

Abstract
The expression of eukaryotic genes is precisely controlled by specific interactions between general transcription initiation factors and gene‐specific transcriptional activators. The general transcription factor TFIID, which plays an essential role in mediating transcriptional activation, is a multisubunit complex comprising the TATA box‐binding protein (TBP) and multiple TBP‐associated factors (TAFs). On the other hand, biochemical and genetic approaches have shown that the promoter‐specific transcriptional activator Sp1 has the ability to interact with one of the components of TFIID, the TBP‐associated factor TAF4. We herein report the structural details of the glutamine‐rich domains (Q‐domains) of Sp1 and TAF4 using circular dichroism (CD) and heteronuclear magnetic resonance (NMR) spectroscopy. We found that the two Q‐domains of Sp1 and four Q‐domains of TAF4 were disordered under physiological conditions. We also quantitatively analyzed the interaction between the Q‐domains of Sp1 and TAF4 by NMR and surface plasmon resonance, and detected a weak but specific association between them. Nevertheless, a detailed analysis of CD spectra suggested that any significant conformational change did not occur concomitantly with this association, at least at the level of the overall secondary structure. These results may represent a prominent and exceptional binding mode for the IDPs, which are not categorized in a well‐accepted concept of “coupled folding and binding.”
Keywords: transcription factor, glutamine‐rich domain, molecular interaction, intrinsically disordered protein, nuclear magnetic resonance
Introduction
The expression of genes is controlled in a temporally and spatially coordinated manner, and this precise regulation is accomplished predominantly at the level of transcription. A gene in a eukaryotic cell typically consists of two types of DNA elements: a common core‐promoter element that is proximal to the transcription start site, and gene‐specific enhancer sequences that are located at more distal positions.1 The core‐promoter element is recognized by the general transcriptional machinery to form a pre‐initiation complex, which contains RNA polymerase II and the general transcription initiation factors TFIIA, ‐B, ‐D, ‐E, ‐F, and ‐H.2, 3
In contrast, enhancer elements are recognized by gene‐specific transcriptional regulators, which typically consist of a DNA‐binding domain and one or more activation domains.4, 5 In the absence of these activators, the expression of most genes in vivo is silenced by the chromatin structure as well as the effects of other repressors. Furthermore, RNA polymerase II and cognate general initiation factors, without the aid of activators, only exhibit a low level of transcriptional activity in vitro. Therefore, the mechanisms by which enhancer selective transcriptional factors stimulate the transcriptional activity of the general transcriptional machinery need to be elucidated to obtain a detailed understanding of the regulation of gene expression.
The basal transcriptional factor TFIID is considered to play a central role during the formation of a pre‐initiation complex by general transcription factors because it is the first to recognize and bind the core promoter, and also provides a scaffold upon which the remaining transcriptional factors assemble.6, 7, 8 TFIID is a multisubunit complex comprising the TATA box‐binding protein (TBP) and multiple TBP‐associated factors (TAFs).9, 10, 11 TAFs have been identified biochemically as stably associating components with TBP and were originally named according to their mobility values in electrophoresis.12 A number of genetic and biochemical approaches have revealed that TAF4 (also known as TAFII130 or TAFII135 in humans, and TAFII110 in Drosophila) interacts with cellular transcriptional activators such as Sp1 and the cAMP‐response element‐binding protein (CREB).13, 14, 15 These interactions are considered to participate in the recruitment of the pre‐initiation complex to the promoter, thereby increasing transcriptional levels.
Although the homologues of TAF4 in humans (hTAFII130) and that in Drosophila (dTAFII110) show limited sequence similarities,16, 17 they share several functional and structural properties.13 Previous studies revealed that Drosophila dTAFII110 directly interacts with the human transcriptional activator Sp1 to enhance the transcriptional levels of the genes located downstream.13, 18 A sequence analysis showed that both proteins contain four glutamine‐rich (Q‐rich) domains, as well as two highly conserved regions, CI and CII with similarities of 68% and 72%, respectively (Fig. 1).15 An in vitro binding analysis and yeast two‐hybrid assays indicated the involvement of one or more of the four glutamine‐rich domains Q1‐Q4 located at the central part of the TAF4 molecule in interactions with transcriptional activators. The central region of human TAF4 (hTAFII130) was found to be sufficient for interactions with the activation domain of human Sp1.17
Figure 1.
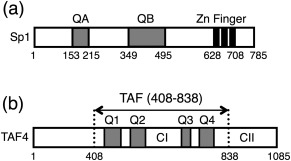
(A) Schematic drawing of the primary structure of the transcriptional factor Sp1. Two Q‐rich domains, QA and QB, and three zinc finger domains are indicated. (B) Schematic representation of human TAF4 (hTAFII130), which is one of the components of the general transcription factor TFIID. Four Q‐domains: Q1[438–485], Q2[526–557], Q3[666–688], and Q4[718–789], are indicated by gray bars. The central region of the molecule TAF4N/C, which includes all four Q‐domains, was prepared and studied here. The highly conserved regions CI and CII, which are located between Q2 and Q3, and at the C‐terminus are also indicated.
The promoter‐specific transcription factor Sp1 is expressed ubiquitously and plays a primary role in regulating the expression of many genes.19, 20, 21 Genetic and biochemical studies on Sp1 have identified two potent activating regions, termed QA and QB, both of which are characterized by an abundance of glutamine residues.22, 23 Sp1 also possesses three C2H2‐type zinc fingers at the C‐terminal, which interact with GC‐boxes on the target gene located distally from the transcriptional initiation site.24 The interaction between Sp1, which recognizes and binds the upstream GC‐box, and TAF4 in the general transcriptional factor via each Q‐rich domain is considered to be important for the recruitment of RNA polymerase II to the transcription initiation site as well as the activation of transcription.
Although previous genetic and biochemical studies showed that Sp1 and TAF4 interacted with each other via Q‐domains in vitro and in vivo, the molecular structures of these proteins remain unclear, and quantitative analyses on this interaction are limited. Therefore, we herein investigated the molecular architecture of the Q‐rich domains of Sp1 and TAF4 using CD and high resolution NMR spectroscopy. We found that the isolated Q‐domains of Sp1 and TAF4 were classified as intrinsically disordered proteins (IDPs), which have neither regular secondary nor tertiary structures under physiological conditions.25 The interaction between proteins was analyzed in detail using surface plasmon resonance, and their dissociation constants were estimated to be in a submillimolar range based on a stoichiometric binding scheme. Nevertheless, a detailed analysis of CD spectra suggested that any significant conformational change did not occur concomitantly with their association, at least at the level of the overall secondary structure. These results may represent a prominent and exceptional binding mode for the IDPs, which are not categorized in a well‐accepted concept of “coupled folding and binding.”
