

Abstract
Mutations in vascular smooth muscle α-actin (SM α-actin), encoded by ACTA2, are the most common cause of familial thoracic aortic aneurysms that lead to dissection (TAAD). The R179H mutation has a poor patient prognosis and is unique in causing multisystemic smooth muscle dysfunction (Milewicz, D. M., Østergaard, J. R., Ala-Kokko, L. M., Khan, N., Grange, D. K., Mendoza-Londono, R., Bradley, T. J., Olney, A. H., Ades, L., Maher, J. F., Guo, D., Buja, L. M., Kim, D., Hyland, J. C., and Regalado, E. S. (2010) Am. J. Med. Genet. A 152A, 2437–2443). Here, we characterize this mutation in expressed human SM α-actin. R179H actin shows severe polymerization defects, with a 40-fold higher critical concentration for assembly than WT SM α-actin, driven by a high disassembly rate. The mutant filaments are more readily severed by cofilin. Both defects are attenuated by copolymerization with WT. The R179H monomer binds more tightly to profilin, and formin binding suppresses nucleation and slows polymerization rates. Linear filaments will thus not be readily formed, and cells expressing R179H actin will likely have increased levels of monomeric G-actin. The cotranscription factor myocardin-related transcription factor-A, which affects cellular phenotype, binds R179H actin with less cooperativity than WT actin. Smooth muscle myosin moves R179H filaments more slowly than WT, even when copolymerized with equimolar amounts of WT. The marked decrease in the ability to form filaments may contribute to the poor patient prognosis and explain why R179H disrupts even visceral smooth muscle cell function where the SM α-actin isoform is present in low amounts. The R179H mutation has the potential to affect actin structure and function in both the contractile domain of the cell and the more dynamic cytoskeletal pool of actin, both of which are required for contraction.
Keywords: actin, cofilin, contractile protein, formin, myosin, profilin, vascular smooth muscle cells
Introduction
Mutations in vascular smooth muscle α-actin (SM α-actin)2 are the most prevalent genetic cause of familial thoracic aortic aneurysms that lead to dissection (TAAD), with more than 40 mutations in ACTA2 identified to date (1–5). TAAD leads to a high mortality rate because it is generally asymptomatic until a dissection occurs. Disease progression in large elastic arteries such as the aorta may be driven by decreased smooth muscle cell contraction, and dissected aortas show a typical pattern of loss and disarray of smooth muscle cells in the medial layer (6). Contraction in smooth muscles occurs by virtue of the cyclic MgATP-dependent interaction of myosin with filamentous SM α-actin, controlled by phosphorylation of the regulatory light chain of smooth muscle myosin, in the “contractile domain” of the cell. Smooth muscles are unique in that force production following agonist activation also requires a small amount of actin polymerization in the more dynamic cytoskeletal domain of a smooth muscle cell, where it strengthens the membrane for force transmission to the extracellular matrix (7).
ACTA2 mutations also cause occlusive vascular diseases such as stroke and premature coronary artery disease, caused by enhanced smooth muscle cell proliferation. Cell proliferation is regulated by actin-associated proteins that regulate cell adhesion, spreading, and movement (8) and by actin-binding proteins that affect transcription and dictate whether the cell has a more contractile or proliferative phenotype (9).
Thus it is predicted that at the molecular level, mutations in SM α-actin may profoundly affect its interaction with myosin, with other actin protomers in the filament, with actin-binding proteins that affect the monomer-polymer equilibrium in the cell, and with binding partners that influence cellular phenotype. Many of these ideas were experimentally confirmed in our recent paper that investigated the effect of the R258C mutation on expressed human SM α-actin function (10). Decreased interaction with myosin, formation of less stable filaments, and altered interactions with profilin and cofilin were observed with R258C SM α-actin.
Here, we investigate a mutation, R179H, that represents the severe end of the disease spectrum. Humans with the R179H mutation in SM α-actin show early onset disease that is highly penetrant, with a median age of onset at <15 years of age (1, 5, 11, 12). A unique feature of this mutation is that it causes multisystemic smooth muscle dysfunction (1), affecting even organs such as the bladder and the gastrointestinal tract, where SM α-actin is not the predominant actin isoform. More recently, a patient with the R179H mutation also showed morphological abnormalities in cerebral arteries of all sizes (12).
The R179H mutation was first observed in a zebrafish embryonic cardiac actin isoform (“cardiofunk”), which caused reduced myocardial function and defects in zebrafish heart development (13). This residue is highly conserved in actin, and the effect of the mutated residue was subsequently characterized biochemically in the backbone of Saccharomyces cerevisiae actin. The primary defects were in filament assembly, with the mutation causing a lower extent of polymerization, a prolonged nucleation phase, and decreased actin filament stability (14). Notably, protein residue Arg-177 (hereafter designated as p.R177 and equivalent to gene numbering Arg-179 due to post-translational processing that removes both the N-terminal Met and Cys residues (15)), is also the target of ADP-ribosylation by various bacterial protein toxins, which results in depolymerization of the actin cytoskeleton and subsequent cell death (16).
The actin monomer has four subdomains. Subdomains 3 and 4 constitute the inner domain in filamentous F-actin, and subdomains 1 and 2 make up the outer domain. ATP is bound in the cleft between the two major domains. Protein residue p.R177, is located in a short β-strand near the base of this inter-domain cleft (Fig. 1A). The amino side chain of p.R177 is ∼5 Å from the carbonyl oxygen of p.H73 (blue) in subdomain 1 and ∼3 Å from the negatively charged side chain of p.D179 (Fig. 1B). Mutation of an Arg to a His decreases the size and positive charge of the side chain and will affect interactions with both residues. Molecular dynamic simulations suggest that following ATP hydrolysis p.H73 and p.R177 may provide an exit pathway for phosphate release (17). Once polymerized, the two helical strands that compose filamentous actin (F-actin) are stabilized by interactions between actin protomers both within and between strands. p.R177 lies at an interface between the two strands and abuts a loop in subdomain 4 of an adjacent protomer (Fig. 1, C and D).
FIGURE 1.
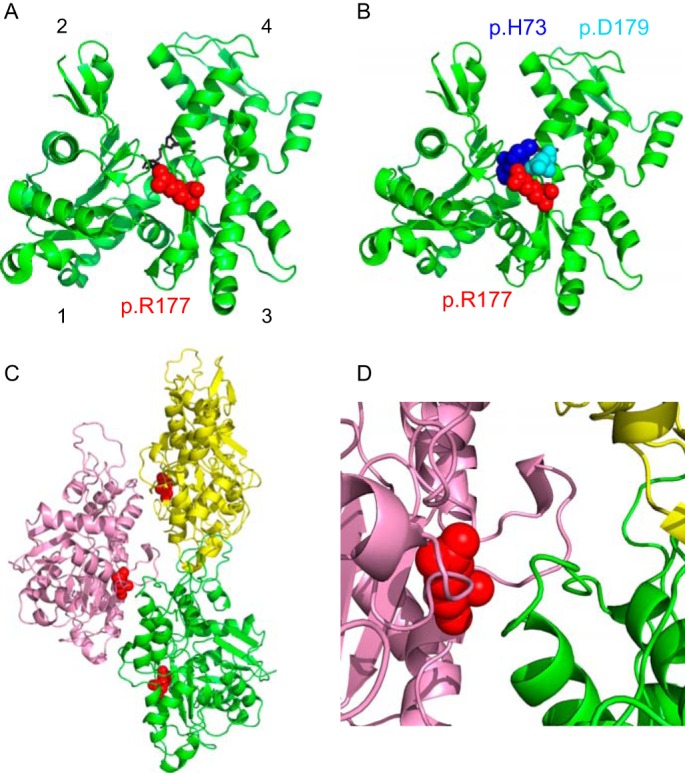
Location of protein residue Arg-177 in the G- and F-actin structure. A, ribbon representation of G-actin (Protein Data Bank code 1NWK). The four subdomains of actin are indicated. Arg-179, which is residue p.R177 in the processed protein, is shown in red space-filling spheres. It is located in a short β-strand in the “backside” of subdomain 3. ATP is shown in black sticks. B, nitrogen on the side chain of residue p.R177 is ∼5 Å from the carbonyl oxygen of p.H73 (blue) in subdomain 1, and ∼3 Å from the negatively charged side chain of p.D179. Neither interaction would occur in the mutant p.H177. C, ribbon representation of three protomers (yellow, pink, and green) from a model of filamentous actin (Protein Data Bank code 3J8A). p.R177 is shown in red spheres at the inter-strand interface. D, close-up of the filament inter-strand interface. Protein residue p.R177 is adjacent to an uncharged loop in subdomain 4 of an adjacent protomer (green) in the filament (protein residues 197–202; Gly-Tyr-Ser-Phe-Val-Thr).
