

To the Editor
The analysis and storage of ever-increasing amounts of sequencing data present a huge computational challenge for the genomics community1. However, only a small proportion of this sequence information varies between individuals, and it is this variation that we hope to identify and understand. Recently, compressive genomics has been introduced as a way of improving the accuracy and efficiency of searching large sequencing databases2. Compressive genomics removes redundancies in genomic sequences and enables compressed data to be analyzed directly—facilitating parsimonious storage and fast access. This approach has been shown to accelerate the performance of standard search tools, such as BLAST and BLAT2. Although some read-mapping tools also remove redundancies in the reference genome to facilitate mapping individual reads3, 4, 5, they do not take full advantage of the redundancy across reads present in large sequencing data sets, which are often much larger and more redundant than the reference genome itself6 (Supplementary Figure S1a).
In this correspondence, we report the development of CORA (compressive read-mapping accelerator; Supplementary Software and http://cora.csail.mit.edu)—a computational tool that utilizes compressive acceleration to boost the performance of existing read mappers3, 4, 7 (Supplementary Figure S1b). CORA takes as input a sequencing read data set in FASTQ format and an off-the-shelf read-mapper. The read data sets are compressed into mid-size (typically ~30-60 bp) k-mer sets that contain only nonredundant sequence information. These representative k-mers are then mapped onto the reference genome using the existing read-mapper by means of a plug-in architecture. A high-resolution homology table is created for the reference sequence by mapping the reference to itself. The homology table contains all homologous pairs of loci in the reference above a similarity threshold, allowing fast, direct access to similar locations in the reference during mapping. The resulting mappings are outputted to a SAM file that can be readily integrated into existing sequence analysis pipelines.
CORA offers tractable mapping of terabyte-sized read data sets and achieves paired-end read mapping up to orders of magnitude faster than existing methods for mapping next-generation sequencing (NGS) reads from the 1000 Genomes Project and Mouse Genomes Project (Figure 1a,b and Supplementary Figure S2). Even for relatively small data sets (~100 GB and ~16× read depth-coverage), when such common mappers as BWA or Bowtie2 are plugged into CORA, our framework provides ~10 to ~4,700 times acceleration for ‘all-mapping’ (each read is mapped to all matching reference loci) with no loss in overall sensitivity, and ~2.7 to ~4.3 times for ‘best-mapping’ with a maximum sensitivity loss of ~1.2%. Mapping reads onto a reference genome in this way overcomes an important computational bottleneck in most sequence analysis pipelines (e.g., GATK)8, 9.
Figure 1.
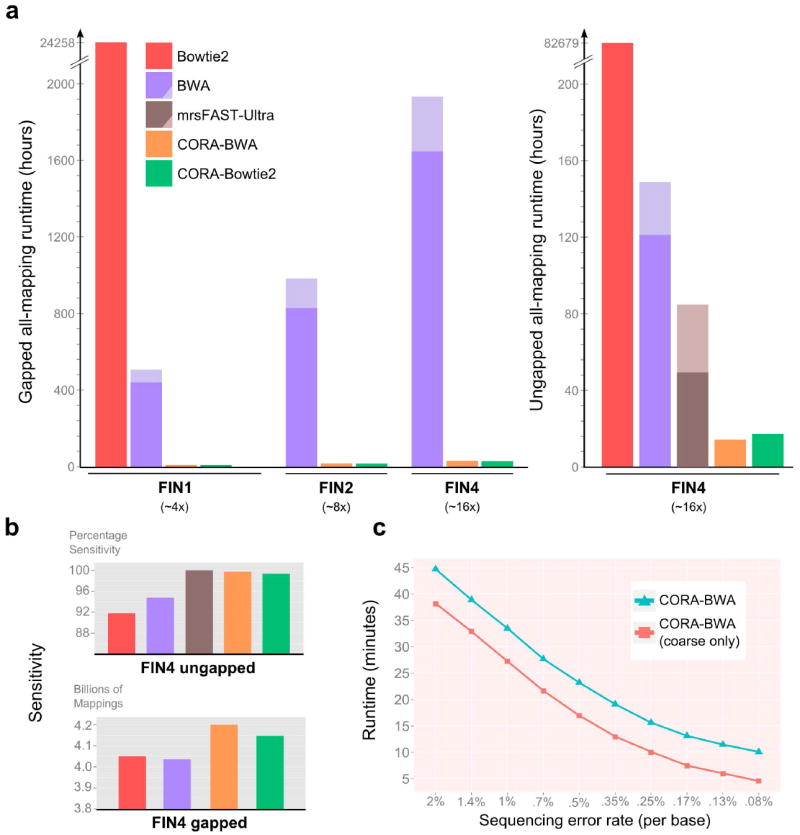
(a) Run-time comparison results between conventional read mapping methods and CORA for whole-genome gapped and ungapped all-mapping of 1000 Genomes Phase 1 Illumina 2 × 108 bp paired-end read data sets of one, two, and four Finnish individuals (FIN1, FIN2 and FIN4, with approximately 4×, 8× and 16× read depth-coverage, respectively; graph at left). The mapping similarity threshold is defined as the Levenshtein (edit) distance of 4 for each 108 bp-long read end. For the FIN4 data set, we additionally performed ungapped mapping experiments with the similarity threshold set as Hamming distance of 4 for each end (graph at right). We compared all-mapping run-times of Bowtie2 v2.1.0 (with ‘–a’ parameter) and BWA aln v0.7.5a (with ‘–N’ parameter) against compressively accelerated versions of each (CORA-Bowtie2 and CORA-BWA); for the ungapped mapping experiment, we also compared against mrsFAST-Ultra v3.3, which does not perform gapped mapping. We included read data set compression in the run-time for CORA mappers, but not time to build the homology table; similarly, we did not include genome indexing for other mappers. To ensure consistency across run-time comparisons, we assumed that all paired-end mappings of a read should be reported individually and consecutively, so that a downstream