

Abstract
MLL1 belongs to the SET1 family of histone H3 lysine 4 (H3K4) methyltransferases, composed of MLL1–4 and SETd1A/B. MLL1 translocations are present in acute leukemias, and mutations in several family members are associated with cancer and developmental disorders. MLL1 associates with a subcomplex containing WDR5, RbBP5, ASH2L, and DPY-30 (WRAD), forming the MLL1 core complex required for H3K4 mono- and dimethylation and transcriptional activation. Core complex assembly requires interaction of WDR5 with the MLL1 Win (WDR5 interaction) motif, which is conserved across the SET1 family. Agents that mimic the SET1 family Win motif inhibit the MLL1 core complex and have become an attractive approach for targeting MLL1 in cancers. Like MLL1, other SET1 family members interact with WRAD, but the roles of the Win motif in complex assembly and enzymatic activity remain unexplored. Here, we show that the Win motif is necessary for interaction of WDR5 with all members of the human SET1 family. Mutation of the Win motif-WDR5 interface severely disrupts assembly and activity of MLL1 and SETd1A complexes but only modestly disrupts MLL2/4 and SETd1B complexes without significantly altering enzymatic activity in vitro. Notably, in the absence of WDR5, MLL3 interacts with RAD and shows enhanced activity. To further probe the role of the Win motif-WDR5 interaction, we designed a peptidomimetic that binds WDR5 (Kd ∼3 nm) and selectively inhibits activity of MLL1 and SETd1A core complexes within the SET1 family. Our results reveal that SET1 family complexes with the weakest Win motif-WDR5 interaction are more susceptible to Win motif-based inhibitors.
Keywords: enzyme, epigenetics, histone methylation, protein structure, protein-protein interaction, MLL1, SETd1A, WDR5, Win motif
Introduction
Mixed Lineage Leukemia 1 (MLL1) protein is a member of the SET1 (or MLL) family of histone methyltransferases. In humans, this family consists of six members: MLL1–4, SETd1A, and SETd1B (1–8). The SET1 family catalyzes methylation of histone 3 lysine 4 (H3K4),3 an epigenetic mark that is associated with active transcription (9–12). The human SET1 family is composed of large proteins with several well characterized functional domains involved in chromatin binding and protein-protein interactions (13, 14) (Fig. 1A). Although some of these domains differ among family members, all share a C-terminal SET (suppressor of variegation, enhancer of Zeste, trithorax) domain that confers H3K4 methyltransferase activity (15). Like many chromatin-modifying enzymes, the SET1 family works as part of multiprotein complexes that contain binding partners involved in enzymatic regulation and gene targeting. Although the majority of isolated SET1 family SET domains catalyze weak H3K4 monomethylation (H3K4me1), enhanced methylation is observed in the context of a “core complex” (16). The minimal core complex required for enhanced methylation is composed of the SET1/MLL SET domain and a subcomplex called WRAD (WD-40 repeat protein 5 (WDR5), retinoblastoma-binding protein 5 (RbBP5), absent small homeotic 2-like (ASH2L), and dumpy-30 (DPY-30)) (17–20). Interestingly, SET1 family core complexes preferentially catalyze different levels of H3K4 methylation in a manner that correlates with their evolutionary lineage (16). Whereas SETd1A/B core complexes catalyze mono-, di-, and trimethylation of H3K4 (H3K4me1, H3K4me2, and H3K4me3, respectively), the MLL1 and MLL4 (also known as MLL2) core complexes predominantly catalyze mono- and dimethylation (16). In contrast, MLL2 and MLL3 core complexes catalyze predominantly H3K4 monomethylation (16). In cells, different levels of H3K4 methylation are localized to distinct genomic regions and are associated with distinct functional outcomes (21–23). Assembly of the MLL1 core complex requires a direct interaction between MLL1 and WDR5, whereby WDR5 acts to stabilize the interaction between the MLL1 SET domain and the RbBP5/ASH2L heterodimer (18, 24). The MLL1-WDR5 interaction occurs via the conserved Win (WDR5 interaction) motif (24), located ∼50 amino acids N-terminal from the SET domain, where a conserved arginine, Arg-3765 (in MLL1), plays a critical role in making contacts with WDR5 (19, 25). Mutation of Arg-3765 leads to severe destabilization of MLL1 core complex and loss of MLL1 H3K4 dimethyltransferase activity in vitro (24, 26).
FIGURE 1.
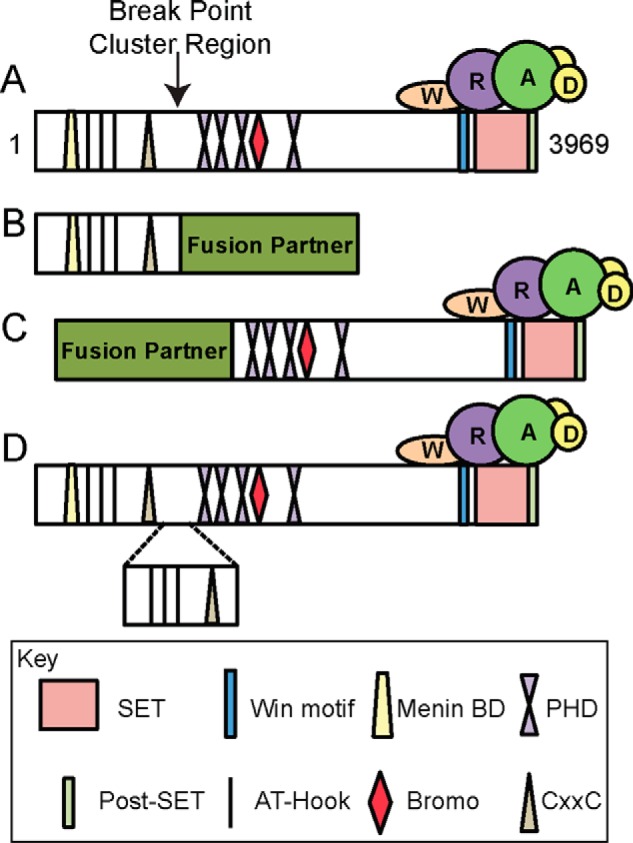
Schematic of common outcomes of MLL1 genetic rearrangements. A, domain map of full-length wild type MLL1 with the breakpoint region denoted by an arrow. MLL1 contains many domains involved in binding chromatin (i.e. AT-Hooks, CxxC domains, PHD, and bromodomains) as well as domains involved in mediating protein-protein interactions (i.e. menin binding domain (Menin BD) and the WDR5 interaction (Win) motif). The SET and post-SET domains are involved in catalysis. The WRAD subcomplex interacts with the C terminus of MLL1. B, the most common outcome of MLL1 genetic rearrangements, which result in replacement of the C terminus of MLL1 with one of ∼70 known fusion partners (i.e. AF9, AF4, and ENL). In this arrangement, the N terminus is retained, but the catalytic SET/post-SET domain is lost. C, a rearrangement that results in the replacement of the N terminus of MLL1 with a fusion partner (i.e. AF4). In this arrangement, the N-terminal domains are lost, but the catalytic domain is retained. D, result of a partial tandem duplication in which a segment of the N terminus (containing the AT-Hooks and CxxC domain) is duplicated and inserted at the break point region.
MLL1 regulates transcriptional activation of genes involved in development, neurogenesis, and fetal and adult hematopoiesis (27–31), including several genes in the HOX cluster (32, 33). Misregulation of HOX genes is commonly observed in leukemias associated with MLL1 genetic rearrangements (34–37). Indeed, the MLL1 gene is a frequent site of genetic rearrangement and accounts for >70% of infant acute lymphocytic leukemia and 10% of de novo acute myeloid leukemia (AML) in adults (38). Most genetic rearrangements of MLL1 result in translocations that fuse the N-terminal fragment of the MLL1 protein (lacking the SET domain) to one of ∼70 known fusion partners (39) (Fig. 1B). One such type of MLL1 fusion, MLL1-AF9, exhibits a dominant gain-of-function phenotype that contributes to AML leukemogenesis in mice (40). Mechanistic studies show that the wild type allele is required for leukemogenesis (41), suggesting that molecules targeting the wild type SET domain may be a useful strategy for treatment (42). Certain MLL1 gene rearrangements result in aberrant MLL1 proteins that retain the catalytic domain (Fig. 1C). For example, AF4 (ALL-1-fused gene on chromosome 4) and NEBL (nebulin) have been reported to form fusions with the MLL1 C terminus; such fusions have been associated with oncogenic roles in leukemia (43–47). Additionally, internal partial tandem duplications (Fig. 1D), which result in duplication of an N-terminal segment of MLL1 retaining the C-terminal SET domain, have been described in ∼10% of AML patients and have been shown to play a dominant gain-of-function role in oncogenesis (48). Thus, targeting wild type MLL1 is a tractable approach for treating these types of cancers.
Since the first demonstration of inhibition of the MLL1 core complex by a peptidomimetic that targets the Win motif-WDR5 interaction (24), several related compounds have been developed (42, 49–54), some of which have been shown to specifically reduce cancer cell growth and elicit genome wide reprograming that is consistent with an MLL1 deletion phenotype (42, 54, 55). However, because the Win motif is conserved among all six SET1 family members, it is unclear the extent to which these changes are due to specific targeting of the MLL1 core complex over other SET1 family complexes. Indeed, the role of the Win motif-WDR5 interaction in other SET1 family complexes remains unexplored.
In this investigation, we characterized the roles of the Win motif in core complex assembly and enzymatic activity for each human SET1 family complex. We found that all SET1 family members interact with WDR5 in a Win motif-dependent manner, but not all complexes are affected by disruption of the interaction in a similar way. Loss of WDR5-Win motif interaction severely destabilizes MLL1 and SETd1A complexes and moderately destabilizes MLL2–4 complexes but does not affect the stability of the MLL3 core complex. To further probe the role of the WDR5-Win motif interaction among family members, we designed a 6-residue Win motif peptidomimetic (Win6mer) that binds to WDR5 with high affinity (Kd ∼3 nm) and found that it inhibits MLL1 and SETd1A complexes but does not inhibit MLL2–4 or SETd1B complexes. This work reveals that the contributions of the Win motif-WDR5 interaction to complex assembly differ among the human SET1 family members and that such differences can be exploited to alter the enzymatic activities of a subset of SET1 family core complexes. In addition, our results reveal that the MLL1 and SETd1A complexes that bind WDR5 with the weakest affinity are most sensitive to inhibition by molecules that mimic the Win motif.
Results
Characterization of the Contributions of the Win Motif to Complex Assembly
The Win motif is a highly conserved region within the SET1 family and is composed of 6 key residues located N-terminal to the catalytic domain (24, 25, 49, 56) (Fig. 2A). This region was previously identified as necessary for the pairwise interaction of MLL1 with WDR5 (24, 25) and for the assembly of the MLL1 core complex (24). However, unlike MLL1, MLL3 does not require interaction with WDR5 to stably interact with RbBP5/ASH2L (16, 42, 57, 58). Furthermore, a recent study suggested that SET1 family members, with the exception of MLL1, do not require WDR5 to assemble a fully functional core complex (57). These results raise questions about the role of the Win motif and why it is so highly conserved among metazoan SET1 family enzymes. Thus, we set out to further characterize the roles of the Win motif in SET1 family core complex assembly.
