amelogenesis and a more incisally located (i.e., delayed) transition from immature to mature enamel in Irf6-cKO animals as compared with other genotypes (Fig. 2A). We next analyzed incisor and molar enamel at multiple ages using coronal sections over the mesial root of the mandibular first molar (region 3, Fig. 2B). Irf6-cKO mice exhibited thinner mandibular incisor enamel versus controls, particularly at P14 (Fig. 2B). Analysis of enamel mineral density revealed that, prior to eruption (P14), Irf6-cKO mandibular molars exhibited significantly decreased density when compared to all other genotypes (P < 0.05). However, at posteruption stages (P28 or P84), the enamel mineral density in Irf6-cKO mice had recovered to levels seen in the heterozygous Cre animals but was still significantly less than wild-type controls (Fig. 2C). The data on Pitx2Cre heterozygotes support the findings of Li et al. (2014) that reduced Pitx2 expression is associated with mild enamel hypoplasia. Nevertheless, we show that loss of Irf6 further affects mineralization. Additional analyses at P28 revealed no differences in dentin density, percentage dentin volume, or percentage enamel volume among any genotypes (data not shown). However, total tooth volume in Irf6-cKO P28 mice was significantly reduced for the upper incisors, upper first molars, and lower first molars when compared with all other genotypes. Third molars were not assessed due to their variable presence in Irf6-cKO mice.
Figure 2.

Delayed enamel maturation in Irf6-cKO (conditional knockout of Irf6) mice. (A) Microcomputed tomography imaging confirmed delayed enamel maturation in postnatal day 28 (P28) Irf6-cKO incisors as compared with controls. Representative cross-sections of incisors show 4 landmark regions (vertical lines), including immature enamel at the distal root of the second molar (4), mesial root of the first molar (3), mental foramen (2), and mature enamel at the alveolar crest (1). The transition between immature and mature enamel is incisally displaced (i.e., delayed) in Irf6-cKO animals, as evident when enamel radiolucency is compared in regions 3 and 4 and, to a lesser degree, in region 2 (see arrows). No significant differences were detected between control genotypes. (B) Microcomputed tomography imaging reveals that Irf6-cKO mice exhibit thinner mandibular incisor enamel (outlined in white) versus controls at P14, although incisor enamel radiolucency was comparable by P84 (data not shown). (C) Quantitative analysis of molar enamel mineral density confirmed a significantly decreased (asterisk; P < 0.05) enamel mineral density in Irf6-cKO mice versus controls at P14. However, by posteruption stages (P28 or P84), the enamel mineral density in Irf6-cKO mice was no longer significantly different from that of the Pitx2-Cre heterozygote controls, although it was still significantly lower than in wild-type mice.
SEM analysis indicated a comparable structure of mature incisor enamel between P28 Irf6-cKO and control mice (Fig. 3A). Even in regions of immature enamel at this age, SEM analysis still showed a comparable structure in Irf6-cKO and control samples (Fig. 3B). However, at higher magnification, shearing of enamel rods was observed in Irf6-cKO samples (Fig. 3A) but not in controls. Taken together, the microCT and SEM analyses suggest that Irf6-cKO mice are able to form prismatic enamel, despite the mineralization process being delayed.
Figure 3.
