

Abstract
Affinity purification coupled with mass spectrometry (AP-MS) is a powerful technique for the identification and quantification of physical interactions. AP-MS requires careful experimental design, appropriate control selection and quantitative workflows to successfully identify bona fide interactors amongst a large background of contaminants. We previously introduced ProHits, a Laboratory Information Management System for interaction proteomics, which tracks all samples in a mass spectrometry facility, initiates database searches and provides visualization tools for spectral counting-based AP-MS approaches. More recently, we implemented Significance Analysis of INTeractome (SAINT) within ProHits to provide scoring of interactions based on spectral counts. Here, we provide an update to ProHits to support Data Independent Acquisition (DIA) with identification software (DIA-Umpire and MSPLIT-DIA), quantification tools (through DIA-Umpire, or externally via targeted extraction), and assessment of quantitative enrichment (through mapDIA) and scoring of interactions (through SAINT-intensity). With additional improvements, notably support of the iProphet pipeline, facilitated deposition into ProteomeXchange repositories and enhanced export and viewing functions, ProHits 4.0 offers a comprehensive suite of tools to facilitate affinity proteomics studies.
Keywords: Laboratory Information Management System, mass spectrometry, Data Independent Acquisition, protein-protein interactions, affinity purification coupled to mass spectrometry, proteomics
Graphical Abstract
Mass spectrometry (MS) data can be a challenge to manage, analyze and subsequently present to an audience in simple, intuitive ways. MS data must be archived, searched, scored, compared and visualized, often requiring a variety of unconnected, non-standardized software tools. These challenges are now compounded by the growing use of data-independent acquisition (DIA) methods [1–9], and more specialized applications such as interaction proteomics [10–14].
In 2010, we introduced an open source Laboratory Information Management System (LIMS) called ProHits [15, 16], designed to handle data generated in a proteomics facility, but which also provided specialized tools for the analysis of AP-MS, a popular proteomics application. ProHits is installed on a LINUX server (behind a firewall); via a web interface, authorized users can access search engines, analysis tools and multiple data visualization options. Through its Data Management module, ProHits automatically backs-up all data acquired in a mass spectrometry facility, converts vendor specific files to common formats (through ProteoWizard [17] or vendor software) and facilitates database searching using both free, open source tools (Comet [18], MSGF+ [19], X!Tandem [20]) and the commercial search engine Mascot [21]. Search results can be evaluated using the PeptideProphet [22] and ProteinProphet [23] components of the Trans Proteomics Pipeline (TPP[24]). The current version of ProHits also enables the use of iProphet [25] for combining the identification results from multiple search engines into a single output (Figure 1a; green boxes).
Figure 1. ProHits organization and protein interaction scoring.

A) The ProHits system consists of two principal modules, a Data Management module and an Analyst module. All mass spectrometers in a facility can be connected to ProHits: scheduled backups of the mass spectrometry data are performed followed by file conversion and database searches. DIA identification is supported by DIA-Umpire and the spectral matching tool MSPLIT-DIA. Peptide and protein identification results are parsed to a Sample defined in the Analyst module. The Samples are defined in a Project → Bait → Experiment → Sample hierarchy. Permissions for different projects are assigned to users in an Admin section. B) Schematic workflow for protein interaction analysis using SAINT through ProHits. Within a project, a user defines which samples should be analyzed and specifies which of those are the controls. The SAINT version (SAINTexpress or standard SAINT) is selected, alongside optional parameters and sample compression level. SAINT uses the quantitative matrix to derive the probability of interactions. Post analysis with SAINT, the data can be visualized or deposited in repositories from ProHits itself.