FIGURE 2.

The Win motif is required for interaction with WDR5. A, sequence alignment of the human SET1 family Win motif generated using Clustal Omega (90). The 6-residue Win motif is highlighted in blue, and the conserved arginine is enclosed in a box. B, GST pull-down of wild type and Win mutant SET1 family members with WDR5. Individual GST-tagged SET domains (wild type or mutant) were incubated with purified WDR5 and glutathione-coated agarose beads. A Coomassie Blue-stained SDS-polyacrylamide gel of the pull-down fractions only is shown. GST was used as a negative control (lane 13), and a sample of purified WDR5 was run on the gel to compare the migration of pull-down bands (lane 15). C, comparison of wild type or mutant GST-SET domain pull-down of WDR5, RbBP5, and ASH2L from MCF-7 cell extracts. WRA components were detected by Western blotting. The top panel shows a Ponceau S-stained PVDF membrane, and the bottom panels show the immunoblots. D, comparison of wild type or Win motif mutant FLAG-tagged full-length SETd1A and SETd1B co-immunoprecipitation with WDR5, RbBP5, ASH2L, CFP1, WDR82, and HCF1 from stably transfected T-REx HEK293 cells.
To determine whether the Win motif is required for direct interaction with WDR5, we first mutated the Win motif arginine to alanine in each SET1 family member and purified each recombinant protein as a GST fusion. We then compared the ability of wild type and mutant SET1 family members to interact with WDR5 using a GST pull-down assay (Fig. 2B). We observed that all SET1 family members pulled down recombinant WDR5 (lanes 1, 3, 5, 7, 9, and 11), whereas the control GST protein lacking a SET1 family member did not (lane 13). Consistent with having the weakest Win motif-WDR5 interaction (49), MLL1 pulled down the least amount of WDR5. In contrast, all SET1 family members containing a mutated Win motif did not pull down WDR5 (lanes 2, 4, 6, 8, 10, and 12). These results suggest that all SET1 family members directly interact with WDR5 in a Win motif-dependent manner.
We then investigated the impact of disruption of Win motif-WDR5 interaction on SET1 family core complex stability. We compared the ability of wild type and Win motif mutant SET1 family GST proteins to pull down endogenous WRAD components from MCF-7 breast cancer cell extracts using GST pull-down assays (Fig. 2C). Although all wild type SET1 family GST-proteins were able to interact with endogenous WRAD components (lanes 1, 3, 5, 7, 9, and 11), we noticed that mutant SET1 family members differed in their ability to interact with WRAD (lanes 2, 4, 6, 8, 10, and 12). Substitution of each Win motif arginine with alanine resulted in disruption of the SET domain-WDR5 interaction in all complexes, consistent with the Win motif playing a role in direct interaction with WDR5. In addition, all complexes, with the exception of the MLL3 core complex, showed reduced interactions with RbBP5 and ASH2L, but to varying degrees. Substitution of the Win motif arginine in the MLL1 and SETd1A constructs nearly abolished core complex assembly, whereas MLL2 and MLL4 showed weak interactions with RbBP5 and ASH2L, respectively. Substitution of the Win motif arginine in SETd1BR1748A pulled down RbBP5 and ASH2L, but to a lesser degree compared with wild type SETd1B (Fig. 2C, lanes 11 and 12). Whereas MLL3R4710A showed a decreased interaction with WDR5, its ability to interact with RbBP5/ASH2L components was similar to that of wild type MLL3 (Fig. 2C, lanes 7 and 8), consistent with previous findings (16, 57, 58). These results demonstrate the importance of the Win motif for the interaction of WDR5 with each SET1 family core complex but also reveal differences in the role that WDR5 plays in complex stability in vitro.
To determine whether the same differences are observed in cells, we selected the closely related SETd1A and SETd1B family members to further probe the role of the WDR5-Win motif interaction in mammalian cells. We stably transfected HEK293 cells with full-length human SETd1A and SETd1B constructs and compared the ability of wild type and Win motif mutant variants to co-immunoprecipitate endogenous WRAD components. Similar to the results of the GST pull-down assays, we found that substitution of the Win motif arginine with alanine abolished the interaction with WDR5 in both complexes (Fig. 2D, compare lanes 3 and 4 and lanes 5 and 6, respectively). In addition, loss of the WDR5-Win motif interaction severely disrupts SETd1A interaction with RbBP5 and ASH2L, whereas the same interactions were only modestly reduced when the SETd1B Win motif arginine was replaced with alanine. Despite these changes, substitution of the Win motif arginine with alanine did not significantly affect the ability of either protein to co-immunoprecipitate with SETd1A/B-interacting proteins CFP1, WDR82, and HCF1. These data confirm the results obtained from the GST pull-down experiments and suggest that amino acid variation between SETd1A and SETd1B proteins accounts for the ability of RbBP5/ASH2L to interact with the SET domain in the absence of WDR5.
Altogether, these results demonstrate that all SET1 family members require the Win motif for interaction with WDR5. Moreover, whereas the Win motif-WDR5 interaction is not required for the interaction between MLL3 and RbBP5/ASH2L, it is required for the stability of the MLL1, MLL2, MLL4, SETd1A, and SETd1B core complexes, but to different degrees.
Contributions of the Win Motif to SET1 Family Histone Methyltransferase Activity
We previously reported that substitution of the MLL1 Win motif arginine with alanine reduces core complex H3K4 dimethylation activity (24), whereas a similar substitution in the MLL3 Win motif increases core complex H3K4 monomethylation activity (16, 58). The contributions of the Win motif to the methyltransferase activity of the other SET1 family core complexes remain to be explored.
To address this knowledge gap, we compared the enzymatic activity of core complexes assembled with wild type or with Win motif mutant SET1 family proteins using histone H3 peptides (residues 1–20) that were unmodified (H3K4me0) or were monomethylated (H3K4me1) or dimethylated (H3K4me2) at lysine 4 as substrates. Reaction mixtures were separated by SDS-PAGE and imaged by fluorography (Fig. 3A). Quantitative measurements were obtained by excising peptide bands from the gels for liquid scintillation counting (Fig. 3B), as described under “Experimental Procedures.” The results upon mutation of each Win motif are highly similar to those of our previously reported assays with wild type complexes in the absence of WDR5 (16). When each complex was incubated with methyl-3H-labeled S-adenosylmethionine ([3H]AdoMet) and the H3K4me0 peptide, we observed that methylation levels were similar between wild type and Win motif variants, with the exception of the mutant MLL3 core complex, which showed ∼2-fold more activity than the wild type MLL3 core complex (Fig. 3, A (lanes 4 and 11) and B (top)). These results suggest that despite the substitution of each Win motif arginine with alanine, the proteins are still folded and enzymatically active.
FIGURE 3.

Substitution of the Win motif arginine alters SET1 core complex-catalyzed H3K4 methyltransferase activity in vitro. A, sample gel showing the comparison of core complex methyltransferase activities among SET1 family members (wild type and mutant) in the presence of WRAD. The top panels show Coomassie Blue-stained SDS-polyacrylamide gels, and the bottom panels show 3H-methyl incorporation after 4 h of exposure as detected by fluorography. The control lane shows the activity of the MLL1WT SET domain with 100 μm H3K4me0 peptide, which is included on each gel. B, quantification of radioactivity from excised histone H3 bands by LSC. Data are normalized to the activity level of the control lane on each gel. Error bars, S.E. of measurement among three independent experiments.
In contrast, when H3K4me1 and H3K4me2 peptides were used as substrates, MLL1 and SETd1A complexes showed significant differences in activity between wild type and Win motif variants. Complexes assembled with MLL1 and SETd1A Win motif variants showed at least a 2-fold reduction in activity compared with their wild type counterparts (Fig. 3, A (compare lanes 1 and 8 and lanes 5 and 12) and B (middle and bottom panels)). Conversely, complexes assembled with MLL4 and SETd1B Win motif variants showed similar amounts of activity compared with that of their wild type counterparts (Fig. 3, A (compare lanes 2 and 9 and lanes 6 and 13) and B (middle)). Interestingly, whereas the wild type MLL2 core complex catalyzes trace amounts of H3K4 dimethylation above background in vitro (Fig. 3, A (lane 3) and B), we observed a 2-fold increase in this activity with the MLL2 Win motif variant (Fig. 3, A (lane 10) and B). These results are similar to the stimulation of the monomethylation activity of MLL3 and MLL2 core complexes in the absence of WDR5 (16). In contrast, both wild type and Win motif mutant MLL3 core complexes showed negligible activity with H3K4me1/2 substrates.
Together, these results demonstrate that the Win motif is required for the full methyltransferase activity of MLL1 and SETd1A complexes, consistent with the central role of WDR5 in their assembly (16). In contrast, despite the moderate destabilization of MLL2, MLL4, and SETd1B core complexes upon mutation of the Win motif, the complexes retain a sufficient amount of interaction with the RbBP5/ASH2L heterodimer to allow nearly full enzymatic activity in the absence of the Win motif-WDR5 interaction under these conditions. These results suggest that targeting the WDR5-Win motif interaction may be a useful strategy for selective inhibition of the MLL1 and SETd1A complexes.
Structure-based Design and Characterization of a New High Affinity Win Motif Peptidomimetic
Our data suggest that SET1 family core complex stability and enzymatic activity are regulated by Win motif-WDR5 interaction to different extents. Thus, molecules designed to disrupt this interface are expected to mainly affect MLL1 and SET1d1A core complexes because they more strongly rely on WDR5 for assembly and function. To test this hypothesis, we designed and characterized a new Win motif peptidomimetic and tested its inhibition properties against all six human SET1 family core complexes. We have previously shown that 14-residue peptides derived from the naturally occurring Win motif sequences of SET1 family members inhibit the dimethylation activity of the MLL1 core complex (49). Structure-function analyses show that each peptide binds to WDR5 in a similar manner but with a wide range of affinities (50–2800 nm) (49) (Table 1). In some peptides with the highest affinity for WDR5, we found an additional hydrogen bond between the fourth residue C-terminal to the Win motif arginine and the conserved Asp-172 in WDR5 that was absent in complexes with lower affinities (49). In addition, we found that valine in the P+3 position (3 residues C-terminal to the crucial arginine residue, denoted as P0) is expected to promote a conformation that places a tyrosine at the P+4 position in an optimal orientation to form this hydrogen bond. Therefore, we synthesized a 6-residue peptidomimetic that was designed to combine the best features of high affinity binding peptides while minimizing its overall size. The sequence contained amino acid residues ARTEVY and was acetylated on the N terminus and amidated on the C terminus to promote stability (Ac-ARTEVY-NH2). This peptidomimetic is referred to here as “Win6mer.” Thermodynamic binding measurements using isothermal titration calorimetry (ITC) revealed that Win6mer binds to WDR5 with a Kd of 2.9 nm (1.7–4.2 nm, 95% confidence interval) (Fig. 4A), which is an ∼18-fold increase in binding affinity over the best naturally occurring Win motif sequence (Table 1).
TABLE 1.