method can directly use the mapping output. Both CORA mappers and Bowtie2 readily satisfied these criteria; the additional computation needed to ensure this for BWA and mrsFAST-Ultra are indicated with a lighter shade (Supplementary Text). (b) Sensitivity comparisons indicate that CORA mappers are substantially more sensitive than BWA and Bowtie2 for both gapped (lower) and ungapped (upper) all-mapping. Though it does not have 100% sensitivity like mrsFAST-Ultra, CORA is able to report mapping results with near-perfect sensitivity (~99.7%) for ungapped all-mapping. Color key as in a. (c) CORA’s compressive framework achieves speed gains inversely related to the sequencing error rate. The graph shows the run-time of full and coarse ungapped mappings of CORA-BWA when aligning 20 million simulated paired-end reads (100 bp) onto hg19 chromosome 20 at varying sequencing error rates and a fixed mutation rate of 0.1%. ‘Coarse only’ run-time stands for the time required to run BWA within the CORA-BWA pipeline.
Because CORA identifies redundancies in both sequencing reads and reference data, its computational cost scales linearly with the size of the nonredundant data, which comprise a smaller portion of the total input data. Furthermore, CORA constructs a reference homology table data structure (Supplementary Text, Supplementary Figures S3 and S4), which also offers general utility beyond read mapping by providing fast access to all pairs of homologous loci in the reference genome (Supplementary Figure S5). Moreover, because CORA’s compressive framework achieves speed gains inversely related to the sequencing error rate, the acceleration it provides will substantially improve as sequencers generate higher-quality reads (Figure 1c, Supplementary Figures S6 and S7).
In contrast, existing read mappers require a costly sequence-comparison step, called seed-extension, which iteratively maps each read onto a reference genome. The computational cost is high even if the seed-matching step (finding exact matches for short seeds in the reference) is performed simultaneously on identical seeds across the read data set10, 11, 12, and the reference is stored, indexed or cached efficiently3, 4, 7. Moreover, as whole-read duplicate removal methods13 require the entire read sequence to be identical, their acceleration is negligible for longer paired-end-read data sets (>75 bp), such as those produced by common sequencing technologies (e.g., San Diego-based Illumina’s HiSeq X and HiSeq 2500; Supplementary Text). Thus, existing methods’ time requirements scale linearly with the size of the full read data set (Figure 1a), and increase each year along with the exponentially growing read data.
CORA accelerates both gapped and ungapped alignment of conventional best-mapping capabilities of state-of-the-art BWA and Bowtie2 (Supplementary Figure S2). However, CORA’s advance is particularly striking for ‘multi-reads’ (reads that map to multiple locations on the reference genome), enabling massively accelerated all-mapping, even in comparison to state-of-the-art all-mappers (e.g., mrsFAST-Ultra10), which achieves improvements in efficiency based on machine architecture, rather than leveraging redundancy in the data themselves. When CORA-BWA (i.e., CORA framework with BWA plugged-in) computes gapped all-mapping results from a ~16× depth-coverage read data set of four Finnish individuals from the 1000 Genomes Project, it is >62 times faster than BWA alone, and CORA-Bowtie2 is three orders of magnitude faster than Bowtie2 alone, both CORA versions demonstrating improvements in sensitivity. Our ungapped all-mapping experiments show that CORA-BWA is approximately sixfold faster than mrsFAST-Ultra for human, and approximately ninefold faster for mouse read data sets with minimal loss in sensitivity (<0.4%) (Figure 1a,b and Supplementary Text). Because of CORA’s use of memory-intensive data structures like the homology table (Supplementary Text), it has relatively higher memory usage (~50 GB) than the other mappers we tested (Figure 1 and Supplementary Figure S2, Supplementary Tables S1–S3). However, the added cost of physical memory is unlikely to be a hindrance to large-scale sequencing studies, given the cost-saving benefits, and high sensitivity of accelerated read-mapping using CORA (Supplementary Tables S4–S6).
