

Abstract
In 2013 there were an estimated 584,000 deaths and 198 million clinical illnesses due to malaria, the majority in sub-Saharan Africa. Vaccines would be the ideal addition to the existing armamentarium of anti-malaria tools. However, malaria is caused by parasites, and parasites are much more complex in terms of their biology than the viruses and bacteria for which we have vaccines, passing through multiple stages of development in the human host, each stage expressing hundreds of unique antigens. This complexity makes it more difficult to develop a vaccine for parasites than for viruses and bacteria, since an immune response targeting one stage may not offer protection against a later stage, because different antigens are the targets of protective immunity at different stages. Furthermore, depending on the life cycle stage and whether the parasite is extra- or intra-cellular, antibody and/or cellular immune responses provide protection. It is thus not surprising that there is no vaccine on the market for prevention of malaria, or any human parasitic infection. In fact, no vaccine for any disease with this breadth of targets and immune responses exists. In this limited review, we focus on four approaches to malaria vaccines, (1) a recombinant protein with adjuvant vaccine aimed at Plasmodium falciparum (Pf) pre-erythrocytic stages of the parasite cycle (RTS,S/AS01), (2) whole sporozoite vaccines aimed at Pf pre-erythrocytic stages (PfSPZ Vaccine and PfSPZ-CVac), (3) prime boost vaccines that include recombinant DNA, viruses and bacteria, and protein with adjuvant aimed primarily at Pf pre-erythrocytic, but also asexual erythrocytic stages, and (4) recombinant protein with adjuvant vaccines aimed at Pf and Plasmodium vivax sexual erythrocytic and mosquito stages. We recognize that we are not covering all approaches to malaria vaccine development, or most of the critically important work on development of vaccines against P. vivax, the second most important cause of malaria. Progress during the last few years has been significant, and a first generation malaria candidate vaccine, RTS,S/AS01, is under review by the European Medicines Agency (EMA) for its quality, safety and efficacy under article 58, which allows the EMA to give a scientific opinion about products intended exclusively for markets outside of the European Union. However, much work is in progress to optimize malaria vaccines in regard to magnitude and durability of protective efficacy and the financing and practicality of delivery. Thus, we are hopeful that anti-malaria vaccines will soon be important tools in the battle against malaria.
Introduction
In 2013 at least $2.6 billion was invested in malaria control programs, which utilize tools including insecticide impregnated bednets, residual insecticide spraying, and early diagnosis and treatment1. These programs have had a tremendous impact on reducing malaria morbidity and mortality during the past decade.1 However, even in the face of this large investment, in 2013 there were an estimated 584,000 deaths and 198 million clinical illnesses due to malaria, the majority in sub-Saharan Africa.1
Vaccines would be the ideal addition to the existing armamentarium of anti-malaria tools. A vaccine was used to eradicate smallpox from the world and polio from the Western hemisphere, and vaccines have had dramatic impacts on many infectious diseases. It is because of these successes that vaccines are considered the most cost effective single intervention for control, prevention, elimination, and eradication of infectious diseases.
However, malaria is caused by parasites, and parasites are much more complex in terms of their biology than the viruses and bacteria for which we have vaccines. For example their genomes are much larger than those of viruses and bacteria (Pf has >5400 genes) and they have multiple stages of their life cycles, which viruses and bacteria do not have (Figure 1). These different stages, (1) must be attacked by different arms of the immune system, depending on whether the parasites are inside or outside of host cells and (2) are in many cases antigenically distinct. For example, protective antibodies against sporozoites injected by mosquitoes do not recognize the asexual erythrocytic stages that cause all disease. Thus, if one sporozoite evades vaccine-induced protective antibodies, a week later 10,000–40,000 merozoites will escape the liver. Each can invade a different erythrocyte, and none of these erythrocytic stage parasites are recognized by antibodies to the circumsporozoite protein (CSP), the major antigen on the sporozoite surface.
Figure 1. Life cycle of P. falciparum and stages at which antibodies and cellular immune responses are primary for protection.
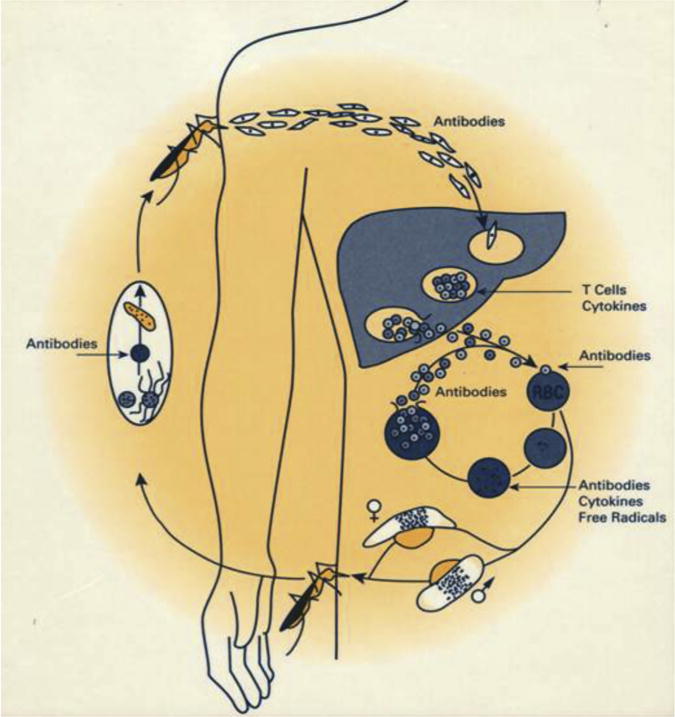
Note: When an infected female Anopheles spp. mosquito feeds on a human, she inoculates uninucleated sporozoites into the tissues or directly into the bloodstream. Sporozoites rapidly pass through the bloodstream to the liver (probably within 2 min, but less than 60 min). In hepatic sinusoids they may penetrate and pass through Kupffer cells and invade hepatocytes. Sporozoites may invade several hepatocytes before finding the correct hepatocyte in which to develop. During a minimum of 5.5 days, the uninucleated P. falciparum sporozoite develops to a mature liver-stage schizont with an average of 30,000 and range of 10,000–40,000 uninucleated merozoites. The hepatocyte containing mature liver-stage schizonts die or rupture releasing “sacks” containing merozoites called merosomes containing thousands of uninucleated merozoites, each of which can invade a normal erythrocyte. In the erythrocyte, the invading merozoite develops during approximately 2 days to a mature asexual erythrocytic-stage schizont with an average of 16 uninucleated merozoites. When fully mature, the infected erythrocyte ruptures, releasing the merozoites, which then invade normal erythrocytes, initiating the cycle of intraerythrocytic-stage development, rupture, and reinvasion that leads to a 10–20-fold increase in the numbers of P. falciparum parasites in the bloodstream approximately every 2 days and to all the clinical manifestations and pathology of malaria. Alternatively, erythrocytic-stage parasites can develop to sexual-stage parasites, gametocyte. In the gut of the mosquito, gametocytes escape from erythrocytes and form gametes. The male gamete fuses with the female, forming a zygote. By 18–24 h, the zygote has transformed into an ookinete. The ookinete traverses the midgut wall by passing through epithelial cells and comes to rest adjacent to the basal lamina. Here it begins to transform into an oocyst in which sporozoites are produced. By day 12, they are released into the hemocele of the mosquito and migrate to the salivary glands. In the salivary glands, they become infectious for humans and are released into the human host when the mosquito feeds.
Source: Figure is from Hoffman, SL. Malaria vaccine development, a multi-immune response approach. 1996. ASM Press, Washington, DC.
This complexity makes it more difficult to develop a vaccine for parasites than for viruses and bacteria. In fact there is no vaccine on the market for prevention of any human parasitic infection. There are five species of Plasmodium that cause malaria in humans, Plasmodium falciparum (Pf), Plasmodium vivax, Plasmodium malariae, Plasmodium ovale, and Plasmodium knowlesi. Pf is responsible for more than 98% of malaria mortality and has the most significant drug resistance of all the human malaria parasites. Furthermore, the most progress has been made on Pf vaccines. For these reasons, in this review we focus primarily on anti-Pf vaccines.
The malaria parasite has a complex life cycle and significant antigenic diversity. Hence, there are many different approaches to the development of malaria vaccines.2 They include targeting the production of antigen specific protective antibodies, CD4+ T cells, and/or CD8+ T cells. Some are focused on the sporozoite and/or liver stages of the life cycle (pre-erythrocytic stages), some on the asexual erythrocytic stages, and some on the sexual erythrocytic and mosquito stages. Some are focused on preventing infection to prevent all disease, some in reducing the morbidity and mortality of the asexual erythrocytic stages by reducing the rate at which individuals become infected and/or reducing asexual erythrocytic stage multiplication, and some aim at reducing or preventing transmission to mosquitoes. With the renewed emphasis on malaria elimination and eradication, it has been suggested that the primary focus of malaria vaccine development should be for vaccines that interrupt malaria transmission (VIMTs).3
In this limited review, we focus on four approaches to malaria vaccine development, including the approach closest to licensure. However, we recognize that we are not covering all approaches to malaria vaccine development, or the critically important work on development of vaccines against P. vivax, the second most important cause of malaria, and a parasite that is expected by many to be more difficult to eliminate than Pf.