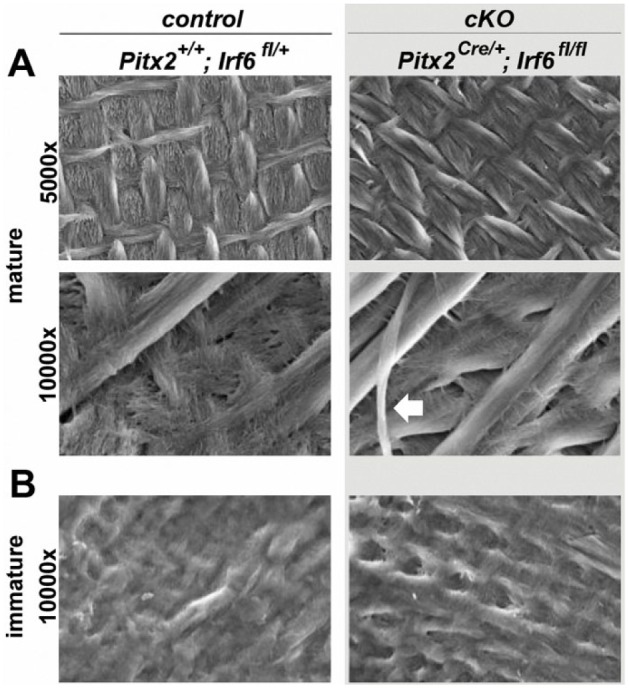
Scanning electron microscopy analysis reveals a normal enamel prismatic structure in Irf6-cKO (conditional knockout of Irf6) mice. Scanning electron microscopy analysis of postnatal day 28 mice reveals a comparable prismatic structure in control and Irf6-cKO mice in regions of mature enamel (A), as well as a similar appearance of immature enamel (B). However, some shearing of enamel rods was observed in Irf6-cKO samples (white arrow), which was not seen in controls.
Irf6-cKO Mice Show Aberrant Ameloblast Organization and Function
To investigate amelogenesis, we examined molar and incisor ameloblasts histologically. At P7, enamel of Irf6-cKO mice was noticeably thinner, particularly around cuspal regions, and the ameloblast layer appeared ragged and discontinuous (Fig. 4A). In situ hybridization for amelogenin (Amel) further highlighted alterations in the ameloblast layer (Fig. 4A). At P14, Irf6-cKO molar ameloblasts appeared less organized, with increased space between cells (Fig. 4B). Epithelial morphology was also perturbed in Hertwig’s epithelial root sheath (HERS), which appeared shortened in Irf6-cKO versus controls. In the Irf6-cKO incisors, ameloblasts appeared to persist in the secretory stage (as compared with the shorter mature ameloblasts of controls), and the enamel matrix layer was reduced. At P28, Irf6-cKO incisors continued to exhibit disrupted ameloblast polarity, reduced enamel space, and persistence of immature enamel matrix (Fig. 4C). In some Pitx2Cre/+;Irf6fl/+ mice, persistence of immature enamel matrix was observed, although ameloblast polarity was not disrupted.
Figure 4.

Irf6-cKO (conditional knockout of Irf6) ameloblasts exhibit disturbances in adhesion, polarity, and enamel formation. (A) In postnatal day 7 (P7) samples, mandibular first molars are in the bell stage, and ameloblasts are actively secreting enamel matrix. In Irf6-cKO P7 mandibular first molars, irregular cusp morphology was observed (yellow arrows, first row). At higher magnification (middle row), the organization of the ameloblast layer appears disrupted near the cusps of Irf6-cKO mice, as observed by hematoxylin and eosin staining. In situ hybridization for amelogenin (Amel; red-brown color) further highlighted the disrupted ameloblast layer and mineralization. To account for variation caused by region and sectioning techniques, multiple samples and tooth regions were examined (n ≥ 4 for each group), and all Irf6-cKO sections exhibited similar ameloblast-layer alterations. (B) In P14 samples, enamel matrix deposition is normally complete in mandibular first molars, and in demineralized sections, the enamel is represented by an empty space. Ameloblasts are in the maturation stages. In the control sample, ameloblasts have shortened but remain polarized. Irf6-cKO ameloblasts appeared less organized, and increased spacing was observed between them (compare control and conditional knockout regions marked by black arrows). Hertwig’s epithelial root sheath (outlined in red) appears shorter in the Irf6-cKO samples versus controls. In incisor sections, the enamel layer was thinner (asterisk). Ameloblasts in control samples appear to have shortened and are approaching maturation stages, whereas in Irf6-cKO samples, the ameloblasts appear to exhibit the morphology of secretory-stage ameloblasts. (C) Enamel space is thinner in Irf6-cKO samples when compared with controls, and immature enamel matrix is present in Irf6-cKO incisors but absent in comparable control sections (asterisk). At high magnification, disturbances are readily apparent in ameloblast morphology and organization in P28 Irf6-cKO mice as compared with control animals. Ab, ameloblasts; D, dentin; E, enamel; Es, enamel space.