All-mapping is an important component of many downstream analyses. It is the most robust way to comprehensively and accurately analyze structural variants, transposons, copy-number variants and other repeat elements within the genome14. There is increasing evidence that using multi-mapped reads enriches the statistical power and accuracy of single-nucleotide polymorphism (SNP) and structural variation discovery15, 16, 17, 18, transcriptome and isoform quantification19, 20, 21, 22, and RNA binding-site prediction23. Presently, all-mapping—in particular gapped all-mapping—is often not used by sequence analysis pipelines because of its high computational cost. CORA’s ability to compressively accelerate existing mappers to achieve sublinear time-scaling enables use of all-mapping in large-scale sequence analysis pipelines (Supplementary Figure S8 and Supplementary Text).
As state-of-the-art NGS technologies continue to improve and generate ever-increasing quantities of data with higher quality, the amount of redundant sequence information within them also increases. Compressive methods such as CORA—which scale sublinearly by operating directly on nonredundant data—may have an important part to play in how the biomedical community handles sequencing data in the upcoming years.
Supplementary Material
Acknowledgments
Special thanks to M.H. Baym and P.-R. Loh for launching compressive genomics with B.B. Thanks to M.H. Baym, P.-R. Loh, N.M. Daniels, R. Daniels, G. Tucker, H. Cho and P. Palmedo for most helpful comments. D.Y., J.P., Y.W.Y. and B.B. are partially supported by the US National Institutes of Health GM108348 (to B.B.). D.Y. is also partially supported by the Howard Hughes Medical Institute and IBM. Y.W.Y. gratefully acknowledges support from the Fannie and John Hertz Foundation.
References
- 1.Berger B, Peng J, Singh M. Nature Reviews Genetics. 2013;14:333–346. doi: 10.1038/nrg3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Loh PR, Baym M, Berger B. Nature Biotechnology. 2012;30:627–630. doi: 10.1038/nbt.2241. [DOI] [PubMed] [Google Scholar]
- 3.Li H, Durbin R. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Langmead B, Salzberg SL. Nature Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang L, Popic V, Batzoglou S. Bioinformatics. 2013;29:i361–i370. doi: 10.1093/bioinformatics/btt215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hsi-Yang Fritz M, Leinonen R, Cochrane G, Birney E. Genome Research. 2011;21:734–740. doi: 10.1101/gr.114819.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hach F, et al. Nature Methods. 2010;7:576–577. doi: 10.1038/nmeth0810-576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DePristo MA, et al. Nature Genetics. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sboner A, et al. Genome Biology. 2011;12:125. doi: 10.1186/gb-2011-12-8-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hach F, et al. Nucleic Acids Research. 2014;42 doi: 10.1093/nar/gku370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Siragusa E, Weese D, Reinert K. Nucleic Acids Research. 2013;41:e78. doi: 10.1093/nar/gkt005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li H, Durbin R. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Veeneman BA, Iyer MK, Chinnaiyan AM. BMC Bioinformatics. 2012;13:297. doi: 10.1186/1471-2105-13-297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Treangen TJ, Salzberg SL. Nature Reviews Genetics. 2012;13:36–46. doi: 10.1038/nrg3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hormozdiari F, et al. Genome Research. 2011;21:840–849. doi: 10.1101/gr.115956.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hormozdiari F, et al. Bioinformatics. 2010;26:i350–i357. doi: 10.1093/bioinformatics/btq216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Simola DF, Kim J. Genome Biology. 2011;12:R55. doi: 10.1186/gb-2011-12-6-r55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jubin C, Serero A, Loeillet S, Barillot E, Nicolas A. G3 (Bethesda) 2014;4:707–715. doi: 10.1534/g3.113.009464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Nature Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 20.Dao P, et al. Bioinformatics. 2014;30:644–51. doi: 10.1093/bioinformatics/btt591. [DOI] [PubMed] [Google Scholar]
- 21.Li B, Dewey CN. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anders S, Pyl PT, Huber W. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kelley DR, et al. Genome Research. 2014;15:537. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.