Materials and Methods
Materials
15N‐Ammonium chloride and deuterium oxide were purchased from SI Science (Saitama, Japan). Other reagents were purchased from Nacalai Tesque (Kyoto, Japan).
Protein expression and purification
There are several GenBank entries for the sequence of human Sp1, and we followed the numbering of amino acid residues for “isoform a” composed of 785 residues (NCBI Reference Sequence: NP_612482.2). The expression plasmids for the glutamine‐rich domains of Sp1 (QA [153–215] and QB [349–495]) were constructed as fusion proteins with glutathione S‐transferase (GST) at the N‐terminal by inserting the genes of interest into the pGEX‐1N (Amrad) or modified pGEX‐3X vector. Each protein contained the three‐residue N‐terminal extension and C‐terminal extension of a FLAG‐octapeptide‐tag followed by a hexahistidine‐tag (Fig. 1). The expression and purification of the glutamine‐rich domains of Sp1, QA, and QB, were performed as described previously.26 Uniformly 15N‐labeled proteins were prepared from cells grown in M9 minimal medium with 0.5 g/L 15NH4Cl.
The gene of human TAF4 is also found as several entries in the database. Previous biochemical studies have referred to the sequence reported by Tanese et al.,15 which corresponds to a protein of 947 amino acid residues (GenBank entry of U75308). By using this sequence, the central region (residues from 270 to 700) of human TAF4 (hTAFII130) was found to be sufficient for interactions with the activation domain of human Sp1.17 We herein prepared the fragment protein corresponding to the same region (from 270 to 700) of human TAF4, and referred to it as TAF4N/C. However, we used a different entry for the sequence encoding a protein with 1085 residues (NCBI Reference Sequence: NP_003176.2), and, thus, the numbering of amino acid residues differs from those in previous studies (from 408 to 838 in this study). The chemically synthesized DNA encoding the central region of TAF4 [408–838], in which codon usage was optimized for Escherichia coli B‐strain, was purchased from Life Technologies (CA). The DNA fragment was inserted into the pET‐Ub expression vector between the Bsp TI and Eco RI sites to produce a fusion protein with human ubiquitin.27 The pET‐UbTAF4N/C vector obtained was introduced into the E. coli strain Rosetta2(DE3)pLysS (Novagen).
Transformed bacteria were grown in Lucia broth medium containing 50 μg/L of kanamycin. To prepare the uniformly 15N‐labeled protein, bacteria were grown in M9 minimal medium with 0.5 g/L 15NH4Cl. Bacterial cells were grown at 37°C, and isopropyl‐β‐d‐thiogalactopyranoside (IPTG) was added to medium when absorbance at 600 nm reached 0.6. After IPTG had been added, cells were incubated for a further 3 h. Cells were harvested by centrifugation at 5000g at 4°C for 15 min. The bacterial cells collected were suspended in buffer A (300 mM NaCl and 50 mM sodium phosphate, pH 7.3) containing 1 mM phenylmethanesulfonyl fluoride, and disrupted by sonication with intermittent pulses for 1 min (pulse of 0.5 s, interval of 0.5 s, output level of 7) three times using the ultrasonic disruptor UD‐201 (Tomy, Tokyo, Japan) equipped with a TP‐012 standard tip. Cell debris was removed by centrifugation at 10,000g at 4ºC for 60 min, and the cell extract was loaded onto TALON resin (GE Healthcare). The column was washed with buffer A containing 5 mM imidazole, and bound protein was eluted with buffer A containing 250 mM imidazole. Solid ammonium sulfate was then added to the eluted protein up to 15% saturation, and the precipitate was collected by centrifugation at 250,000g for 60 min. Precipitated TAF4N/C was dissolved in buffer B (140 mM NaCl and 20 mM sodium phosphate, pH 7.3), and dialyzed against buffer B.
To remove the hexahistidine‐tag and ubiquitin‐tag at the N‐terminus, the expressed His‐Ubq‐TAF4N/C was digested by the yeast ubiquitin carboxyl‐terminal hydrolase (Refs. 28, 29). The enzyme was added to the dialyzed substrate protein at a ratio of 0.5 mol of enzyme per 1 mol of substrate, and the protein mixture was incubated at 37°C for 4 h. The digested protein was purified by Ni‐NTA and then by gel‐filtration chromatography using a column of Superdex 75 10/300 GL (GE Healthcare) equilibrated with buffer B. The purity of the protein was confirmed to be >95% by gel‐filtration chromatography and SDS‐PAGE. Purified proteins were collected, concentrated with Amicon Ultra (Millipore), and stored at 4°C.
Circular dichroism (CD)
CD spectra were measured on a Jasco J‐820 spectropolarimeter. An assembling cell composed of a pair of quartz plates with a 0.1‐mm path length was used to record spectra at each protein concentration of 50 μM in buffer B at 4°C. The results obtained were expressed as apparent ellipticity with the unit of millidegrees (mdeg). Sixteen scans were averaged for each sample.
Nuclear magnetic resonance (NMR)
NMR spectra were measured on a Bruker DMX600 spectrometer at 4°C equipped with a triple‐axis gradient and triple‐resonance TXI probe. The sample concentration was 50 μM 15N‐labeled protein in buffer B containing 10% D2O. Spectra were processed with nmrPipe and analyzed with nmrDraw and PIPP.30, 31
In the titration experiment, we assumed 1:1 binding between ((Sp1–QB and TAF4N/C as
where QB, TAF, and QB•TAF represent a monomeric Sp1‐QB, TAF4N/C, and the complex between them, respectively. This defines the dimeric association constant K as
By using K, and the total concentrations of QB and TAF, [QB]0 and [TAF4]0, the concentration of the monomeric form of QB was represented as,
| (1) |
Small‐angle X‐ray scattering (SAXS)
SAXS measurements were performed with the spectrometer installed at BL‐10C of Photon Factory, a synchrotron radiation facility of Institute of Materials Structure Science, High Energy Accelerator Research Organization (Tsukuba, Japan). The scattering intensity in a q‐range between 0.02 and 0.2 Å−1 was recorded with PILATUS 2M detector. The q‐range is estimated by the diffraction from the standard sample, silver behenate. Here, (q = 4πsinθ/λ, where 2θ and λ (= 0.1488 nm) is the scattering angle and the X‐ray wavelength, respectively. Measured two‐dimensional pattern was converted into one‐dimensional curve by circulation average and then the scattering profile, I(q), of protein was obtained by the standard procedure of data correction for cell and background scatterings, transmission, and beam intensity.