Here, we compare the biochemical properties of expressed WT and R179H human SM α-actin to understand how the mutation affects the basic functions of actin, as this initial insult culminates in smooth muscle dysfunction. The results suggest that the R179H mutation has the potential to decrease smooth muscle contraction via impaired interaction with myosin, to increase the relative amount of monomeric actin in the cell, and to alter the distribution of actin bound to various actin-binding proteins. The severity of the observed biochemical defects with R179H may explain why this mutation has the highest cumulative risk of aortic disease of ACTA2 mutations identified to date.
Results
R179H Mutant Filaments Have a Very High Critical Concentration for Assembly
The R179H mutation was expressed in the human SM α-actin backbone (ACTA2 gene) using the baculovirus/insect cell expression system, as described previously (10). The expressed SM α-actin was His-tagged, which allows us to purify it free of contamination with endogenous Sf9 cell actin. The thymosin and the His tag were removed prior to functional analysis of the actin as described in Ref. 10. The properties of the R179H mutant were compared with wild-type (WT) SM α-actin expressed and purified by the same procedure. An important aspect of this work is that the effect of the mutation was studied in the actin isoform that is expressed in human vascular smooth muscle cells.
Given the severity of the R179H phenotype in patients, we first tested whether the mutant actin could assemble into filaments. Total internal reflection fluorescence microscopy was used to follow total filament length as a function of time. To illustrate filament growth, a series of TIRF images are shown as a function of time (Fig. 2A). At the light microscope level, R179H polymerizes into filaments that are indistinguishable from WT. The rate of polymerization as a function of actin concentration is plotted in Fig. 2C. The assembly rate (slope of the rate versus actin concentration graph) is decreased 40% for the mutant filaments, whereas the disassembly rate (y intercept) is increased 27-fold compared with WT. This results in a critical concentration (x intercept) of ∼2 μm actin, 40-fold greater than the 50 nm critical concentration for WT SM α-actin. Kinetic data obtained from multiple experiments using independent preparations of R179H are summarized in Table 1. These changes in polymerization suggest that monomers of R179H are less able to form a stable nucleus that allows the filament to elongate. Once filament elongation begins, the net rate of growth is slower than WT partly because of the high rate of disassembly.
FIGURE 2.
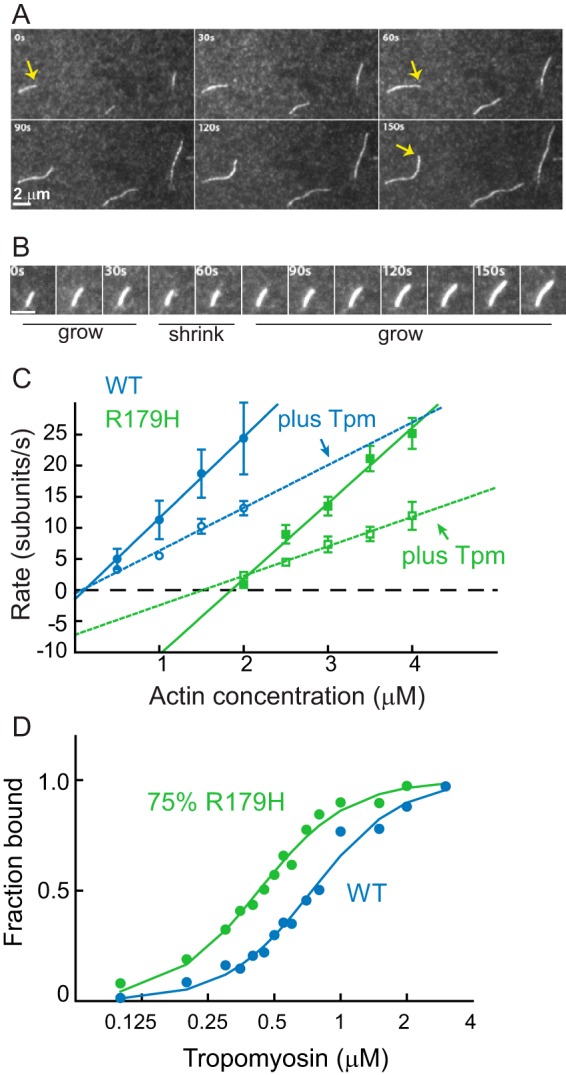
Quantification of R179H actin filament growth observed by TIRF microscopy and tropomyosin binding affinity. A, growth of filaments from 4 μm R179H G-actin as a function of time. Each panel is separated by 30 s. The yellow arrow marks the growth of one filament. Bar in bottom left corner is 2 μm. B, time series images showing the growing and shrinking of R179H filaments at 2.5 μm actin. The white bar in the lower left corner is 2 μm long. C, rate of filament growth as a function of actin concentration for WT SM α-actin (blue) and R179H (green) actin, in the absence (solid line) or presence (dashed line) of smooth muscle tropomyosin. Error bars are S.E. Parameters obtained from these data (assembly rate, disassembly rate, and critical concentration) with multiple independent preparations are given in Table 1. D, affinity of WT (blue) and R179H (green, filaments formed from 25%WT and 75% R179H actin) F-actin for smooth muscle tropomyosin determined by an actin pelleting assay. Maximal binding was normalized to 1. Data were fit to the Hill equation. Kapp for WT is 1.3 × 106 m−1 and for R179H is 2.3 × 106 m−1. The Hill coefficient is 2.2 for WT and 2.1 for R179H.
TABLE 1.
Polymerization parameters for R179H and WT actin
| Tpm | Assembly rate (subunits per μm·s) | Disassembly rate (subunits per s) | Critical concentration | |
|---|---|---|---|---|
| nm | ||||
| R179Ha | − | 9.5 ± 2.4 | 19 ± 3.9 | 2057 ± 422 |
| WTb | − | 15.9 ± 3.4 | 0.7 ± 0.6 | 47.8 ± 44.2 |
| R179Hc | + | 4.7 ± 0.2 | 7.2 ± 0.6 | 1514 |
| WTb,c | + | 6.9 ± 0.7 | 0.5 ± 0.9 | 76 |
a Average ± S.D. from 11 experiments using seven independent preparations of R179H is shown.
b WT actin data were from our previous publication (10).
c Data in the presence of tropomyosin were obtained from one experiment. Errors are standard error of the fit.
At actin concentrations slightly higher than the critical concentration, we occasionally observed a growing R179H filament that suddenly shortened before it started to grow again (Fig. 2B). The shortening occurred at the fast-growing barbed end, which distinguishes it from the proposed actin treadmilling mechanism. This phenomenon was not observed at higher actin concentration where the assembly rate is faster, indicating that it is caused by depolymerization that occurs while waiting for the next monomer to be added onto the filament end and not filament cleavage. Filament breakage was rarely observed during experiments.
Tropomyosin Slows Polymerization Kinetics
Actin filaments in smooth muscle cells are predominantly associated with tropomyosin (Tpm), an α-helical coiled-coil that binds along the length of the actin filament. In addition to its regulatory role, Tpm also plays a structural role in stabilizing the actin filament. TIRF polymerization assays carried out in the presence of smooth muscle Tpm showed that tropomyosin slowed both the assembly and disassembly rates of WT SM α-actin, with little change in critical concentration. The same trend was seen with R179H actin (Fig. 2C and Table 1). Although Tpm stabilizes the R179H filaments by slowing disassembly, subunits still dissociated ∼14-fold faster from R179H actin-Tpm filaments than from WT actin-Tpm filaments (7 versus 0.5 subunits/s, respectively).
A cosedimentation assay was used to measure the binding of smooth muscle Tpm to WT and mutant filaments (Fig. 2D). The binding constant (Kapp) for Tpm to filaments composed of 75% R179H/25% WT was slightly higher (2.0 × 106 m−1) than to WT filaments alone (1.0 × 106 m−1).
Copolymers of WT and R179H Are More “WT-like”
A heterozygous human carrying the R179H mutation will express WT and R179H actin in approximately equal amounts. To mimic this condition, the polymerization of equimolar amounts of WT and R179H actin was followed as a function of varying total actin concentration. As shown in Fig. 3A, the polymerization properties of the mixture more closely resembled WT actin than R179H actin. The assembly rate for the 50:50 mixture, obtained from the slope of the polymer elongation rate versus actin concentration, is greater than half of the WT rate, providing evidence that the WT and mutant actin copolymerize. In a cellular context, this result suggests that WT actin will dampen the adverse effect of the R179H mutation on filament stability. Note that the filament will likely not contain equal amounts of the two actins.
FIGURE 3.