To enable sample tracking, an “Analyst” module helps users organize their data into projects to which different user permissions can be assigned (Figure 1a). Due to the strong focus of our team on affinity proteomics, the flow within each project is organized according to a “bait” protein. The bait is defined by its gene name, species, protein accession number and epitope tag(s), as appropriate. Note however, that the system can be used for any type of enrichment approach (e.g. with a nucleic acid or chemical compound as a bait), or even for more generic profiling by simply considering the “bait” level as part of an organizational hierarchy (there are 4 layers: “project” → “bait” → “experiment” → “sample”). Once a “bait” is entered, “experiments” can be associated to it, and annotated with text-based protocols (accessible via drop-down menus), controlled vocabularies, and free text notes. Under the “experiment” hierarchy, “samples” can be created (Figure 1a; orange boxes). Each sample is associated with a unique identifier: creating “samples” first in the Analyst module and naming the files on the mass spectrometry acquisition computer following this nomenclature ensures automatic links between the raw mass spectrometry data and the metadata associated with the sample description.
Besides providing a number of generic functions for organizing, browsing, searching, comparing and exporting data, ProHits provides a user-friendly interface for the analysis of interaction proteomics data via SAINT tools (both the standard SAINT [12, 13] and the computationally efficient SAINTexpress [10] algorithms are enabled; Figure 1b). Visualization of SAINT results directly through the interface or (after downloading) through publication-quality visualization tools [26] enables rapid exploration of the data. When a sufficient number of control experiments are unavailable or alternative scoring and visualization is desired, the user can download a CRAPome-compatible [11] file through the “export functions” within the Analyst module, facilitating re-analysis of the data.
We have redesigned the ProHits data management search functions to streamline standard data-dependent acquisition (DDA) searches (Supplementary Fig. 1), and enable peptide identification from DIA runs (Figure 1a; blue boxes). For DIA, untargeted identification is performed either through the generation of pseudo-MS/MS spectra from clusters of co-eluting MS1 and MS2 peaks using DIA-Umpire [27], or the spectral matching tool designed for peptide identification directly from DIA data, MSPLIT-DIA [28]. DIA-Umpire pseudo-MS/MS spectra can be analyzed with all search engines available in the local implementation of ProHits (currently, three open source tools, X!Tandem [20], Comet [18] and MS-GF+ [19] and the commercial tool Mascot [21] are supported), or a combination of search engines that can be combined into a single output using iProphet (Supplementary Fig. 2–4). MSPLIT-DIA identification requires a spectral library; this can be a pre-built generic spectral collection, such as the SWATHAtlas [29], or a library generated in-house using DDA files (currently, these files must be searched with MSGFDB [30]). Both functions (searching a preexisting or building a custom library) are supported by ProHits and can be further combined in a single search task (Supplementary Fig. 5,6). DIA-Umpire and MSPLIT-DIA support both fixed or variable DIA window designs and a generic file format (mzXML [31]), and in the case of DIA-Umpire, have been benchmarked on data from different instrument vendors. Search results from DIA-Umpire and MSPLIT-DIA can be directly parsed through the Analyst module, as is the case for DDA, and the ability to collapse protein results to genes is now supported within ProHits (Supplementary Fig. 7).
Arguably the most popular benefit of DIA is robust quantification. This is also supported through ProHits. For MSPLIT-DIA, besides extraction of spectral counts (which are more meaningful in DIA compared to DDA since there is a cyclic acquisition of each DIA window and no exclusion list [29]), outputs are prepared for quantification of peptides via targeted extraction through OpenSWATH [32], Skyline [33] and PeakView [34] (Figure 2a; Supplementary Fig. 6). Running MSPLIT-DIA prior to targeted extraction restricts the search space both by providing actual retention times and a list of the peptides expected to be in the sample: this was recently demonstrated to greatly facilitate the performance of these targeted extraction tools [29]. For DIA-Umpire [27], ProHits facilitates the selection of samples for targeted re-extraction, which improves the sensitivity of quantification (Figure 2b; Supplementary Fig. 4; note that DIA-Umpire results can also be further analyzed in Skyline outside of ProHits). In this mode, which is accessed through the Analyst module of ProHits, selected DIA-Umpire untargeted search tasks can be reviewed and selected data files can be re-extracted in the semi-targeted DIA-Umpire mode. Parameters such as the false discovery rates (at the peptide and protein levels) and the number of peptides and fragments to be extracted can be selected, and targeted re-extraction initiated. DIA-Umpire quantification results are available in several formats, including quantification at the MS1 and MS2 levels as direct output [27]. Alternatively, for interaction proteomics studies, the results can be further analyzed using the SAINT intensity model (initially built for MS1 data [35]). Results for SAINT quantification at either the MS1 or MS2 levels (benchmarked in the DIA-Umpire manuscript [27]) can be downloaded or viewed online.