During the X‐ray exposure, the protein solutions were continuously flowed in a 1 mm thickness cell with the rate of 0.3 μL/s to avoid the radiation damage by the X‐ray beam. It is confirmed that all samples were free from the radiation damage in the exposure time of 120 s.
Surface plasmon resonance (SPR)
SPR measurements were performed using a ProteOn XPR36 system (Bio‐Rad) at 15°C (the lowest temperature that is able to be set by the instrument) with an HTG sensor chip equilibrated with buffer B as a running buffer at a flow rate of 100 μL/min. The HTG sensor chip was first activated with 10 mM NiSO4 for 4 min. One hundred and twenty five microliters of the ligand protein (20 μM Sp1‐QA and Sp1‐QB with hexahistidine at the C‐terminal) was injected and immobilized on the HTG chip. The analyte protein (TAF4N/C) was then injected for 150 s at a constant flow rate of 100 μL/min, and sensorgrams were recorded as the association phase. The concentration of analyte loaded was 2.0, 1.2, and 0.4 μM for Sp1‐QA, and 2.0, 1.6, 0.8, and 0.4 μM for Sp1‐QB. The dissociation of the bound analyte was subsequently followed for 300 s at the same flow rate. Kinetic parameters were calculated using a global fitting analysis with the assumption of the 1:1 Langmuir binding model using Eqs. (2) and (3) for the association and dissociation phases, respectively.
| (2) |
| (3) |
where t is time, R is the time‐dependent response of the SPR signal, and R 0 and R max are the initial (just after the dissociation phase) and the maximum (reaches at the infinite time in the association phase) values of the SPR signal, respectively, C is the concentration of the analyte, and k a and k d are the association and dissociation rate constants for the analyte and ligand, respectively.
Results
1H‐15N HSQC spectra of 15N‐TAF4
To elucidate the structural features of the glutamine‐rich domains of TAF4N/C expressed in E. coli, we measured the 1H‐15N HSQC spectrum under nearly physiological conditions at 4°C in buffer B (140 mM NaCl and 20 mM sodiumphosphate, pH 7.3). The phrase “nearly physiological conditions” was used because the temperature was not physiological (4°C). All resonance peaks were poorly dispersed along the 1H chemical shift axis and appeared within the range of 8.5 and 7.5 ppm, indicating that the protein had neither secondary nor tertiary structures that were stabilized by regular hydrogen bonds and the rigid packing of side chains (Fig. 2). This result suggests that the central region of TAF4, which contains all the Q‐domains, is intrinsically disordered under physiological conditions. We measured HSQC spectra at various temperatures, and found that signal intensity markedly decreased at higher temperatures (Supporting Information Fig. S1A). The temperature dependence of the signal intensity was similarly observed in another IDP, the QB domain from Sp1 (Sp1‐QB).26 We partly attributed these behaviors to the rapid exchange of amide hydrogen with solvent water at higher temperatures because the decrease in intensity was more significant for amide signals than aliphatic protons (Supporting Information Fig. S1B). This was reasonable if rigid hydrogen bonds were lacking in these proteins. Therefore, we performed all NMR experiments at 4°C.
Figure 2.
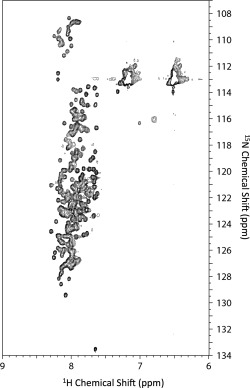
1H‐15N HSQC spectra of TAF4N/C measured at pH 7.3 and 4°C.
We also prepared fragment proteins, in which TAF4N/C was divided into three parts, and measured their 1H‐15N HSQC spectra under the same conditions. The superposition of HSQC spectra obtained by three fragment proteins was almost identical to the spectrum of whole TAF4N/C (Supporting Information Fig. S2). This result indicates the absence of any significant interaction between the fragment proteins, which was consistent with TAF4N/C being largely disordered under the conditions examined.
Interaction between Q‐rich domains of Sp1 and TAF4 elucidated by 1H‐15N HSQC spectra
The intensity and position (chemical shift value) of each NMR peak sensitively reflects changes in the environment surrounding the nuclear spin. Therefore, we expected a detailed analysis of NMR spectra to reveal the intermolecular interaction between the Q‐rich domains of Sp1 and TAF4. To detect these interactions, the 1H‐15N HSQC spectra of the QA and QB domains of Sp1 (Sp1‐QA and Sp1‐QB) were measured in the absence and presence of unlabeled TAF4N/C. We initially measured the 1H‐15N HSQC spectra of Sp1‐QA. These spectra revealed the poor dispersion of each resonance signal, suggesting that Sp1‐QA is also intrinsically disordered [Fig. 3(A)]. We then measured the spectrum under the same conditions in the presence of a three molar excess amount of unlabeled TAF4N/C. The two spectra recorded in the absence and presence of the excess amount of TAF4N/C were identical. This result suggests that Sp1‐QA and TAF4N/C do not interact with each other, or that the affinity of the two proteins is too weak to be detected under the conditions examined.
Figure 3.
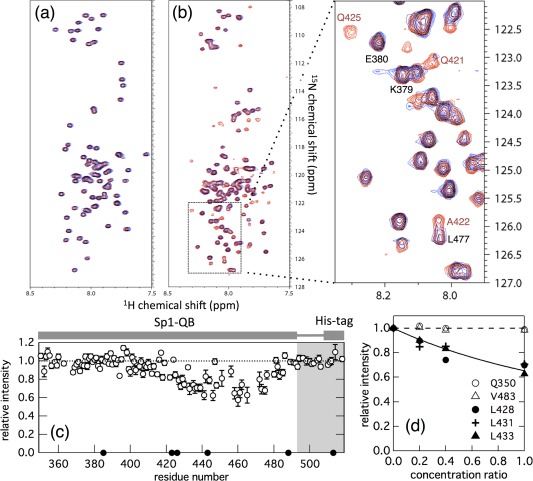
(A) Overlay of the 1H‐15N HSQC spectra of 15N‐labeled Sp1‐QA in the absence (red) and presence (blue) of a three molar excess amount of unlabeled TAF4N/C measured at 4°C. The concentrations of 15N‐ Sp1‐QA in both spectra were 50 μM, and 150 μM unlabeled TAF4N/C was added. The peaks colored black indicate that both chemical shifts and the intensities of the peaks were not changed. (B) Overlay of the 1H‐15N HSQC spectra of 15N‐Sp1‐QB domains in the absence (red) and presence (blue) of an equimolar amount of unlabeled TAF4N/C measured at 4°C. Concentrations of 15N‐Sp1‐QB in both spectra were 50 μM and the same amount of unlabeled TAF4N/C was added. The region indicated by the dashed box in the spectrum is expanded in the right panel. The intensities of several peaks of 15N‐Sp1‐QB markedly decreased with the addition of unlabeled TAF4N/C. (C) The relative peak intensity of 1H‐15N HSQC spectra plotted against the residue number of Sp1‐QB. Intensity in the presence of the same concentration of unlabeled TAF4N/C relative to that in its absence is shown. The closed circle shows residues that could not be analyzed due to signal overlap. (D) Changes in the relative peak intensities of several representative residues in 15N‐Sp1‐QB in the presence of various concentrations (10, 20, and 50 μM) of unlabeled TAF4. Peak intensity in the presence of unlabeled TAF4N/C relative to its absence was plotted against the concentration ratio of TAF4N/C relative to that of 15N‐Sp1‐QB. Open and closed symbols represent residues with intensities that showed little or strong dependency on the concentration of TAF4N/C added, respectively. The solid line indicates the fraction of monomeric Sp1‐QB calculated on the assumption of a 1:1 binding model [Eq. (1)] at the binding constant K a = 4.0 × 103 M−1.