Purified Recombinant Circumsporozoite Protein Vaccine, RTS,S/AS01
The RTS,S malaria candidate vaccine antigen targets the Pf circumsporozoite protein (CSP), a 412 amino acid protein (7G8 clone sequence) present on the sporozoite surface, expressed by early liver forms and exported into the cytoplasm of hepatocytes. It has a characteristic central NANP repeat region and non-repeat flanking regions with both conserved and variable segments. Functional properties have been unraveled, with conformational status determining biological activity related to parasite binding, motility, cell traversal and biochemical properties.4
CSP was the first malaria protein to be cloned and has been an early target of malaria vaccine research. It was shown to be a key target of protective immunity induced by irradiated sporozoite vaccines in animal models.5 Passive transfer of CSP-specific antibodies and CD4+ and CD8+ T cells can protect rodents from experimental infection.6,7
Initial candidate vaccine constructs targeting the central repeat region of the PfCSP failed to provide substantial protection.8,9 Based on experience acquired during the development of the first genetically engineered hepatitis B vaccine, a new construct was generated at GSK and tested through a collaboration with the Walter Reed Army Institute of Research. The hepatitis B surface antigen (HBsAg) was used as carrier matrix. The repetitive immunodominant B-cell epitope from the central region (NANP repeats) and T-cell epitope containing C-terminal flanking region (CSP amino acids 207–395 of the 3D7 clone of the NF54 strain of Pf) were fused to the HBsAg protein, and expressed together with free HBsAg in Saccharomyces cerevisiae. The new vaccine was designated RTS,S to indicate the presence of the CSP repeat region (R), T-cell epitopes (T) fused to the HBsAg (S) and assembled with unfused copies of HBsAg. The resulting proteins assembled into virus-like particles (VLPs), similar to those formed by the unfused HBsAg proteins.10
The first study to establish RTS,S as a promising malaria vaccine candidate also showed the key role of adjuvantation.11 Three different vaccine formulations were tested and one (RTS,S/AS02) protected 6 out of 7 individuals against controlled human malaria infection (CHMI) by the bite of mosquitoes infected with Pf 3D7 sporozoites. The RTS,S/AS02 formulation was first selected for further development, before a switch to the RTS,S/AS01 formulation with superior immunogenicity and efficacy.12,13 Both AS01 and AS02 include the immune-enhancers monophosphoryl lipid A (MPL) and QS21 (Stimulon® adjuvant licensed from Antigenics Inc.). MPL consists of a chemically detoxified form of the parent lipopolysaccharide (LPS) from the Gram negative bacterium Salmonella minnesota. QS21 is a natural saponin molecule purified from the bark of the South American tree, Quillaja saponaria. AS02 contains in addition an oil in water emulsion while AS01 contains a liposomal suspension.14
The targeted vaccine’s mechanism of action is to induce a specific anti-PfCSP immune response that prevents initiation of patent blood-stage infection by killing parasites at the pre-erythrocytic stage. There is currently no established correlate of protection, but associations between anti-PfCSP antibody levels and CD4+ T cell activation have been shown.15,16
The GSK-sponsored RTS,S pediatric development program has been under the leadership of a public– private product development partnership between GSK and the PATH Malaria Vaccine Initiative (MVI), with support from the Bill & Melinda Gates Foundation, since 2001, in collaboration with multiple African, European and American research institutions. The overall objective of the program is to reduce the burden of disease related to P. falciparum malaria in infants and children residing in sub-Saharan African malaria-endemic countries. The vaccine would ideally be implemented through the existing infant vaccine delivery program, the Expanded Program on Immunization, in conjunction with other malaria control interventions.
Phase 2 studies in conditions of natural exposure in various countries in Africa confirmed that the vaccine conferred partial protection against malaria in children, and demonstrated favorable safety down to infants in the EPI age-range and HIV-infected children. Immunogenicity of 0, 1, 2 and 0, 1, 7-months, regimens was shown, and it was demonstrated that three doses were better than two doses when assessed for immunogenicity or vaccine efficacy.17,18
In 2009, a multicenter phase 3 trial of RTS,S/AS01 was undertaken in 11 research centers across 7 countries with various patterns of malaria transmission. The study population included children aged 5–17 months at first experimental immunization, and infants first immunized at 6–12 weeks of age together with routine vaccines from the Expanded Program on Immunization (EPI).19 The phase 3 study, analyzed as per the WHO malaria vaccine advisory committee recommendations20 showed across all study centers a vaccine efficacy against clinical malaria (primary case definition was fever and parasitemia >5000/μL blood) over 14 months after first vaccination of 50.4% (95% CI=45.8–54.6) in 6000 children in the older age category, and 30.1% (95% CI=23.6–36.1) in 6537 infants of the younger age category (intent-to-treat analysis (ITT)). Peak anti-PfCSP antibody levels were also lower in the younger infants.21,22 A site-specific analysis of vaccine efficacy over 20 months of follow-up post first immunization showed some evidence of heterogeneity in VE across study centers, but no demonstration that VE was associated to intensity of transmission. Point estimates of vaccine efficacy ranged between 41% and 70% (ITT, p < 0.05 in all study sites) in children in the older age category. In infants of the youngest age category, point estimates of VE ranged from −57% to 49% ITT.21–23
Analyses of both phase 2 and phase 3 studies showed reduced point estimates of vaccine efficacy in consecutive post-RTS,S/AS01 vaccination time windows, suggesting waning of efficacy.23,24 In the phase 3 trial, over 18 months of follow-up post dose 3 vaccination (according-to-protocol [ATP] analysis, follow-up for efficacy starting 14 days post dose 3), the reduction in incidence of all malaria events attributed to vaccination in 3 successive 6-month periods in the older age category was 68% (95% CI=64–72), 41% (95% CI=36–46) and 26% (95% CI=19–33), and in the younger age category 47% (95% CI=39–54), 23% (95% CI=15–31) and 12% (95% CI=1–21).23 Results from further follow-up in the study, including the effect of a booster dose at Month 20 have now been published.25
Without a booster dose, when considering children in the older age category over the whole follow-up period (average 48 months), primary RTS,S/AS01 vaccination provided 28% (95% CI=23–33) protection against clinical malaria, and the total number of cases averted ranged in different study sites between 215 and 4,443 per 1,000 children vaccinated. No protection against severe malaria was seen when considering the total follow-up period. The reduction of early exposure to blood stage infection associated with vaccination may have delayed acquisition of blood-stage immunity, with a displacement of incidence of severe malaria toward older age as suggested by a negative point estimate of vaccine efficacy (−41.0%, (95% CI=−98.5 to −0.8) between Month 21 and study end. When a booster dose at Month 20 was given, overall protection against clinical malaria and severe malaria were 36% (95% CI=32–41) and 32% (95% CI=14–47), respectively, and depending on study site, 205–6565 cases of malaria were averted per 1000 children vaccinated. When considering other endpoints of public health interest, vaccination with a booster dose provided overall partial protection against incident severe malaria anemia and blood transfusion, malaria hospitalization and all-cause hospitalization. Primary vaccination without a booster dose reduced malaria hospitalizations and all-cause hospitalizations, but the reductions in incident severe malaria anemia and blood transfusion were not statistically significant.
When considering the younger age category, point estimates of vaccine efficacy, when significant, were lower than in the older age category. Vaccine efficacy against clinical malaria, over the whole study (average follow-up 38 months) was 18% (95% CI=12–24) without a booster dose, and 26% (95% CI=20–32) with a booster dose. There was no evidence for protection against severe malaria. Vaccination with a booster dose reduced malaria hospitalizations by 25% (95% CI=6–40) but there was no evidence for the reduction of other endpoints of public health interest.
There is presently no evidence in favor of a specific biological mechanism explaining the difference in immunogenicity of RTS,S/AS01 in young infants and older children, but the role of immune immaturity, inhibition by passively transferred maternal anti-PfCSP or anti-HBs antibodies, existing anti-hepatitis B immunity, and an inhibitory effect of the co-administration of other vaccines from the immunization program are considered.
Safety results were favorable, although a risk of simple febrile seizures was seen when children were vaccinated at a susceptible age. In children aged 5–17 months upon first vaccination, simple febrile seizures occurred at an incidence of about 1 per 1,000 doses of primary immunization and about 2.5 per 1,000 Month 20 booster doses. An unexplained, increased reporting of cases of meningitis due to a heterogeneous group of pathogens, with no cluster in time-to-event, was reported in children in the older age category (11 in the boosted group, ten in the non-boosted group, and one in the control group), but not in the younger age category.25
The RTS,S/AS01 candidate vaccine is currently under review for its quality, safety and efficacy by the European Medicine Agency (EMA) under article 58, which allows the EMA to give a scientific opinion about products exclusively for markets outside of the European Union, before consideration for WHO pre-qualification. Submission for review by national regulatory authorities in sub-Saharan African countries would follow.
Although RTS,S/AS01 phase 3 results showed the potential for a considerable impact on the malaria burden in some settings, a vaccine with higher efficacy levels could fulfill more ambitious goals expressed by the global health community, such as improved control including a broader age target, reduction of transmission and containment of drug-resistant malaria. The degree and duration of vaccination-induced protection will be key factors determining the ability of vaccine-based approaches to contribute to elimination in areas with different transmission patterns, from very low to high endemicity, and from regions with highly seasonal to perennial malaria transmission.
Efforts toward the generation of higher vaccine efficacy are ongoing. Various approaches are being considered in the context of multi-institutional collaborations, including increasing immunogenicity using RTS, S/AS01-only based alternative immunization regimens, the use of other PfCSP-based platforms, and combination of RTS,S/AS01 with alternative antigen targets of the pre-erythrocytic, blood or sexual stages.
Whole Plasmodium falciparum (Pf) Sporozoite (SPZ) Vaccines
It has been known for four decades that it is possible to induce highly effective pre-erythrocytic stage immunity in humans using attenuated whole parasites. Studies in which volunteers were immunized by exposure to the bites of mosquitoes carrying radiation attenuated Pf sporozoites (PfSPZ) showed that when subjects were exposed to the bites of >1,000 mosquitoes carrying radiation attenuated PfSPZ, 13/14 (93%) were protected against first CHMI by the bite of infected mosquitoes from 2 to 10 weeks after last exposure, 5/6 were protected for at least 10 months, and there was protection against heterologous strains of Pf in 7/7 CHMIs by mosquito bite.26–28 These results in humans are supported by prior and subsequent studies in animal models, which likewise have shown high-grade protection,29 and which indicate that protection induced by radiation-attenuated sporozoites is based primarily on the induction of CD8+ T cell responses targeting antigens expressed during liver stage development.30–32 In 2009 and 2011 it was reported that when subjects who were taking chloroquine chemoprophylaxis were exposed to the bites of 45 mosquitoes carrying fully infectious PfSPZ, 10/10 (100%) were protected against CHMI by mosquito bite 8 weeks after the last exposure to PfSPZ and 4/6 were protected against CHMI by mosquito bite 28 months after last immunizing exposure to mosquitoes.33,34 This second approach, which induces potent, long-lasting immunity using a fraction of the PfSPZ dose required for irradiated PfSPZ, is called “chemoprophylaxis with sporozoites (CPS).” The protection induced by CPS appears to be based on cell-mediated immunity, like that of radiation-attenuated sporozoites; a variety of immune cell populations have been identified as correlates.35,36
The company Sanaria was founded to translate the mosquito-based immunization approach into injectable whole PfSPZ malaria vaccines, and to use the vaccine(s) to eliminate malaria from geographically defined areas through mass immunization and to prevent malaria in non-immune visitors to malarious areas. To be considered for these indications such a vaccine must be highly effective (>85–90% protective efficacy sustained for at least 6 months), and amenable to mass administration. In 2013 it was reported that intravenous (IV) immunization of humans with radiation attenuated, aseptic, purified, cryopreserved PfSPZ, Sanaria® PfSPZ Vaccine, protected 6/6 (100%) subjects receiving the highest dose regimen tested in that trial against CHMI by mosquito bite.32 To achieve this milestone and to eventually achieve licensure, Sanaria and its collaborators have had or will have to overcome a series of challenges outlined below.