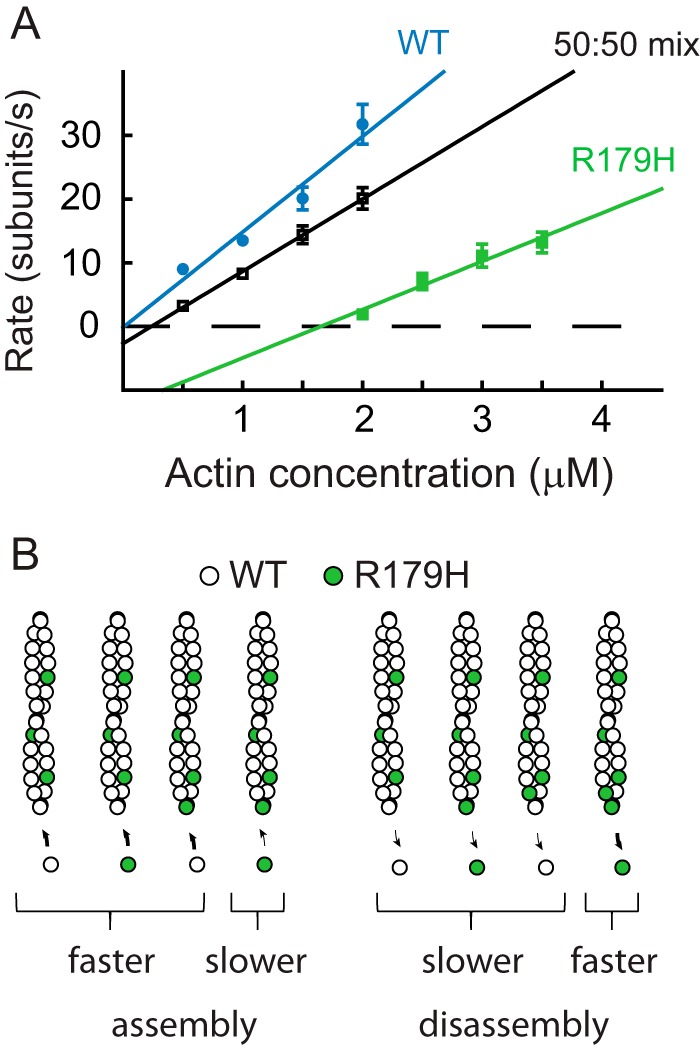
Copolymerization of equal amounts of WT and R179H actin. A, rate of filament growth versus actin concentration graph for WT SM α-actin (blue), R179H (green), and an equimolar mixture of WT and R179H actin (black). Error bars are S.E. B, schematic of a simple model to explain the copolymerization data (see “Experimental Procedures” for details). The slower assembly rate of the mutant actin occurs only when R179H adds onto a filament with a mutant protomer at the end. Disassembly only occurs at the fast mutant rate if both end protomers are R179H actin. Table 2 tabulates the polymerization parameters and values calculated from the model.
Using a simplified model similar to that described earlier (10), the experimental data were compared with the value calculated from the model (Table 2 and Fig. 3B). The basic premise is that the slower assembly rate seen for R179H occurs only when a mutant monomer adds onto a filament end that has a mutant protomer. If a mutant monomer adds onto a filament end containing a WT protomer, the assembly rates will be the faster rate seen with WT actin alone. The calculated assembly rate from this simple model is close to the experimental result, suggesting that a mutant-WT copolymer adopts an overall filament conformation that is more similar to that of a polymer composed of WT actin alone. The model also predicts that the fast disassembly rate seen with R179H only occurs when both end protomers are R179H. The lower experimental disassembly rate compared with this simple model prediction suggests that longer range changes propagated by nearby WT protomers slow the R179H dissociation rate.
TABLE 2.
Polymerization rates of mixtures of WT and R179H
Conditions used as follows: 10 mm imidazole, pH 7.5, 50 mm KCl, 4 mm MgCl2, 1 mm EGTA, 37 °C.
| Assembly rate (subunits per μm·s) | Disassembly rate (subunits per s) | Critical concentration | |
|---|---|---|---|
| nm | |||
| WT SM α-actin | 15 | 0.15 | 9.9 |
| R179H | 7.6 | 16.34 | 2453.6 |
| 50% WT α-actin, 50% R179H | 11.4 | 2.7 | 237.6 |
| Calculated from modela | 13.15 | 4.2 | 319.4 |
| WT SM γ-actin | 13.0 | 0.99 | 76.8 |
| R179H | 8.6 | 19.43 | 2249.9 |
| 50% WT γ-actin, 50% R179H | 11.4 | 1.65 | 144.7 |
| Calculated from modela | 11.9 | 5.6 | 470 |
a Details of the model are described under “Experimental Procedures.”
The R179H mutation in SM α-actin is unique among the actin mutations known to cause TAAD, because it also affects the function of smooth muscles such as the bladder and stomach that predominantly express SM γ-actin from the ACTG2 gene. A possible explanation is that the combination of WT SM γ-actin and R179H SM α-actin in the same copolymer forms a particularly unstable filament. However, when we formed copolymers from WT SM γ-actin and R179H mutant SM α-actin, the result was very similar to that seen when the mutant actin was copolymerized with WT SM α-actin (Table 2).
Profilin Binds More Tightly to R179H Actin than to WT
Profilin is an abundant actin-sequestering protein that binds monomeric actin and regulates the pool of available free actin. The affinity of profilin for actin is therefore important for all actin-related processes. When R179H actin was polymerized in the presence of 1 μm profilin, the polymerization curve shifted rightward, resulting in a further increase in the already high critical concentration (Fig. 4). Note that addition of a 3-fold higher concentration of profilin (3 μm) to SM α-actin results in a much smaller shift to the right than addition of only 1 μm profilin to R179H actin. The fit to a model as described in Ref. 10 yields a Kd of 0.65 μm profilin for R179H actin versus 3 μm for WT, indicating that the mutant binds profilin with ∼4-fold higher affinity (Table 3).
FIGURE 4.
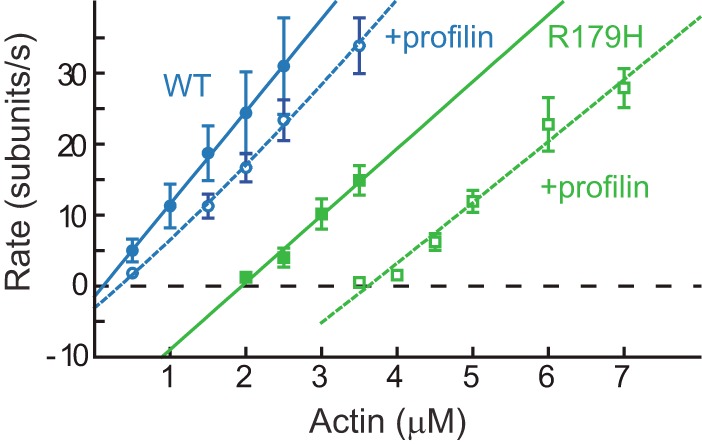
R179H actin binds more strongly to profilin than WT SM α-actin. Rate of polymerization of WT SM α-actin (blue) or R179H actin (green) in the absence (solid line) or presence (dashed line) of profilin. Error bars are S.E. Profilin concentration was 1 μm for R179H actin and 3 μm for WT. The dashed lines are fits to the data as described under “Experimental Procedures.” Profilin binds to R179H actin ∼4-fold tighter than to WT actin (see Table 3).
TABLE 3.
Polymerization data in the presence of profilin
WT actin is plus 3 μm profilin; R179H actin is plus 1 μm profilin.
| Fit parameters | WT | R179H |
|---|---|---|
| Assembly rate (subunits per μm·s) | 3.5 | 0 |
| Disassembly rate (subunits per s) | 3.1 | 24.8 |
| Kd (μm) | 3 | 0.65 |
| Critical concentration (nm) | 330 | 3,610 |
R179H Shows a Defect in Formin-mediated Nucleation and Polymerization
Formin is a key actin nucleation factor for linear filaments in vivo, and it acts processively to facilitate elongation of actin filaments at the barbed end. The conserved formin-homology domain 1 (FH1) binds profilin, and formin-homology domain 2 (FH2) binds actin (18, 19). We compared the interaction of WT and R179H actin with a constitutively active FH1-FH2 fragment of mDia (10, 20, 21). Interestingly, when 50 nm mDia was included during polymerization, the average number of filaments formed and the rate of polymerization were both slightly reduced with R179H actin (Fig. 5A). This observation was in contrast to WT actin, which showed an increased number of filaments and an unchanged polymerization rate. Thus, mDia not only failed to enhance R179H nucleation but was detrimental to it. The decreased filament number and polymerization rate may be inter-related, i.e. the slower polymerization rate with mDia could slow elongation of the actin seed, thus increasing the possibility that the nucleation complex disintegrates.
FIGURE 5.

R179H actin has a defect in formin-mediated polymerization. A, number of filaments (in 54 × 54 μm2 area after 2 min) and the polymerization rate for WT SM α-actin (blue bars) and R179H actin (green bars) in the presence of 50 nm formin (FH1-FH2 fragment of mDia1) and/or 1 μm profilin. B, number of filaments and polymerization rate versus formin concentration for WT and R179H actin. The curve was fit to r = (r2 − r1)a + r1, in which r1 is the assembly rate of formin-free actin filaments (14.4 subunits/s); r2 is the assembly rate of formin-capped filaments (1 subunit/s), and a is the percentage of filaments that are formin-capped. C, polymerization rate histogram of R179H in the presence of increasing amounts of formin. Two distinct rates were observed; the slower rate is due to polymerization with formin bound to the growing barbed end, and the faster rate is polymerization onto a formin-free barbed end.