We performed a similar experiment to examine the interaction between Sp1‐QB and TAF4N/C. The 1H‐15N HSQC spectrum of Sp1‐QB in the absence of TAF4N/C showed the poor dispersion of resonance peaks, as observed previously.26 In contrast to Sp1‐QA, the intensity of several peaks of Sp1‐QB markedly decreased when an equimolar amount of unlabeled TAF4N/C was added [Fig. 3(B)]. Although we do not know the exact reason why the peak intensity was decreased for several residues at present, severe line broadening may be resulted from a large molecular weight of the complex with unlabeled TAF4N/C and/or chemical exchange in the conformation of 15N‐Sp1‐QB due to the equilibrium between free and bound forms. In any cases, the result clearly demonstrates that Sp1‐QB and TAF4N/C interact with each other. To identify the region of this interaction, peak intensity in the presence of unlabeled TAF4N/C relative to that recorded in its absence was plotted against the residue number of Sp1‐QB [Fig. 3(C)]. The results obtained revealed that the residue, the intensity of which significantly decreased, was located in the region from the center to the C‐terminal part of the molecule, indicating that the decrease observed was site‐specific and also that these residues in Sp1‐QB may represent the binding site for TAF4N/C.
To analyze the interaction between Sp1‐QB and TAF4N/C in more detail, we measured several 1H‐15N HSQC spectra of 15N‐Sp1‐QB in the presence of various concentrations of unlabeled TAF4N/C. We found that it was possible to classify the residues of Sp1‐QB into two groups: one independent of the concentrations of TAF4N/C and the other showing a significant decrease in intensity. The results for several representative residues are shown in Figure 3(D). These plots corresponded well among residues belonging to the same group, suggesting that the interaction between Sp1‐QB and TAF4N/C is consistent to the two‐state binding model. These plots also corresponded well to the fraction of free Sp1‐QB that did not form a complex with TAF4N/C calculated based on Eq. (1) with an estimated K a of 4 × 103 M−1 and the concentrations of TAF4N/C at each point.
Changes in the secondary structure monitored by CD spectroscopy
Although changes in the peak intensities of NMR spectra provided evidence for Sp1‐QB interacting with TAF4N/C, no information on the structure of the complex was obtained because the resonance peak of Sp1‐QB simply disappeared when it formed a complex with TAF4N/C. We measured the CD spectra of these proteins to elucidate whether any conformational change in Sp1‐QB and/or TAF4N/C was triggered by their association. We measured initially the CD spectra of Sp1‐QB and TAF4N/C separately (Fig. 4). The spectrum of Sp1‐QB showed a deep minimum at 200 nm, indicating that this protein did not form any well‐defined secondary structure. The spectrum of TAF4N/C was also represented by a large minimum at 205 nm, suggesting that the molecule was largely disordered. However, a careful observation revealed that the negative minimum in the spectrum of TAF4N/C was broader and shallower than that of Sp1‐QB. Furthermore, the spectrum of TAF4N/C showed a small, but significant shoulder at approximately 220 nm. These results suggest that some part of TAF4N/C may adopt an α‐helical conformation. Nevertheless, the spectra of both proteins indicated that most of the molecule was largely disordered, consistent with NMR observations.
Figure 4.
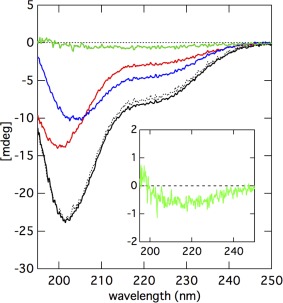
Far UV‐CD spectra of Sp1‐QB (red), TAF4N/C (blue), and their mixture (black) measured at 4°C. Each sample contains 50 μM of protein. The dashed black line indicates a simple sum of the spectra of Sp1‐QB and TAF4N/C that were recorded separately. The green line indicates the difference spectrum between the observed and calculated spectra for the mixture of Sp1‐QB and TAF4N/C. The inset shows the expansion of the difference spectrum between the observed and calculated spectra for the mixture of Sp1‐QB and TAF4N/C.
We then measured the spectrum of an equimolar mixture of Sp1‐QB and TAF4N/C, and compared it with the sum of two separately measured spectra of these two proteins. Note that the intensity of CD was expressed as an apparent ellipticity value with the unit of mdeg because two proteins consisted of different numbers of amino acid residues (172 and 431). By using the apparent ellipticity value, a direct comparison was possible between the summed spectrum and that obtained from an equimolar mixture because all spectra were measured at the same concentration of proteins (50 μM each). We expected the spectrum measured for the mixed sample to be different from that of the simple sum of two separately measured spectra if any change occurred in the conformation of Sp1‐QB and/or TAF4N/C molecules. A careful comparison of the two spectra revealed that they were not identical and the spectra of the mixed sample showed a slightly larger negative value at approximately 220 nm [Fig. 4(B)]. This result suggests that an α‐helical conformation is induced in some part of Sp1‐QB and/or TAF4N/C by their interaction. However, this difference was not very large, and thus, we considered the overall secondary structures of QB and TAF4 did not change significantly.