Manufacture and delivery of PfSPZ
The first challenge was to produce an injectable vaccine that could be transported to all places and which was aseptic, pure, adequately attenuated, and potent. It took several years refining manufacturing processes to produce the first lots of radiation attenuated, aseptic purified, vialed PfSPZ that met these standards; these were cryopreserved in liquid nitrogen vapor phase (LNVP) enabling long term stability and transport without the need for electricity to support a cold chain. In 2009 a successful Investigational New Drug application (IND) was submitted to the U.S. FDA.30,37 Since then PfSPZ have been shipped successfully to more than 12 clinical sites in the USA, Europe, and Africa; the criteria for success were safety and infectivity of PfSPZ Challenge,38–42 and safety, immunogenicity, and and/or protective efficacy of PfSPZ Vaccine and PfSPZ-CVac.30,32,38–43 The manufacturing team at Sanaria is now moving toward scale-up and manufacture suitable for phase 3 clinical trials, licensure, and launch, and the logistics team is working with partners to establish a LNVP delivery system for PfSPZ products.44 Their contention is that an electricity-free LNVP delivery system will have significant advantages over current cold chains.
Administration of PfSPZ
When feeding, mosquitoes inoculate PfSPZ directly into the circulation by cannulating microcirculatory vessels, but the majority of PfSPZ are deposited in the dermis and subcutaneous tissue. For the first clinical trial, PfSPZ Vaccine was administered by subcutaneous (ID) or intradermal (SC) injection. PfSPZ Vaccine was safe and well tolerated, but poorly immunogenic, and the best protective efficacy against homologous CHMI by mosquito bite was 2/16 (12.5%).30 Two explanations were considered for the poor immunogenicity and protective efficacy, (1) the vaccine was not potent and (2) the SC and ID routes of administration were not efficient. Studies in non-human primates (NHPs) demonstrated that when radiation attenuated PfSPZ were administered by IV injection they were highly potent, but not when administered SC. Four months after the last immunization with the highest doses given to humans, ∼3% of all CD8+ T cells in the livers of NHPs immunized IV were specific for PfSPZ, whereas less than 0.1% were specific for PfSPZ in NHPs immunized SC.30 All current and planned trials, including a trial planned for ∼450 African infants, will use direct venous inoculation (DVI) of PfSPZ, the method of administration that is expected to be used for the first licensed PfSPZ-based vaccine.
Clinical development of PfSPZ Vaccine
With these results in hand, a dose escalation trial of PfSPZ Vaccine administered by IV injection was initiated. There was a dose response for antibody and T cell responses.32 4–6 doses of 3 × 104 PfSPZ (maximum total of 1.8 × 105) protected only one of the 11 subjects. However, 6/9 subjects immunized with 4 doses of 1.35 × 105 PfSPZ (total of 5.4 × 105) were protected, and 6/6 (100%) subjects immunized with 5 doses of 1.35 × 105 PfSPZ (total of 6.75 × 105) were protected against CHMI by mosquito bite.32 It was clear that as with subjects immunized by the bite of PfSPZ-infected mosquitoes,28 there was a threshold that had to be passed to achieve protection. Based on the data from mosquito bite immunization studies,28,33,36,45 it was hypothesized that increased numbers of PfSPZ will be needed to achieve protection at 6 months and for protection against heterologous (non-vaccine) strains of Pf.
Based on the published results, some unpublished data and the mosquito bite immunization results, a four-stage clinical development plan (CDP) was developed and is being implemented by an international PfSPZ consortium. The first stage was designed to establish, (1) reproducibility of the tolerability, safety, immunogenicity and protective efficacy of the previous trial,30 (2) durability of high level protection for at least 6 months, (3) protection against heterologous strains of Pf after CHMI by mosquito bite and natural exposure in Africa, (4) immunogenicity and protective efficacy in Africans with previous exposure to malaria, (5) an immunological assay that predicts protection, (6) operational feasibility of mass administration of PfSPZ Vaccine, (7) projective efficacy of reduced numbers of doses of vaccine, and (8) equivalence of IV injection through an in-dwelling IV catheter of PfSPZ in 1.0 mL,30 and direct venous inoculation (DVI) by injection into a vein of PfSPZ in 0.5 mL through a 25 gauge needle in regard to safety, tolerability, immunogenicity and protective efficacy. Clinical trials based on this CDP are now underway at 3 sites in the USA (NCT02015091, NCT02215707), and in Mali (NCT01988636), Tanzania (NCT02132299), and Equatorial Guinea (NCT02418962) and additional trials are planned to begin in 7 countries in 2015–16, including in children and infants. In stage 2 of the CDP, the intent is to test the vaccine in the elderly and HIV seropositive populations and to determine a final dosing regimen that will provide high level sterile protection against parasitemia for at least 6 months duration, in malaria-exposed and malaria-naive individuals 6 months of age and older. Thus far, 388 subjects have received 1577 doses of PfSPZ Vaccine ranging from 2 × 103 to 2.2 × 106 PfSPZ, with the testing of higher doses anticipated in the next set of trials. Based on the results of completed and ongoing and planned trials, phase 3 clinical trials will be conducted (stage 3 of the CDP). A Biologics License Application (BLA) to the U.S. FDA and other regulatory agencies would follow their completion. In stage 4 (post-licensure) the vaccine would be used in mass administration campaigns to halt transmission of Pf malaria and to eliminate the parasite from geographically defined areas. It will also be used to prevent malaria in multiple populations, including travelers and infants in endemic areas.
PfSPZ Challenge
Sanaria has also developed a product called Sanaria® PfSPZ Challenge, which is composed of aseptic, purified, cryopreserved, infectious PfSPZ. PfSPZ Challenge is identical to PfSPZ Vaccine, except that the PfSPZ are not attenuated. Sanaria and its collaborators have been assessing different routes and doses of PfSPZ Challenge.38–42,46 8 clinical trials have been conducted in the Netherlands,38 UK,39 Tanzania,40 USA (NCT01546389), Kenya,41 Germany,42 Spain (NCT01771848), and Gabon (NCT02237586). They were designed to demonstrate, (1) 100% infection rates, (2) a pre-patent period comparable to the bite of five PfSPZ-infected mosquitoes (≤11.5 days), (3) a dose response in regard to infectivity and pre-patent period, and (4) comparability of IV and DVI injection. It has been established that the administration of 3200 PfSPZ by IV and DVI injection42,46 induces 100% infection rates in non-immune individuals with a pre-patent period of 11.2–11.4 days, and there is a dose response. Injection of ∼23 times more PfSPZ by the IM route (75,000 PfSPZ) gives a similar outcome,46 and 100% infection rates have been achieved in non-immune Europeans42,46 and semi-immune Africans.41 Results will soon be available from studies using doses of 10,000–50,000 PfSPZ by the ID route in non-immunes (NCT01546389), and studies of a dose of 3200 PfSPZ by DVI in semi-immunes with sickle cell trait (NCT02237586).
PfSPZ-CVac
The PfSPZ-Chemoprophylaxis Vaccine (Sanaria® PfSPZ-CVac) utilizes PfSPZ Challenge administered to individuals taking malaria chemoprophylaxis to duplicate the results of the CPS approach to vaccination. It has been studied by the ID (NCT01728701)43 and DVI routes (NCT02115516). Unpublished results demonstrate that PfSPZ-CVac requires fewer parasites than PfSPZ Vaccine to achieve high-level protective efficacy (NCT02115516). This is thought to be due to the fact that the parasites replicate 10,000–40,000 times and express more than 80% of the Pf proteome, thus presenting the immune system with much more of the same antigens and thousands of more antigens than do irradiated PfSPZ. Clinical trials of PfSPZ-CVac are being planned for sites in the US, Ghana, Germany and Indonesia.
PfSPZ-GA1 and other genetically attenuated PfSPZ Vaccines
The PfSPZ in PfSPZ Vaccine are attenuated by irradiation and the PfSPZ in PfSPZ-CVac are attenuated by an antimalarial drug. Another approach is to attenuate the parasites by creating mutant parasites that are deficient in specific genes. This was first attempted for Pf by knocking out the genes encoding P52 and P36.47,48 However, this did not fully attenuate the parasites, and one of the first 6 volunteers exposed to PfΔp52Δp36 genetically attenuated parasites (GAP) developed Pf parasitemia.49 This clinical result was predicted by in vitro studies.50 In addition to p52 and p36 a third gene, sap1 (also known as slarp), which is involved in the regulation of transcription, has been knocked out in Pf, and these parasites are moving toward a clinical trial administered by mosquito bite.51 Recently another gene, B9, like P52 and P36, a member of the Plasmodium 6-Cys family of proteins that is involved in liver stage parasitophorous vacuole membrane formation, has been identified as a potential target for creating a GAP.52 In the rodent malaria parasite, P. berghei (Pb), a PbΔb9Δslarp parasite is 100% attenuated and more protective than a PbΔslarp p52Δp36 GAP.52 PfΔb9Δslarp has been made and shown to be fully attenuated in vitro.53 It has now been manufactured by Sanaria as an aseptic, purified, cryropreserved product called PfSPZ-GA1 (genetically attenuated 1), and is intended to move into clinical trials in the next year. The PfΔp52Δp36Δsap1 and the PfΔb9Δslarp parasites arrest early, more or less at the same stage as irradiated PfSPZ, and are expected to give comparable immunity. It would be ideal to produce a genetically attenuated Pf that fully develops in the liver, and that arrests either at the late liver stage or after invading erythrocytes. Such a knockout would hopefully mimic the CPS/PfSPZ-CVac approach and produce much more efficient protection than PfSPZ-GA1.
Whole SPZ approach: summary and conclusion
The parasites that cause malaria are complex. The subunit vaccine approach seeks to unravel that complexity and identify a few important targets among the greater than 5000 proteins in the parasite’s proteome, and induce protective immune responses against these selected targets. The whole PfSPZ approach does not try to distinguish among the potential targets, and allows the parasite to present potential targets. It required development of a new technology platform to move the whole PfSPZ approach forward. Both PfSPZ Vaccine and PfSPZ-CVac have now been show to induce high-level protective efficacy against CHMI by mosquito bite (PfSPZ Vaccine) and injection of PfSPZ Challenge by DVI (PfSPZ-CVac) in humans, and PfSPZ-GA1 will soon move into clinical trials. In the next few years the team at Sanaria and its many collaborators will undertake the studies required for licensure applications with the goal to prevent Pf malaria in individuals and to halt transmission and eliminate Pf malaria in communities. However, many challenges remain that must be overcome to realize this grand vision.
Heterologous Prime Boost with DNA, Recombinant Viruses and Bacteria and Recombinant Protein with Adjuvant
RTS,S/AS01 and the PfSPZ vaccines represent two different approaches to inducing protective pre-erythrocytic immune responses, based primarily on antibody-mediated and cell-mediated immune mechanisms, respectively (as discussed above). A third approach to pre-erythrocytic Pf vaccine development is to attempt to induce high levels of antibodies, CD4+ T cells responses, and CD8+ T cell responses in the same individual, and to do this using subunit vaccine approaches, meaning that just one or a few proteins are targeted. Such a multi-armed immune response could provide two lines of defense that together result in high grade protection – antibodies to block sporozoite entry into hepatocytes, and for those remaining sporozoites that still invade hepatocytes, CD8+ T cell responses to kill developing parasites within hepatocytes. Recombinant proteins with adjuvants can induce extremely high levels of antibodies (e.g. RTS,S/AS01), and thereby muster the first line of defense, but do not induce protective CD8+ T cell responses. Thus there has been a major effort to develop vaccine platforms able to induce high levels of protective CD8+ T cell responses against one or a small number of target antigens.