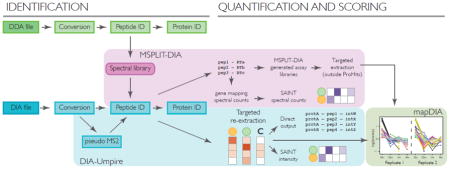
Figure 2. Handling DIA data in ProHits.

A) MSPLIT-DIA workflow in ProHits. In the Data Management module (green boxes) a spectral library can be built from parallel DDA runs (in the current release, only MSGFDB is supported for library generation); all previous analyses within a facility are assembled into an “Archive”. Public repositories (e.g. SWATHAtlas) are also enabled. ProHits uses the spectrum with the highest MSGFDB score as the library spectrum for peptide identification. Identified peptides can be used to generate an assay library to be fed into targeted extraction tools outside of ProHits; alternatively, ProHits has implemented a simple inference mapping of unique peptides to “genes” (here, identified as prot*) for spectral counting and SAINT scoring in the ProHits Analyst module (yellow boxes). B) DIA-Umpire workflow in ProHits. Untargeted identification is performed in the Data Management module, while semi-targeted re-extraction is performed in the Analyst module. Note that SAINT and SAINTexpress intensity models can be used to analyze DIA-Umpire results; alternatively, the results can be analyzed by mapDIA. C) mapDIA workflow in ProHits. The current version has fully implemented the statistical tool mapDIA as a downstream analysis tool post quantification by Umpire Quant; mapDIA also accepts data extracted by targeted extraction tools. mapDIA analysis parameters and experimental design options are selected in ProHits. Visualization of the mapDIA results (here, an example of a time course is shown) can be performed outside of ProHits on the ProHits-viz server (grey boxes).
To provide scoring of differentially abundant peptides and proteins across various workflows, including time-course experiments, we have also integrated the recently introduced statistical tool mapDIA [36], which is fully compatible with the DIA-Umpire quantification workflow. mapDIA can be used for statistical analysis of differential protein expression directly using fragment-level quantification data. A graphical user interface implemented in ProHits enables the user to specify which samples should be grouped and to select analysis parameters (Supplementary Fig. 8). The complete workflow of mapDIA is then performed, including normalization of fragment level intensities (total ion sum or local intensity sum in retention time space), outlier rejection, selection of the best fragments and peptides for quantification, and model-based statistical significance analysis of protein-level differential expression between the specified groups of samples (Figure 2c; Supplementary Fig. 8). Results are downloaded as a standard mapDIA output folder [36] with some basic visualization capability, though we have also implemented new visualization tools that can be used for figure generation as part of an expanded ProHits-viz suite of tools ([26]; a manuscript by Knight et al. describing new functionalities in ProHits-viz is in prep).
ProHits 4.0 also introduces several new functionalities, notably facilitated dataset submissions (including spectrum files, metadata and processed results) to ProteomeXchange [37] through MassIVE (massive.ucsd.edu) (Supplementary Fig. 9). Simply selecting the files used for an analysis (e.g. all files included in a SAINT task, including negative controls) and opting to transfer the data to MassIVE via the ProHits FTP server will package the dataset into different folders, namely raw files, peak lists (mzML, mzXML) and results files (mzid), named for direct association in MassIVE. Selection of the FASTA file associated with these files (via a dropdown menu) will create a fourth folder (“other”) that will also be submitted to MassIVE, facilitating complete submission to the ProteomeXchange. With other improvements including links to the reagent management system OpenFreezer [38], direct export to a quantitative interaction repository (ProHits-web.lunenfeld.ca)[39], and customizable export functions (Supplementary Fig. 10), ProHits 4.0 offers a comprehensive yet user friendly interaction proteomics Laboratory Information Management System.