Small‐angle X‐ray scattering (SAXS)
To further elucidate the structure of Sp1‐QA, Sp1‐QB and TAF4N/C, we performed SAXS experiments that provide overall conformational properties of protein molecules. We first plot the logarithm of scattering intensity (I(q)) against the square of magnitude of scattering vector (q 2) [Fig. 5(A)]. It is known that in the very narrow q‐region (<1.3/R g), the linear dependency of I(q) against q 2 (Guinier plot) provides the radius of gyration (R g) of the solute. From the Guinier plot shown in Figure 5(A), the R g values for Sp1‐QA, Sp1‐QB and TAF4N/C were estimated to be 25.6 ± 1.5, 36.2 ± 0.3 and 49.0 ± 7.4 Å, respectively. In the case of Sp1‐QA and Sp1‐QB, these experimental values were in good agreement with those expected for the urea denatured states of ∼30 and ∼40 Å, which were estimated for proteins with 92 and 172 amino acid residues.32 On the other hand, the R g value of TAF4N/C was significantly smaller than that is expected for the urea‐denatured state of protein with 431 amino acid residues (∼70 Å).32 The result is consistent with that of CD spectrum, suggesting possible formation of α‐helical structure in some part of TAF4N/C.
Figure 5.
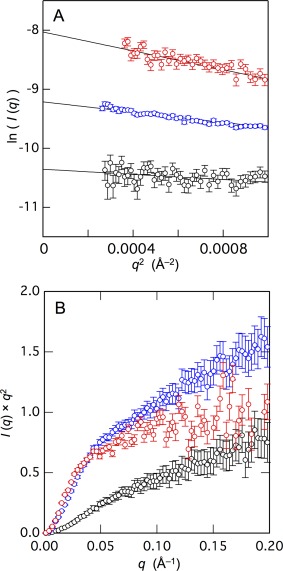
(A) The Guinier plots of small‐angle X‐ray scattering of Sp1‐QA (black), Sp1‐QB (blue), and TAF4N/C (red) shown in open circles. The lines indicate the results of linear curve fitting for the data within the Guinier‐region (q × R g < 1.3). (B) The Kratky plots of Sp1‐QA (black), Sp1‐QB (blue), and TAF4N/C (red).
We then expressed the scattering profile of these proteins by the Kratky plot, which provides the information on the overall shape of the protein [Fig. 5(B)]. The Kratky plot for the native conformation has a distinct peak, the position of which is dependent on R g, while the plot for a chain molecule gives a plateau and then rises monotonically.33 None of the plot for Sp1‐QA, Sp1‐QB, and TAF4N/C showed any clear peaks, suggesting that these proteins are largely disordered.
Surface plasmon resonance (SPR)
Although the changes observed in the spectra of NMR and CD were considered to reflect the interaction between Sp1‐QB and TAF4N/C, the results were somewhat ambiguous. Therefore, we measured SPR to obtain more direct evidence for binding between Sp1‐QB and TAF4N/C. An SPR analysis requires the immobilization of the “ligand” molecule on the sensor chip. To minimize the effects of immobilization on the structure and function of the ligand protein, we utilized a hexahistidine‐tag attached at the C‐terminus of the Sp1‐QB protein via a flexible linker sequence. Injections of various concentrations of TAF4N/C as the “analyte” resulted in marked increases in resonance, demonstrating the association between immobilized Sp1‐QB and TAF4N/C (Fig. 6). After the injection of TAF4N/C for 150 s, the same buffer B without the analyte was loaded for 300 s. Gradual decreases were observed in resonance, suggesting the slow dissociation of the bound analytes from the immobilized ligand protein.
Figure 6.
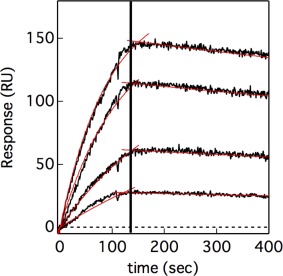
Kinetics of the association between Sp1‐QB and TAF4N/C, and their dissociation measured by SPR. Sp1‐QB was immobilized on an HTG chip via the hexahistidine‐tag at the C‐terminus. Black lines show sensorgrams recorded after the loading of TAF4N/C at various concentrations for 150 s, followed by the flow of its absence. Red lines indicate the results of global fitting of four sensorgrams by Eqs. (2) and (3).
All sensorgrams were globally fit to Eqs. (2) and (3) for the association and dissociation phases, respectively. The fit converged well, and single association and dissociation rate constants were estimated to be 4.40 ± 0.08 M−1 s−1 and (3.03 ± 0.06) × 10−4 s−1, respectively. The equilibrium association constant between Sp1‐QB and TAF4N/C was estimated to be (1.45 ± 0.04) × 104 M−1 from these association and dissociation rate constants. These results suggest a significant interaction between Sp1‐QB and TAF4N/C. On the other hand, there were no detectable interactions between Sp1‐QA and TAF4N/C by SPR experiments performed under the same conditions (Supporting Information Fig. S3).
Discussion
Structural features of glutamine‐rich domains
We previously reported that one of the Q‐rich domains, the QB domain, in the human transcription factor Sp1 was largely disordered under nearly physiological conditions at 4°C and pH 7.3.26 We herein showed that the other Q‐rich domain in Sp1, QA, as well as those located at the central region of TAF4, Q1–Q4, were also intrinsically disordered under the same conditions. The reason why we performed NMR characterizations of these domains at 4°C is because the peak intensities in the 1H‐15N HSQC spectra were markedly decreased at higher temperatures. Although we currently do not know the exact conformations of these Q‐rich domains at physiological temperatures, their CD spectra at higher temperatures were not significantly different from those recorded at 4°C (data not shown). Therefore, we concluded that these Q‐rich domains are also intrinsically disordered under physiological conditions at 37°C and pH 7.3.
Glutamine‐rich domains, which are often found in transcription factors, are characterized by a high degree (25% or more) of glutamine residues in the primary sequence. The Q‐rich domains examined here, two in Sp1 and four in TAF4, were intrinsically disordered. The native structure of a globular protein is characterized by well‐ordered secondary structures stabilized by hydrogen bonds, which are typically buried inside the protein molecule, otherwise they are destabilized by non‐specific hydrogen bonding with the surrounding water molecules. The interior of a protein molecule mostly consists of hydrophobic amino acids, which is referred to as a “hydrophobic core,” and its formation is involved in the specific and rigid packing of side chains of amino acids. Therefore, a certain number of hydrophobic residues is necessary for the formation of a typical “native” structure.34 The Q‐rich domains studied here may not have had a sufficient number of hydrophobic residues because of the large number of glutamine residues.
A detailed analysis of the CD spectrum of TAF4N/C indicated that the molecule was largely disordered; however, a significant amount of the secondary structure was also present. The central region of TAF4 contains four Q‐rich domains, as well as the highly conserved region I (CI, residues 558–669; Ref. 15). A relatively long stretch of residues intervening the Q2 and Q3 domains may contribute significantly to the ellipticity of TAF4N/C.