We performed qPCR and immunohistochemistry to assess ameloblast markers in molars and incisors, respectively, at P4 and P13. qPCR confirmed almost complete elimination of Irf6 mRNA in Irf6-cKO samples (Fig. 5A; P < 0.05). At P4, control molars exhibited high relative expression of Amel and low levels of Klk4, as expected for primarily secretory-stage ameloblasts (Fig. 5A). At P13, Amel was decreased and Klk4 mRNA increased when compared with P4, consistent with more maturation-stage ameloblasts. Loss of Irf6 did not significantly affect expression of Amel or Klk4 mRNAs at either stage, although expression of both Amel and Klk4 tended to be slightly lower in P4 Irf6-cKO animals. Other enamel genes (ameloblastin, enamelin, Mmp20) were also unaffected by loss of Irf6 (data not shown). However, expression of Wnt10b, a gene involved in enamel knot signaling (Liu et al. 2008), was decreased in P4 Irf6-cKO molars, although levels had recovered by P13 (Fig. 5A).
Figure 5.

Expression of amelogenesis markers in control and Irf6-cKO mice. (A) Reverse transcription quantitative polymerase chain reaction was performed with RNA harvested from postnatal day 4 (P4) and P13 molars. As expected, Irf6 was significantly reduced in P4 and P13 samples (P < 0.05). Expression of genes encoding enamel matrix proteins (Amel, Klk4) was not significantly altered. However, Wnt10b, an enamel knot marker, exhibited a statistically significant decrease in response to loss of Irf6 in P4 molars but not P13 molars. (B) Immunohistochemical analysis of various enamel proteins in P14 and P28 incisors revealed higher levels of Amel (red-brown staining) in ameloblasts of Irf6-cKO mice as compared with controls. In P14 Irf6-cKO sections, Klk4 (red-brown staining) was expressed at much lower levels when compared with controls. Nme2 localization was altered in Irf6-cKO samples. In control sections, Nme2 (red-brown staining) was distributed throughout the cytoplasm of the ameloblasts, with some localization also at the proximal and distal ends of the ameloblasts. Irf6-cKO P14 ameloblasts exhibited lower amounts of Nme2 in the cytoplasm, with a distinct concentration at the apical surface of the ameloblasts.
Through immunohistochemistry, amelogenin was detected in secretory-stage ameloblasts adjacent to the developing enamel matrix of control and mutant P14 incisors but at notably higher levels in Irf6-cKO ameloblasts (Fig. 5B). In maturation-stage P14 ameloblasts, Klk4 protein was detected adjacent to the immature enamel matrix with decreased levels in Irf6-cKO mice (Fig. 5B). At P28, high levels of amelogenin were still evident in Irf6-cKO ameloblasts as well as throughout the immature enamel matrix layer (which, at this age, is not present in controls). Wnt10b levels were too low to be detected in P1 and P4 incisors and molars (data not shown). We also examined Nme2, a component of a complex that regulates RhoA and Rac1 GTPase activation and epithelial adhesion (Otsuki et al. 2001) and that physically interacts with IRF6 (Parada et al. unpublished data). Strikingly, a distinct apical surface localization of Nme2 was observed in Irf6-cKO ameloblasts, which contrasted with the more cytoplasmic and basal localization in control ameloblasts (Fig. 5B). A similar apical distribution of a fraction of the Irf6 protein pool was observed in control ameloblasts but was lost in conditional knockout ameloblasts (compare Appendix Fig. 1 with Fig. 5B, bottom panel), suggesting that Irf6 has a nonnuclear function in limiting Nme2 and, therefore, Rho-GTPase activation at the apical membrane.