The first platform explored was “naked DNA,” in which the gene encoding the selected pre-erythrocytic stage protein is spliced into a plasmid and expression of the protein driven by an upstream promoter. The recombinant DNA is injected into the muscle where passive uptake leads to intracellular expression of the malarial antigen in vivo to engage Class I major histocompatibility complex (MHC) presentation and induce cytotoxic or IFNγ producing T cell responses. Protective CD8+ T cell responses using this approach were first induced in a murine model using DNA plasmids encoding Plasmodium yoelii CSP.54 A follow-on human trial of pDNA encoding PfCSP likewise induced cytotoxic, CD8+ T cell responses, but these were of relatively low magnitude55 and a subsequent clinical trial in which pDNA encoding PfCSP was combined with plasmids encoding four additional pre-erythrocytic stage antigens plus escalating doses of a sixth plasmid encoding human granulocyte macrophage-colony stimulating factor (hGM-CSF) showed no protection against CHMI by mosquito bite.56
In a second gene-based approach, the selected transgene is spliced into the genome of viral vectors rendered non-replicating through the deletion of one or more essential genes, or the genome of replicating vectors that are non-pathogenic. Viral vectors add efficiency and potency, enabling targeted transfection of host cells (e.g., the receptor-mediated entry of serotype 5 adenovirus vectors using the coxsackie adenovirus receptor, or CAR), rather than relying upon passive uptake. They also provide an adjuvant effect by presenting pathogen associated molecular patterns that stimulate the innate immune system, potentiating the adaptive response. Viral vectors can be engineered to express the recombinant antigens on the surface of the capsid, strengthening the induction of humoral immunity.57 However, like DNA plasmids, viral vectors used alone have failed to induce significant protection against CHMI by mosquito bite even when two or more antigens have been tested in combination.58,59,97
The most promising approach to induce strong antibody and T cell responses to malaria has been heterologous prime boost approaches in which different vaccine platforms are given in sequence.60–62 For reasons that remain somewhat unclear, this approach improves immunogenicity and protective efficacy relative to either construct given one or more times by itself. One factor may be that viral vectors express viral proteins in addition to the transgene product, and these likewise induce antibody and T cell responses. If a second dose of the same vector is given, this “anti-backbone” immunity may partially neutralize the vaccine or may kill antigen presenting cells during antigen processing, thereby aborting the anamnestic response. However, if the second administration of the antigen uses a heterologous platform, the only antigenic determinant that is common to both the prime and the boost is the targeted malaria transgene, removing antigen competition as the boosted response develops. This may explain the phenomenal responses, especially for CD8+ T cell responses, that are seen in some prime boost combinations.63
The benefit of DNA priming was first demonstrated in mice more than 15 years ago with a DNA prime and recombinant virus boost60–62 and was also demonstrated in non-human primate models.64,65 It was first assessed clinically with PfCSP DNA prime and RTS,S/AS02 boost, and this achieved the simultaneous induction of antibody, CD4+ and CD8+ responses in the same individuals, although the T cell responses were considered to be only modest.66,67 Since then additional subunit malaria vaccines have employed the DNA prime/heterologous boost technique. The immune responses are of greater magnitude and/or characterized by better focus on inducing responses from protective epitopes than when either vector is given alone.68,69 In the most successful application of the DNA prime/heterologous boost combination to date, a mixture of two plasmids encoding the pre-erythrocytic stage antigens PfCSP and PfAMA1 was administered in three priming doses, followed by a single boost with an adenovirus vector (Ad); this regimen, developed by the US Navy, sterilely protected 4/15 (28%) of malaria-naive volunteers against CHMI by mosquito bite.70 Protection was associated with CD8+ T cell responses, a first for gene-based vaccine approaches to curtailing human infections. Subsequent mapping implicated effector memory CD8+ T cells targeting specific PfAMA1 class I epitopes as the basis of protection.69
Another approach is to combine different viral vectors in sequence. The best example is under development at the University of Oxford, and is now being tested in multiple sites in Africa: a single priming injection is given, comprising an Ad vector encoding the pre-erythrocytic stage antigen TRAP (which is fused to a multi-epitope (ME) string derived from several pre-erythrocytic stage antigens), followed by a single boost with a poxvirus vector (MVA) encoding the same ME-TRAP antigen.71 The combination of Ad followed by pox induced very strong CD8+ T cell responses in humans and protected 3/14 malaria-naive volunteers against CHMI by mosquito bite58; however, when second vector constructs expressing CSP were added to those expressing TRAP, protection was not improved,72 and interference was observed when MSP1 and AMA1 were combined.73 As with the DNA/Ad vaccine, protection induced by the Ad/MVA vaccine correlated with CD8+ T cell responses.58
Immunization with recombinant bacteria, such as Listeria monocytogenes (Lm) has also shown promise for induction of CD8+ T cells that are protective against malaria infection.74,75 Recombinant Lm home to the liver, the primary target of pre-erythrocytic stage malaria vaccines, and preferentially enter dendritic cells in vivo.76 The use of Lm to effectively stimulate robust, multifunctional, cell-mediated immunity against malaria harnesses recent advances made in humans in the cancer vaccine field.77,78
While the improved immunogenicity and protection resulting from heterologous prime-boost approaches have been encouraging, the need for further progress is widely recognized, and consequently a great deal of research has been conducted to strengthen subunit vaccine approaches. For example, the ability to manufacture complex proteins in native configuration has been improved through codon harmonization, aiming to induce higher affinity antibodies; likewise the expression of transgenes has been optimized by appropriate synonymous codon substitutions.79,80 The sometimes limited immunogenicity of protein or peptide-based vaccines has been improved by new adjuvants and by particle-based delivery systems.13 The challenge of antigenic heterogeneity has been addressed by combining representative antigenic types into a single formulation, or by constructing immunogens consisting of conserved, shuffled or consensus sequences.56,81 The immunogenicity of DNA plasmids has been improved by permeabilizing host cell membranes to increase uptake using electroporation82 and by the addition of auxiliary plasmids encoding immunostimulatory cytokines.56 The limitations faced by some viral vectors due to pre-existing immunity (induced by prior immunization or prior infections with naturally occurring, cross-reactive viruses) has been addressed by the selection of vector backbones showing a low degree of serorecognition in the human population63 or by modifications to capsid proteins.83 Finally, aiming for synergy, antigens have been combined into pairs or more complex cocktails,56 allowing the targeting of several parasite stages simultaneously, although antigen combination can also result in interference and reduced immunogenicity for one or more of the components. These and many other improvements to subunit technologies have resulted in significant, if incremental, progress.
Several recent efforts in malaria vaccines are directed at combining the strongest inducers of antibodies – recombinant proteins in adjuvant – with the strongest inducers of CD8+ T cells – Ad vectors or Ad/MVA prime-boost combinations.84–87 For example, RTS,S/AS01 and Ad/MVA ME-TRAP combined regimens are being evaluated in collaborative efforts (NCT01883609). While such regimens add complexity to the regimen – as many as three vaccine platforms administered together or in sequence, ongoing studies will hopefully confirm the potential to induce a multi-armed immune response targeting multiple antigens and potentially multiple parasite stages. Whether or not this newest development in subunit vaccine design can achieve the simultaneous induction of strong antibody and T cell responses against multiple targets using a practical approach remains to be seen.
Most work on heterologous prime boost approaches has focused on pre-erythrocytic stage vaccines. However, there are now exciting efforts which are moving this approach into the development of asexual erythrocytic stage vaccines such as those focused on RH5 or other blood stage antigens.88 It is envisioned that eventually an effective pre-erythrocytic stage vaccine can be combined with an effective erythrocytic stage vaccine to provide multiple layers of defense against disease and death.
In parallel with the efforts to simultaneously induce protective humoral and cellular immune responses must come identification of new protein/epitope targets of protective immunity and the incorporation of those proteins/epitopes into subunit vaccines without reduction of the immunogenicity of other proteins/epitopes through antigen competition. Many groups are using the tools of genomics, transcriptomics, and proteomics to identify these new targets.89 The next decade should see the systematic evaluation of large numbers of potential candidate antigens and the down-selection of combinations interacting synergistically to induct protective responses targeting multiple stages of the parasite life cycle. Hopefully this will result in highly protective subunit vaccine candidates.
Sexual Erythrocytic and Mosquito Stage Transmission Blocking Vaccines
Transmission-blocking vaccines (TBV) target sexual erythrocytic and early mosquito stage antigens as the parasite passes from a human host to a mosquito, and require herd immunity to reduce the incidence of infections in a community. TBV are especially attractive in campaigns to eliminate malaria from an area, and therefore can be referred to as a Vaccine that Interrupts Malaria Transmission (VIMT),3 but also can play a role to reduce the spread of escape mutant parasites when added to vaccines that are targeting other parasite stages. TBV were originally conceived as “an approach to malaria control based on immunization of the host against extracellular malarial gametes, the stage in the mosquito guts, in order to block transmission by the mosquito vector”.90
TBV emerged as a concept in the 1970s with two seminal papers providing proof of the concept using animal models. In studies of the avian parasite Plasmodium gallinaceum,91 chickens that were immunized with formalin- or radiation-treated parasites developed antibody that immobilized microgametes and blocked transmission to mosquitoes; this activity could be reproduced by immunizing with purified gametes.92 In monkeys, immunization with P. knowlesi gamete preparations required immunopotentiators such as Freund’s complete adjuvant in order to induce antibodies that successfully blocked infection of mosquitoes. However, after successful immunization, the spleen was not required for effective immunity, since splenectomized animals equally blocked transmission. Further, transmission-blocking activity induced by gamete immunization also blocked transmission of parasite strains other than the strain used to vaccinate the monkeys.90 In these initial animal studies, TBV acted against the extracellular gamete to prevent fertilization, and did not have activity against the gametocyte stages that reside within erythrocytes.90
These seminal studies established several principles that continue to guide TBV development. (1) Gamete antigens are one source of TBV candidates; subsequent studies have shown that antigens expressed later in development and after fertilization can be targeted (such as Pfs25 and Pfs28 in P. falciparum), as can antigens expressed by the mosquito itself (e.g., AgAPN1).93 (2) Aside from the mosquito antigens, the leading TBV candidate antigens have been identified using monoclonal antibodies prepared from B cells of mice after immunization with whole parasites. (3) The activity of TBV is mediated by IgG, and therefore inducing and sustaining an adequate titer of functional antibody over 1–2 years of malaria transmission remains the primary goal of candidate TBV products. (4) Epitopes recognized by transmission-blocking antibodies can be conserved between different parasite isolates, although sequence variation exists to greater or lesser degrees with all existing TBV candidates. (5) The leading antigens under consideration as vaccine targets are poorly immunogenic in primates, and therefore the recent focus of TBV development has dwelled on enhancing immunogenicity of candidate vaccines without incurring undue reactogenicity.