Interaction between Q‐rich domains of Sp1 and TAF4
Titration experiments monitored by the 1H‐15N HSQC spectra of Sp1‐QB revealed that the intensities of several residues were markedly decreased in the presence of unlabeled TAF4N/C in a concentration‐dependent manner. Moreover, the plot of relative peak intensities against residue numbers shown in Figure 3(C) suggests that the interaction site is located between the center and C‐terminus of QB. These results are consistent with previous findings, in which a fragment protein of Sp1 corresponding to the C‐terminal half of the QB domain (residues 428–549) was suggested to interact with dTAFII110, a Drosophila homologue of human hTAFII130 (TAF4).14, 15 Furthermore, a mutational study suggested that the residues located at the C‐terminal of the QB domain (464WQTLQLQNL472) were of significant importance in the interaction with dTAFII110.18 Because the interaction between the proteins is not so strong, the “true” binding site might be obscured by a possible equilibrium between bound and free state.
In contrast to the QB domain, neither the chemical shift nor peak intensity of the 15N‐QA domain changed in the presence of unlabeled TAF4N/C up to a three molar excess amount. On the other hand, Hoey et al.13 reported that the QA and QB domains interacted with dTAFII110. In addition, the central region of human hTAFII130 (TAF4) has also been suggested to interact with the QA and QB domains.17 Both studies analyzed the interaction between the Q domains of Sp1 and TAF using a yeast two‐hybrid system, in which many other cellular components may have contributed to the interaction. Therefore, we concluded that the QA domain did not interact by itself with TAF4N/C, at least under the conditions examined.
Quantitative analysis of the interaction between Q‐rich domains of Sp1 and TAF4
We examined the interaction between Sp1‐QB and TAF4N/C using SPR, which provides the association and dissociation rate constants, k a and k d. Sensorgrams were analyzed by the standard Langmuir model, which assumes 1:1 binding between the ligand and analyte. The binding constant for Sp1‐QB and TAF4N/C was calculated to be 1.5 × 104 M−1, indicating a significant interaction between these proteins.
We found that the peak intensities for several residues of 15N‐Sp1‐QB were markedly decreased in the presence of unlabeled TAF4N/C in a concentration‐dependent manner. We analyzed changes in the relative peak intensity of the 1H‐15N HSQC spectra of Sp1‐QB recorded in the presence of various concentrations of unlabeled TAF4N/C on the basis of the following assumptions. (1) Binding stoichiometry between Sp1‐QB and TAF4N/C is 1:1. (2) The peak intensity is proportional to the monomeric fraction of 15N‐Sp1‐QB, namely, the heterodimer composed of Sp1‐QB and TAF4N/C does not contribute to the peak intensity. By using these assumptions, relative peak intensity as a function of the concentration of TAF4N/C added was analyzed with Eq. (1). As shown in Figure 3(D), a semi‐quantitative analysis appeared to reproduce the observed peak intensity. However, the estimated value of the binding constant was 4 × 103 M−1, which was slightly smaller than that obtained by the SPR analysis. Although the reason for this difference in binding constants estimated by two methods remains unclear, the assumption that “only the monomeric fraction contributes to peak intensity” may not be valid. In addition, the temperature difference between NMR (4°C) and SPR (15°C) experiments may also contribute to the difference.
Self‐association of QB versus the heterogeneous interaction with TAF4
We previously found that Sp1‐QB self‐associated with itself under the same conditions examined here, namely, at pH 7.3 and 4°C. This process was analyzed using analytical ultracentrifugation, and the binding constant was estimated to be 4.5 × 103 M−1 on the basis of a simple homo‐dimerization model. We also examined the signal intensities of the 1H‐15N HSQC spectra of Sp1‐QB at various concentrations, and found that the binding site was localized from the center to the C‐terminal of the molecule. The binding site for the self‐association of Sp1‐QB was similar to that for the interaction with TAF4N/C. This result suggests that the self‐association of Sp1‐QB domains and the heterogeneous interaction between Sp1‐QB and TAF4N/C compete with each other. It is important to note that the binding constant for the interaction with TAF4N/C was stronger than that for the self‐association of Sp1‐QB, suggesting that a heterodimer composed of Sp1‐QB and TAF4N/C is predominantly formed even if the two processes compete.
Glutamine‐rich domains are found in many nuclear proteins such as transcriptional factors, and are considered to be important for molecular recognition. The promoter‐specific transcriptional factor Sp1 also possesses two Q‐rich domains, QA and QB, which have been suggested to be responsible for the self‐association by Sp1 as well as the interaction with TAF4. Biochemical studies indicated that the self‐association of Sp1 enhances its transcriptional activity synergistically, and this effect is known as “superactivation.”23, 35 On the other hand, the interaction between Sp1 and TAF4 (hTFIID130), which is one of the components of the general transcription factor TFIID, is considered to recruit RNA polymerase II to the transcription initiation site and, thus, activate transcription.
In the present study, we found that the same region in the QB domain, from the center to the C‐terminal, was responsible for the self‐association of Sp1‐QB as well as the interaction with TAF4N/C. Therefore, we propose the following interaction model as possible events during the process of activation of certain DNA sequences by Sp1. [1] The Sp1 molecule first recognizes and binds via Zn‐fingers to the GC‐box located upstream of the transcriptional initiation site.36 [2] Sp1 molecules that bind to the same DNA will self‐associate with each other via their QB regions, and this process will synergistically enhance affinity for the other transcriptional factors. [3] TAF4 preferentially interacts with multiple Sp1 molecules, resulting in the recruitment of RNA polymerase as well as the dissociation of the preformed Sp1 homo‐oligomer. [4] The dissociation of the Sp1 homo‐oligomer will reduce affinity for TAF4 and may contribute to preventing prolonged transcriptional activation. Such a negative‐feedback mechanism is considered to be important for the proper regulation of the timing of gene activation.
A novel interaction mode of the disordered region
Significant advances in bioinformatics and genome sequencing have revealed the prevalence of disordered proteins in the eukaryotic proteome. Notably, nearly half of the eukaryotic proteome was found to contain regions of a significant size (> 50 residues) that were predicted to be disordered under physiological conditions,37 and a large proportion of these proteins were involved in crucial and complex cellular processes such as transcriptional regulation, translation, and signal transduction. The inherent flexibility of IDPs has been suggested to enable access to a broad conformational space for interactions with a wide array of macromolecular targets. Many IPDs undergo a disorder‐to‐order transition to form well‐defined structures upon binding to their cellular targets.38, 39 This process is called “coupled folding and binding,” and is suggested to be a common mechanism by which IDPs interact with target molecules.