We further investigated these effects as a function of formin concentration. As the formin concentration was increased, filament number decreased for R179H but increased for WT actin (Fig. 5B). The rate of R179H filament growth also decreased with formin concentration but was constant for WT actin (Fig. 5B). A closer look at the distribution of polymerization rates for R179H showed that there were two distinct rates, one fast and one slow (Fig. 5C). The relative percentage of the slow rate population increased with an increase in formin concentration, while the relative amount of the fast rate population decreased. If we assume that the slower rate is due to polymerization with formin bound to the growing barbed end, and the fast rate is polymerization onto a formin-free barbed end, the raw data can be fit to a curve (Fig. 5B). The fit result shows that FH1-FH2 formin has a Kd of ∼190 nm for the R179H filament barbed end, which is in the same range as reported for WT actin in the literature (20, 21). The rate of elongation onto a formin-capped filament is ∼1 subunit/s at 4 μm R179H.
Formin and profilin generally work in concert to enhance actin polymerization. However, the presence of both profilin and mDia significantly reduced the rate of R179H actin polymerization (Fig. 5A). Because R179H binds very tightly to profilin, the lower amount of free actin could explain this slower rate. However, the rate of polymerization in the presence of formin and profilin was not enhanced compared with that of profilin alone, implying that the defect in formin binding to R179H primarily causes the slow rate. These data do not exclude the possibility that R179H-profilin may bind weakly to the formin FH1 domain or that the high affinity of the mutant actin for profilin might slow the transfer of actin from profilin to the formin-capped barbed end for polymerization.
R179H Filaments Are More Susceptible to Cofilin Severing
Cofilin is an actin severing protein that plays a role in the remodeling and turnover of actin filaments. Given that filaments composed of pure R179H mutant actin are less stable than WT filaments, we speculated that R179H filaments would also be more susceptible to cofilin cleavage. This expectation was confirmed experimentally. R179H filaments were severed (Fig. 6A) and shortened (Fig. 6B) at lower cofilin concentrations than WT. Smooth muscle tropomyosin protected both WT and mutant actin against cofilin cleavage and shortening by shifting the effect of cofilin to higher concentrations.
FIGURE 6.

R179H filaments are more susceptible to cofilin-induced shortening and severing than WT SM α-actin, even in the presence of tropomyosin. A, frequency of severing actin filaments by cofilin for WT SM α-actin (blue bar), R179H (green bar), and mixtures of WT and mutant actin (50% R179H, darker gray bars; 90% R179H, lighter gray bars). Lighter shaded blue and green bars are in the presence of smooth muscle tropomyosin. Frequency is reported as the number of events per 1 min per 1 μm of actin filament. Error bars are S.E. Equimolar copolymers of R179H and WT are more resistant to cofilin severing than R179H homopolymers. B, rate of actin filament shortening induced by cofilin. Same color scheme as A. Error bars are S.E. Equimolar copolymers of R179H and WT are more resistant to cofilin shortening than R179H homopolymers. C, affinity of WT (blue) and R179H (green, formed from 90% R179H actin and 10% WT) filaments for cofilin determined by an actin pelleting assay. Maximal binding was normalized to 1. Data were fit to the Hill equation. Kapp for WT filaments is 0.6 × 106 m−1 and 0.4 × 106 m−1 for R179H filaments. The Hill coefficient is 2.2 for WT and 6.9 for R179H.
The enhanced cleavage of R179H filaments by cofilin may also be a result of enhanced binding of cofilin to R179H filaments. We carried out an actin-cofilin pelleting assay to test this possibility. To reduce the amount of actin in the supernatant, the assay was performed with filaments formed from a mixture of 90% R179H and 10% WT monomers. The affinity of the R179H copolymer for cofilin was slightly lower than for WT filaments (Fig. 6C). The copolymer formed from 90% R179H and 10% WT monomers showed almost the same sensitivity to cofilin as R179H homopolymers (Fig. 6, A and B). These results rule out the possibility that enhanced susceptibility of the mutant filaments to cofilin cleavage is due to tighter binding of cofilin.
We also investigated the effect of cofilin on actin filaments polymerized from an equimolar ratio of WT and R179H monomers. Based on polymerization kinetics (Fig. 3), these 50:50 copolymers are more stable than R179H homopolymers. Similarly, the 50:50 heteropolymer was less susceptible to cofilin severing than the mutant homopolymer (Fig. 6A).
R179H Binds Non-cooperatively to Myocardin-related Transcription Factor-A
Myocardin-related transcription factor-A (MRTF-A) is a transcriptional coactivator whose cellular localization is controlled by its interaction with G-actin (22). To determine whether the mutation affects the affinity of MRTF-A for actin, polymerization experiments were performed in the presence of MRTF-A. Fig. 7A shows that the presence of MRTF-A shifts the polymerization curve to higher actin concentration. In addition, the assembly rate no longer increases linearly with actin concentration. The data were fit to the Hill equation, which describes cooperative binding. Each MRTF-A can bind up to five actin monomers (22). The fits yield similar affinities for both actins (Kd = 1.8 μm for WT; Kd = 1.1 μm for R179H actin). More interestingly, the Hill coefficient for WT versus R179H is 3 and 1.5, respectively. This result shows that R179H actin binds less cooperatively to MRTF-A than WT. This lower cooperativity could be due to weak interactions between the R179H monomers bound to MRTF-A, consistent with weaker interactions between protomers in the R179H filament. When a 50:50 mix of mutant and WT actin was polymerized in the presence of MRTF-A (Fig. 7B), and the data fit to the Hill equation, the Hill coefficient was 1.7, more similar to that seen with R179H alone. With only five actin monomers in the complex, the tempering effect of WT actin may not be as strong as for a filament that is composed of hundreds of protomers.
FIGURE 7.

R179H actin binds less cooperatively to the transcription factor MRTF-A than WT SM α-actin. A, rate of polymerization of WT SM α-actin (blue) or R179H actin (green) in the absence (solid line) or presence (dashed line) of MRTF-A (3 μm for WT, 1 μm for R179H). Error bars are S.E. The dashed curves are fits to the Hill equation as described under “Experimental Procedures.” The fits yield a Kd of 1.8 μm for WT and 1.1 μm for R179H. Hill coefficients are 3 for WT and 1.5 for R179H. B, rate of polymerization of WT SM α-actin (blue, repeated from A) or a 50:50 mixture of WT and R179H actin (gray) in the presence (dashed line) of 3 μm MRTF-A. Error bars are S.E. The dashed curves are fits to the Hill equation as described under “Experimental Procedures.” The fits yield a Kd of 1.7 μm for the 50:50 mix of WT and R179H, and a Hill coefficient of 1.7.
R179H Actin Is Moved More Slowly by Smooth Muscle Myosin
An in vitro motility assay was used to measure the speed at which phosphorylated human smooth muscle myosin moves R179H actin, in the presence or absence of smooth muscle tropomyosin. Speed was slowed even when the mutant filaments were stabilized with rhodamine-phalloidin (Fig. 8A). In the absence of tropomyosin, homopolymers of R179H moved at ∼60% the speed of WT, but speeds were the same with filaments polymerized from an equimolar mixture of WT and R179H. Tropomyosin enhanced the speed of both WT and mutant filaments. Filaments polymerized from mixtures containing 50–100% R179H all showed a small but statistically significant decrease in speed in the presence of tropomyosin when compared with WT actin-Tpm.
FIGURE 8.