Binding affinities of Win motif-based MLL1 inhibitors toward WDR5 as determined by isothermal titration calorimetry
| Win motif mimetic | Dissociation constant Kd ± S.D. |
|---|---|
| nm | |
| MLL1a (49) | 2762 ± 338 |
| MLL4a (49) | 88 ± 6 |
| MLL2a (49) | 75 ± 5 |
| MLL3a (49) | 54 ± 5 |
| SETd1Aa (49) | 541 ± 46 |
| SETd1Ba (49) | 103 ± 14 |
| Win6mer | 2.9 (1.7–4.2)b |
| WDR5-0103 (53) | 450 ± 0.02 |
| OICR-9429 (50) | 93 ± 28 |
a Peptides derived from the native Win motif sequence of each SET1 family member.
b 95% confidence interval of the determined dissociation constant.
FIGURE 4.

Co-crystal structure of Win6mer and WDR5 at 2.0 Å resolution. A, thermodynamic characterization of the WDR5-Win6mer interaction by ITC. The binding affinity Kd is reported with a confidence interval of 2σ or 95%. B, simulated annealing Fo − Fc omit map contoured at 3σ unambiguously shows electron density corresponding to the Win6mer peptide. C, intra-Win6mer and Win6mer-WDR5 bonding network. Hydrogen bonds are represented by dashed lines. Participating residues are labeled in dark yellow (Win6mer) and purple (WDR5). Position P0 corresponds to the conserved arginine residue. Positions P− and P+ correspond to residues N- and C-terminal of P0, respectively. D, cut-away of a surface rendition of the Win6mer/WDR5 structure. WDR5 is shown in gray, and the Win6mer peptide is shown in yellow. The conserved arginine is inserted into the Win motif binding pocket in WDR5. E, overlay of the MLL1 Win motif peptide (green) (Protein Data Bank code 3EG6 (56)) and Win6mer peptide (yellow). The conserved arginines are oriented in a highly similar manner within the central cavity in WDR5 (magenta). The P+4 residue in both peptides binds the A-pocket (orange dashed circle) and not the B-pocket (blue dashed pocket) (49).
To determine whether Win6mer binds WDR5 in a similar manner to other Win motif peptides, we determined the co-crystal structure of the Win6mer-WDR5 complex at 2.0 Å resolution (Fig. 4, B–E). Data collection and refinement statistics are summarized in Table 2. The overall structure of WDR5 was highly similar to previously reported structures (25, 56, 59–62), consisting of a seven-blade β-propeller with a cavity through the center of the protein (Fig. 4, D and E). This central cavity is denoted as the “Win motif-binding pocket” because previous co-crystal structures of WDR5 with SET1 family Win motif peptides have shown it to be the binding site of the conserved Win motif arginine (25, 49, 56, 62). A simulated annealing Fo − Fc omit map contoured at 3σ unambiguously shows density for the peptide in the co-crystal structure in the Win motif-binding pocket (Fig. 4B), indicating that it does indeed bind in a manner similar to that of other Win motif peptides.
TABLE 2.
X-ray data collection and refinement statistics
| Data collection | |
| Space group | P31 |
| Cell | |
| a, b, c (Å) | 74.9, 74.9, 93.5 |
| α, β, γ (degrees) | 90.0, 90.0, 120.0 |
| X-ray source | MacCHESS F1 |
| Wavelength (Å) | 0.977 |
| Resolution (Å) | 50.0–2.00 (2.07–2.00) |
| Total reflections | 330,393 |
| Unique reflections | 39482 |
| Completeness (%) | 100.0 (100.0) |
| Rsym (%) | 11.7 (56.8) |
| 〈I/σ(I)〉 | 32.2 (5.0) |
| Multiplicity | 8.4 (7.9) |
| Wilson B factor (Å2) | 26.1 |
| Refinement | |
| Resolution (Å) | 37.95–2.00 (2.05–2.00) |
| R factor (%) | 21.4 (30.4) |
| Free R factor (%) | 24.3 (36.3) |
| Free R reflections (%) | 4.9 |
| No. of free R reflections | 1955 |
| Molecules in asymmetric unit | 2 |
| Residue range built | A/31–334, B/32–334, C/1–6, D/1–6 |
| No. of non-hydrogen atoms | 5143 |
| No. of water molecules | 272 |
| Model quality | |
| Root mean square deviations | |
| Bond lengths (Å) | 0.003 |
| Bond angles (degrees) | 0.884 |
| Mean B factors (Å2) | |
| Overall | 31.3 |
| Chain A | 32.9 |
| Chain B | 32.8 |
| Chain C (Win6mer) | 26.6 |
| Chain D (Win6mer) | 26.5 |
| Water | 37.5 |
| Ramachandran plot (%) | |
| Favored | 96.5 |
| Allowed | 3.2 |
| Disallowed | 0.3 |
Win6mer binds to WDR5 in a 310-helical conformation with the conserved arginine (P0) inserted into the Win motif-binding pocket (Fig. 4, B and C). The 310-helical conformation is stabilized by two sets of intrapeptide i to i+3 hydrogen bonds: one between the acetyl-capping group at the N terminus of the peptide and the main and side chain of the P+1 threonine (Fig. 4C) and the other between the main chain of the P−1 alanine and the main chain of the P+2 glutamate. The P−1 alanine amino group also hydrogen-bonds with Asp-107 of WDR5, whereas the main chain of the P+2 glutamate hydrogen-bonds with the main chain of the P0 arginine (Fig. 4C). The side chain of the P0 arginine showed extensive hydrogen bonds within the Win motif-binding pocket of WDR5. Furthermore, the position of the P0 arginine guanidinium is sandwiched between two conserved phenylalanines (Phe-133 and -263) in WDR5 and is probably stabilized by cation-π interactions. This feature is nearly identical to all previously published Win motif peptide-WDR5 structures (25, 49, 56, 62). The P+3 valine side chain is solvent-exposed and orients the P+4 tyrosine side chain in a region in WDR5 known as the A pocket (Fig. 4E) (49). The A pocket in WDR5 contains residues Tyr-191, Pro-173, Phe-149, and Asp-172. As predicted, the P+4 tyrosine hydrogen bonds with the side chain of Asp-172 from WDR5 (Fig. 4E). Overall, the structure reveals that all 6 residues in Win6mer play important roles in binding WDR5, which probably explains its improved binding affinity for WDR5 when compared with the previous SET1 family Win motif peptides.
Win6mer Alters the Methyltransferase Activities of a Subset of SET1 Family Core Complexes
Given that different SET1 family members differ in their requirement for the Win motif-WDR5 interaction for complex assembly and function, we hypothesized that treatment of the SET1 family of core complexes with Win6mer would result in methyltransferase activity patterns that mimic loss of WDR5 or mutation of the Win motif (Fig. 3). To test this hypothesis, we compared the methyltransferase activity of SET1 core complexes that were treated with increasing concentrations of Win6mer with that of untreated complexes using radiometric assays. When H3K4me0 was the substrate, we observed that Win6mer reduced the activity of the MLL1 and SETd1A core complexes in a dose-dependent manner (Fig. 5A), whereas complexes assembled with MLL2–4 and SETd1B were uninhibited. The Win6mer peptide shows similar inhibition efficiency for MLL1 and SETd1A core complexes, with average IC50 values of 2.2 and 2.5 μm, respectively (2 μm enzyme complex assayed) (Table 3). Similar inhibition patterns were observed when the H3K4me1 and H3K4me2 peptides were used as substrates (Fig. 5, B and C, respectively). Together, these results show that MLL1 and SETd1A core complexes are specifically inhibited by the Win6mer peptidomimetic that targets the Win motif-WDR5 interaction.
FIGURE 5.

Inhibition of SET1 family core complex activity by Win6mer. The activity of SET1 family core complexes upon titration of Win6mer was assessed via scintillation proximity assay. SET1 family core complexes were assayed according to their substrate specificity as follows (16). A, all SET1 core complexes were assayed for monomethylation (H3K4me0 substrate). B, MLL1, MLL4, SETd1A, and SETd1B core complexes were assayed for dimethylation (H3K4me1 substrate). C, SETd1A and SETd1B core complexes were assayed for trimethylation (H3K4me2 substrate). IC50 values are reported on Table 3. D, efficiency of MLL1 core complex inhibition by Win6mer is dependent on enzyme concentration. IC50 values are shown in the inset. Activity data for each SET1 family member were normalized to the activity of uninhibited core complex. Data were fit to a dose response with variable slope equation (Equation 1). Shown is monomethyltransferase activity of MLL1-RAD (E) and SETd1A-RAD (F) upon titration of WDR5. Activity data were normalized to the activity of MLL1-RAD or SETd1A-RAD in the absence of WDR5. Data were fit to a dose response with variable slope equation (Equation 1). Error bars, S.E. of measurement between two independent experiments.
TABLE 3.
Win6mer inhibition efficiency of SET1 family core complexes
| Core complex | Inhibition efficiency, IC50 (95% confidence interval) |
||
|---|---|---|---|
| H3K4me0 | H3K4me1 | H3K4me2 | |
| μm | |||
| MLL1 | 2.3 (1.6–3.3) | 2.2 (1.7–2.8) | NAa |
| MLL4 | —b | — | NA |
| MLL2 | — | NA | NA |
| MLL3 | — | NA | NA |
| SETd1A | 3.2 (2.0–5.2) | 2.9 (1.7–5.1) | 2.4 (1.4–4.1) |
| SETd1B | — | — | — |
a NA, not applicable due to substrate specificity (16).
b —, not inhibited at tested Win6mer concentrations.
These results raise the question of why MLL1 and SETd1A complexes are specifically targeted over complexes assembled with MLL2–4 and SETd1B. We hypothesize that two key variables account for the differences: 1) affinity of the Win motif for WDR5 and 2) affinity of the RbBP5/ASH2L heterodimer for the SET domain in the absence of WDR5, which influences overall complex stability. We previously found that Win motif peptides derived from the human MLL1 and SETd1A sequences bind WDR5 with significantly weaker affinity (Kd = 2.8 and 0.5 μm, respectively) compared with that of peptides derived from human MLL2–4 and SETd1B sequences (Kd = 0.05–0.1 μm) (Table 1) (49). These data suggest that the Win motif-WDR5 interaction is more easily disrupted in MLL1 and SETd1A complexes compared with the other complexes. Consistent with the second hypothesis, we previously found that complexes assembled with MLL1 and SETd1A rely more heavily on WDR5 for interaction with the RbBP5/ASH2L heterodimer than the other complexes for enzymatic activity (16). Indeed, we found in this investigation that titration of WDR5 into the MLL1-RAD and SETd1A-RAD complexes showed that stoichiometric amounts of WDR5 are required for full activity (Fig. 5, E and F). Together, these results suggest that WDR5 plays a more crucial role in overall stability of the MLL1 and SETd1A core complexes and are therefore more susceptible to inhibition by molecules that target the WDR5-Win motif interaction. Last, our data suggest that the MLL1 and SETd1A complexes have lower overall stabilities compared with other complexes. Consistent with this hypothesis, we observed that the IC50 values for the Win6mer were highly dependent on enzyme complex concentration. We found that Win6mer inhibited MLL1 core complex with IC50 values of 2.2, 0.8, and 0.3 μm when 1.8, 1, and 0.5 μm of enzyme complex were assayed, respectively (Fig. 5D). Lower concentrations of complex shift the equilibrium toward the unassembled complex, making it easier to gain access to the Win motif-binding pocket on WDR5. Overall, these results suggest that Win6mer preferentially inhibits the methylation activities of MLL1 and SETd1A core complexes due to lower overall complex stability and easier access to the Win motif-binding pocket on WDR5.