Discussion
A significantly elevated incidence of dental anomalies has been reported in all CLP cohorts studied to date. The most common anomalies involve hypodontia and supernumerary teeth around the cleft region, although variation in tooth number has also been observed outside the cleft region. Additional anomalies include altered crown size and morphology, taurodontism, and enamel hypoplasia. Notably, unaffected siblings and parents of children with overt clefts also exhibit elevated incidence of these dental anomalies (Schroeder and Green 1975; Eerens et al. 2001; Aspinall et al. 2014), which implies that these are subclinical phenotypes of the CLP spectrum sharing a common underlying etiology. However, few studies have addressed the underlying mechanistic basis, likely because of the perinatal mortality in most mouse CLP models.
We focused our efforts on IRF6 not only because common variants in IRF6 are major contributors to the incidence of nonsyndromic CLP but because patients with van der Woude syndrome harboring IRF6 loss-of-function mutations also exhibit an elevated frequency of hypodontia, taurodontism, and abnormally shaped teeth (Ranta and Rintala 1982; Oberoi and Vargervik 2005; Nawa et al. 2008). To investigate the role of Irf6 in the presentation of dental phenotypes, we generated a unique conditional knockout strategy to bypass its essential role in lip and palate morphogenesis and skin differentiation. Remarkably, Irf6-cKO mice recapitulate the full range of CLP-associated dental defects, including hypodontia, occasional supernumerary incisors and molars, disturbances in crown and root morphologies (including peg-shaped molars, taurodontism, and C-shaped roots), and enamel defects.
Small, irregular-shaped cusps and taurodontic molars have been described in Wnt10a-null mice (Yang et al. 2015), and mutations in WNT10A cause oligodontia in humans (van den Boogaard et al. 2012). Wnt10a and its paralog, Wnt10b, are similarly expressed in the enamel knot, which determines molar cuspal position and shape, and we find that Wnt10b mRNA levels, which were still detectable in P4 molars, were reduced in Irf6-cKO mice. A feedback loop between Irf6 and Wnt signaling regulates morphologic changes in the oral epithelium required for fusion of the primary palate (Ferretti et al. 2011). While definitively demonstrating a similar regulatory loop between Irf6 and Wnt10b was beyond the scope of the current study, a similar mechanism operating to regulate tooth morphogenesis is tantalizing, as it could reasonably explain the morphologic diversity (variation in tooth number as well as crown shape and size) in Irf6-cKO dentition. Interestingly, Blackburn et al. (2012) showed that Irf6 was required for proper invagination of the incisor tooth bud epithelium, with canonical Wnt activity increased in mutants. Notably, they did not find similar molecular or cellular changes in the molar tooth bud epithelium. As molar crown anomalies, but not incisor shape defects, are commonly seen in van der Woude syndrome, Blackburn et al. postulated that differences in regulation of tooth epithelial invagination might exist between mice and humans. However, differences between their and our conditional knockout lines may offer an intriguing alternate explanation, with our knockout line also being haploinsufficient for Pitx2 (due to the insertional inactivation by Cre integration). A 50% reduction in Pitx2 expression in molar tooth bud epithelia, although not sufficient to produce crown shape anomalies, may have unmasked a sensitivity to loss of Irf6. In support of this, we noted a moderate level of taurodontism in heterozygous Pitx2Cre controls that was exacerbated in Irf6-cKO mice. It is unknown whether a similar additional genetic susceptibility in humans (e.g., a common subpathogenic variant in another epithelial expressed gene) is needed to manifest dental anomalies in CLP patients; however, this may be clarified as genome sequencing data are acquired from families segregating for CLP. With respect to the taurodontic and peg-shaped molars, we also noted an altered HERS structure in Irf6-cKO mice. In multirooted teeth, folding of HERS—a bilayered epithelial structure—directs the size, shape, and furcation of the developing tooth root (Kumakami-Sakano et al. 2014). Together, these observations strongly support a previously unrecognized role for IRF6 in HERS function.