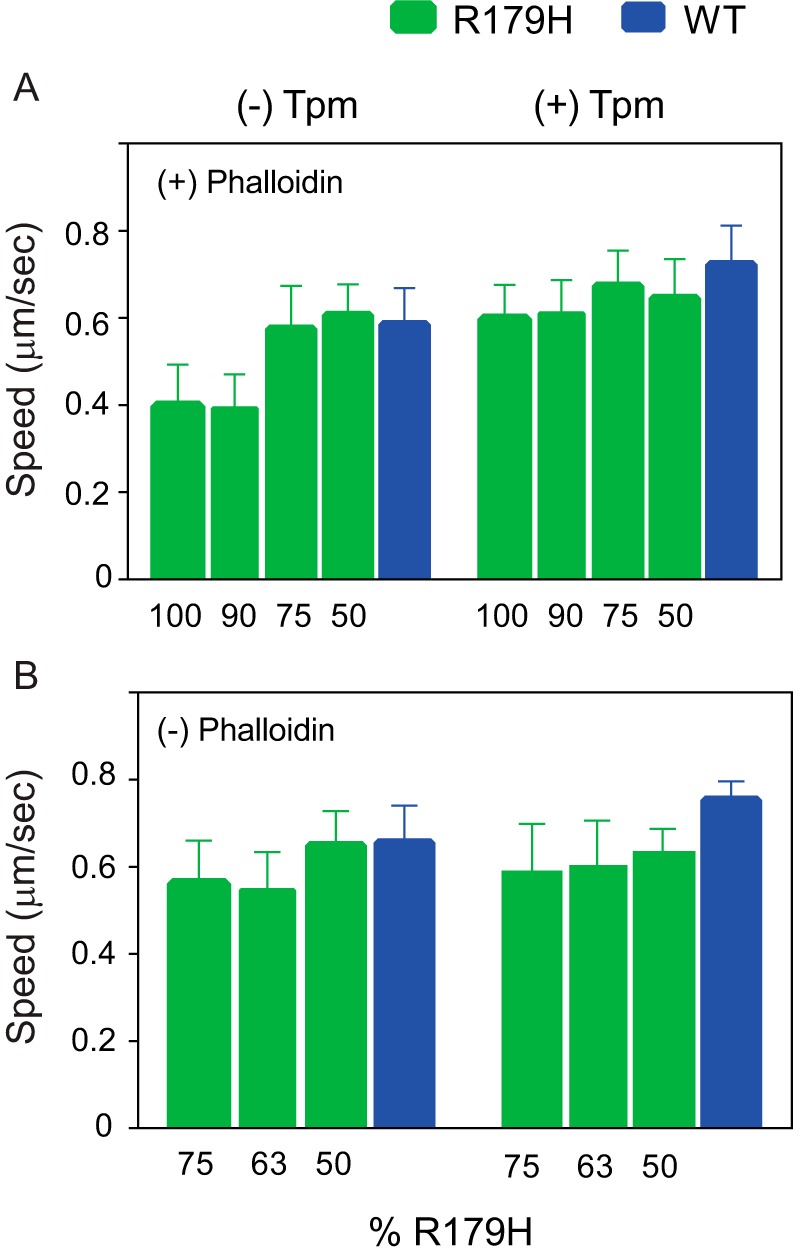
In vitro motility performed with R179H or WT SM α-actin in the absence or presence of tropomyosin. A, speed at which phosphorylated human smooth muscle myosin moves phalloidin-stabilized actin filaments. Filaments were polymerized from mixtures containing the indicated percentage of R179H actin. All pairs showed a statistically significant decrease compared with WT, except for the 50 and 75% R179H mixtures in the absence of tropomyosin. With bare actin, speeds were as follows: 0.585 ± 0.083 (n = 100) for WT; 0.606 ± 0.071 (n = 41) for 50% R179H; 0.574 ± 0.100 (n = 41) for 75% R179H; 0.392 ± 0.079 (n = 41) for 90% R179H; and 0.397 ± 0.096 (n = 58) for 100% R179H. In the presence of Tpm1.4, speeds were as follows: 0.722 ± 0.090 (n = 100) for WT; 0.645 ± 0.090 (n = 40) for 50% R179H; 0.673 ± 0.082 (n = 40) for 75% R179H; 0.610 ± 0.077 (n = 40) for 90% R179H; and 0.597 ± 0.079 (n = 40) for 100% R179H. B, speed at which phosphorylated human smooth muscle myosin moves actin filaments that were not stabilized with phalloidin. Filaments were polymerized from mixtures containing the indicated percentage of R179H actin. All pairs showed a statistically significant decrease compared with WT, except for the 50% R179H mixtures with bare actin. Bare actin speeds were as follows: 0.616 ± 0.084 (n = 40) for WT; 0.591 ± 0.089 (n = 40) for 50% R179H; 0.490 ± 0.106 (n = 40) for 63% R179H; and 0.494 ± 0.111 (n = 40) for 75% R179H. In the presence of Tpm1.4, speeds were as follows: 0.732 ± 0.052 (n = 40) for WT; 0.613 ± 0.048 (n = 40) for 50% R179H; 0.584 ± 0.087 (n = 40) for 63% R179H; and 0.571 ± 0.092 (n = 40) for 75% R179H. Data were obtained using three independent protein preparations each of WT and R179H.
A similar trend was seen when the filaments were not stabilized with phalloidin (Fig. 8B). When polymerized from mixtures containing more than 50% R179H actin, the speed of bare actin was slowed. In the presence of tropomyosin, the slowing was more pronounced, with mutant filaments moving at ∼75% the speed of WT actin, even with filaments polymerized from equimolar mixtures of WT and mutant actin.
Discussion
The R179H mutation in SM α-actin shows severe defects in many facets of actin function that can account for its poor prognosis for patients. A notable defect is its inability to readily polymerize, with R179H requiring 40-fold more actin (∼2 μm) to form filaments than is needed with WT actin. The location of protein residue p.R177 at the interface between the two strands that comprise the actin filament is consistent with the observed polymerization defect. The difficulty in forming a stable seed to initiate polymerization results in filaments that can grow and then shrink around the critical concentration, reminiscent of dynamic instability that is a property generally associated with microtubules (23). The polymerization defect is primarily driven by a very fast disassembly rate of ∼20 subunits/s, compared with ∼1 subunit/s for WT actin. The observation that the disassembly rate increased by a much larger factor than the assembly rate suggests that the interactions critical for assembly and disassembly differ. Multiple changes occur once a monomer is incorporated into the filament, including hydrolysis of MgATP and structural changes in the protomer. In F-actin, the two major domains become flatter with respect to one another compared with that observed in monomeric G-actin (24–27). Identification of the structural differences between the R179H and WT filaments should provide insight into the structural basis for the extremely fast depolymerization of R179H filaments.
Modeling studies have suggested that protein residues p.H73 and p.R177, which form a hydrogen bond, may be part of a back door mechanism that facilitates phosphate release following ATP hydrolysis (17). Mutation of this residue thus has the potential to increase the rate of phosphate release. Although we did not directly measure this parameter, a faster rate of phosphate release would result in more protomers with ADP bound, which reduces filament stability. ADP-bound G-actin has a higher critical concentration and a slower rate of polymerization (28), consistent with the defects we observed.
Binding of smooth muscle tropomyosin or copolymerization with WT SM α-actin stabilizes the R179H filament. Once R179H monomers are incorporated into a filament with WT actin, the mutant protomers may be forced to take the canonical filamentous conformation by virtue of their interaction with neighboring WT actin protomers. We would predict that the structure of a mixed WT-mutant heteropolymer will be more similar to WT homopolymers. Our observations imply that the extremely detrimental effect of the R179H mutation will be partially attenuated in heterozygous patient cells that express both mutant and WT actin.
In the cell, formin collaborates with actin-profilin to enhance polymerization of linear actin filaments such as those found in the contractile domain of a smooth muscle cell (18, 19). In the presence of formin alone, WT actin forms more filaments because formin acts as a nucleator, but the filaments grow at approximately the same rate as without formin. In contrast, with R179H actin, not only is there no increase in filament number, but the rate of polymerization of formin-capped filaments slows ∼14-fold compared with the rate of assembly of a formin-free end. Formin is thought to exist in an open conformation where actin incorporation is allowed, and a closed conformation where both FH2 monomers form a ring-like structure that binds the terminal actin subunits and blocks further actin incorporation (18). Our results suggest that a mutation in actin can shift the formin equilibrium toward a closed state.
Other Defects of Filaments Composed of R179H Protomers
The instability of R179H filaments also manifests as an enhanced susceptibility to being severed and shortened by cofilin. Cofilin binds with a slightly lower affinity to mutant filaments and thus cannot be the reason for the enhanced susceptibility. Tropomyosin binding partly suppressed this sensitivity, either by competing for binding sites with cofilin or by stabilizing the R179H filament. It has been suggested that cofilin induces cleavage of actin by targeting non-uniformity in the filaments (29). Our result showing that heteropolymers are less readily cleaved by cofilin than the R179H homopolymer implies that a mixture of R179H and WT protomers within the filament does not introduce discontinuities in the filament.
The effect of an increased susceptibility to cofilin severing may be manifested as a decrease in force by compromising the structural integrity of actin filaments in the contractile domain of the cell. Cofilin activation, which occurs upon dephosphorylation of Ser-3 by slingshot phosphatase (30), has also been shown to be necessary for the dynamic reorganization of cortical actin that is required for smooth muscle contraction (31). However, the amount of actin that is remodeled likely needs to be tightly regulated, and an increased susceptibility of filaments to cofilin cleavage is the functional equivalent of having too much activated cofilin. Immunofluorescence studies and differential centrifugation suggest that the non-muscle β- and γ-actin isoforms may be primarily responsible for agonist-induced polymerization (32). However, given that the R179H mutation creates an actin that is considerably more dynamic than WT SM α-actin, we speculate that the mutant actin comprises a significant amount of the pool of cortical actin that is re-organized prior to contraction.