The Win6mer Peptide Disrupts MLL1 and SETd1A Core Complex Assembly but Does Not Inhibit Isolated SET Domain Activity
Activity assays show that Win6mer selectively down-regulates the methyltransferase activity of MLL1 and SETd1A core complexes. This inhibition is most likely due to disruption of the Win motif-WDR5 interaction and destabilization of complex assembly. However, the amino acid sequences of SET1 family Win motifs and Win6mer peptides are somewhat similar to that of the histone H3 N-terminal tail (24), a SET domain substrate. Thus, it is also possible that the Win6mer may inhibit core complex activity by binding to the SET domain active site. To distinguish between these potential mechanisms of core complex inhibition, we assessed the effects of Win6mer treatment on isolated SET domain activity and on SET1 family core complex assembly.
First, we tested whether Win6mer inhibits the isolated SET1 family catalytic domains. For this purpose, we treated isolated SET domains with 100 μm Win6mer and assessed their enzymatic activity toward H3 via a radiometric assay. We found that Win6mer treatment did not affect the intrinsic monomethyltransferase activity of SET1 family members (Fig. 6, A and B). This suggests that Win6mer does not bind to the SET domain and does not interfere with SET domain-catalyzed H3K4 methylation.
FIGURE 6.

The Win6mer peptide interferes with core complex assembly for a subset of SET1 family members. A, representative gel of a comparison of monomethyltransferase activities among isolated SET1 family SET domains in the absence of WRAD treated with (100 μm) or without Win6mer. The top panel shows the Coomassie Blue-stained SDS-polyacrylamide gel, and the bottom panel shows 3H-methyl incorporation into H3K4 peptide after 4 h of exposure, as detected by fluorography. The control lane shows the activity of the MLL1WT SET domain on 100 μm H3K4me0 peptide. B, quantification of radioactivity from excised histone H3 peptide bands by LSC. Data are normalized to the activity level of the control lane on each gel. Error bars, S.E. of measurement among three independent experiments. C, comparison of core complex assembly by SET1 family members from pull-down experiments from MCF-7 cell extracts in the presence (+) (10 μm) or absence (−) of Win6mer. Individual GST-tagged SET domains were incubated with cell extracts and pulled down with glutathione-agarose beads. WRA components were detected by Western blotting. The top panel shows a Ponceau S-stained PVDF membrane, and the bottom panels show the immunoblots. GST (not treated with Win6mer) was used as a negative control (lane 14).
Next, we tested whether Win6mer inhibits core complex activity via disruption or destabilization of the core complex assembly. We compared the ability of SET1 family members to interact with endogenous WRAD components from MCF-7 cell extracts in the presence and absence of Win6mer by GST pull-down experiments. Treatment with Win6mer greatly reduced the ability of all six SET1 family members to interact with endogenous WDR5 compared with untreated samples (Fig. 6C). However, we found that Win6mer treatment resulted in a nearly total loss of RbBP5/ASH2L interaction only with MLL1 and SETd1A complexes (Fig. 6C, compare lanes 1 and 2 and lanes 9 and 10). Other SET1 family members (MLL2, MLL3, MLL4, and SETd1B) showed only a modest reduction in RbBP5/ASH2L binding upon Win6mer treatment when compared with the untreated set (Fig. 6C, lanes 3–8, 11, and 12). These results are largely consistent with those observed upon mutation of each SET1 family Win motif (Fig. 2C) and confirm that MLL1 and SETd1A core complexes are more sensitive to complex destabilization by inhibition of the WDR5-Win motif interaction. These results also reveal that the mechanism of inhibition by Win6mer is due to MLL1 and SETd1A core complex destabilization and not competition with histone H3 for binding to the SET domain.
Discussion
WDR5 was first associated with accelerating osteogenic differentiation (63) before its identification as a member of the SET1 family core complexes (8, 64). WDR5 is required to maintain H3K4 methylation levels in yeast and in vertebrates (10, 18, 65–68), but the molecular mechanisms involved are debatable. Previous studies implicate WDR5 in bridging interactions between SET domain and the RbBP5/ASH2L heterodimer within the MLL1 core complex (18, 24), but other studies suggest that it has additional roles in gene targeting. For example, WDR5 has been shown to be preferentially pulled down by H3K4me2 peptides, leading to the hypothesis that it acts as a histone effector domain that recruits the MLL1 core complex to chromatin to catalyze H3K4 trimethylation (66). However, crystal structures of WDR5 bound to H3K4 peptides reveal that it specifically recognizes Arg-2 instead of Lys-4 of histone H3 (59–61, 69), leading to the hypothesis that WDR5 functions as a presenter domain that positions lysine 4 for methylation by the SET domain active site (60). Missing is an unambiguous demonstration that the MLL1 core complex can be pulled down in a manner that depends on the Arg-2 binding site of WDR5. In an effort to better understand the role of WDR5, we and others have found that WDR5 recognizes Arg-3765 of the MLL1 Win motif using the same site that it uses for binding H3R2 (24, 25, 56), indicating that WDR5 cannot simultaneously interact with MLL1 and histone H3 when it is assembled into the MLL1 core complex. These results argue against the effector or presenter domain hypotheses and suggest that WDR5 functions mainly as a bridge for stabilizing the interaction between MLL1 and the RbBP5/ASH2L heterodimer. Because the Win motif is highly conserved (24), it has been hypothesized that WDR5 plays a similar role in all six human SET1 family complexes.
In this investigation, we show that all six human SET1 family members indeed require the Win motif for interaction with WDR5. However, we also observed differences in the role of WDR5 in stabilizing complex assembly and enzymatic activity, suggesting that the Win motif-WDR5 interaction may be conserved for additional functions. In particular, mutation of the Win motif severely disrupts assembly and enzymatic activity of MLL1 and SETd1A complexes but only modestly disrupts assembly of MLL2/4 and SETd1B complexes, without significant changes in their activity. In contrast, disruption of the Win motif-WDR5 interface does not affect MLL3 core complex assembly but enhances its enzymatic activity, consistent with previous work (16, 58). These results raise the question of why the Win motif is highly conserved among metazoan SET1 family members. Our results suggest that WDR5 plays, to varying degrees, a role in complex stabilization for most SET1 family members. Alternatively, it is possible that the Win motif-WDR5 interaction has been retained throughout evolution for additional roles, such as for interaction with other proteins or in gene targeting. Indeed, WDR5 interacts with several transcription factors (e.g. OCT4 and MYC) (70, 71), with long non-coding RNAs (e.g. HOTTIP) (72) and PIWI-interacting RNAs (e.g. GAS5) (73), and has been implicated in recruiting the MLL1 core complex to specific genomic loci.
Our results also reveal differences in SET1 family SET domains that allow varying degrees of interaction with RbBP5/ASH2L in the absence of WDR5. MLL1 and SETd1A both require WDR5 for complex assembly but come from separate phylogenetic subclades (16) within the SET1 family, each containing members that do not rely as much on WDR5 for interaction with RbBP5/ASH2L. This observation suggests that the requirement of WDR5 for complex assembly may be a convergent property within each subclade, which raises the possibility that different mechanisms may be involved. For example, it was recently shown that substituting MLL1 residues at both positions Asn-3861 and Gln-3867 with the corresponding amino acids in the MLL2 and MLL3 SET domains results in increased interaction with an RbBP5 peptide/ASH2L SPRY domain heterodimer and increased core complex activity in the absence of WDR5 (57). However, variation in the same amino acid positions does not explain the differences in the requirement for WDR5 in the assembly of the human SETd1A and SETd1B complexes, because the amino acids are highly similar in those positions. These results suggest that variation in other amino acid positions accounts for the differences in the requirement for WDR5 in human SETd1A and SETd1B complexes. Although it was previously suggested that a 4-residue basic patch in yeast Set1 and human SETd1A SET domains stabilizes interaction with RbBP5/ASH2L in the absence of WDR5 (74), this basic patch is conserved in both SETd1A and SETd1B, but not in MLL1–4, and therefore cannot account for the differences. Likewise, we previously reported the identification of a common cluster of Kabuki syndrome missense variants that disrupt interaction between the SET domain and RbBP5/ASH2L in a WDR5-independent manner (26). However, the amino acids in this Kabuki interaction surface are conserved in all SET1 family SET domains and therefore cannot explain the variation in complex stability among family members. Identification of the amino acid residues that do account for those differences in future studies may allow development of inhibitors that more specifically target unique protein-protein interactions in the MLL1 or SETd1A core complexes.
Our observation that MLL1 and SETd1A require WDR5 for enzymatic activity and complex assembly contrasts with previous reports suggesting that only MLL1 requires WDR5 for these purposes (42, 57). A possible reason for this discrepancy is that in previous studies, enzymatic activity was measured with only the H3K4me0 substrate using an assay that does not distinguish among different methylation states. Indeed, we observed little differences in activity among family members with Win motif mutants in assays using the H3K4me0 substrate with a relatively long incubation period (Fig. 3). Assays conducted with shorter incubation periods within the linear range do show that both MLL1 and SETd1A, but not MLL2–4 and SETd1B core complexes, are sensitive to Win6mer inhibition when H3K4me0 is used as a substrate (Fig. 5A). However, rates of H3K4 di- and trimethylation are most affected by disruption of the Win motif-WDR5 interaction in the human MLL1 and SETd1A complexes, respectively, because both show significantly reduced activity upon mutation of the Win motif or Win6mer treatment under both assay conditions (Figs. 3 and 5 (B and C)). The results emphasize the importance of examining the impact of SET1 family inhibitors on each H3K4 methylation state.
In addition, a recent report suggests that all SET1 family members, with the exception of MLL1, interact with RbBP5/ASH2L in the absence of WDR5 (42, 57). This contrasts with the results of this study, which show that all human SET1 family core complexes, with the exception of the MLL3 core complex, are destabilized to varying degrees in the absence of WDR5. This discrepancy is probably due to differences in the stringency in binding and wash conditions in GST pull-down experiments. Indeed, it was previously shown that both MLL1 and SETd1A, but not MLL2–4 and SETd1B, lose interaction with RbBP5/ASH2L when higher ionic strength wash conditions (300 mm NaCl) are used (57) as in this study.
Although a previous report suggests that targeting the Win motif-WDR5 interaction inhibits only the MLL1 core complex (42), several independent lines of evidence suggest that both MLL1 and SETd1A complexes are inhibited. A peptidomimetic called MM-401 was shown to cause G1/S cell cycle arrest, reduce H3K4 trimethylation levels, and alter gene expression in a manner that resembles in part the phenotype of the MLL1 knock-out (42). Although the effect of MM-401 on the H3K4 methylation activity of the human SETd1A complex was unclear in that study (42), a more recent investigation showed that a similar phenotype is observed in SETd1A knock-out mouse embryonic stem cells (75). In addition, we have previously shown that the MLL1 core complex catalyzes predominantly H3K4 mono- and dimethylation, whereas the SETd1A core complex catalyzes H3K4 mono-, di-, and trimethylation in in vitro assays, posing the possibility that inhibition of SETd1A accounts for loss of H3K4 trimethylation in response to MM-401. This is consistent with reports suggesting that SETd1A and SETd1B account for the bulk of H3K4 trimethylation in cells (76, 77). However, because both complexes contribute to the cellular pool of H3K4 dimethylation, the substrate for the trimethylation reaction, it is possible that inhibition of both enzyme complexes contributes to the overall loss of H3K4 trimethylation in cells.