The most notable overt phenotype in the Irf6-cKO mice was the white translucent appearance of their incisors. A similar incisor appearance is seen in the epithelial-specific knockout of Rac1, which encodes a GTPase that regulates the actin cytoskeleton and cell-cell and cell-matrix adhesion (Huang et al. 2011). This prompted us to analyze ameloblast morphology and gene/protein expression throughout amelogenesis. Persistence of immature enamel matrix in the proximal incisor, seen histologically, was confirmed by increased amelogenin and reduced Klk4 protein, with the balance of these factors being major determinants in enamel maturation (Bartlett 2013). KLK4 mutations are associated with human enamel maturation defects (Wright et al. 2009; Bartlett and Simmer 2014; Wright et al. 2015), consistent with the Irf6-cKO enamel phenotype. Of note, neither Amel nor Klk4 mRNA was significantly changed in Irf6-cKO molars, suggesting instead that the changes reflect an alteration in the timing of processing of enamel matrix proteins (i.e., delayed mineralization). Although no alterations in presecretory-stage ameloblasts were noted, we found that secretory-phase ameloblasts were less tightly packed, less columnar, and the position of the nuclei more irregular. Secretory-stage ameloblasts are normally recognized by their tall columnar morphology and the characteristic prismatic structure of enamel rods that they apically deposit. It has been proposed that the distinctive decussating enamel structure assembles through the dynamic regulation of lateral adhesion complexes that permits distinct rows of secretory ameloblasts to slide against each other as they secrete mineral (Nishikawa et al. 1990). Despite the slightly abnormal ameloblast morphology, SEM revealed a largely normal prismatic enamel structure in Irf6-cKO mice, in contrast to Rac1-cKO incisors, although shearing of some enamel rods was observed. Nevertheless, microCT revealed reduced enamel mineral density at preeruption ages. Collectively, these findings lead us to conclude that loss of Irf6 delays ameloblast maturation. In addition, we found that Nme2, a component of a complex that negatively regulates Rac1 and RhoA activation (Otsuki et al. 2001) and a newly identified Irf6 binding partner (Parada et al. unpublished data), accumulates at the apical end in Irf6-cKO secretory-phase ameloblasts—the location occupied by a pool of Irf6 protein in control secretory-phase ameloblasts. This suggests that Irf6 also has a nonnuclear role in limiting the Nme complex at the apical end, where it presumably regulates adhesion strength via activation of the Rho-type GTPases. Because of a lack of high-specificity antibodies to the active form of Rac1 and RhoA, we did not demonstrate an increase in the active form of these GTPases at the apical end of ameloblasts. However, increased activation of RhoA was recently found following knockdown of Irf6 in cultured keratinocytes (Biggs et al. 2014). Despite the fairly subtle changes in ameloblast morphology, Irf6-cKO teeth exhibited significant and rapid attrition upon eruption into the oral cavity. Overall, our studies provide some of the first insights into the molecular and cellular basis of tooth anomalies in patients with CLP and reinforce the notion that various tooth defects should be considered part of the CLP spectrum.
Author Contributions
E.Y. Chu, contributed to conception, design, data acquisition, analysis, and interpretation, drafted, critically read, and edited the manuscript; B. Tamasas, contributed to conception, design, data acquisition, analysis, and interpretation, critically read and edited the manuscript; H. Fong, contributed to data acquisition and analysis, critically read and edited the manuscript; B.L. Foster, M.R. LaCourse, and A.B. Tran, contributed to data acquisition, critically read and edited the manuscript; J.F. Martin and B.C. Schutte, contributed to data interpretation, critically read and edited the manuscript; M.J. Somerman, contributed to conception, design, data analysis, and interpretation, critically revised the manuscript; T.C. Cox, conceived and designed, contributed to data analysis and interpretation, drafted and critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Supplementary Material
Acknowledgments
The authors thank members of the Somerman and Cox laboratories for their support and Thuy Tien Tran (Baylor College of Medicine) and Youssef Kousa (Michigan State University) for providing assistance with genotyping. Additionally, the authors thank the Light Imaging Core Facility (National Institute of Arthritis and Musculoskeletal and Skin Diseases / National Institutes of Health), James Schmitz (University of Texas Health Sciences Center at San Antonio), Dr. Yong-Hee Chun (University of Texas Health Science Center at San Antonio), and Drs. Daniel Chan, Michael Cunningham, Tracy Popowics, and Avina Paranjpe (University of Washington) for their support and advice.