Interaction with Myosin
In the contractile domain of smooth muscle cells, phosphorylated smooth muscle myosin interacts with SM α-actin to produce force. Mutant actin filaments that were not stabilized by phalloidin showed an ∼20% decrease in the speed at which they are moved by myosin in an in vitro motility assay. The slower speed could be due to a slowing of the rate of ADP release from myosin, although this mechanism was ruled out for the R258C mutation that we recently characterized (10). Alternatively, it is possible that the powerstroke of myosin may not be fully translated into motion because the mutant actin filament is too compliant, another mechanism that could lead to a decrease in speed. This slowing persisted in the presence of Tpm1.4, the predominant smooth muscle tropomyosin isoform that decorates actin in the contractile domain (33). The observed slower velocity will decrease power output, assuming that the force produced by myosin is the same or lower than that produced with WT actin (power = force × velocity). Thus, these data are consistent with the hypothesis that a decrease in smooth muscle contraction is one of the initial insults that leads to aortic aneurysms.
Defects in Interactions of Monomeric R179H with Actin-binding Partners
Given the decreased propensity for R179H to be part of a polymer, we explored how mutant monomeric G-actin interacts with several of its binding partners. Changes in these interactions will impact the amount of filamentous actin in the cell needed for contraction, and it may indirectly affect the level of expression of contractile proteins via altered interaction with the transcription factor MRTF-A.
Profilin is an abundant actin-binding protein that binds monomeric actin with high affinity. We show that R179H actin binds profilin ∼4-fold more tightly than WT actin, which will increase the monomeric pool of actin in the cell. Based on the crystal structure of profilin-actin (PDB code 1HLU), actin protein residue Arg-177 is at least 15 Å away from two main areas of contact between profilin and actin that include profilin residues 119–122 (α-helix) and 69–74 (β-sheet). Thus actin protein residue Arg-177 does not interact directly with profilin. However, p.R177 resides in a small β-strand that follows a loop-helix that is in direct contact with profilin. It is possible that the mutation causes the chain to shift position in a way that enhances the affinity between the mutant actin and profilin.
Profilin-actin also normally works in concert with formin in the cell to efficiently polymerize linear actin filaments. However, profilin-R179H actin and formin-profilin-R179H actin showed the same rate of polymerization, rather than the expected enhancement with the latter ternary complex. The lack of rate enhancement could be mainly due to formin existing in the closed conformation that slows/blocks monomer addition (discussed above). It is also possible that R179H-profilin binds weakly to the formin FH1 domain. In all cases, the result will be more monomeric and less filamentous mutant actin.
A second binding partner is MRTF-A, a transcriptional coactivator whose localization is controlled by its interaction with G-actin (22). MRTF-A is localized in the cytoplasm where it forms a complex with five actin monomers. Responding to upstream signals, actin polymerization causes release of MRTF-A, which allows it to be transferred to and accumulated in the nucleus. In the nucleus, MRTF-A binds with serum response factor to CArG elements and induces expression of serum response factor-dependent, smooth muscle-specific genes, including SM α-actin. The affinity of MRTF-A for actin is thus an important parameter in this feedback control scheme that regulates the expression of smooth muscle genes and the amount of actin in vivo. Our data show that R179H actin binds with similar affinity, but less cooperativity, to MRTF-A. However, balancing the lower cooperativity is the fact that there is more mutant actin available in the G-form to bind MRTF-A. If MRTF-A is mainly sequestered in the cytoplasm, it will favor a proliferative phenotype in the vascular smooth muscle cells that may contribute to the occlusive vascular diseases. Distinct cerebrovascular pathologic changes were observed in a patient harboring the R179H mutation who died of an ischemic stroke; the changes were characterized by marked proliferation of smooth muscle-like cells in small and large arteries in the brain (12).
Why Is This Mutation Unique in Showing Global Smooth Muscle Failure?
Essentially all of the other ACTA2 mutations implicated in TAAD restrict their effects to vascular smooth muscle. R179H is an exception in that it shows multisystemic smooth muscle dysfunction (1), i.e. the effect of this mutation also extends to visceral smooth muscles that predominantly express ACTG2, the SM γ-actin isoform. Despite being present in only small amounts in visceral tissue, it was shown that lack of expression of SM α-actin results in decreased contractile function of the mouse bladder, providing evidence that the SM α-actin isoform contributes to visceral smooth muscle function (34). At present, our data favor the idea that because the molecular defects with R179H are so severe, particularly with regard to polymerization, even small amounts of the mutant actin can exert a negative effect on tissue function.
Interestingly, mutation of Arg-178 in ACTG2 (equivalent to Arg-179 in ACTA2), the predominant actin isoform in visceral smooth tissue (SM γ-actin), has recently been shown to cause megacystis-microcolon-intestinal hypoperistalsis syndrome, a rare disorder of enteric smooth muscle function that affects the intestine and bladder (35). The TAAD mutations we previously characterized in ACTA2, R258C, and R258H (10), also causes megacystis-microcolon-intestinal hypoperistalsis syndrome when mutated in ACTG2. Patients with this disease are dependent on total parenteral nutrition and urinary catheterization. The fact that actin mutations have been implicated in both diseases highlights the importance of the actin cytoskeleton in the maintenance of proper smooth muscle function.
Comparison of Two Mutations That Cause TAAD
We have now investigated the effect of the two most severe ACTA2 mutations, R258C (10) and R179H, which have the poorest patient prognosis. There are similarities in many of the defects but differences in severity. Although both mutations form less stable filaments (i.e. the mutant filaments polymerize more slowly than WT, depolymerize faster, and have higher critical concentration compared with WT), the defects are more pronounced in R179H, which has poorer patient outcomes (5). Particularly with regard to polymerization defects, the R179H mutation may define the extreme end of the spectrum. Both mutants copolymerize with WT, and when copolymerized the defects are suppressed but not eradicated. Both mutants have a higher affinity toward profilin than WT actin, implying that the pool of monomeric actin will be higher in the cell. The two mutants interact differently with formin; R258C interaction with formin is more WT-like, but formin bound to R179H slows polymerization and interferes with nucleation. Both mutant filaments are more readily severed and shortened by cofilin. Smooth muscle myosin moves both mutant actin filaments more slowly than WT actin.
It will be interesting to see the extent to which a more benign mutation, such as R185Q (5), shows the same molecular defects. It is quite possible that other TAAD related actin mutations may share these traits but with varying degrees of severity. The similarity in phenotypes, despite the different locations of the mutation in the actin structure, suggests that the effect of the mutation propagates throughout the monomer, and it affects binding sites distant from the mutation site. A simple prediction of defects based on the location of the mutation is thus not feasible with actin.
Conclusions
The defects observed for both R258C and R179H are consistent with the idea that mutations that lead to thoracic aneurysms and aortic dissections are caused by a “loss of function” that leads to smooth muscle cell weakness, a failure of tension sensing, and medial layer degeneration. In a cell containing mutant actin, filaments would be expected to be sparser because they are less stable and more readily severed, and the pool of monomeric actin larger because the mutant actin binds more tightly to profilin than WT actin. The molecular motor myosin also interacts more poorly with the mutant filamentous actin. The mutant actins are more labile and thus may show a greater tendency to mingle with the cytoplasmic γ-actin at the cell cortex. Compromising polymerization of this pool would be another path to compromise force production, as a small pool of cortical actin must polymerize for contraction to occur (7). Cellular studies with fibroblasts and smooth muscle cells derived from patients, as well as mouse models, will establish how these molecular defects are manifested in a cellular context.
Experimental Procedures
Protein Cloning, Expression, and Purification
The methods used for human SM α-actin cloning, expression, and purification are essentially as described previously (10). Lifeact-GFP, cofilin, profilin, formin, and smooth muscle myosin were expressed and purified as described previously (10).
MRTF-A
DNA for the RPEL domain of Mus musculus MRTF-A, residues 67–199 (GenBankTM EDL04588), was synthesized by Integrated DNA Technologies. The DNA, with a C-terminal His tag, was cloned into pET24b (Novagen). The plasmid was transformed into the Escherichia coli strain Rosetta (Novagen) and cultured in enriched broth. One liter of bacterial culture was pelleted and lysed by sonication in 50 ml of a buffer containing 50 mm Tris, pH 8 (4 °C), 0.3 m NaCl, 1 mm EDTA, 7 mm β-ME, 0.5 mm 4-(2-aminoethyl)benzenesulfonyl fluoride-HCl, 0.5 mm Nα-tosyl-l-lysine chloromethyl ketone hydrochloride, 5 μg/ml leupeptin, 15 μg/ml benzamidine, and 1% Triton X-100. The supernatant, following clarification, was applied to a 5-ml nickel affinity gel (Sigma HIS-Select®). The column was washed with 50 mm Tris, pH 8, 0.3 m NaCl, 1 mm EDTA, 7 mm β-ME, and MRTF-A was eluted from the column with 10 mm imidazole, pH 8, 0.3 m NaCl, 50 mm Tris, pH 8, 1 mm EDTA, 7 mm β-ME. Peak fractions were pooled and dialyzed overnight against 50 mm Tris, pH 8, 0.3 m NaCl, 1 mm EDTA, 1 mm DTT, 1 μg/ml leupeptin, and then into 20 mm Tris, pH 8.0, 20 mm NaCl, 1 mm DTT and flash-frozen for storage at −80 °C. Yield was ∼6 mg from 1 liter of culture.