Despite conservation of the Win motif, WDR5 recognizes SET1 family Win motifs with significantly different affinities (49), suggesting a therapeutic window in which to selectively target individual family members. In this work, we utilized a new Win motif peptidomimetic (Win6mer) that exploits a unique set of hydrogen bonds that significantly increases affinity for WDR5 compared with other inhibitors. Indeed, Win6mer has the highest reported affinity for the Win motif-binding site on WDR5 when comparing dissociation constants among inhibitors obtained by a direct binding ITC assay (Table 1). Although it is unclear whether Win6mer can cross cell membranes, it has proven to be a useful tool in dissecting mechanistic differences in the assembly of SET1 family core complexes and should serve as the basis for the development of the next round of Win motif inhibitors with improved specificity and pharmacodynamic properties.
We propose that the basis of Win6mer selectivity for MLL1 and SETd1A core complexes lies in the disruption of the Win motif-WDR5 interface, which is required for stabilizing contacts with the RbBP5/ASH2L heterodimer (Fig. 7). MLL1 and SETd1A share the properties that they have the lowest binding affinities for WDR5 (49) and for the RbBP5/ASH2L heterodimer, the combination of which makes them susceptible to inhibition of the Win motif-WDR5 interface. Uncoupling this combination, through amino acid variation that increases affinity either for WDR5 or for the RbBP5/ASH2L heterodimer, renders those complexes less susceptible to inhibition. This explains why the MLL2–4 and SETd1B can retain catalytic activity in the presence of the Win6mer, because they have higher binding affinities for WDR5 or they retain to varying degrees the ability to interact with RbBP5/ASH2L. It is likely that the same mechanism accounts for the inhibition properties of other molecules that target the Win motif-WDR5 interaction.
FIGURE 7.

Model for down-regulation of MLL1 and SETd1A core complex activity by Win6mer. A, MLL1 and SETd1A SET domains require interaction with WDR5 for stabilizing assembly with the components of RAD. Like all SET1 family members, MLL1 and SETd1A utilize the Win motif to interact with WDR5. Stably assembled MLL1 and SETd1A core complexes exhibit full H3K4 methyltransferase activity. B, treatment with Win6mer, a Win motif peptidomimetic, competes with MLL1 and SETd1A SET domains for WDR5 binding, thus destabilizing core complex assembly. This, in turn, leads to down-regulation of MLL1 and SETd1A core complex activity.
Experimental Procedures
Materials
WDR5 antibody was obtained from Abcam (ab22512). RbBP5 and ASH2L antibodies were obtained from Bethyl (A300-109A and A300-498A, respectively). An HRP-conjugated donkey anti-rabbit antibody was obtained from GE Healthcare. Anti-FLAG M2 agarose beads and anti-FLAG (mouse monoclonal M2) antibody were obtained from Sigma. Custom antisera directed against WDR5, CFP1, and WDR82 were prepared as described (1). HCF1 antiserum was a generous gift from Dr. Winship Herr.
Histone H3 peptides were synthesized by GenScript and contained residues 1–20 followed by GGK-biotin and were either unmodified (H3K4me0), monomethylated (H3K4me1), or dimethylated (H3K4me2) at H3K4. All peptides were purified to >95% purity. All H3K4 peptides were modified by amidation of the C terminus. The 6-mer Win motif peptidomimetic (Win6mer), of sequence Ac-ARTEVY-NH2, was synthesized by GenScript. Win6mer was acetylated on the N terminus and amidated on the C terminus to remove charge and improve peptide stability. MCF-7 cell extracts were obtained from Santa Cruz Biotechnology, Inc. (sc-24793).
Protein Expression/Purification
Human SET1 family Win-SET cDNAs encoding residues MLL1(3745–3969) (UniProtKB ID Q03164), MLL2(5319–5537) (UniProtKB ID O14686), MLL3(4689–4911) (UniProtKB ID Q8NEZ4), MLL4(2490–2715) (UniProtKB ID Q9UMN6), SETd1A(1474–1708) (UniProtKB ID O15047), and SETd1B(1727–1966) (UniProtKB ID Q9UPS6) were subcloned into pGST parallel expression vectors (78) and individually expressed in Escherichia coli (Rosetta II (DE3) pLysS; Novagen) and purified as described previously (16). Briefly, transformed E. coli were grown at 37 °C, shaking at 200–220 rpm for ∼2.5 h until the A600 reached 0.75. Protein expression was induced with 1 mm isopropyl β-d-1-thiogalactopyranoside for 26 h at 16 °C, shaking at 200–220 rpm. Cell pellets were resuspended in lysis buffer consisting of Buffer 1 (50 mm Tris (pH 7.5), 300 mm NaCl, 10% glycerol, 3 mm DTT, 1 μm ZnCl2), supplemented with cOmpleteTM protease inhibitor tablets (Roche Applied Science) and 0.1 mm PMSF. Cells were mechanically lysed using a microfluidizer. Proteins were purified from cleared lysates using a GSTrap-FF column (GE Healthcare) and eluted over a gradient of 0–10 mm reduced glutathione. Pooled fractions were dialyzed with three changes into Buffer 1. Full-length WRAD constructs in pHis (78) parallel expression vectors were individually expressed in E. coli (Rosetta II (DE3) pLysS; Novagen) and purified as described previously (24). WRAD components were further purified and buffer-exchanged by gel filtration chromatography (Superdex 200 HiLoad 16/60, GE Healthcare) pre-equilibrated with 20 mm Tris (pH 7.5), 300 mm NaCl, 1 mm tris(2-carboxyethyl)phosphine, and 1 μm ZnCl2 (Buffer 2). SET1 family mutants were prepared by site-directed mutagenesis (QuikChange II XL, Agilent). DNA sequencing was performed to verify that only the intended mutation was introduced. Expression and purification of SET1 family mutants was carried out as described above.
Methyltransferase Assays
MLL Core Complex Win Motif Mutants
Histone H3 methyltransferase assays were performed by incubating GST-tagged SET domains (wild type or Win motif mutants) with a stoichiometric amount of WRAD (3 μm final), 1 μCi of [3H]AdoMet (PerkinElmer Life Sciences), and 100 μm histone H3 peptides that were unmodified or previously mono- or dimethylated at H3K4 in Assay Buffer (50 mm Tris, pH 8.5, 200 mm NaCl, 5 mm MgCl2, 5% glycerol) at 15 °C for 6 h. 15 °C was chosen as the incubation temperature due to SET domain instability at higher temperatures.4 Reactions were quenched with SDS-loading buffer and separated by SDS-PAGE using a 4–12% BisTris gel (Life Technologies) run at 200 V for 30 min. The gels were enhanced at room temperature for 30 min (Enlightning, PerkinElmer Life Sciences) and then dried for 2.5 h at 72 °C under constant vacuum. The dried gels were exposed to film (Eastman Kodak Co. Biomax MS Film) at −80 °C for 4–24 h before developing. Liquid scintillation counting (LSC) was performed by excising gel bands corresponding to histone H3 peptides, which were dissolved in 750 μl of Solvable (PerkinElmer Life Sciences), incubated at room temperature for 30 min, followed by incubation at 50 °C for 3 h. The solubilized volume of each sample was transferred to liquid scintillation vials containing 10 ml of Ultima Gold XL liquid scintillation mixture (PerkinElmer Life Sciences). Samples were dark-adapted for 1 h and then counted for 5 min each with a 2σ error cut-off using an all purpose scintillation counter (Beckman Coulter).
MLL Core Complex Inhibition by Win6mer
Assay Conditions
MLL core complexes were reconstituted by mixing purified GST-SET domains (MLL1, MLL4, MLL2, MLL3, SETd1A, and SETd1B) with WDR5, RbBP5, ASH2L, and DPY-30 in stoichiometric amounts (GST-SET/W/R/A/D, 1:1:1:1:2). The activity of each core complex was assessed under the following conditions: 2 μm SET1 core complex, 80 μm biotinylated H3 substrate peptide, 0.68 μm [3H]AdoMet (0.5 μCi) at 15 °C in Assay Buffer, in a total volume of 10 μl. Core complex- or H3 peptide-only reactions incubated with [3H]AdoMet served as background controls. Reactions were quenched with 167 mm EDTA. Each sample was diluted in 50 μl of Buffer 2 containing 0.2 mg/ml BSA and then transferred to 96-well streptavidin-coated FlashPlate® microplates (PerkinElmer Life Sciences). Samples were incubated overnight at 4 °C to allow binding of the biotinylated H3 peptide to the streptavidin-coated surface before scintillation counting in a Hidex Sense microplate reader (LabLogic).
Determination of Linear Ranges
The activity of MLL core complexes toward H3K4me0, H3K4me1, or H3K4me2 substrate peptides was assayed as described above. Reactions were quenched at varying time points with 167 mm EDTA and prepared for scintillation counting on Streptavidin FlashPlates. A plot of counts/min versus time was constructed, from which the linear range was determined. Single time points within the linear range were selected for each SET1 core complex for performing inhibition studies: for H3K4me0: MLL1, MLL2*, SETd1A, and SETd1B*, 5 min; MLL4, 3 min; MLL3, 15 min; for H3K4me1: MLL1, 5 min; MLL4, 3 min; SETd1A, 15 min; SETd1B, 45 min; for H3K4me2: SETd1A, 2 h; SETd1B, 1 h (where an asterisk indicates that 1 μm of MLL2 and SETd1B core complex were used for the H3K4me0 reactions).
Dose-Response Curves
Inhibition studies were performed in order to determine the efficiency of Win6mer as an MLL core complex inhibitor. The activity of each MLL core complex toward H3 substrate peptides was assessed (as described above) with increasing doses of Win6mer (0, 0.005, 0.05, 0.10, 0.5, 1.0, 2.5, 5.0, 10.0, and 50 μm). Reactions were quenched with 167 mm EDTA at the determined time points and prepared for scintillation counting on streptavidin-coated FlashPlates as described above.
The activity of SET1 core complexes at each Win6mer concentration point was normalized to the activity of the uninhibited core complex to obtain the relative methyltransferase activity. Inhibition data were plotted as relative methyltransferase activity versus log[Win6mer] (nm). The Win6mer IC50 values for MLL1 and SETd1A core complexes were determined by fitting the data to a dose response with variable slope equation in SigmaPlot 11 as follows.
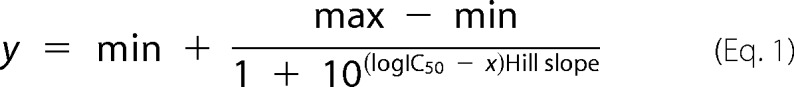 |
WDR5 Titration into MLL1- and SETd1A-RAD
GST-MLL1 and GST-SETd1A were mixed with RbBP5, ASH2L, and DPY-30 in stoichiometric amounts (SET/R/A/D, 1:1:1:2). WDR5 was then titrated into the GST-SET-RAD mixtures at the following final concentrations: 0.1, 0.5, 0.75, 1.0, 1.5, 2.0, and 5.0 μm. Methyltransferase activity toward H3K4me0 substrate was assessed as described under “Assay Conditions.”