Footnotes
This research was supported in part by the Laurel Foundation Endowment for Craniofacial Research (T.C.C.), a University of Washington National Institute of Dental and Craniofacial Research T32 Institutional Trainee Award (DE007132; E.Y.C.), an National Institute of Diabetes and Digestive and Kidney Diseases Ruth L. Kirschstein F30 fellowship (DK100280; E.Y.C.), National Institutes of Health DE023177 (J.F.M.), a Cleft Palate Foundation Junior Investigator Research Grant (E.Y.C.), a Libyan Government scholarship (B.T.), and the Internal Research Program of the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health (M.J.S.).
The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
A supplemental appendix to this article is published electronically only at http://jdr.sagepub.com/supplemental.
References
- Al Jamal GA, Hazza’a AM, Rawashdeh MA. 2010. Prevalence of dental anomalies in a population of cleft lip and palate patients. Cleft Palate Craniofac J. 47(4):413–420. [DOI] [PubMed] [Google Scholar]
- Aspinall A, Raj S, Jugessur A, Marazita M, Savarirayan R, Kilpatrick N. 2014. Expanding the cleft phenotype: the dental characteristics of unaffected parents of Australian children with non-syndromic cleft lip and palate. Int J Paediatr Dent. 24(4):286–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett JD. 2013. Dental enamel development: proteinases and their enamel matrix substrates. ISRN Dent. 2013:684607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett JD, Simmer JP. 2014. Kallikrein-related peptidase-4 (KLK4): role in enamel formation and revelations from ablated mice. Front Physiol. 5:240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggs LC, Naridze RL, DeMali KA, Lusche DF, Kuhl S, Soll DR, Schutte BC, Dunnwald M. 2014. Interferon regulatory factor 6 regulates keratinocyte migration. J Cell Sci. 127(Pt 13): 2840–2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggs LC, Rhea L, Schutte BC, Dunnwald M. 2012. Interferon regulatory factor 6 is necessary, but not sufficient, for keratinocyte differentiation. J Invest Dermatol. 132(1):50–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn J, Ohazama A, Kawasaki K, Otsuka-Tanaka Y, Liu B, Honda K, Rountree RB, Hu Y, Kawasaki M, Birchmeier W, et al. 2012. The role of Irf6 in tooth epithelial invagination. Dev Biol. 365(1):61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon MJ, Marazita ML, Beaty TH, Murray JC. 2011. Cleft lip and palate: understanding genetic and environmental influences. Nat Rev Genet. 12(3):167–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eerens K, Vlietinck R, Heidbuchel K, Van Olmen A, Derom C, Willems G, Carels C. 2001. Hypodontia and tooth formation in groups of children with cleft, siblings without cleft, and nonrelated controls. Cleft Palate Craniofac J. 38(4):374–378. [DOI] [PubMed] [Google Scholar]
- Ferretti E, Li B, Zewdu R, Wells V, Hebert JM, Karner C, Anderson MJ, Williams T, Dixon J, Dixon MJ, et al. 2011. A conserved Pbx-Wnt-p63–Irf6 regulatory module controls face morphogenesis by promoting epithelial apoptosis. Developmental Cell. 21(4):627–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster BL, Soenjaya Y, Nociti FH Jr, Holm E, Zerfas PM, Wimer HF, Holdsworth DW, Aubin JE, Hunter GK, Goldberg HA, et al. 2013. Deficiency in acellular cementum and periodontal attachment in bsp null mice. J Dent Res. 92(2):166–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes AC, Neves LT, Gomide MR. 2009. Enamel defects in maxillary central incisors of infants with unilateral cleft lip. Cleft Palate Craniofac J. 46(4):420–424. [DOI] [PubMed] [Google Scholar]