We herein revealed that the two Q‐rich domains in the cellular‐specific transcriptional factor Sp1 were intrinsically disordered under the physiological conditions examined. In addition, the central region of TAF4, which is one of the components of general transcriptional factors and is suggested to interact with Sp1, was also largely disordered under the same conditions. We also found that one of the Q‐rich domains of Sp1, QB, interacted with the central region of TAF4 in NMR titration experiments as well as SPR measurements. The CD spectrum of a 1:1 mixture of these proteins was almost identical to the sum of two separately measured spectra of two proteins. This result suggests that that the interaction between Sp1‐QB and TAF4N/C is not accompanied by the significant conformational changes that are typically expected for the coupled folding and binding mechanism.25 Furthermore, in a previous study, we found that the homo‐oligomerization of Sp1‐QBs was also not accompanied by any significant conformational change.26 Therefore, the interaction mode of Sp1‐QB may be distinct from other IDPs that often change their conformations upon binding to the partner. Such a phenomenon may not be a major interaction mode of IDPs, but similar examples have been reported. Sigalov et al. showed that one of the IPDs, T Cell receptor ζ chain, formed a homodimer by itself, as well as a heterodimer with the SIV Nef protein.40, 41 Very similar to the case of Sp1‐QB, the homo‐ and heterodimerization of T Cell receptor ζ chain were not accompanied by any significant conformational change. Another example has also been found in the interaction between the C‐terminal domain of Caldesmon.42 These results may suggest a novel mode of interaction for IDPs that enables them to recognize many different cellular target molecules.
Supporting information
Figure S1. 1H‐15N HSQC (A) and one‐dimensional 1H (B) spectra of 15N‐TAF4(270‐700) recorded at 4°C, 25°C and 37°C.
Figure S2. (A) 1H‐15N HSQC spectra of 15N‐TAF4(270‐700) recorded at 4°C. (B) Overlay of the 1H‐15N HSQC spectra of 15N‐Q1Q2 (red), 15N‐CI (green), and 15N‐Q3Q4 (blue) recorded separately at 4°C.
Figure S3. The interaction between Sp1‐QA and TAF4N/C measured by the SPR. Sp1–QA was immobilized on an HTG chip via the hexahistidine‐tag at the C‐terminus. The lines represent the sensorgrams recorded after loading TAF4N/C for 150 s at a concentration of 2 (red), 1.2 (green) and 0.4 μM (blue), respectively, followed by the flow of its absence for 300 s.
Acknowledgment
This work was supported by Grant‐in‐Aid for Scientific Research, 26440025 to MH, 15H02042, 26102524 to MS, 15K18515 to RI. SAXS experiments at Photon Factory were performed under Proposal No. 2014G0162, No. 2014G0127, No. 2015G0658 and No. 2016G0174, respectively. Preliminary SAXS experiments were performed using Small and wide angle X‐ray scattering instrument (NANOPIX, RIGAKU) at Research Reactor Institute, Kyoto University.
References
- 1. Hampsey M (1998) Molecular genetics of the RNA polymerase II general transcriptional machinery. Microbiol Mol Biol Rev 62:465–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Roeder RG (1996) The role of general initiation factors in transcription by RNA polymerase II. Trends Biochem Sci 21:327–335. [PubMed] [Google Scholar]
- 3. Albright SR, Tjian R (2000) TAFs revisited: more data reveal new twists and confirm old ideas. Gene 242:1–13. [DOI] [PubMed] [Google Scholar]
- 4. Blackwood EM, Kadonaga JT (1998) Going the distance: a current view of enhancer action. Science 281:60–63. [DOI] [PubMed] [Google Scholar]
- 5. Bulger M, Groudine M (1999) Looping versus linking: toward a model for long‐distance gene activation. Genes Dev 13:2465–2477. [DOI] [PubMed] [Google Scholar]
- 6. Hoey T, Dynlacht BD, Peterson MG, Pugh BF, Tjian R (1990) Isolation and characterization of the Drosophila gene encoding the TATA box binding protein, TFIID. Cell 61:1179–1186. [DOI] [PubMed] [Google Scholar]
- 7. Verrijzer CP, Chen JL, Yokomori K, Tjian R (1995) Binding of TAFs to core elements directs promoter selectivity by RNA polymerase II. Cell 81:1115–1125. [DOI] [PubMed] [Google Scholar]
- 8. Burke TW, Kadonaga JT (1997) The downstream core promoter element, DPE, is conserved from Drosophila to humans and is recognized by TAFII60 of Drosophila . Genes Dev 11:3020–3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Burley SK, Roeder RG (1996) Biochemistry and structural biology of transcription factor IID (TFIID). Annu Rev Biochem 65:769–799. [DOI] [PubMed] [Google Scholar]
- 10. Tanese N, Pugh BF, Tjian R (1991) Coactivators for a proline‐rich activator purified from the multisubunit human TFIID complex. Genes Dev 5:2212–2224. [DOI] [PubMed] [Google Scholar]
- 11. Chen JL, Attardi LD, Verrijzer CP, Yokomori K, Tjian R (1994) Assembly of recombinant TFIID reveals differential coactivator requirements for distinct transcriptional activators. Cell 79:93–105. [DOI] [PubMed] [Google Scholar]
- 12. Dynlacht BD, Hoey T, Tjian R (1991) Isolation of coactivators associated with the TATA‐binding protein that mediate transcriptional activation. Cell 66:563–576. [DOI] [PubMed] [Google Scholar]
- 13. Hoey T, Weinzierl RO, Gill G, Chen JL, Dynlacht BD, Tjian R (1993) Molecular cloning and functional analysis of Drosophila TAF110 reveal properties expected of coactivators. Cell 72:247–260. [DOI] [PubMed] [Google Scholar]
- 14. Ferreri K, Gill G, Montminy M (1994) The cAMP‐regulated transcription factor CREB interacts with a component of the TFIID complex. Proc Natl Acad Sci USA 91:1210–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tanese N, Saluja D, Vassallo MF, Chen JL, Admon A (1996) Molecular cloning and analysis of two subunits of the human TFIID complex: hTAFII130 and hTAFII100. Proc Natl Acad Sci USA 93:13611–13616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mengus G, May M, Carré L, Chambon P, Davidson I (1997) Human TAF(II)135 potentiates transcriptional activation by the AF‐2s of the retinoic acid, vitamin D3, and thyroid hormone receptors in mammalian cells. Genes Dev 11:1381–1395. [DOI] [PubMed] [Google Scholar]