Tpm1.4
Human smooth muscle tropomyosin (Tpm1.4, previously called Tm6) was cloned into pAcSG2 for recombinant baculovirus production. Sf9 cells (2 × 109) were infected with recombinant virus encoding for Tpm1.4 and harvested after 3 days. The cells were lysed by sonication in 50 ml of 10 mm imidazole, pH 7.2, 0.1 m NaCl, 2 mm EDTA, 1 mm DTT, and protease inhibitors 0.5 mm 4-(2-aminoethyl)benzenesulfonyl fluoride HCl, 5 μg/ml leupeptin, 0.5 mm phenylmethanesulfonyl fluoride, and 5 mm benzamidine. The lysate was clarified at 20,000 × g for 30 min. The supernatant was boiled for 7 min, cooled on ice for 25 min, and clarified again. The pH of the supernatant was slowly lowered to pH 4.5 with 0.3 m HCl to precipitate the tropomyosin. The suspension was pelleted by centrifugation for 30 min at 20,000 × g. The pellet was resuspended in 10 mm imidazole, pH 7.2, 50 mm NaCl, 1 mm EGTA, 1 mm DTT, and 1 μg/ml leupeptin and dialyzed against this buffer overnight. Tpm1.4 was bound to a 6-ml Mono Q column (GE Healthcare) and eluted with a gradient from 50 mm to 1 m NaCl. Peak fractions were pooled, and the protein was stored at −20 °C in a buffer containing 55% glycerol, 10 mm imidazole, pH 7.4, 50 mm NaCl, 1 mm EGTA, 1 mm DTT. Yield was ∼40 mg per 109 cells.
Actin Polymerization Visualized by TIRF Microscopy
Actin polymerization was visualized with Lifeact GFP at 37 °C as described previously (10). Final polymerization buffer was 10 mm imidazole, pH 7.5, 50 mm KCl, 2 mm MgCl2, 1 mm EGTA, 0.25% methylcellulose, 2 mm MgATP, 0.13 mg/ml glucose oxidase, 50 μg/ml catalase, 3 mg/ml glucose, and 10 mm DTT. Experiments with profilin and cofilin were as described (10). For experiments with MRTF-A, 1 μm MRTF-A was added to the final buffer.
Fluorescence Data Processing
Polymerization and depolymerization rates and cofilin severing and shortening rates were obtained as described previously (10).
Modeling of Polymerization of Mixtures of WT and R179H
As modeled previously for the R258C SM α-actin mutant, the composition of the barbed end of the F-actin polymer determines association and dissociation rates (10). Equations describing the association rates are as described in Ref. 10. For the dissociation reaction, if any one of the two barbed end promoters in F-actin is WT, the end protomer dissociates with rate of rdw (WT dissociation rate). If both end protomers are R179H, it dissociates with the fast rate of rdm (R179H mutant dissociation rate). Equations 1–4 describe the dissociation reaction.
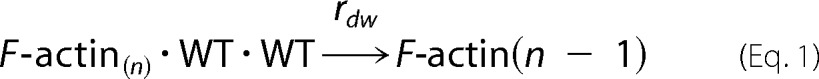 |
 |
 |
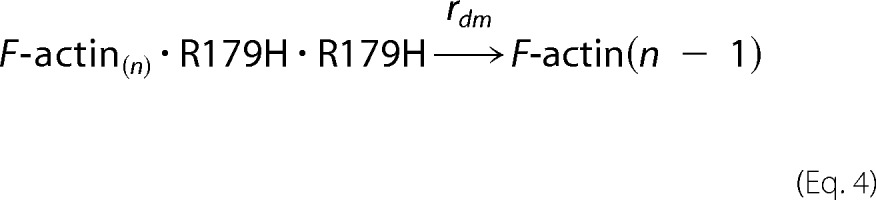 |
Modeling of Polymerization Data in the Presence of Profilin
Assumptions and equations describing polymerization of G-actin in the presence of profilin are as described in Ref. 10.
Fits to Polymerization Data in the Presence of MRTF-A
The data were fit to the Hill equation θ = 1/((Kd/[actin]free)n + 1), with the assumption that each MRTF-A can bind up to five monomers. θ is the percentage of occupied binding sites; n is the Hill coefficient; Kd is the dissociation constant; [actin]free is the free actin monomer concentration that was not bound to MRTF-A. For 3 μm MRTF-A, θ = ([actin]total − [actin]free)/(3·5). The [actin]free was calculated from the polymerization rate versus actin concentration equation obtained on the same day.
Tropomyosin and Cofilin Binding Assays
Tropomyosin binding curves were obtained and analyzed as described in Ref. 10.
Cofilin (0.5–7 μm) was incubated with 4 μm F-actin in 10 mm MOPS, pH 6.8, 50 mm KCl, 4 mm MgCl2, 1 mm EGTA, and 1 mm DTT at 4 °C and then sedimented for 25 min at 350,00 × g (4 °C). Pelleted fractions were analyzed on 12% SDS-polyacrylamide gels stained with Coomassie Blue, and band intensity was quantified using ImageJ. The binding constant was determined by fitting the data to the Hill equation as described in Ref. 36.
In Vitro Motility
Motility was performed at 30 °C as described in Ref. 10. Motility buffer contained 90 mm KCl, 25 mm imidazole, pH 7.5, 4 mm MgCl2, 1 mm EGTA, and 10 mm DTT. Full-length human smooth muscle myosin was used as the motor.
Author Contributions
H. L. and K. M. T. designed the experiments; H. L. and P. M. F conducted the experiments and analyzed the data; P. M. F. and E. B. K cloned, expressed, and purified the proteins; H. L. and K. M. T wrote the manuscript.
Acknowledgments
We thank Kristine Kamm, Susan Lowey, and Dianna Milewicz for critical reading of the manuscript and Jason Stumpff for use of the TIRF microscope.
Note Added in Proof
There was some overlap in text in the Introduction and Experimental Procedures between this article that was published as a Paper in Press on August 22, 2016, and Lu, H., Fagnant, P. M., Bookwalter, C. S., Joel, P., and Trybus, K. M. (2015) Vascular disease-causing mutation R258C in ACTA2 disrupts actin dynamics and interaction with myosin. Proc. Natl. Acad. Sci. U.S.A. 112, E4168–E4177. The text overlap has now been removed.
This work was supported by National Institutes of Health Grant P01 HL110869. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- SM α-actin
- smooth muscle α-actin expressed from the ACTA2 gene
- TAAD
- thoracic aortic aneurysm and dissection
- FH1
- formin-homology domain 1
- FH2
- and formin-homology domain 2
- MRTF-A
- myocardin related transcription factor-A
- p.R177
- protein residue Arg-177, equivalent to gene numbering Arg-179
- SM γ-actin
- smooth muscle γ-actin expressed from the ACTG2 gene
- Tpm
- tropomyosin
- β-ME
- β-mercaptoethanol
- PDB
- Protein Data Bank
- TIRF
- total internal reflection fluorescence.