- Huang Z, Kim J, Lacruz RS, Bringas P Jr, Glogauer M, Bromage TG, Kaartinen VM, Snead ML. 2011. Epithelial-specific knockout of the Rac1 gene leads to enamel defects. Eur J Oral Sci. 119 Suppl 1:168–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingraham CR, Kinoshita A, Kondo S, Yang B, Sajan S, Trout KJ, Malik MI, Dunnwald M, Goudy SL, Lovett M, et al. 2006. Abnormal skin, limb and craniofacial morphogenesis in mice deficient for interferon regulatory factor 6 (Irf6). Nat Genet. 38(11):1335–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight AS, Schutte BC, Jiang R, Dixon MJ. 2006. Developmental expression analysis of the mouse and chick orthologues of IRF6: the gene mutated in Van der Woude syndrome. Dev Dyn. 235(5):1441–1447. [DOI] [PubMed] [Google Scholar]
- Kondo S, Schutte BC, Richardson RJ, Bjork BC, Knight AS, Watanabe Y, Howard E, de Lima RL, Daack-Hirsch S, Sander A, et al. 2002. Mutations in IRF6 cause Van der Woude and popliteal pterygium syndromes. Nat Genet. 32(2):285–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumakami-Sakano M, Otsu K, Fujiwara N, Harada H. 2014. Regulatory mechanisms of Hertwigs epithelial root sheath formation and anomaly correlated with root length. Exp Cell Res. 325(2):78–82. [DOI] [PubMed] [Google Scholar]
- Li X, Venugopalan SR, Cao H, Pinho FO, Paine ML, Snead ML, Semina EV, Amendt BA. 2014. A model for the molecular underpinnings of tooth defects in Axenfeld-Rieger syndrome. Hum Mol Genet. 23(1):194–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limaye A. 2012. Drishti: a volume exploration and presentation tool. In: Proceedings of SPIE 8506: developments in x-ray tomography VIII, 85060X, San Diego, CA. [Google Scholar]
- Liu F, Chu EY, Watt B, Zhang Y, Gallant NM, Andl T, Yang SH, Lu MM, Piccolo S, Schmidt-Ullrich R, et al. 2008. Wnt/beta-catenin signaling directs multiple stages of tooth morphogenesis. Dev Biol. 313(1):210–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Selever J, Lu MF, Martin JF. 2003. Genetic dissection of Pitx2 in craniofacial development uncovers new functions in branchial arch morphogenesis, late aspects of tooth morphogenesis and cell migration. Development. 130(25):6375–6385. [DOI] [PubMed] [Google Scholar]
- Nawa H, Oberoi S, Vargervik K. 2008. Taurodontism and Van der Woude syndrome: is there an association? Angle Orthod. 78(5):832–837. [DOI] [PubMed] [Google Scholar]
- Nishikawa S, Tsukita S, Tsukita S, Sasa S. 1990. Localization of adherens junction proteins along the possible sliding interface between secretory ameloblasts of the rat incisor. Cell Struct Funct. 15(5):245–249. [DOI] [PubMed] [Google Scholar]
- Oberoi S, Vargervik K. 2005. Hypoplasia and hypodontia in Van der Woude syndrome. Cleft Palate Craniofac J. 42(5):459–466. [DOI] [PubMed] [Google Scholar]
- Otsuki Y, Tanaka M, Yoshii S, Kawazoe N, Nakaya K, Sugimura H. 2001. Tumor metastasis suppressor nm23H1 regulates Rac1 GTPase by interaction with Tiam1. Proc Natl Acad Sci U S A. 98(8):4385–4390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pegelow M, Peyrard-Janvid M, Zucchelli M, Fransson I, Larson O, Kere J, Larsson C, Karsten A. 2008. Familial non-syndromic cleft lip and palate: analysis of the IRF6 gene and clinical phenotypes. Eur J Orthod. 30(2):169–175. [DOI] [PubMed] [Google Scholar]