Supplementary Material
Significance.
It remains challenging to score, annotate and analyze proteomics data in a transparent manner. ProHits was previously introduced as a LIMS to enable storing, tracking and analysis of standard AP-MS data. In this revised version, we expand ProHits to include integration with a number of identification and quantification tools based on Data-Independent Acquisition (DIA). ProHits 4.0 also facilitates data deposition into public repositories, and the transfer of data to new visualization tools.
Highlights.
Upgrade to the ProHits Laboratory Information System for proteomics
Support for Data Independent Acquisition through DIA-Umpire, MSPLIT-DIA, and mapDIA
Facilitation of deposition in ProteomeXchange through MassIVE
Visualization and export of proteomics data
Analytical pipeline for affinity purification coupled with mass spectrometry
Acknowledgments
We are grateful to all members of the Gingras lab for critical feedback on ProHits and on this manuscript and to Jeremy Carver for help with MassIVE submissions. We acknowledge funding from the Government of Canada through the Genome Canada Bioinformatics and Computational Biology LAP program (to A.-C.G., M.T., A.I.N and H.C), the Genome Canada Genome Innovation (GIN) network (through the Ontario Genomics Institute OGI-069 to A.-C.G.) and the Canadian Institutes of Health Research (Foundation grant FDN143301 to A.-C.G.); the US National Institutes of Health (to A.I.N. and A.-C.G.; 5R01GM94231; to N.B. 2 P41 GM103484-06A1; to M.T. R01RR024031); and the Singapore Ministry of Education (to H.C.; Tier 2 R-608-000-088-112). A.-C.G. is the Canada Research Chair in Functional Proteomics and the Lea Reichmann Chair in Cancer Proteomics; N.B. is an Alfred P. Sloan Research Fellow; B.R. is the Canada Research Chair in Molecular Medicine; M.T. is the Canada Research Chair in Systems and Synthetic Biology. J.-P.L. was supported by a TD Bank Health Research Fellowship at the Lunenfeld-Tanenbaum Research Institute and by a Scholarship for the Next Generation of Scientists from the Cancer Research Society.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gillet LC, Navarro P, Tate S, Rost H, Selevsek N, Reiter L, et al. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Molecular & cellular proteomics: MCP. 2012;11:O111 016717. doi: 10.1074/mcp.O111.016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Venable JD, Dong MQ, Wohlschlegel J, Dillin A, Yates JR. Automated approach for quantitative analysis of complex peptide mixtures from tandem mass spectra. Nature methods. 2004;1:39–45. doi: 10.1038/nmeth705. [DOI] [PubMed] [Google Scholar]
- 3.Silva JC, Gorenstein MV, Li GZ, Vissers JP, Geromanos SJ. Absolute quantification of proteins by LCMSE: a virtue of parallel MS acquisition. Molecular & cellular proteomics: MCP. 2006;5:144–56. doi: 10.1074/mcp.M500230-MCP200. [DOI] [PubMed] [Google Scholar]
- 4.Panchaud A, Scherl A, Shaffer SA, von Haller PD, Kulasekara HD, Miller SI, et al. Precursor acquisition independent from ion count: how to dive deeper into the proteomics ocean. Analytical chemistry. 2009;81:6481–8. doi: 10.1021/ac900888s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Geiger T, Cox J, Mann M. Proteomics on an Orbitrap benchtop mass spectrometer using all-ion fragmentation. Molecular & cellular proteomics: MCP. 2010;9:2252–61. doi: 10.1074/mcp.M110.001537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Egertson JD, Kuehn A, Merrihew GE, Bateman NW, MacLean BX, Ting YS, et al. Multiplexed MS/MS for improved data-independent acquisition. Nature methods. 2013;10:744–6. doi: 10.1038/nmeth.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Distler U, Kuharev J, Navarro P, Levin Y, Schild H, Tenzer S. Drift time-specific collision energies enable deep-coverage data-independent acquisition proteomics. Nature methods. 2014;11:167–70. doi: 10.1038/nmeth.2767. [DOI] [PubMed] [Google Scholar]