- 17. Saluja D, Vassallo MF, Tanese N (1998) Distinct subdomains of human TAFII130 are required for interactions with glutamine‐rich transcriptional activators. Mol Cell Biol 18:5734–5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gill G, Pascal E, Tseng ZH, Tjian R (1994) A glutamine‐rich hydrophobic patch in transcription factor Sp1 contacts the dTAFII110 component of the Drosophila TFIID complex and mediates transcriptional activation. Proc Natl Acad Sci USA 91:192–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pugh BF, Tjian R (1990) Mechanism of transcriptional activation by Sp1: evidence for coactivators. Cell 61:1187–1197. [DOI] [PubMed] [Google Scholar]
- 20. Kadonaga JT, Jones KA, Tjian R (1986) Promoter‐specific activation of RNA polymerase II transcription by Sp1. Trends Biochem Sci 11:20–23. [Google Scholar]
- 21. Dynan WS, Tjian R (1983) Isolation of transcription factors that discriminate between different promoters recognized by RNA polymerase II. Cell 32:669–680. [DOI] [PubMed] [Google Scholar]
- 22. Courey AJ, Tjian R (1988) Analysis of Sp1 in vivo reveals multiple transcriptional domains, including a novel glutamine‐rich activation motif. Cell 55:887–898. [DOI] [PubMed] [Google Scholar]
- 23. Courey AJ, Holtzman DA, Jackson SP, Tjian R (1989) Synergistic activation by the glutamine‐rich domains of human transcription factor Sp1. Cell 59:827–836. [DOI] [PubMed] [Google Scholar]
- 24. Kadonaga JT, Carner KR, Masiarz FR, Tjian R (1987) Isolation of cDNA encoding transcription factor Sp1 and functional analysis of the DNA binding domain. Cell 51:1079–1090. [DOI] [PubMed] [Google Scholar]
- 25. Dyson HJ, Wright PE (2005) Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol 6:197–208. [DOI] [PubMed] [Google Scholar]
- 26. Hiramatsu N, Hibino E, Matsuzaki K, Kuwahara J, Hoshino M (2012) Interaction between isolated transcriptional activation domains of Sp1 revealed by heteronuclear magnetic resonance. Protein Sci 21:1481–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yamaguchi T, Yagi H, Goto Y, Matsuzaki K, Hoshino M (2010) A disulfide‐linked amyloid‐β peptide dimer forms a protofibril‐like oligomer through a distinct pathway from amyloid fibril formation. Biochemistry 49:7100–7107. [DOI] [PubMed] [Google Scholar]
- 28. Kohno T, Kusunoki H, Sato K, Wakamatsu K (1998) A new general method for the biosynthesis of stable isotope‐enriched peptides using a decahistidine‐tagged ubiquitin fusion system: an application to the production of mastoparan‐X uniformly enriched with 15N and 15N/13C. J Biomol NMR 12:109–121. [DOI] [PubMed] [Google Scholar]
- 29. Kusunoki H, Wakamatsu K, Sato K, Miyazawa T, Kohno T (1998) G protein‐bound conformation of mastoparan‐X: heteronuclear multidimensional transferred nuclear overhauser effect analysis of peptide uniformly enriched with 13C and 15N. Biochemistry 37:4782–4790. [DOI] [PubMed] [Google Scholar]
- 30. Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 6:277–293. [DOI] [PubMed] [Google Scholar]
- 31. Garrett DS, Powers R, Gronenborn AM, Clore GM (1991) A common sense approach to peak picking in two‐, three‐, and four‐dimensional spectra using automatic computer analysis of contour diagrams. J Magn Reson 95:214–220. [DOI] [PubMed] [Google Scholar]
- 32. Kohn JE, Millett IS, Jacob J, Zagrovic B, Dillon TM, Cingel N, Dothager RS, Seifert S, Thiyagarajan P, Sosnick TR, Hasan MZ, Pande VS, Ruczinski I, Doniach S, Plaxco KW (2004) Random‐coil behavior and the dimensions of chemically unfolded proteins. Proc Natl Acad Sci USA 101:12491–12496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kataoka M, Goto Y (1996) X‐ray solution scattering studies of protein folding. Fold Des 1:R107–R114. [DOI] [PubMed] [Google Scholar]
- 34. Uversky VN, Gillespie JR, Fink AL (2000) Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins 41:415–427. [DOI] [PubMed] [Google Scholar]
- 35. Pascal E, Tjian R (1991) Different activation domains of Sp1 govern formation of multimers and mediate transcriptional synergism. Genes Dev 5:1646–1656. [DOI] [PubMed] [Google Scholar]
- 36. Kuwahara J, Yonezawa A, Futamura M, Sugiura Y (1993) Binding of transcription factor Sp1 to GC box DNA revealed by footprinting analysis: different contact of three zinc fingers and sequence recognition mode. Biochemistry 32:5994–6001. [DOI] [PubMed] [Google Scholar]
- 37. Dunker AK, Brown CJ, Lawson JD, Iakoucheva LM, Obradović Z (2002) Intrinsic disorder and protein function. Biochemistry 41:6573–6582. [DOI] [PubMed] [Google Scholar]
- 38. Berlow RB, Dyson HJ, Wright PE (2015) Functional advantages of dynamic protein disorder. FEBS Lett 589:2433–2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Uversky VN (2015) The multifaceted roles of intrinsic disorder in protein complexes. FEBS Lett 589:2498–2506. [DOI] [PubMed] [Google Scholar]
- 40. Sigalov AB, Zhuravleva AV, Orekhov VY (2007) Binding of intrinsically disordered proteins is not necessarily accompanied by a structural transition to a folded form. Biochimie 89:419–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sigalov AB, Kim WM, Saline M, Stern LJ (2008) The intrinsically disordered cytoplasmic domain of the T cell receptor ζ chain binds to the Nef protein of simian immunodeficiency virus without a disorder‐to‐order transition. Biochemistry 47:12942–12944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Permyakov SE, Millett IS, Doniach S, Permyakov EA, Uversky VN (2003) Natively unfolded C‐terminal domain of caldesmon remains substantially unstructured after the effective binding to calmodulin. Proteins 53:855–862. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. 1H‐15N HSQC (A) and one‐dimensional 1H (B) spectra of 15N‐TAF4(270‐700) recorded at 4°C, 25°C and 37°C.
Figure S2. (A) 1H‐15N HSQC spectra of 15N‐TAF4(270‐700) recorded at 4°C. (B) Overlay of the 1H‐15N HSQC spectra of 15N‐Q1Q2 (red), 15N‐CI (green), and 15N‐Q3Q4 (blue) recorded separately at 4°C.
Figure S3. The interaction between Sp1‐QA and TAF4N/C measured by the SPR. Sp1–QA was immobilized on an HTG chip via the hexahistidine‐tag at the C‐terminus. The lines represent the sensorgrams recorded after loading TAF4N/C for 150 s at a concentration of 2 (red), 1.2 (green) and 0.4 μM (blue), respectively, followed by the flow of its absence for 300 s.