Four antigens have been the primary focus of TBV development. The P. falciparum antigens now known as Pfs230 and Pfs48/45 are expressed by gametocytes, appear on the surface of the gametes and newly fertilized zygotes, and are then shed as zygotes transform94: the orthologues of these were identified using fertilization blocking antibodies induced by immunization with P. gallinaceum gametes.95,96 Two other proteins, whose P. falciparum orthologues are known as Pfs25 and Pfs28, are expressed on the surface of zygotes during their development to ookinetes, and were initially identified using mAbs prepared from mice that had been immunized with P. gallinaceum ookinete preparations.94
Among these antigens, Pfs25 has advanced furthest as a product and, along with its P. vivax orthologue Pvs25, are the only TBV antigens evaluated in human trials to date. Pfs25 was included among 7 P. falciparum antigens incorporated into an attenuated vaccinia virus vector to create a multi-stage vaccine called NYVAC-Pf7, but antibody responses against most of the antigens were poor.97 Interestingly, the highest level of antibody response to any antigen in NYVAC-Pf7 was elicited against the Pfs25 antigen, but no transmission blocking activity was detected for immune sera in the membrane feeding assay that is used to measure parasite transmission to mosquitoes in the laboratory setting. Early clinical grade recombinant protein products were prepared in S. cerevisiae,98,99 but trials of a Pfs25 candidate expressed in this system and formulated with alum were terminated early owing to reactogenicity, likely due to a unbound antigen in the formulation. Conversely, Pvs25 expressed in S. cerevisiae and formulated with alum was found to be well-tolerated in humans100; antibody responses were modest, but encouragingly functional transmission blocking activity was detected and correlated with antibody concentration. Unfortunately, efforts to enhance responses to S. cerevisiae-expressed Pvs25 and Pichia pastoris-expressed Pfs25 by formulation with Montanide ISA51, a water-in-oil emulsion, were halted early due to unexpected reactogenicity, including erythema nodosum seen in 2 vaccinees who received the Pvs25 product.101
The current emphasis in TBV development has been on increasing immunogenicity against the lead candidate antigens without unduly raising safety concerns. Safety concerns undergo particular scrutiny for TBV products, because these will not directly protect a vaccinee from becoming infected but will only benefit the vaccinee when herd immunity brings malaria transmission down across the entire community. Several research groups are exploring virus-like particles or carrier-conjugated particles as strategies to enhance immunogenicity without increasing reactogenicity or adverse event risks. Conjugation of Pfs25 to the outer membrane protein complex of Neisseria meningitidis (OMPC) produced impressive results in rhesus monkeys: alum-adjuvanted Pfs25-OMPC increased antibody titers by 1–3 logs over Pfs25 monomer in Montanide or alum adjuvants, and antibody levels were sustained for 18 months after Pfs25-OMPC immunization, with potent transmission blocking activity observed in membrane feeding assays.102 Pfs25 has also been conjugated to individual carrier proteins as an alternative to OMPC. When Pfs25 is chemically conjugated to the carrier protein EPA (the detoxified recombinant form of the Pseudomonas aeruginosa exotoxin A), antibody responses are significantly increased.103 The Pfs25-EPA polymer appears as a nanoparticle with average diameter 20 nm,104 and the particle properties of this entity may contribute in part to its immunogenicity. Carrier proteins used in conjugated vaccines can also function to provide T cell help to poorly immunogenic antigens that lack potent T cell epitopes. Researchers are pursuing other particle-delivery technologies in addition to chemical conjugates, such as fusion proteins that form VLPs.105
Thus far, the Pfs25 candidates prepared as particles that have entered phase 1 clinical trials include the Pfs25-EPA conjugate formulated with Alhydrogel, and a purified plant-derived Pfs25 VLP also combined with Alhydrogel.106 Results from these trials have not yet been announced. The efficacy requirements for a TBV that will eliminate malaria from any particular area remain uncertain, and therefore the criteria by which clinical trial results will be assessed for further advancement remain a subject of discussion. Initial efforts to define criteria using mathematical models estimated that TBV should reduce mosquito infections by ≥85% to have the desired impact,107 but more knowledge is needed to understand how TBV will reduce human infections and not only mosquito infections.108 Further, an animal model of transmission suggests that even lower levels of activity can contribute to elimination.109 Thus, TBVs will also need to be assessed in conjunction with other interventions for their potential contribution to elimination efforts. For example TBV may be used in combination with vaccines against other parasite stages, or in combination with drugs and vector control measures.
In light of this background, current TBV efforts are focused on establishing the proof of concept that vaccines delivered to humans can induce functional antibodies that reduce mosquito infections. During early phase trials, the endpoint measurements are made in membrane feeding assays, which are performed in the laboratory by adding sera to cultured parasites and then feeding this material to mosquitoes to demonstrate functional antibody activity. As trials progress in target populations in endemic areas, the goal will be to show that these vaccines can completely prevent infection of mosquitoes that feed on naturally infected vaccinees. In practice, the activity of TBV should last for at least an entire malaria season, and a single additional dose given a year later should be able to boost this response for a second malaria season during an elimination campaign.
Future steps for enhancing vaccine activity will likely include combining 2 or more TBV antigens to assess additive or synergistic effects, combining TBV with malaria vaccines against other parasite stages to assess their combined activity for interrupting malaria transmission, and re-examining the potential for adjuvants other than alum to enhance and sustain the antibody response induced by TBV. While most effort at present focuses on P. falciparum TBV, progress against this parasite species provides a path to develop TBV for P. vivax. Because P. vivax is not tractable to in vitro culture, improved tools are needed for proxy assays of vaccine activity, such as chimeric rodent malaria parasites engineered to express P. vivax TBV antigens,110 or more robust monkey models of P. vivax transmission to assess TBV activity.
Summary
In this paper we have described four of the major approaches to malaria vaccine development. Progress during the last few years has been significant. A phase 3 clinical trial for one has been completed and efficacy, albeit not optimal, has been demonstrated. Because vaccines are the most efficient tools for controlling and eliminating infectious diseases, development efforts should be accelerated. However, much work will be needed in the future to optimize vaccines in regard to magnitude and durability of protective efficacy and financing and practicality of delivery. We are hopeful that anti-malaria vaccines will soon be important tools in the battle against malaria.
Acknowledgments
This article is being published concurrently in the American Journal of Preventive Medicine and Vaccine. The articles are identical except for stylistic changes in keeping with each journal’s style. Either of these versions may be used in citing this article. Publication of this article was supported by Merck and Novartis.
SLH and TLR are supported by Sanaria Inc., JV by GSK Vaccines, and PED by the Intramural Research Program of NIAID, NIH.
We are grateful to the many institutions and individuals supporting the research programs outlined above and wish to address our special thanks to the study participants and their families.
SLH and TLR are supported by Sanaria Inc., which manufactures PfSPZ Vaccine, PfSPZ Challenge, PfSPZ-Cvac and PfSPZ-GA1. Support for JV comes from GSK Vaccines, which manufactures RTS,S/AS01 Vaccine.
No other financial disclosures were reported.
References
- 1.World Health Organization. World malaria report 2014. Geneva: World Health Organization; 2014. [Google Scholar]
- 2.Hoffman SL, Miller LH. Perspectives on malaria vaccine development. In: Hoffman SL, editor. Malaria vaccine development: a multi-immune response approach. Washington, DC: ASM Press; 1996. pp. 1–13. [Google Scholar]
- 3.Alonso PL, Ballou R, Brown G, Chitnis C, Loucq C, Moorthy V, et al. A research agenda for malaria eradication: vaccines. PLoS Med. 2011;8:e1000398. doi: 10.1371/journal.pmed.1000398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coppi A, Natarajan R, Pradel G, Bennett BL, James ER, Roggero MA, et al. The malaria circumsporozoite protein has two functional domains, each with distinct roles as sporozoites journey from mosquito to mammalian host. J Exp Med. 2011;208:341–356. doi: 10.1084/jem.20101488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kumar KA, Sano G, Boscardin S, Nussenzweig RS, Nussenzweig MC, Zavala F, et al. The circumsporozoite protein is an immunodominant protective antigen in irradiated sporozoites. Nature. 2006;444:937–940. doi: 10.1038/nature05361. [DOI] [PubMed] [Google Scholar]
- 6.Egan JE, Weber JL, Ballou WR, Hollingdale MR, Majarian WR, Gordon DM, et al. Efficacy of murine malaria sporozoite vaccines: implications for human vaccine development. Science. 1987;236:453–456. doi: 10.1126/science.3551073. [DOI] [PubMed] [Google Scholar]
- 7.Schofield L, Villaquiran J, Ferreira A, Schellekens H, Nussenzweig RS, Nussenzweig V. Gamma-interferon, CD8+ T cells and antibodies required for immunity to malaria sporozoites. Nature. 1987;330:664–666. doi: 10.1038/330664a0. [DOI] [PubMed] [Google Scholar]