The activity of MLL1- and SETd1A-RAD at each WDR5 concentration point was normalized to the activity of the MLL1- and SETd1A-RAD without WDR5 to obtain the relative methyltransferase activity. Data points were plotted as relative methyltransferase activity versus log[WDR5] (nm).
MLL SET Domains Treated with Win6mer
Isolated SET1 family SET domains (5 μm) were incubated with 1 μCi of [3H]AdoMet (PerkinElmer Life Sciences) and 100 μm H3K4me0, H3K4me1, or H3K4me2 peptides, with and without 100 μm Win6mer, in Assay Buffer at 15 °C for 8 h. Reactions were quenched with SDS-loading buffer. Fluorography and LSC were carried out as described above.
GST Pull-downs and Immunoblots
GST tagged SET1 family proteins were preincubated with a stoichiometric amount of purified WDR5 (3 μm) for 1 h at 4 °C before being added to prewashed agarose beads coated with glutathione (Thermo Fisher) and incubated for an additional 2 h at 4 °C with rotation. The beads were washed three times with Buffer 2 supplemented with 0.05% Triton X-100 and 0.05% sodium deoxycholate. The complexes were eluted from beads by boiling the samples at 95 °C in SDS-loading buffer for 10 min. Samples of the supernatant were run on a 4–12% BisTris gel (Life Technologies) and either stained with Coomassie Brilliant Blue or transferred to a PVDF membrane (Life Technologies) for 1 h at 30 V.
For pull-down assays using cell extracts, a 3 μm concentration of each GST-tagged SET domain was incubated with 100 μg of MCF-7 cell extract for 16 h at 4 °C. Following the initial incubation, 20 μl of a 50:50 slurry of glutathione-agarose beads was added to each sample and incubated for an additional 2 h at 4 °C. Samples were washed three times in radioimmunoprecipitation buffer. Samples were eluted from beads by boiling the samples at 95 °C in SDS-loading buffer for 10 min. Samples of the supernatant were separated on a 4–12% BisTris gel and transferred to a PVDF membrane at 30 V for 1 h. PVDF membranes were blocked for 1 h with a 5% nonfat milk solution and then incubated with primary antibody (1:2000) for 1 h at room temperature. Blots were washed four times and then incubated with an HRP-conjugated anti-rabbit secondary antibody (1:5000) for 1 h at room temperature. Blots were washed an additional four times and then visualized by chemiluminescence (Clarity Western, Bio-Rad) on a Bio-Rad ChemiDoc MP Imager using the chemiluminescence setting.
Co-immunoprecipitation from Mammalian Cells
Inducible human embryonic kidney (T-REx HEK293) cells were transfected with pcDNA5/TO-FLAG-tagged SETd1A or pcDNA5/TO-FLAG-tagged SETd1B constructs expressing either the wild type or Win motif mutants, and stably transfected cells were selected with hygromycin B, as described previously (1). Following induction with doxycycline, nuclear extracts were prepared as described (1) and incubated with anti-FLAG M2 agarose beads (Sigma) for 3 h. Bound proteins were eluted with SDS sample buffer after extensive washing and analyzed by Western blotting.
ITC
Purified full-length WDR5 and Win6mer peptide were extensively dialyzed in separate Micro Float-A-Lyzer® dialysis devices, molecular mass cut-off 500–1000 Da (Spectrum Labs) against sample buffer consisting of 20 mm Tris, pH 7.5, 300 mm NaCl, 1 mm tris(2-carboxyethyl)phosphine, 1 μm ZnCl2 buffer. ITC experiments were carried out at 20 °C using a VP-ITC microcalorimeter (MicroCal). After an initial delay of 120 s, Win6mer (200 μm) was titrated into the experimental cell containing full-length WDR5 (20 μm) over the course of 45 injections, 5 μl each. Reference power was set to 10 μcal/s, and stirring speed was set to 295 rpm. Data were integrated using NITPIC (79). SEDPHAT was used to fit the integrated data to a single-site heterogeneous association model, using simulated annealing and Maxquardt-Levenberg algorithms (80). An automatic confidence interval search with projection method was applied to estimate the error of the determined thermodynamic parameters. The confidence interval was set to 2σ (95%). The ITC figure was generated using GUSSI (81).
Crystallization and Structure Determination
A WDR5ΔN construct (residues 23–334) was purified as described previously (49). The final preparation buffer contained 20 mm Tris (pH 7.5), 150 mm NaCl, 1 mm tris(2-carboxyethyl)phosphine, and 1 μm ZnCl2. Crystals of the WDR5ΔN-Win6mer binary complex were obtained by hanging drop vapor diffusion. Before crystallization, a 45 mg/ml stock solution of WDR5ΔN was mixed with a 10 mm stock solution of Win6mer dissolved in water. The final concentrations of WDR5ΔN and Win6mer were 350 and 700 μm, respectively. The initial crystals were obtained from screening with the JCSG Core I Suite in a condition consisting of 0.1 m sodium citrate, pH 5.6, 20% (v/v) isopropyl alcohol, and 20% (w/v) PEG 4000. Crystals were reproduced by manual screening of initial conditions and were observed in a condition containing 0.1 m sodium citrate, pH 5.6, 15% (v/v) isopropyl alcohol, and 17% (w/v) PEG 4000. Hampton Research Additive Screen was used for further optimization. The final crystallization condition contained 0.1 m sodium citrate, pH 5.6, 15% 2-propanol, 17% PEG 4000, and 10 mm ATP. The crystals were flash-frozen in the final mother liquor containing 25% ethylene glycol as a cryoprotectant.
X-ray diffraction data were collected on the F1 beamline at the Cornell High Energy Synchrotron Source (Ithaca, NY) using an ADSC Quantum 270 CCD detector. The data set was indexed, reduced, and scaled with HKL-2000 (82). Data were originally scaled to a resolution limit of 1.9 Å and then rescaled to 2.0 Å as the resolution limit cut-off was set to 2.0 Å during refinement. Data collection statistics of the rescaled data set are reported in Table 2. Initial phases were obtained by molecular replacement with Phaser (83) using the coordinates from the WDR5 apo-structure (Protein Data Bank code 2H14) (59) as the search model. After an initial rigid body refinement, autobuilding was performed using ARP/WARP (84). Standard structural modeling and refinement were performed with Coot (85) and PHENIX (86), respectively. Refinement statistics are summarized in Table 2. Validation of the model quality was assessed with MolProbity (87). All images were made using CCP4mg (88). Crystallographic software was accessed through SBGrid (89).
Author Contributions
N. L. A.-V., S. A. S., J.-H. L., D. G. S., and M. S. C. designed the experiments; N. L. A.-V., S. A. S., D. M. L., and J.-H. L. performed the experiments and together with D. G. S. and M. S. C. interpreted the results. N. L. A.-V., S. A. S., and M. S. C. prepared the manuscript.
Acknowledgments
We thank the staff at the Macromolecular Diffraction Facility at the Cornell High Energy Synchrotron Source (MacCHESS) facilities for assistance in training and x-ray data collection. We are thankful to Dr. Stephan Wilkens and Nicholas J. Stam at SUNY Upstate for providing access to and assistance with the VP-ITC instrument. We thank Vicki Lyle at the core DNA sequencing facility at SUNY Upstate for plasmid sequencing services. We are grateful to Dr. Winship Herr for providing HCF1 antiserum. We also thank Dr. Steven Hanes for helpful comments on the manuscript.
This work was supported, in whole or in part, by the National Institutes of Health Grant R01CA140522 (to M. S. C.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The atomic coordinates and structure factors (code 5SXM) have been deposited in the Protein Data Bank (http://wwpdb.org/).
Nilda L. Alicea-Velázquez, S. A. Shinsky, D. M. Loh, J.-H. Lee, D. G. Skalnik, and M. S. Cosgrove, unpublished observation.
- H3K4
- histone H3 lysine 4
- H3K4me0
- unmethylated H3K4
- H3K4me1
- H3K4me2, and H3K4me3, H3K4 mono-, di-, and trimethylation, respectively
- AML
- acute myeloid leukemia
- ITC
- isothermal titration calorimetry
- LSC
- liquid scintillation counting
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol.