- Peyrard-Janvid M, Leslie EJ, Kousa YA, Smith TL, Dunnwald M, Magnusson M, Lentz BA, Unneberg P, Fransson I, Koillinen HK, et al. 2014. Dominant mutations in GRHL3 cause Van der Woude Syndrome and disrupt oral periderm development. Am J Hum Genet. 94(1):23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranta R. 1986. A review of tooth formation in children with cleft lip/palate. Am J Orthod Dentofacial Orthop. 90(1):11–18. [DOI] [PubMed] [Google Scholar]
- Ranta R, Rintala A. 1982. Tooth anomalies associated with congenital sinuses of the lower lip and cleft lip/palate. Angle Orthod. 52(3):212–221. [DOI] [PubMed] [Google Scholar]
- Richardson RJ, Dixon J, Malhotra S, Hardman MJ, Knowles L, Boot-Handford RP, Shore P, Whitmarsh A, Dixon MJ. 2006. Irf6 is a key determinant of the keratinocyte proliferation-differentiation switch. Nat Genet. 38(11):1329–1334. [DOI] [PubMed] [Google Scholar]
- Rullo R, Festa VM, Rullo R, Addabbo F, Chiodini P, Vitale M, Perillo L. 2015. Prevalence of dental anomalies in children with cleft lip and unilateral and bilateral cleft lip and palate. Eur J Paediatr Dent. 16(3):229–232. [PubMed] [Google Scholar]
- Schroeder DC, Green LJ. 1975. Frequency of dental trait anomalies in cleft, sibling and non-cleft groups. J Dent Res. 54:802–807. [DOI] [PubMed] [Google Scholar]
- van den Boogaard MJ, Créton M, Bronkhorst Y, van der Hout A, Hennekam E, Lindhout D, Cune M, Ploos van Amstel HK. 2012. Mutations in WNT10A are present in more than half of isolated hypodontia cases. J Med Genet. 49(5):327–331. [DOI] [PubMed] [Google Scholar]
- Wang F, Flanagan J, Su N, Wang LC, Bui S, Nielson A, Wu X, Vo HT, Ma Xj, Luo Y. 2012. RNAscope: a novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. J Mol Diagn. 14(1):22–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang KH, Heike CL, Clarkson MD, Mejino JL, Brinkley JF, Tse RW, Birgfeld CB, Fitzsimons DA, Cox TC. 2014. Evaluation and integration of disparate classification systems for clefts of the lip. Front Physiol. 5:163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washbourne BJ, Cox TC. 2006. Expression profiles of cIRF6, cLHX6 and cLHX7 in the facial primordia suggest specific roles during primary palatogenesis. BMC Dev Biol. 6:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright JT, Carrion IA, Morris C. 2015. The molecular basis of hereditary enamel defects in humans. J Dent Res. 94(1) 52–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright JT, Hart TC, Hart PS, Simmons D, Suggs C, Daley B, Simmer J, Hu J, Bartlett JD, Li Y, et al. 2009. Human and mouse enamel phenotypes resulting from mutation or altered expression of AMEL, ENAM, MMP20 and KLK4. Cells Tissues Organs. 189(1–4): 224–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Wang SK, Choi M, Reid BM, Hu Y, Lee YL, Herzog CR, Kim-Berman H, Lee M, Benke PJ, et al. 2015. Taurodontism, variations in tooth number, and misshapened crowns in Wnt10a null mice and human kindreds. Mol Genet Genomic Med. 3(1):40–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucchero TM, Cooper ME, Maher BS, Daack-Hirsch S, Nepomuceno B, Ribeiro L, Caprau D, Christensen K, Suzuki Y, Machida J, et al. 2004. Interferon regulatory factor 6 (IRF6) gene variants and the risk of isolated cleft lip or palate. N Engl J Med. 351(8):769–780. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.