- 8.Ballou WR, Hoffman SL, Sherwood JA, Hollingdale MR, Neva FA, Hockmeyer WT, et al. Safety and efficacy of a recombinant DNA Plasmodium falciparum sporozoite vaccine. Lancet. 1987;1:1277–1281. doi: 10.1016/s0140-6736(87)90540-x. [DOI] [PubMed] [Google Scholar]
- 9.Herrington DA, Clyde DF, Losonsky G, Cortesia M, Murphy JR, Davis J, et al. Safety and immunogenicity in man of a synthetic peptide malaria vaccine against Plasmodium falciparum sporozoites. Nature. 1987;328:257–259. doi: 10.1038/328257a0. [DOI] [PubMed] [Google Scholar]
- 10.Gordon DM, McGovern TW, Krzych U, Cohen JC, Schneider I, LaChance R, et al. Safety, immunogenicity, and efficacy of a recombinantly produced Plasmodium falciparum circumsporozoite protein-hepatitis B surface antigen subunit vaccine. J Infect Dis. 1995;171:1576–1585. doi: 10.1093/infdis/171.6.1576. [DOI] [PubMed] [Google Scholar]
- 11.Stoute JA, Slaoui M, Heppner DG, Momin P, Kester KE, Desmons P, et al. A preliminary evaluation of a recombinant circumsporozoite protein vaccine against Plasmodium falciparum malaria. N Engl J Med. 1997;336:86–91. doi: 10.1056/NEJM199701093360202. [DOI] [PubMed] [Google Scholar]
- 12.Kester KE, McKinney DA, Tornieporth N, Ockenhouse CF, Heppner DG, Hall T, et al. Efficacy of recombinant circumsporozoite protein vaccine regimens against experimental Plasmodium falciparum malaria. J Infect Dis. 2001;183:640–647. doi: 10.1086/318534. [DOI] [PubMed] [Google Scholar]
- 13.Kester KE, Cummings JF, Ofori-Anyinam O, Ockenhouse CF, Krzych U, Moris P, et al. Randomized, double-blind, phase 2a trial of falciparum malaria vaccines RTS,S/AS01B and RTS,S/AS02A in malaria-naive adults: safety, efficacy, and immunologic associates of protection. J Infect Dis. 2009;200:337–346. doi: 10.1086/600120. [DOI] [PubMed] [Google Scholar]
- 14.Garcon N, Chomez P, Van Mechelen M. GlaxoSmithKline adjuvant systems in vaccines: concepts, achievements and perspectives. Expert Rev Vaccines. 2007;6:723–739. doi: 10.1586/14760584.6.5.723. [DOI] [PubMed] [Google Scholar]
- 15.White MT, Bejon P, Olotu A, Griffin JT, Riley EM, Kester KE, et al. The relationship between RTS,S vaccine-induced antibodies, CD4(+) T cell responses and protection against Plasmodium falciparum infection. PLOS ONE. 2013;8:e61395. doi: 10.1371/journal.pone.0061395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.White MT, Bejon P, Olotu A, Griffin JT, Bojang K, Lusingu J, et al. A combined analysis of immunogenicity, antibody kinetics and vaccine efficacy from phase 2 trials of the RTS,S malaria vaccine. BMC Med. 2014;12:117. doi: 10.1186/s12916-014-0117-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Agnandji ST, Asante KP, Lyimo J, Vekemans J, Soulanoudjingar SS, Owusu R, et al. Evaluation of the safety and immunogenicity of the RTS, S/AS01E malaria candidate vaccine when integrated in the expanded program of immunization. J Infect Dis. 2010;202:1076–1087. doi: 10.1086/656190. [DOI] [PubMed] [Google Scholar]
- 18.Asante KP, Abdulla S, Agnandji S, Lyimo J, Vekemans J, Soulanoudjingar S, et al. Safety and efficacy of the RTS,S/AS01(E) candidate malaria vaccine given with expanded-programme-on-immunisation vaccines: 19 month follow-up of a randomised, open-label, phase 2 trial. Lancet Infect Dis. 2011;11:741–749. doi: 10.1016/S1473-3099(11)70100-1. [DOI] [PubMed] [Google Scholar]
- 19.Leach A, Vekemans J, Lievens M, Ofori-Anyinam O, Cahill C, Owusu-Agyei S, et al. Design of a phase III multicenter trial to evaluate the efficacy of the RTS,S/AS01 malaria vaccine in children across diverse transmission settings in Africa. Malar J. 2011;10:224. doi: 10.1186/1475-2875-10-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moorthy VS, Reed Z, Smith PG. MALVAC 2008: measures of efficacy of malaria vaccines in phase 2b and phase 3 trials – scientific, regulatory and public health perspectives. Vaccine. 2009;27:624–628. doi: 10.1016/j.vaccine.2008.11.034. [DOI] [PubMed] [Google Scholar]
- 21.Agnandji ST, Lell B, Soulanoudjingar SS, Fernandes JF, Abossolo BP, Conzelmann C, et al. First results of phase 3 trial of RTS,S/AS01 malaria vaccine in African children. N Engl J Med. 2011;365:1863–1875. doi: 10.1056/NEJMoa1102287. [DOI] [PubMed] [Google Scholar]
- 22.Agnandji S, Lell B, Fernandes J, Abossolo B, Methogo B, Kabwende A, et al. A phase 3 trial of RTS,S/AS01 malaria vaccine in African infants. N Engl J Med. 2012;367:2284–2295. doi: 10.1056/NEJMoa1208394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.RTSS Clinical Trials Partnership. Efficacy and safety of the RTS,S/AS01 malaria vaccine during 18 months after vaccination: a phase 3 randomized, controlled trial in children and young infants at 11 African sites. PLoS Med. 2014;11:e1001685. doi: 10.1371/journal.pmed.1001685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Olotu A, Fegan G, Wambua J, Nyangweso G, Awuondo KO, Leach A, et al. Four-year efficacy of RTS,S/AS01E and its interaction with malaria exposure. N Engl J Med. 2013;368:1111–1120. doi: 10.1056/NEJMoa1207564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.RTSS Clinical Trials Partnership. Efficacy and safety of RTS,S/AS01 malaria vaccine with or without a booster dose in infants and children in Africa: final results of a phase 3, individually randomised, controlled trial. Lancet. 2015;386:31–45. doi: 10.1016/S0140-6736(15)60721-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Clyde DF, Most H, McCarthy VC, Vanderberg JP. Immunization of man against sporozoite-induced falciparum malaria. Am J Med Sci. 1973;266:169–177. doi: 10.1097/00000441-197309000-00002. [DOI] [PubMed] [Google Scholar]
- 27.Rieckmann KH, Carson PE, Beaudoin RL, Cassells JS, Sell KW. Sporozoite induced immunity in man against an Ethiopian strain of Plasmodium falciparum. Trans R Soc Trop Med Hyg. 1974;68:258–259. doi: 10.1016/0035-9203(74)90129-1. [DOI] [PubMed] [Google Scholar]
- 28.Hoffman SL, Goh LM, Luke TC, Schneider I, Le TP, Doolan DL, et al. Protection of humans against malaria by immunization with radiation-attenuated Plasmodium falciparum sporozoites. J Infect Dis. 2002;185:1155–1164. doi: 10.1086/339409. [DOI] [PubMed] [Google Scholar]
- 29.Nussenzweig RS, Vanderberg J, Most H, Orton C. Protective immunity produced by the injection of X-irradiated sporozoites of Plasmodium berghei. Nature. 1967;216:160–162. doi: 10.1038/216160a0. [DOI] [PubMed] [Google Scholar]
- 30.Epstein JE, Tewari K, Lyke KE, Sim BK, Billingsley PF, Laurens MB, et al. Live attenuated malaria vaccine designed to protect through hepatic CD8+ T cell immunity. Science. 2011;334:475–480. doi: 10.1126/science.1211548. [DOI] [PubMed] [Google Scholar]
- 31.Luke TC, Hoffman SL. Rationale and plans for developing a non-replicating, metabolically active, radiation-attenuated Plasmodium falciparum sporozoite vaccine. J Exp Biol. 2003;206:3803–3808. doi: 10.1242/jeb.00644. [DOI] [PubMed] [Google Scholar]
- 32.Seder RA, Chang LJ, Enama ME, Zephir KL, Sarwar UN, Gordon IJ, et al. Protection against malaria by intravenous immunization with a nonreplicating sporozoite vaccine. Science. 2013;341:1359–1365. doi: 10.1126/science.1241800. [DOI] [PubMed] [Google Scholar]
- 33.Roestenberg M, McCall M, Hopman J, Wiersma J, Luty AJ, van Gemert GJ, et al. Protection against a malaria challenge by sporozoite inoculation. N Engl J Med. 2009;361:468–477. doi: 10.1056/NEJMoa0805832. [DOI] [PubMed] [Google Scholar]
- 34.Roestenberg M, Teirlinck AC, McCall MB, Teelen K, Makamdop KN, Wiersma J, et al. Long-term protection against malaria after experimental sporozoite inoculation: an open-label follow-up study. Lancet. 2011;377:1770–1776. doi: 10.1016/S0140-6736(11)60360-7. [DOI] [PubMed] [Google Scholar]
- 35.Nganou-Makamdop K, van Gemert GJ, Arens T, Hermsen CC, Sauerwein RW. Long term protection after immunization with P. berghei sporozoites correlates with sustained IFNgamma responses of hepatic CD8+ memory T cells. PLoS ONE. 2012;7:e36508. doi: 10.1371/journal.pone.0036508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bijker EM, Teirlinck AC, Schats R, van Gemert GJ, van de Vegte-Bolmer M, van Lieshout L, et al. Cytotoxic markers associate with protection against malaria in human volunteers immunized with Plasmodium falciparum sporozoites. J Infect Dis. 2014;210:1605–1615. doi: 10.1093/infdis/jiu293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hoffman SL, Billingsley P, James E, Richman A, Loyevsky M, Li T, et al. Development of a metabolically active, non-replicating sporozoite vaccine to prevent Plasmodium falciparum malaria. Hum Vaccines. 2010;6:97–106. doi: 10.4161/hv.6.1.10396. [DOI] [PubMed] [Google Scholar]
- 38.Roestenberg M, Bijker EM, Sim BK, Billingsley PF, James ER, Bastiaens GJ, et al. Controlled human malaria infections by intradermal injection of cryopreserved Plasmodium falciparum sporozoites. Am J Trop Med Hyg. 2013;88:5–13. doi: 10.4269/ajtmh.2012.12-0613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sheehy SH, Spencer AJ, Douglas AD, Sim BK, Longley RJ, Edwards NJ, et al. Optimising controlled human malaria infection studies using cryopreserved parasites administered by needle and syringe. PLOS ONE. 2013;8:e65960. doi: 10.1371/journal.pone.0065960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shekalaghe S, Rutaihwa M, Billingsley PF, Chemba M, Daubenberger CA, James ER, et al. Controlled human malaria infection of Tanzanians by intradermal injection of aseptic, purified, cryopreserved Plasmodium falciparum sporozoites. Am J Trop Med Hyg. 2014;91:471–480. doi: 10.4269/ajtmh.14-0119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hodgson SH, Juma EA, Salim A, Magiri C, Kimani D, Njenga D, et al. Evaluating controlled human malaria infection in Kenyan adults with varying degrees of prior exposure to Plasmodium falciparum using sporozoites administered by intramuscular injection. Front Microbiol. 2014:2014. doi: 10.3389/fmicb.2014.00686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mordmüller B, Supan C, Sim KL, Gómez-Pérez GP, Ospina Salazar CL, Held J, et al. Direct venous inoculation of Plasmodium falciparum sporozoites for controlled human malaria infection: a dose-finding trial in two centres. Malar J. 2015;14:117. doi: 10.1186/s12936-015-0628-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bastiaens GJ, van Meer MP, Scholzen A, Obiero JM, Vatanshenassan M, van Grinsven T, et al. Safety, immunogenicity and protective efficacy after intradermal immunization with aseptic, purified, cryopreserved Plasmodium falciparum sporozoites in volunteers under chloroquine prophylaxis: a randomized controlled trial. doi: 10.4269/ajtmh.15-0621. unpublished results. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Garcia CR, Manzi F, Tediosi F, Hoffman SL, James ER. Comparative cost models of a liquid nitrogen vapor phase (LNVP) cold chain-distributed cryopreserved malaria vaccine vs. a conventional vaccine. Vaccine. 2013;31:380–386. doi: 10.1016/j.vaccine.2012.10.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bijker EM, Bastiaens GJ, Teirlinck AC, van Gemert GJ, Graumans W, van de Vegte-Bolmer M, et al. Protection against malaria after immunization by chloroquine prophylaxis and sporozoites is mediated by preerythrocytic immunity. Proc Natl Acad Sci USA. 2013;110:7862–7867. doi: 10.1073/pnas.1220360110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gómez-Pérez GP, Legarda A, Muñoz J, Sim BKL, Ballester MR, Dobaño C, et al. Controlled human malaria infection by intramuscular and direct venous inoculation of cryopreserved Plasmodium falciparum sporozoites in malaria-naïve volunteers: effect of injection volume and dose on infectivity rates. Malar J. 2015;14:306. doi: 10.1186/s12936-015-0817-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Aly AS, Vaughan AM, Kappe SH. Malaria parasite development in the mosquito and infection of the mammalian host. Annu Rev Microbiol. 2009;63:195–221. doi: 10.1146/annurev.micro.091208.073403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van Buskirk KM, O’Neill MT, De La Vega P, Maier AG, Krzych U, Williams J, et al. Preerythrocytic, live-attenuated Plasmodium falciparum vaccine candidates by design. Proc Natl Acad Sci USA. 2009;106:13004–13009. doi: 10.1073/pnas.0906387106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Spring M, Murphy J, Nielsen R, Dowler M, Bennett JW, Zarling S, et al. First-in-human evaluation of genetically attenuated Plasmodium falciparum sporozoites administered by bite of Anopheles mosquitoes to adult volunteers. Vaccine. 2013;31:4975–4983. doi: 10.1016/j.vaccine.2013.08.007. [DOI] [PubMed] [Google Scholar]
- 50.Annoura T, Ploemen IH, van Schaijk BC, Sajid M, Vos MW, van Gemert GJ, et al. Assessing the adequacy of attenuation of genetically modified malaria parasite vaccine candidates. Vaccine. 2012;30:2662–2670. doi: 10.1016/j.vaccine.2012.02.010. [DOI] [PubMed] [Google Scholar]
- 51.Mikolajczak SA, Lakshmanan V, Fishbaugher M, Camargo N, Harupa A, Kaushansky A, et al. A next-generation genetically attenuated Plasmodium falciparum parasite created by triple gene deletion. Mol Ther. 2014;22:1707–1715. doi: 10.1038/mt.2014.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Annoura T, van Schaijk BC, Ploemen IH, Sajid M, Lin JW, Vos MW, et al. Two Plasmodium 6-Cys family-related proteins have distinct and critical roles in liver-stage development. FASEB J. 2014;28:2158–2170. doi: 10.1096/fj.13-241570. [DOI] [PubMed] [Google Scholar]
- 53.van Schaijk BC, Ploemen IH, Annoura T, Vos MW, Lander F, van Gemert GJ, et al. A genetically attenuated malaria vaccine candidate based on gene-deficient sporozoites. Elife. 2014;3 doi: 10.7554/eLife.03582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sedegah M, Hedstrom R, Hobart P, Hoffman SL. Protection against malaria by immunization with plasmid DNA encoding circumsporozoite protein. Proc Natl Acad Sci USA. 1994;91:9866–9870. doi: 10.1073/pnas.91.21.9866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang R, Doolan DL, Le TP, Hedstrom RC, Coonan KM, Charoenvit Y, et al. Induction of antigen-specific cytotoxic T lymphocytes in humans by a malaria DNA vaccine. Science. 1998;282:476–480. doi: 10.1126/science.282.5388.476. [DOI] [PubMed] [Google Scholar]
- 56.Richie TL, Charoenvit Y, Wang R, Epstein JE, Hedstrom RC, Kumar S, et al. Clinical trial in healthy malaria-naïve adults to evaluate the safety, tolerability, immunogenicity and efficacy of MuStDO5, a five-gene, sporozoite/hepatic stage Plasmodium falciparum DNA vaccine combined with escalating dose human GM-CSF DNA. Hum Vaccines Immunother. 2012;8:1564–1584. doi: 10.4161/hv.22129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Krause A, Joh JH, Hackett NR, Roelvink PW, Bruder JT, Wickham TJ, et al. Epitopes expressed in different adenovirus capsid proteins induce different levels of epitope-specific immunity. J Virol. 2006;80:5523–5530. doi: 10.1128/JVI.02667-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ewer KJ, O’Hara GA, Duncan CJ, Collins KA, Sheehy SH, Reyes-Sandoval A, et al. Protective CD8(+) T-cell immunity to human malaria induced by chimpanzee adenovirus-MVA immunisation. Nat Commun. 2013;4:2836. doi: 10.1038/ncomms3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tamminga C, Sedegah M, Maiolatesi S, Fedders C, Reyes S, Reyes A, et al. Human adenovirus 5-vectored Plasmodium falciparum NMRC-M3V-Ad-PfCA vaccine encoding CSP and AMA1 is safe, well-tolerated and immunogenic but does not protect against controlled human malaria infection. Hum Vaccine Immunother. 2013;9:2165–2177. doi: 10.4161/hv.24941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li S, Rodrigues M, Rodriguez D, Rodriguez JR, Esteban M, Palese P, et al. Priming with recombinant influenza virus followed by administration of recombinant vaccinia virus induces CD8+ T-cell-mediated protective immunity against malaria. Proc Natl Acad Sci USA. 1993;90:5214–5218. doi: 10.1073/pnas.90.11.5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Plebanski M, Gilbert SC, Schneider J, Hannan CM, Layton G, Blanchard T, et al. Protection from Plasmodium berghei infection by priming and boosting T cells to a single class I-restricted epitope with recombinant carriers suitable for human use. J Immunol. 1998;28:4345–4355. doi: 10.1002/(SICI)1521-4141(199812)28:12<4345::AID-IMMU4345>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 62.Sedegah M, Jones TR, Kaur M, Hedstrom R, Hobart P, Tine JA, et al. Boosting with recombinant vaccinia increases immunogenicity and protective efficacy of malaria DNA vaccine. Proc Natl Acad Sci USA. 1998;95:7648–7653. doi: 10.1073/pnas.95.13.7648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Reyes-Sandoval A, Rollier CS, Milicic A, Bauza K, Cottingham MG, Tang CK, et al. Mixed vector immunization with recombinant adenovirus and MVA can improve vaccine efficacy while decreasing antivector immunity. Mol Ther. 2012;20:1633–1647. doi: 10.1038/mt.2012.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rogers WO, Weiss WR, Kumar A, Aguiar JC, Tine JA, Gwadz R, et al. Protection of rhesus macaques against lethal Plasmodium knowlesi malaria by a heterologous DNA priming and poxvirus boosting immunization regimen. Infect Immun. 2002;70:4329–4335. doi: 10.1128/IAI.70.8.4329-4335.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jiang G, Shi M, Conteh S, Richie N, Banania G, Geneshan H, et al. Sterile protection against Plasmodium knowlesi in rhesus monkeys from a malaria vaccine: comparison of heterologous prime boost strategies. PLoS ONE. 2009;4:e6559. doi: 10.1371/journal.pone.0006559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Epstein JE, Charoenvit Y, Kester KE, Wang R, Newcomer R, Fitzpatrick S, et al. Safety, tolerability, and antibody responses in humans after sequential immunization with a PfCSP DNA vaccine followed by the recombinant protein vaccine RTS,S/AS02A. Vaccine. 2004;22:1592–1603. doi: 10.1016/j.vaccine.2004.01.031. [DOI] [PubMed] [Google Scholar]
- 67.Wang R, Epstein J, Charoenvit Y, Baraceros FM, Rahardjo N, Gay T, et al. Induction in humans of CD8+ and CD4+ T cell and antibody responses by sequential immunization with malaria DNA and recombinant protein. J Immunol. 2004;172:5561–5569. doi: 10.4049/jimmunol.172.9.5561. [DOI] [PubMed] [Google Scholar]
- 68.McConkey SJ, Reece WH, Moorthy VS, Webster D, Dunachie S, Butcher G, et al. Enhanced T-cell immunogenicity of plasmid DNA vaccines boosted by recombinant modified vaccinia virus Ankara in humans. Nat Med. 2003;9:729–735. doi: 10.1038/nm881. [DOI] [PubMed] [Google Scholar]
- 69.Sedegah M, Hollingdale MR, Farooq F, Ganeshan H, Belmonte M, Kim Y, et al. Sterile immunity to malaria after DNA prime/adenovirus boost immunization is associated with effector memory CD8+ T cells targeting AMA1 class I epitopes. PLOS ONE. 2014;9:e106241. doi: 10.1371/journal.pone.0106241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chuang I, Sedegah M, Cicatelli S, Spring M, Polhemus M, Tamminga C, et al. DNA prime/adenovirus boost malaria vaccine encoding P. falciparum CSP and AMA1 induces sterile protection associated with cell-mediated immunity. PLOS ONE. 2013;8:e55571. doi: 10.1371/journal.pone.0055571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.O’Hara GA, Duncan CJ, Ewer KJ, Collins KA, Elias SC, Halstead FD, et al. Clinical assessment of a recombinant simian adenovirus ChAd63: a potent new vaccine vector. J Infect Dis. 2012;205:772–781. doi: 10.1093/infdis/jir850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hodgson SH, Ewer KJ, Bliss CM, Edwards NJ, Rampling T, Anagnostou NA, et al. Evaluation of the efficacy of ChAd63-MVA vectored vaccines expressing circumsporozoite protein and ME-TRAP against controlled human malaria infection in malaria-naive individuals. J Infect Dis. 2014;211:1076–1086. doi: 10.1093/infdis/jiu579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sheehy SH, Duncan CJ, Elias SC, Choudhary P, Biswas S, Halstead FD, et al. ChAd63-MVA-vectored blood-stage malaria vaccines targeting MSP1 and AMA1: assessment of efficacy against mosquito bite challenge in humans. Mol Ther. 2012;20:2355–2368. doi: 10.1038/mt.2012.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pham NL, Pewe LL, Fleenor CJ, Langlois RA, Legge KL, Badovinac VP, et al. Exploiting cross-priming to generate protective CD8 T-cell immunity rapidly. Proc Natl Acad Sci USA. 2010;107:12198–12203. doi: 10.1073/pnas.1004661107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schmidt NW, Butler NS, Harty JT. CD8 T cell immunity to Plasmodium permits generation of protective antibodies after repeated sporozoite challenge. Vaccine. 2009;27:6103–6106. doi: 10.1016/j.vaccine.2009.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Neuenhahn M, Kerksiek KM, Nauerth M, Suhre MH, Schiemann M, Gebhardt FE, et al. CD8alpha+ dendritic cells are required for efficient entry of Listeria monocytogenes into the spleen. Immunity. 2006;25:619–630. doi: 10.1016/j.immuni.2006.07.017. [DOI] [PubMed] [Google Scholar]