- 8.Purvine S, Eppel JT, Yi EC, Goodlett DR. Shotgun collision-induced dissociation of peptides using a time of flight mass analyzer. Proteomics. 2003;3:847–50. doi: 10.1002/pmic.200300362. [DOI] [PubMed] [Google Scholar]
- 9.Weisbrod CR, Eng JK, Hoopmann MR, Baker T, Bruce JE. Accurate peptide fragment mass analysis: multiplexed peptide identification and quantification. Journal of proteome research. 2012;11:1621–32. doi: 10.1021/pr2008175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Teo G, Liu G, Zhang J, Nesvizhskii AI, Gingras AC, Choi H. SAINTexpress: improvements and additional features in Significance Analysis of INTeractome software. Journal of proteomics. 2014;100:37–43. doi: 10.1016/j.jprot.2013.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mellacheruvu D, Wright Z, Couzens AL, Lambert JP, St-Denis NA, Li T, et al. The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nature methods. 2013;10:730–6. doi: 10.1038/nmeth.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Choi H, Liu G, Mellacheruvu D, Tyers M, Gingras AC, Nesvizhskii AI. Analyzing protein-protein interactions from affinity purification-mass spectrometry data with SAINT. In: Baxevanis Andreas D, et al., editors. Current protocols in bioinformatics. Unit8. Chapter 8. 2012. p. 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choi H, Larsen B, Lin ZY, Breitkreutz A, Mellacheruvu D, Fermin D, et al. SAINT: probabilistic scoring of affinity purification-mass spectrometry data. Nature methods. 2011;8:70–3. doi: 10.1038/nmeth.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Breitkreutz A, Choi H, Sharom JR, Boucher L, Neduva V, Larsen B, et al. A global protein kinase and phosphatase interaction network in yeast. Science. 2010;328:1043–6. doi: 10.1126/science.1176495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu G, Zhang J, Larsen B, Stark C, Breitkreutz A, Lin ZY, et al. ProHits: integrated software for mass spectrometry-based interaction proteomics. Nature biotechnology. 2010;28:1015–7. doi: 10.1038/nbt1010-1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu G, Zhang J, Choi H, Lambert JP, Srikumar T, Larsen B, et al. Using ProHits to store, annotate, and analyze affinity purification-mass spectrometry (AP-MS) data. In: Baxevanis Andreas D, et al., editors. Current protocols in bioinformatics. Unit8. Chapter 8. 2012. p. 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chambers MC, Maclean B, Burke R, Amodei D, Ruderman DL, Neumann S, et al. A cross-platform toolkit for mass spectrometry and proteomics. Nature biotechnology. 2012;30:918–20. doi: 10.1038/nbt.2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eng JK, Fischer B, Grossmann J, Maccoss MJ. A fast SEQUEST cross correlation algorithm. Journal of proteome research. 2008;7:4598–602. doi: 10.1021/pr800420s. [DOI] [PubMed] [Google Scholar]
- 19.Kim S, Pevzner PA. MS-GF+ makes progress towards a universal database search tool for proteomics. Nature communications. 2014;5:5277. doi: 10.1038/ncomms6277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Craig R, Beavis RC. TANDEM: matching proteins with tandem mass spectra. Bioinformatics. 2004;20:1466–7. doi: 10.1093/bioinformatics/bth092. [DOI] [PubMed] [Google Scholar]
- 21.Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–67. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 22.Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Analytical chemistry. 2002;74:5383–92. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 23.Nesvizhskii AI, Keller A, Kolker E, Aebersold R. A statistical model for identifying proteins by tandem mass spectrometry. Analytical chemistry. 2003;75:4646–58. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- 24.Deutsch EW, Mendoza L, Shteynberg D, Farrah T, Lam H, Tasman N, et al. A guided tour of the Trans-Proteomic Pipeline. Proteomics. 2010;10:1150–9. doi: 10.1002/pmic.200900375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shteynberg D, Deutsch EW, Lam H, Eng JK, Sun Z, Tasman N, et al. iProphet: multi-level integrative analysis of shotgun proteomic data improves peptide and protein identification rates and error estimates. Molecular & cellular proteomics: MCP. 2011;10:M111 007690. doi: 10.1074/mcp.M111.007690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Knight JD, Liu G, Zhang JP, Pasculescu A, Choi H, Gingras AC. A web-tool for visualizing quantitative protein-protein interaction data. Proteomics. 2015;15:1432–6. doi: 10.1002/pmic.201400429. [DOI] [PubMed] [Google Scholar]