References
- 1. Lee J. H., Tate C. M., You J. S., and Skalnik D. G. (2007) Identification and characterization of the human Set1B histone H3-Lys4 methyltransferase complex. J. Biol. Chem. 282, 13419–13428 [DOI] [PubMed] [Google Scholar]
- 2. Lee J. H., and Skalnik D. G. (2005) CpG-binding protein (CXXC finger protein 1) is a component of the mammalian Set1 histone H3-Lys4 methyltransferase complex, the analogue of the yeast Set1/COMPASS complex. J. Biol. Chem. 280, 41725–41731 [DOI] [PubMed] [Google Scholar]
- 3. Ruault M., Brun M. E., Ventura M., Roizès G., and De Sario A. (2002) MLL3, a new human member of the TRX/MLL gene family, maps to 7q36, a chromosome region frequently deleted in myeloid leukaemia. Gene 284, 73–81 [DOI] [PubMed] [Google Scholar]
- 4. Huntsman D. G., Chin S. F., Muleris M., Batley S. J., Collins V. P., Wiedemann L. M., Aparicio S., and Caldas C. (1999) MLL2, the second human homolog of the Drosophila trithorax gene, maps to 19q13.1 and is amplified in solid tumor cell lines. Oncogene 18, 7975–7984 [DOI] [PubMed] [Google Scholar]
- 5. Prasad R., Zhadanov A. B., Sedkov Y., Bullrich F., Druck T., Rallapalli R., Yano T., Alder H., Croce C. M., Huebner K., Mazo A., and Canaani E. (1997) Structure and expression pattern of human ALR, a novel gene with strong homology to ALL-1 involved in acute leukemia and to Drosophila trithorax. Oncogene 15, 549–560 [DOI] [PubMed] [Google Scholar]
- 6. Hughes C. M., Rozenblatt-Rosen O., Milne T. A., Copeland T. D., Levine S. S., Lee J. C., Hayes D. N., Shanmugam K. S., Bhattacharjee A., Biondi C. A., Kay G. F., Hayward N. K., Hess J. L., and Meyerson M. (2004) Menin associates with a trithorax family histone methyltransferase complex and with the hoxc8 locus. Mol. Cell 13, 587–597 [DOI] [PubMed] [Google Scholar]
- 7. Dou Y., Milne T. A., Tackett A. J., Smith E. R., Fukuda A., Wysocka J., Allis C. D., Chait B. T., Hess J. L., and Roeder R. G. (2005) Physical association and coordinate function of the H3 K4 methyltransferase MLL1 and the H4 K16 acetyltransferase MOF. Cell 121, 873–885 [DOI] [PubMed] [Google Scholar]
- 8. Nakamura T., Mori T., Tada S., Krajewski W., Rozovskaia T., Wassell R., Dubois G., Mazo A., Croce C. M., and Canaani E. (2002) ALL-1 is a histone methyltransferase that assembles a supercomplex of proteins involved in transcriptional regulation. Mol. Cell 10, 1119–1128 [DOI] [PubMed] [Google Scholar]
- 9. Strahl B. D., Ohba R., Cook R. G., and Allis C. D. (1999) Methylation of histone H3 at lysine 4 is highly conserved and correlates with transcriptionally active nuclei in Tetrahymena. Proc. Natl. Acad. Sci. U.S.A. 96, 14967–14972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Krogan N. J., Dover J., Khorrami S., Greenblatt J. F., Schneider J., Johnston M., and Shilatifard A. (2002) COMPASS, a histone H3 (lysine 4) methyltransferase required for telomeric silencing of gene expression. J. Biol. Chem. 277, 10753–10755 [DOI] [PubMed] [Google Scholar]
- 11. Roguev A., Schaft D., Shevchenko A., Pijnappel W. W., Wilm M., Aasland R., and Stewart A. F. (2001) The Saccharomyces cerevisiae Set1 complex includes an Ash2 homologue and methylates histone 3 lysine 4. EMBO J. 20, 7137–7148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Briggs S. D., Bryk M., Strahl B. D., Cheung W. L., Davie J. K., Dent S. Y., Winston F., and Allis C. D. (2001) Histone H3 lysine 4 methylation is mediated by Set1 and required for cell growth and rDNA silencing in Saccharomyces cerevisiae. Genes Dev. 15, 3286–3295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Smith E., Lin C., and Shilatifard A. (2011) The super elongation complex (SEC) and MLL in development and disease. Genes Dev. 25, 661–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cosgrove M. S., and Patel A. (2010) Mixed lineage leukemia: a structure-function perspective of the MLL1 protein. FEBS J. 277, 1832–1842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dillon S. C., Zhang X., Trievel R. C., and Cheng X. (2005) The SET-domain protein superfamily: protein lysine methyltransferases. Genome Biol. 6, 227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shinsky S. A., Monteith K. E., Viggiano S., and Cosgrove M. S. (2015) Biochemical reconstitution and phylogenetic comparison of human SET1 family core complexes involved in histone methylation. J. Biol. Chem. 290, 6361–6375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cho Y. W., Hong T., Hong S., Guo H., Yu H., Kim D., Guszczynski T., Dressler G. R., Copeland T. D., Kalkum M., and Ge K. (2007) PTIP associates with MLL3- and MLL4-containing histone H3 lysine 4 methyltransferase complex. J. Biol. Chem. 282, 20395–20406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dou Y., Milne T. A., Ruthenburg A. J., Lee S., Lee J. W., Verdine G. L., Allis C. D., and Roeder R. G. (2006) Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat. Struct. Mol. Biol. 13, 713–719 [DOI] [PubMed] [Google Scholar]
- 19. Patel A., Dharmarajan V., Vought V. E., and Cosgrove M. S. (2009) On the mechanism of multiple lysine methylation by the human mixed lineage leukemia protein-1 (MLL1) core complex. J. Biol. Chem. 284, 24242–24256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Steward M. M., Lee J. S., O'Donovan A., Wyatt M., Bernstein B. E., and Shilatifard A. (2006) Molecular regulation of H3K4 trimethylation by ASH2L, a shared subunit of MLL complexes. Nat. Struct. Mol. Biol. 13, 852–854 [DOI] [PubMed] [Google Scholar]
- 21. Santos-Rosa H., Schneider R., Bannister A. J., Sherriff J., Bernstein B. E., Emre N. C., Schreiber S. L., Mellor J., and Kouzarides T. (2002) Active genes are tri-methylated at K4 of histone H3. Nature 419, 407–411 [DOI] [PubMed] [Google Scholar]
- 22. Pokholok D. K., Harbison C. T., Levine S., Cole M., Hannett N. M., Lee T. I., Bell G. W., Walker K., Rolfe P. A., Herbolsheimer E., Zeitlinger J., Lewitter F., Gifford D. K., and Young R. A. (2005) Genome-wide map of nucleosome acetylation and methylation in yeast. Cell 122, 517–527 [DOI] [PubMed] [Google Scholar]
- 23. Liu C. L., Kaplan T., Kim M., Buratowski S., Schreiber S. L., Friedman N., and Rando O. J. (2005) Single-nucleosome mapping of histone modifications in S. cerevisiae. PLoS Biol. 3, e328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Patel A., Vought V. E., Dharmarajan V., and Cosgrove M. S. (2008) A conserved arginine-containing motif crucial for the assembly and enzymatic activity of the mixed lineage leukemia protein-1 core complex. J. Biol. Chem. 283, 32162–32175 [DOI] [PubMed] [Google Scholar]
- 25. Song J. J., and Kingston R. E. (2008) WDR5 interacts with mixed lineage leukemia (MLL) protein via the histone H3-binding pocket. J. Biol. Chem. 283, 35258–35264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shinsky S. A., Hu M., Vought V. E., Ng S. B., Bamshad M. J., Shendure J., and Cosgrove M. S. (2014) A non-active-site SET domain surface crucial for the interaction of MLL1 and the RbBP5/Ash2L heterodimer within MLL family core complexes. J. Mol. Biol. 426, 2283–2299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Artinger E. L., Mishra B. P., Zaffuto K. M., Li B. E., Chung E. K., Moore A. W., Chen Y., Cheng C., and Ernst P. (2013) An MLL-dependent network sustains hematopoiesis. Proc. Natl. Acad. Sci. U.S.A. 110, 12000–12005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jude C. D., Climer L., Xu D., Artinger E., Fisher J. K., and Ernst P. (2007) Unique and independent roles for MLL in adult hematopoietic stem cells and progenitors. Cell Stem Cell 1, 324–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. McMahon K. A., Hiew S. Y., Hadjur S., Veiga-Fernandes H., Menzel U., Price A. J., Kioussis D., Williams O., and Brady H. J. (2007) Mll has a critical role in fetal and adult hematopoietic stem cell self-renewal. Cell Stem Cell 1, 338–345 [DOI] [PubMed] [Google Scholar]
- 30. Yu B. D., Hess J. L., Horning S. E., Brown G. A., and Korsmeyer S. J. (1995) Altered Hox expression and segmental identity in Mll-mutant mice. Nature 378, 505–508 [DOI] [PubMed] [Google Scholar]
- 31. Lim D. A., Huang Y. C., Swigut T., Mirick A. L., Garcia-Verdugo J. M., Wysocka J., Ernst P., and Alvarez-Buylla A. (2009) Chromatin remodelling factor Mll1 is essential for neurogenesis from postnatal neural stem cells. Nature 458, 529–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Milne T. A., Briggs S. D., Brock H. W., Martin M. E., Gibbs D., Allis C. D., and Hess J. L. (2002) MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol. Cell 10, 1107–1117 [DOI] [PubMed] [Google Scholar]
- 33. Wang P., Lin C., Smith E. R., Guo H., Sanderson B. W., Wu M., Gogol M., Alexander T., Seidel C., Wiedemann L. M., Ge K., Krumlauf R., and Shilatifard A. (2009) Global analysis of H3K4 methylation defines MLL family member targets and points to a role for MLL1-mediated H3K4 methylation in the regulation of transcriptional initiation by RNA polymerase II. Mol. Cell Biol. 29, 6074–6085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zeisig B. B., Milne T., García-Cuéllar M. P., Schreiner S., Martin M. E., Fuchs U., Borkhardt A., Chanda S. K., Walker J., Soden R., Hess J. L., and Slany R. K. (2004) Hoxa9 and Meis1 are key targets for MLL-ENL-mediated cellular immortalization. Mol. Cell Biol. 24, 617–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yeoh E. J., Ross M. E., Shurtleff S. A., Williams W. K., Patel D., Mahfouz R., Behm F. G., Raimondi S. C., Relling M. V., Patel A., Cheng C., Campana D., Wilkins D., Zhou X., Li J., et al. (2002) Classification, subtype discovery, and prediction of outcome in pediatric acute lymphoblastic leukemia by gene expression profiling. Cancer Cell 1, 133–143 [DOI] [PubMed] [Google Scholar]
- 36. Armstrong S. A., Staunton J. E., Silverman L. B., Pieters R., den Boer M. L., Minden M. D., Sallan S. E., Lander E. S., Golub T. R., and Korsmeyer S. J. (2002) MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat. Genet. 30, 41–47 [DOI] [PubMed] [Google Scholar]
- 37. Rozovskaia T., Feinstein E., Mor O., Foa R., Blechman J., Nakamura T., Croce C. M., Cimino G., and Canaani E. (2001) Upregulation of Meis1 and HoxA9 in acute lymphocytic leukemias with the t(4:11) abnormality. Oncogene 20, 874–878 [DOI] [PubMed] [Google Scholar]
- 38. Krivtsov A. V., and Armstrong S. A. (2007) MLL translocations, histone modifications and leukaemia stem-cell development. Nat. Rev. Cancer 7, 823–833 [DOI] [PubMed] [Google Scholar]
- 39. Muntean A. G., and Hess J. L. (2012) The pathogenesis of mixed-lineage leukemia. Annu. Rev. Pathol. 7, 283–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dobson C. L., Warren A. J., Pannell R., Forster A., Lavenir I., Corral J., Smith A. J., and Rabbitts T. H. (1999) The mll-AF9 gene fusion in mice controls myeloproliferation and specifies acute myeloid leukaemogenesis. EMBO J. 18, 3564–3574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Thiel A. T., Blessington P., Zou T., Feather D., Wu X., Yan J., Zhang H., Liu Z., Ernst P., Koretzky G. A., and Hua X. (2010) MLL-AF9-induced leukemogenesis requires coexpression of the wild-type Mll allele. Cancer Cell 17, 148–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cao F., Townsend E. C., Karatas H., Xu J., Li L., Lee S., Liu L., Chen Y., Ouillette P., Zhu J., Hess J. L., Atadja P., Lei M., Qin Z. S., Malek S., Wang S., and Dou Y. (2014) Targeting MLL1 H3K4 methyltransferase activity in mixed-lineage leukemia. Mol. Cell 53, 247–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Benedikt A., Baltruschat S., Scholz B., Bursen A., Arrey T. N., Meyer B., Varagnolo L., Müller A. M., Karas M., Dingermann T., and Marschalek R. (2011) The leukemogenic AF4-MLL fusion protein causes P-TEFb kinase activation and altered epigenetic signatures. Leukemia 25, 135–144 [DOI] [PubMed] [Google Scholar]
- 44. Bursen A., Schwabe K., Rüster B., Henschler R., Ruthardt M., Dingermann T., and Marschalek R. (2010) The AF4.MLL fusion protein is capable of inducing ALL in mice without requirement of MLL. AF4. Blood 115, 3570–3579 [DOI] [PubMed] [Google Scholar]