- 77.Le DT, Brockstedt DG, Nir-Paz R, Hampl J, Mathur S, Nemunaitis J, et al. A live-attenuated Listeria vaccine (ANZ-100) and a live-attenuated Listeria vaccine expressing mesothelin (CRS-207) for advanced cancers: phase I studies of safety and immune induction. Clin Cancer Res. 2012;18:858–868. doi: 10.1158/1078-0432.CCR-11-2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wood LM, Paterson Y. Attenuated Listeria monocytogenes: a powerful and versatile vector for the future of tumor immunotherapy. Front Cell Infect Microbiol. 2014;4:51. doi: 10.3389/fcimb.2014.00051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Angov E, Hillier CJ, Kincaid RL, Lyon JA. Heterologous protein expression is enhanced by harmonizing the codon usage frequencies of the target gene with those of the expression host. PLoS ONE. 2008;3:e2189. doi: 10.1371/journal.pone.0002189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Narum DL, Kumar S, Rogers WO, Fuhrmann SR, Liang H, Oakley M, et al. Codon optimization of gene fragments encoding Plasmodium falciparum merzoite proteins enhances DNA vaccine protein expression and immunogenicity in mice. Infect Immun. 2001;69:7250–7253. doi: 10.1128/IAI.69.12.7250-7253.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Terheggen U, Drew DR, Hodder AN, Cross NJ, Mugyenyi CK, Barry AE, et al. Limited antigenic diversity of Plasmodium falciparum apical membrane antigen 1 supports the development of effective multi-allele vaccines. BMC Med. 2014;12:183. doi: 10.1186/s12916-014-0183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dobano C, Widera G, Rabussay D, Doolan DL. Enhancement of antibody and cellular immune responses to malaria DNA vaccines by in vivo electroporation. Vaccine. 2007;25:6635–6645. doi: 10.1016/j.vaccine.2007.06.036. [DOI] [PubMed] [Google Scholar]
- 83.Bruder JT, Semenova E, Chen P, Limbach K, Patterson NB, Stefaniak ME, et al. Modification of Ad5 hexon hypervariable regions circumvents pre-existing Ad5 neutralizing antibodies and induces protective immune responses. PLoS ONE. 2012;7:e33920. doi: 10.1371/journal.pone.0033920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hutchings CL, Birkett AJ, Moore AC, Hill AV. Combination of protein and viral vaccines induces potent cellular and humoral immune responses and enhanced protection from murine malaria challenge. Infect Immun. 2007;75:5819–5826. doi: 10.1128/IAI.00828-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Draper SJ, Biswas S, Spencer AJ, Remarque EJ, Capone S, Naddeo M, et al. Enhancing blood-stage malaria subunit vaccine immunogenicity in rhesus macaques by combining adenovirus, poxvirus, and protein-in-adjuvant vaccines. J Immunol. 2010;185:7583–7595. doi: 10.4049/jimmunol.1001760. [DOI] [PubMed] [Google Scholar]
- 86.Biswas S, Choudhary P, Elias SC, Miura K, Milne KH, de Cassan SC, et al. Assessment of humoral immune responses to blood-stage malaria antigens following ChAd63-MVA immunization, controlled human malaria infection and natural exposure. PLOS ONE. 2014;9:e107903. doi: 10.1371/journal.pone.0107903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hodgson SH, Choudhary P, Elias SC, Milne KH, Rampling TW, Biswas S, et al. Combining viral vectored and protein-in-adjuvant vaccines against the blood-stage malaria antigen AMA1: report on a phase 1a clinical trial. Mol Ther. 2014;22:2142–2154. doi: 10.1038/mt.2014.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Reddy KS, Pandey AK, Singh H, Sahar T, Emmanuel A, Chitnis CE, et al. Bacterially expressed full-length recombinant Plasmodium falciparum RH5 protein binds erythrocytes and elicits potent strain-transcending parasite-neutralizing antibodies. Infect Immun. 2014;82:152–164. doi: 10.1128/IAI.00970-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Duffy PE, Sahu T, Akue A, Milman N, Anderson C. Pre-erythrocytic malaria vaccines: identifying the targets. Expert Rev Vaccines. 2012;11:1261–1280. doi: 10.1586/erv.12.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gwadz RW, Carter R, Green I. Gamete vaccines and transmission-blocking immunity in malaria. Bull World Health Organ. 1979;57(Suppl. 1):175–180. [PMC free article] [PubMed] [Google Scholar]
- 91.Gwadz RW. Successful immunization against the sexual stages of Plasmodium gallinaceum. Science. 1976;193:1150–1151. doi: 10.1126/science.959832. [DOI] [PubMed] [Google Scholar]
- 92.Carter R, Chen DH. Malaria transmission blocked by immunisation with gametes of the malaria parasite. Nature. 1976;263:57–60. doi: 10.1038/263057a0. [DOI] [PubMed] [Google Scholar]
- 93.Dinglasan RR, Kalume DE, Kanzok SM, Ghosh AK, Muratova O, Pandey A, et al. Disruption of Plasmodium falciparum development by antibodies against a conserved mosquito midgut antigen. Proc Natl Acad Sci USA. 2007;104:13461–13466. doi: 10.1073/pnas.0702239104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Carter R, Miller LH, Rener J, Kaushal DC, Kumar N, Graves PM, et al. Target antigens of malaria transmission blocking immunity. Philos Trans R Soc Lond Ser B: Biol Sci. 1984;307:201–213. doi: 10.1098/rstb.1984.0120. [DOI] [PubMed] [Google Scholar]
- 95.Rener J, Carter R, Rosenberg Y, Miller LH. Anti-gamete monoclonal antibodies synergistically block transmission of malaria by preventing fertilization in the mosquito. Proc Natl Acad Sci USA. 1980;77:6797–6799. doi: 10.1073/pnas.77.11.6797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kaushal DC, Carter R, Rener J, Grotendorst CA, Miller LH, Howard RJ. Monoclonal antibodies against surface determinants on gametes of Plasmodium gallinaceum block transmission of malaria parasites to mosquitoes. J Immunol. 1983;131:2557–2562. [PubMed] [Google Scholar]
- 97.Ockenhouse CF, Sun PF, Lanar DE, Wellde BT, Hall BT, Kester K, et al. Phase I/IIa safety, immunogenicity and efficacy of NYVAC-Pf7, a pox-vectored, multiantigen, multistage vaccine candidate for Plasmodium falciparum malaria. J Infect Dis. 1998;177:1664–1673. doi: 10.1086/515331. [DOI] [PubMed] [Google Scholar]
- 98.Gozar MM, Price VL, Kaslow DC. Saccharomyces cerevisiae-secreted fusion proteins Pfs25 and Pfs28 elicit potent Plasmodium falciparum transmission-blocking antibodies in mice. Infect Immun. 1998;66:59–64. doi: 10.1128/iai.66.1.59-64.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gozar MM, Muratova O, Keister DB, Kensil CR, Price VL, Kaslow DC. Plasmodium falciparum: immunogenicity of alum-adsorbed clinical-grade TBV25-28, a yeast-secreted malaria transmission-blocking vaccine candidate. Exp Parasitol. 2001;97:61–69. doi: 10.1006/expr.2000.4580. [DOI] [PubMed] [Google Scholar]
- 100.Malkin EM, Durbin AP, Diemert DJ, Sattabongkot J, Wu Y, Miura K, et al. Phase 1 vaccine trial of Pvs25H: a transmission blocking vaccine for Plasmodium vivax malaria. Vaccine. 2005;23:3131–3138. doi: 10.1016/j.vaccine.2004.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wu Y, Ellis RD, Shaffer D, Fontes E, Malkin EM, Mahanty S, et al. Phase 1 trial of malaria transmission blocking vaccine candidates Pfs25 and Pvs25 formulated with montanide ISA 51. PLoS ONE. 2008;3:e2636. doi: 10.1371/journal.pone.0002636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wu Y, Przysiecki C, Flanagan E, Bello-Irizarry SN, Ionescu R, Muratova O, et al. Sustained high-titer antibody responses induced by conjugating a malarial vaccine candidate to outer-membrane protein complex. Proc Natl Acad Sci USA. 2006;103:18243–18248. doi: 10.1073/pnas.0608545103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Qian F, Wu Y, Muratova O, Zhou H, Dobrescu G, Duggan P, et al. Conjugating recombinant proteins to Pseudomonas aeruginosa Exo-Protein A: a strategy for enhancing immunogenicity of malaria vaccine candidates. Vaccine. 2007;25:3923–3933. doi: 10.1016/j.vaccine.2007.02.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Shimp RL, Jr, Rowe C, Reiter K, Chen B, Nguyen V, Aebig J, et al. Development of a Pfs25-EPA malaria transmission blocking vaccine as a chemically conjugated nanoparticle. Vaccine. 2013;31:2954–2962. doi: 10.1016/j.vaccine.2013.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Jones RM, Chichester JA, Mett V, Jaje J, Tottey S, Manceva S, et al. A plant-produced Pfs25 VLP malaria vaccine candidate induces persistent transmission blocking antibodies against Plasmodium falciparum in immunized mice. PLOS ONE. 2013;8:e79538. doi: 10.1371/journal.pone.0079538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.WHO. Malaria vaccine rainbow tables. Available from: http://www.who.int/vaccine_research/links/Rainbow/en/index.html [accessed 04.01.15]
- 107.Penny MA, Maire N, Studer A, Schapira A, Smith TA. What should vaccine developers ask? Simulation of the effectiveness of malaria vaccines. PLoS ONE. 2008;3:e3193. doi: 10.1371/journal.pone.0003193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nunes JK, Woods C, Carter T, Raphael T, Morin MJ, Diallo D, et al. Development of a transmission-blocking malaria vaccine: progress, challenges, and the path forward. Vaccine. 2014;32:5531–5539. doi: 10.1016/j.vaccine.2014.07.030. [DOI] [PubMed] [Google Scholar]
- 109.Blagborough AM, Churcher TS, Upton LM, Ghani AC, Gething PW, Sinden RE. Transmission-blocking interventions eliminate malaria from laboratory populations. Nat Commun. 2013;4:1812. doi: 10.1038/ncomms2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Blagborough AM, Yoshida S, Sattabongkot J, Tsuboi T, Sinden RE. Intranasal and intramuscular immunization with Baculovirus dual expression system-based Ps25 vaccine substantially blocks Plasmodium vivax transmission. Vaccine. 2010;28:6014–6020. doi: 10.1016/j.vaccine.2010.06.100. [DOI] [PubMed] [Google Scholar]