- 27.Tsou CC, Avtonomov D, Larsen B, Tucholska M, Choi H, Gingras AC, et al. DIA-Umpire: comprehensive computational framework for data-independent acquisition proteomics. Nature methods. 2015;12:258–64. doi: 10.1038/nmeth.3255. 7 p following 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang J, Tucholska M, Knight JDR, Lambert JP, Tate S, Larsen B, et al. MSPLIT-DIA: sensitive peptide identification for Data Independent Acquisition. Nature methods. 2015 doi: 10.1038/nmeth.3655. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rosenberger G, Koh CC, Guo T, Rost HL, Kouvonen P, Collins BC, et al. A repository of assays to quantify 10,000 human proteins by SWATH-MS. Scientific data. 2014;1:140031. doi: 10.1038/sdata.2014.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim S, Mischerikow N, Bandeira N, Navarro JD, Wich L, Mohammed S, et al. The generating function of CID, ETD, and CID/ETD pairs of tandem mass spectra: applications to database search. Molecular & cellular proteomics: MCP. 2010;9:2840–52. doi: 10.1074/mcp.M110.003731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pedrioli PG, Eng JK, Hubley R, Vogelzang M, Deutsch EW, Raught B, et al. A common open representation of mass spectrometry data and its application to proteomics research. Nature biotechnology. 2004;22:1459–66. doi: 10.1038/nbt1031. [DOI] [PubMed] [Google Scholar]
- 32.Rost HL, Rosenberger G, Navarro P, Gillet L, Miladinovic SM, Schubert OT, et al. OpenSWATH enables automated, targeted analysis of data-independent acquisition MS data. Nature biotechnology. 2014;32:219–23. doi: 10.1038/nbt.2841. [DOI] [PubMed] [Google Scholar]
- 33.MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010;26:966–8. doi: 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lambert JP, Ivosev G, Couzens AL, Larsen B, Taipale M, Lin ZY, et al. Mapping differential interactomes by affinity purification coupled with data-independent mass spectrometry acquisition. Nature methods. 2013;10:1239–45. doi: 10.1038/nmeth.2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Choi H, Glatter T, Gstaiger M, Nesvizhskii AI. SAINT-MS1: protein-protein interaction scoring using label-free intensity data in affinity purification-mass spectrometry experiments. Journal of proteome research. 2012;11:2619–24. doi: 10.1021/pr201185r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Teo G, Kim S, Tsou CC, Collins B, Gingras AC, Nesvizhskii AI, et al. mapDIA: Preprocessing and Statistical Analysis of Quantitative Proteomics Data from Data Independent Acquisition Mass Spectrometry. Journal of proteomics. 2015 doi: 10.1016/j.jprot.2015.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vizcaino JA, Deutsch EW, Wang R, Csordas A, Reisinger F, Rios D, et al. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nature biotechnology. 2014;32:223–6. doi: 10.1038/nbt.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Olhovsky M, Williton K, Dai AY, Pasculescu A, Lee JP, Goudreault M, et al. OpenFreezer: a reagent information management software system. Nature methods. 2011;8:612–3. doi: 10.1038/nmeth.1658. [DOI] [PubMed] [Google Scholar]
- 39.Couzens AL, Knight JD, Kean MJ, Teo G, Weiss A, Dunham WH, et al. Protein interaction network of the mammalian Hippo pathway reveals mechanisms of kinase-phosphatase interactions. Science signaling. 2013;6:rs15. doi: 10.1126/scisignal.2004712. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.