- 45. Emerenciano M., Kowarz E., Karl K., de Almeida Lopes B., Scholz B., Bracharz S., Meyer C., Pombo-de-Oliveira M. S., and Marschalek R. (2013) Functional analysis of the two reciprocal fusion genes MLL-NEBL and NEBL-MLL reveal their oncogenic potential. Cancer Lett. 332, 30–34 [DOI] [PubMed] [Google Scholar]
- 46. Pless B., Oehm C., Knauer S., Stauber R. H., Dingermann T., and Marschalek R. (2011) The heterodimerization domains of MLL–FYRN and FYRC–are potential target structures in t(4;11) leukemia. Leukemia 25, 663–670 [DOI] [PubMed] [Google Scholar]
- 47. Wilkinson A. C., Ballabio E., Geng H., North P., Tapia M., Kerry J., Biswas D., Roeder R. G., Allis C. D., Melnick A., de Bruijn M. F., and Milne T. A. (2013) RUNX1 is a key target in t(4;11) leukemias that contributes to gene activation through an AF4-MLL complex interaction. Cell Rep. 3, 116–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dorrance A. M., Liu S., Chong A., Pulley B., Nemer D., Guimond M., Yuan W., Chang D., Whitman S. P., Marcucci G., and Caligiuri M. A. (2008) The Mll partial tandem duplication: differential, tissue-specific activity in the presence or absence of the wild-type allele. Blood 112, 2508–2511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dharmarajan V., Lee J. H., Patel A., Skalnik D. G., and Cosgrove M. S. (2012) Structural basis for WDR5 interaction (Win) motif recognition in human SET1 family histone methyltransferases. J. Biol. Chem. 287, 27275–27289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Getlik M., Smil D., Zepeda-Velázquez C., Bolshan Y., Poda G., Wu H., Dong A., Kuznetsova E., Marcellus R., Senisterra G., Dombrovski L., Hajian T., Kiyota T., Schapira M., Arrowsmith C. H., et al. (2016) Structure-based optimization of a small molecule antagonist of the interaction between WD repeat-containing protein 5 (WDR5) and mixed-lineage leukemia 1 (MLL1). J. Med. Chem. 59, 2478–2496 [DOI] [PubMed] [Google Scholar]
- 51. Karatas H., Townsend E. C., Bernard D., Dou Y., and Wang S. (2010) Analysis of the binding of mixed lineage leukemia 1 (MLL1) and histone 3 peptides to WD repeat domain 5 (WDR5) for the design of inhibitors of the MLL1-WDR5 interaction. J. Med. Chem. 53, 5179–5185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Karatas H., Townsend E. C., Cao F., Chen Y., Bernard D., Liu L., Lei M., Dou Y., and Wang S. (2013) High-affinity, small-molecule peptidomimetic inhibitors of MLL1/WDR5 protein-protein interaction. J. Am. Chem. Soc. 135, 669–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Senisterra G., Wu H., Allali-Hassani A., Wasney G. A., Barsyte-Lovejoy D., Dombrovski L., Dong A., Nguyen K. T., Smil D., Bolshan Y., Hajian T., He H., Seitova A., Chau I., Li F., et al. (2013) Small-molecule inhibition of MLL activity by disruption of its interaction with WDR5. Biochem. J. 449, 151–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Grebien F., Vedadi M., Getlik M., Giambruno R., Grover A., Avellino R., Skucha A., Vittori S., Kuznetsova E., Smil D., Barsyte-Lovejoy D., Li F., Poda G., Schapira M., Wu H., et al. (2015) Pharmacological targeting of the Wdr5-MLL interaction in C/EBPα N-terminal leukemia. Nat. Chem. Biol. 11, 571–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhu J., Sammons M. A., Donahue G., Dou Z., Vedadi M., Getlik M., Barsyte-Lovejoy D., Al-awar R., Katona B. W., Shilatifard A., Huang J., Hua X., Arrowsmith C. H., and Berger S. L. (2015) Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth. Nature 525, 206–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Patel A., Dharmarajan V., and Cosgrove M. S. (2008) Structure of WDR5 bound to mixed lineage leukemia protein-1 peptide. J. Biol. Chem. 283, 32158–32161 [DOI] [PubMed] [Google Scholar]
- 57. Li Y., Han J., Zhang Y., Cao F., Liu Z., Li S., Wu J., Hu C., Wang Y., Shuai J., Chen J., Cao L., Li D., Shi P., Tian C., et al. (2016) Structural basis for activity regulation of MLL family methyltransferases. Nature 530, 447–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Shinsky S. A., and Cosgrove M. S. (2015) Unique role of the WD-40 repeat protein 5 (WDR5) subunit within the mixed lineage leukemia 3 (MLL3) histone methyltransferase complex. J. Biol. Chem. 290, 25819–25833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Couture J. F., Collazo E., and Trievel R. C. (2006) Molecular recognition of histone H3 by the WD40 protein WDR5. Nat. Struct. Mol. Biol. 13, 698–703 [DOI] [PubMed] [Google Scholar]
- 60. Ruthenburg A. J., Wang W., Graybosch D. M., Li H., Allis C. D., Patel D. J., and Verdine G. L. (2006) Histone H3 recognition and presentation by the WDR5 module of the MLL1 complex. Nat. Struct. Mol. Biol. 13, 704–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Schuetz A., Allali-Hassani A., Martín F., Loppnau P., Vedadi M., Bochkarev A., Plotnikov A. N., Arrowsmith C. H., and Min J. (2006) Structural basis for molecular recognition and presentation of histone H3 by WDR5. EMBO J. 25, 4245–4252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zhang P., Lee H., Brunzelle J. S., and Couture J. F. (2012) The plasticity of WDR5 peptide-binding cleft enables the binding of the SET1 family of histone methyltransferases. Nucleic Acids Res. 40, 4237–4246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gori F., Divieti P., and Demay M. B. (2001) Cloning and characterization of a novel WD-40 repeat protein that dramatically accelerates osteoblastic differentiation. J. Biol. Chem. 276, 46515–46522 [DOI] [PubMed] [Google Scholar]
- 64. Miller T., Krogan N. J., Dover J., Erdjument-Bromage H., Tempst P., Johnston M., Greenblatt J. F., and Shilatifard A. (2001) COMPASS: a complex of proteins associated with a trithorax-related SET domain protein. Proc. Natl. Acad. Sci. U.S.A. 98, 12902–12907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Schneider J., Wood A., Lee J. S., Schuster R., Dueker J., Maguire C., Swanson S. K., Florens L., Washburn M. P., and Shilatifard A. (2005) Molecular regulation of histone H3 trimethylation by COMPASS and the regulation of gene expression. Mol. Cell 19, 849–856 [DOI] [PubMed] [Google Scholar]
- 66. Wysocka J., Swigut T., Milne T. A., Dou Y., Zhang X., Burlingame A. L., Roeder R. G., Brivanlou A. H., and Allis C. D. (2005) WDR5 associates with histone H3 methylated at K4 and is essential for H3 K4 methylation and vertebrate development. Cell 121, 859–872 [DOI] [PubMed] [Google Scholar]
- 67. Roguev A., Schaft D., Shevchenko A., Aasland R., Shevchenko A., and Stewart A. F. (2003) High conservation of the Set1/Rad6 axis of histone 3 lysine 4 methylation in budding and fission yeasts. J. Biol. Chem. 278, 8487–8493 [DOI] [PubMed] [Google Scholar]
- 68. Nagy P. L., Griesenbeck J., Kornberg R. D., and Cleary M. L. (2002) A trithorax-group complex purified from Saccharomyces cerevisiae is required for methylation of histone H3. Proc. Natl. Acad. Sci. U.S.A. 99, 90–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Han Z., Guo L., Wang H., Shen Y., Deng X. W., and Chai J. (2006) Structural basis for the specific recognition of methylated histone H3 lysine 4 by the WD-40 protein WDR5. Mol. Cell 22, 137–144 [DOI] [PubMed] [Google Scholar]
- 70. Thomas L. R., Wang Q., Grieb B. C., Phan J., Foshage A. M., Sun Q., Olejniczak E. T., Clark T., Dey S., Lorey S., Alicie B., Howard G. C., Cawthon B., Ess K. C., Eischen C. M., et al. (2015) Interaction with WDR5 promotes target gene recognition and tumorigenesis by MYC. Mol. Cell 58, 440–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ang Y. S., Tsai S. Y., Lee D. F., Monk J., Su J., Ratnakumar K., Ding J., Ge Y., Darr H., Chang B., Wang J., Rendl M., Bernstein E., Schaniel C., and Lemischka I. R. (2011) Wdr5 mediates self-renewal and reprogramming via the embryonic stem cell core transcriptional network. Cell 145, 183–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wang K. C., Yang Y. W., Liu B., Sanyal A., Corces-Zimmerman R., Chen Y., Lajoie B. R., Protacio A., Flynn R. A., Gupta R. A., Wysocka J., Lei M., Dekker J., Helms J. A., and Chang H. Y. (2011) A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature 472, 120–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. He X., Chen X., Zhang X., Duan X., Pan T., Hu Q., Zhang Y., Zhong F., Liu J., Zhang H., Luo J., Wu K., Peng G., Luo H., Zhang L., et al. (2015) An Lnc RNA (GAS5)/SnoRNA-derived piRNA induces activation of TRAIL gene by site-specifically recruiting MLL/COMPASS-like complexes. Nucleic Acids Res. 43, 3712–3725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Mersman D. P., Du H. N., Fingerman I. M., South P. F., and Briggs S. D. (2012) Charge-based interaction conserved within histone H3 lysine 4 (H3K4) methyltransferase complexes is needed for protein stability, histone methylation, and gene expression. J. Biol. Chem. 287, 2652–2665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Bledau A. S., Schmidt K., Neumann K., Hill U., Ciotta G., Gupta A., Torres D. C., Fu J., Kranz A., Stewart A. F., and Anastassiadis K. (2014) The H3K4 methyltransferase Setd1a is first required at the epiblast stage, whereas Setd1b becomes essential after gastrulation. Development 141, 1022–1035 [DOI] [PubMed] [Google Scholar]
- 76. Wu M., Wang P. F., Lee J. S., Martin-Brown S., Florens L., Washburn M., and Shilatifard A. (2008) Molecular regulation of H3K4 trimethylation by Wdr82, a component of human Set1/COMPASS. Mol. Cell Biol. 28, 7337–7344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Lee J. H., and Skalnik D. G. (2008) Wdr82 is a C-terminal domain-binding protein that recruits the Setd1A histone H3-Lys4 methyltransferase complex to transcription start sites of transcribed human genes. Mol. Cell Biol. 28, 609–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Sheffield P., Garrard S., and Derewenda Z. (1999) Overcoming expression and purification problems of RhoGDI using a family of “parallel” expression vectors. Protein Expr. Purif. 15, 34–39 [DOI] [PubMed] [Google Scholar]
- 79. Scheuermann T. H., and Brautigam C. A. (2015) High-precision, automated integration of multiple isothermal titration calorimetric thermograms: new features of NITPIC. Methods 76, 87–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Zhao H., Piszczek G., and Schuck P. (2015) SEDPHAT: a platform for global ITC analysis and global multi-method analysis of molecular interactions. Methods 76, 137–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Brautigam C. A., Zhao H., Vargas C., Keller S., and Schuck P. (2016) Integration and global analysis of isothermal titration calorimetry data for studying macromolecular interactions. Nat. Protoc. 11, 882–894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Otwinowski Z., and Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 83. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Perrakis A., Morris R., and Lamzin V. S. (1999) Automated protein model building combined with iterative structure refinement. Nat. Struct. Biol. 6, 458–463 [DOI] [PubMed] [Google Scholar]
- 85. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 86. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Chen V. B., Arendall W. B. 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. McNicholas S., Potterton E., Wilson K. S., and Noble M. E. (2011) Presenting your structures: the CCP4mg molecular-graphics software. Acta Crystallogr. D Biol. Crystallogr. 67, 386–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Morin A., Eisenbraun B., Key J., Sanschagrin P. C., Timony M. A., Ottaviano M., and Sliz P. (2013) Collaboration gets the most out of software. Elife 2, e01456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Sievers F., Wilm A., Dineen D., Gibson T. J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J., Thompson J. D., and Higgins D. G. (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]