References
- 1. Milewicz D. M., Østergaard J. R., Ala-Kokko L. M., Khan N., Grange D. K., Mendoza-Londono R., Bradley T. J., Olney A. H., Ades L., Maher J. F., Guo D., Buja L. M., Kim D., Hyland J. C., and Regalado E. S. (2010) De novo ACTA2 mutation causes a novel syndrome of multisystemic smooth muscle dysfunction. Am. J. Med. Genet. A 152A, 2437–2443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Guo D. C., Pannu H., Tran-Fadulu V., Papke C. L., Yu R. K., Avidan N., Bourgeois S., Estrera A. L., Safi H. J., Sparks E., Amor D., Ades L., McConnell V., Willoughby C. E., Abuelo D., et al. (2007) Mutations in smooth muscle α-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat. Genet. 39, 1488–1493 [DOI] [PubMed] [Google Scholar]
- 3. Guo D. C., Papke C. L., Tran-Fadulu V., Regalado E. S., Avidan N., Johnson R. J., Kim D. H., Pannu H., Willing M. C., Sparks E., Pyeritz R. E., Singh M. N., Dalman R. L., Grotta J. C., Marian A. J., et al. (2009) Mutations in smooth muscle α-actin (ACTA2) cause coronary artery disease, stroke, and Moyamoya disease, along with thoracic aortic disease. Am. J. Hum. Genet. 84, 617–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Morisaki H., Akutsu K., Ogino H., Kondo N., Yamanaka I., Tsutsumi Y., Yoshimuta T., Okajima T., Matsuda H., Minatoya K., Sasaki H., Tanaka H., Ishibashi-Ueda H., and Morisaki T. (2009) Mutation of ACTA2 gene as an important cause of familial and nonfamilial nonsyndromatic thoracic aortic aneurysm and/or dissection (TAAD). Hum Mutat 30, 1406–1411 [DOI] [PubMed] [Google Scholar]
- 5. Regalado E. S., Guo D. C., Prakash S., Bensend T. A., Flynn K., Estrera A., Safi H., Liang D., Hyland J., Child A., Arno G., Boileau C., Jondeau G., Braverman A., Moran R., et al. (2015) Aortic disease presentation and outcome associated with ACTA2 mutations. Circ. Cardiovasc. Genet. 8, 457–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Milewicz D. M., Guo D. C., Tran-Fadulu V., Lafont A. L., Papke C. L., Inamoto S., Kwartler C. S., and Pannu H. (2008) Genetic basis of thoracic aortic aneurysms and dissections: focus on smooth muscle cell contractile dysfunction. Annu. Rev. Genomics Hum. Genet. 9, 283–302 [DOI] [PubMed] [Google Scholar]
- 7. Gunst S. J., and Zhang W. (2008) Actin cytoskeletal dynamics in smooth muscle: a new paradigm for the regulation of smooth muscle contraction. Am. J. Physiol. Cell Physiol 295, C576–C587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tang D. D. (2015) Critical role of actin-associated proteins in smooth muscle contraction, cell proliferation, airway hyperresponsiveness and airway remodeling. Respir. Res. 16, 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Olson E. N., and Nordheim A. (2010) Linking actin dynamics and gene transcription to drive cellular motile functions. Nat. Rev. Mol. Cell Biol. 11, 353–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lu H., Fagnant P. M., Bookwalter C. S., Joel P., and Trybus K. M. (2015) Vascular disease-causing mutation R258C in ACTA2 disrupts actin dynamics and interaction with myosin. Proc. Natl. Acad. Sci. U.S.A. 112, E4168–E4177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Munot P., Saunders D. E., Milewicz D. M., Regalado E. S., Ostergaard J. R., Braun K. P., Kerr T., Lichtenbelt K. D., Philip S., Rittey C., Jacques T. S., Cox T. C., and Ganesan V. (2012) A novel distinctive cerebrovascular phenotype is associated with heterozygous Arg179 ACTA2 mutations. Brain 135, 2506–2514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Georgescu M. M., Pinho Mda C., Richardson T. E., Torrealba J., Buja L. M., Milewicz D. M., Raisanen J. M., and Burns D. K. (2015) The defining pathology of the new clinical and histopathologic entity ACTA2-related cerebrovascular disease. Acta Neuropathol. Commun. 3, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bartman T., Walsh E. C., Wen K. K., McKane M., Ren J., Alexander J., Rubenstein P. A., and Stainier D. Y. (2004) Early myocardial function affects endocardial cushion development in zebrafish. PLos Biol. 2, E129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wen K. K., and Rubenstein P. A. (2003) Biochemical consequences of the cardiofunk (R177H) mutation in yeast actin. J. Biol. Chem. 278, 48386–48394 [DOI] [PubMed] [Google Scholar]
- 15. Strauch A. R., and Rubenstein P. A. (1984) A vascular smooth muscle α-isoactin biosynthetic intermediate in BC3H1 cells. Identification of acetylcysteine at the NH2 terminus. J. Biol. Chem. 259, 7224–7229 [PubMed] [Google Scholar]
- 16. Aktories K., Schwan C., Papatheodorou P., and Lang A. E. (2012) Bidirectional attack on the actin cytoskeleton. Bacterial protein toxins causing polymerization or depolymerization of actin. Toxicon 60, 572–581 [DOI] [PubMed] [Google Scholar]
- 17. Wriggers W., and Schulten K. (1999) Investigating a back door mechanism of actin phosphate release by steered molecular dynamics. Proteins 35, 262–273 [PubMed] [Google Scholar]
- 18. Campellone K. G., and Welch M. D. (2010) A nucleator arms race: cellular control of actin assembly. Nat. Rev. Mol. Cell Biol. 11, 237–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chesarone M. A., DuPage A. G., and Goode B. L. (2010) Unleashing formins to remodel the actin and microtubule cytoskeletons. Nat. Rev. Mol. Cell Biol. 11, 62–74 [DOI] [PubMed] [Google Scholar]
- 20. Moseley J. B., Sagot I., Manning A. L., Xu Y., Eck M. J., Pellman D., and Goode B. L. (2004) A conserved mechanism for Bni1- and mDia1-induced actin assembly and dual regulation of Bni1 by Bud6 and profilin. Mol. Biol. Cell 15, 896–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zigmond S. H. (2004) Formin-induced nucleation of actin filaments. Curr. Opin. Cell Biol. 16, 99–105 [DOI] [PubMed] [Google Scholar]
- 22. Mouilleron S., Langer C. A., Guettler S., McDonald N. Q., and Treisman R. (2011) Structure of a pentavalent G-actin*MRTF-A complex reveals how G-actin controls nucleocytoplasmic shuttling of a transcriptional coactivator. Sci. Signal. 4, ra40. [DOI] [PubMed] [Google Scholar]
- 23. Mitchison T., and Kirschner M. (1984) Dynamic instability of microtubule growth. Nature 312, 237–242 [DOI] [PubMed] [Google Scholar]
- 24. Fujii T., Iwane A. H., Yanagida T., and Namba K. (2010) Direct visualization of secondary structures of F-actin by electron cryomicroscopy. Nature 467, 724–728 [DOI] [PubMed] [Google Scholar]
- 25. Galkin V. E., Orlova A., Vos M. R., Schröder G. F., and Egelman E. H. (2015) Near-atomic resolution for one state of F-actin. Structure 23, 173–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Oda T., Iwasa M., Aihara T., Maéda Y., and Narita A. (2009) The nature of the globular- to fibrous-actin transition. Nature 457, 441–445 [DOI] [PubMed] [Google Scholar]
- 27. von der Ecken J., Müller M., Lehman W., Manstein D. J., Penczek P. A., and Raunser S. (2015) Structure of the F-actin-tropomyosin complex. Nature 519, 114–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pollard T. D. (1984) Polymerization of ADP-actin. J. Cell Biol. 99, 769–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. De La Cruz E. M., Martiel J. L., and Blanchoin L. (2015) Mechanical heterogeneity favors fragmentation of strained actin filaments. Biophys J 108, 2270–2281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Torres R. A., Drake D. A., Solodushko V., Jadhav R., Smith E., Rocic P., and Weber D. S. (2011) Slingshot isoform-specific regulation of cofilin-mediated vascular smooth muscle cell migration and neointima formation. Arterioscler. Thromb. Vasc. Biol. 31, 2424–2431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhao R., Du L., Huang Y., Wu Y., and Gunst S. J. (2008) Actin depolymerization factor/cofilin activation regulates actin polymerization and tension development in canine tracheal smooth muscle. J. Biol. Chem. 283, 36522–36531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yamin R., and Morgan K. G. (2012) Deciphering actin cytoskeletal function in the contractile vascular smooth muscle cell. J. Physiol. 590, 4145–4154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gallant C., Appel S., Graceffa P., Leavis P., Lin J. J., Gunning P. W., Schevzov G., Chaponnier C., DeGnore J., Lehman W., and Morgan K. G. (2011) Tropomyosin variants describe distinct functional subcellular domains in differentiated vascular smooth muscle cells. Am. J. Physiol. Cell Physiol 300, C1356–C1365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zimmerman R. A., Tomasek J. J., McRae J., Haaksma C. J., Schwartz R. J., Lin H. K., Cowan R. L., Jones A. N., and Kropp B. P. (2004) Decreased expression of smooth muscle α-actin results in decreased contractile function of the mouse bladder. J. Urol. 172, 1667–1672 [DOI] [PubMed] [Google Scholar]
- 35. Wangler M. F., Gonzaga-Jauregui C., Gambin T., Penney S., Moss T., Chopra A., Probst F. J., Xia F., Yang Y., Werlin S., Eglite I., Kornejeva L., Bacino C. A., Baldridge D., Neul J., et al. (2014) Heterozygous de novo and inherited mutations in the smooth muscle actin (ACTG2) gene underlie megacystis-microcolon-intestinal hypoperistalsis syndrome. PLoS Genet. 10, e1004258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Barua B., Fagnant P. M., Winkelmann D. A., Trybus K. M., and Hitchcock-DeGregori S. E. (2013) A periodic pattern of evolutionarily conserved basic and acidic residues constitutes the binding interface of actin-tropomyosin. J. Biol. Chem. 288, 9602–9609 [DOI] [PMC free article] [PubMed] [Google Scholar]