

Summary
Acute myeloid leukemia (AML) is an aggressive cancer with a poor prognosis, for which mainstream treatments have not changed for decades. To identify additional therapeutic targets in AML, we optimize a genome-wide clustered regularly interspaced short palindromic repeats (CRISPR) screening platform and use it to identify genetic vulnerabilities in AML cells. We identify 492 AML-specific cell-essential genes, including several established therapeutic targets such as DOT1L, BCL2, and MEN1, and many other genes including clinically actionable candidates. We validate selected genes using genetic and pharmacological inhibition, and chose KAT2A as a candidate for downstream study. KAT2A inhibition demonstrated anti-AML activity by inducing myeloid differentiation and apoptosis, and suppressed the growth of primary human AMLs of diverse genotypes while sparing normal hemopoietic stem-progenitor cells. Our results propose that KAT2A inhibition should be investigated as a therapeutic strategy in AML and provide a large number of genetic vulnerabilities of this leukemia that can be pursued in downstream studies.
Keywords: CRISPR, genetic screen, genetic vulnerability, acute myeloid leukemia, AML, KAT2A, MB-3
Graphical Abstract
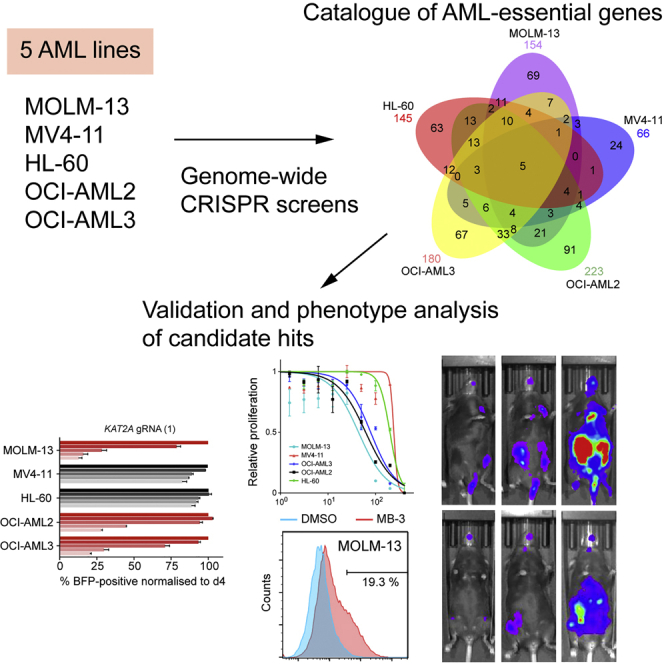
Highlights
-
•
Optimized CRISPR platform for identification of genome-wide genetic vulnerabilities
-
•
Catalog of genetic vulnerabilities in acute myeloid leukemia cell lines
-
•
KAT2A inhibition induces myeloid differentiation and apoptosis
-
•
KAT2A inhibition arrests the growth of primary AML cells, but not of normal progenitors
Tzelepis et al. optimize a CRISPR-Cas9-based platform for the performance of genome-wide recessive screens and apply it to identify genetic vulnerabilities of human AML cells. They identify several known therapeutic targets including BRD4, DOT1L, and MEN1, and numerous additional candidates. They provide data proposing KAT2A as a potential therapeutic target.
Introduction
The successful adaptation of the Streptococcus pyogenes-derived type II clustered regularly interspaced short palindromic repeats (CRISPR)-Cas system for genome editing is transforming the landscape of genetic research in many organisms (Cho et al., 2013, Cong et al., 2013, Jinek et al., 2012, Mali et al., 2013). Furthermore, the system’s high efficiency and flexibility make it ideal for use in genome-wide recessive genetic screens. In fact, recent proof-of-principle studies have demonstrated the potential of this technology to identify cell-essential genes in mammalian cells (Koike-Yusa et al., 2014, Shalem et al., 2014, Shi et al., 2015, Wang et al., 2014). Previously, this was typically conducted using RNA interference (RNAi) in the form of short interfering RNA (siRNA) or short hairpin RNA (shRNA) libraries (Boutros and Ahringer, 2008, Luo et al., 2009, Schlabach et al., 2008, Silva et al., 2008, Zuber et al., 2011). Such screens have made important contributions to biology, but their success has been moderated by the varying efficiencies of siRNAs/shRNAs for the stringent and specific suppression of target genes required for genome-wide studies (Boutros and Ahringer, 2008). CRISPR-Cas9-based functional genomics may be able to overcome such limitations and, therefore, hold great promise in re-shaping cell-essentiality screens. In cancer research, such screens can be applied to identify genetic vulnerabilities of cancer cells that can be used to develop new anti-cancer treatments. Recent reports on CRISPR screens on several cancer cell lines have demonstrated their power (Hart et al., 2015, Wang et al., 2015).
A human malignancy in urgent need of additional therapies is acute myeloid leukemia (AML), a devastating disorder with a long-term survival rate of less than 30% (Ferrara and Schiffer, 2013). Steady progress in deciphering its molecular pathogenesis has been made over the last few decades with a dramatic acceleration in recent years, particularly as a consequence of advances in cancer genomics (Cancer Genome Atlas Research Network., 2013, Welch et al., 2012). Despite such progress, the therapeutic landscape of AML has changed little for 40 years, with cytarabine still representing the last significant advance (Evans et al., 1961). Although the improved molecular understanding of AML permits some optimism that progress may be forthcoming, an alternative approach for the identification of therapeutic targets is the agnostic interrogation of AML genomes for genetic vulnerabilities using the CRISPR-Cas9 technology. Here, we make significant improvements in this technology and apply these to perform such a screen in AML.
Results
Optimization of Genome-wide CRISPR-Cas9 Dropout Screens
We and others have demonstrated that the CRISPR-Cas9 system can be adapted for use in functional genetic screens in the form of pooled guide RNA (gRNA) libraries, and that enrichment screens for genes whose inactivation confers resistance to toxins, chemotherapeutics, and targeted cancer treatments can be successfully conducted (Koike-Yusa et al., 2014, Shalem et al., 2014, Wang et al., 2014, Zhou et al., 2014). However, when we applied statistical analyses (Li et al., 2014) to our own genome-wide screen data in mouse embryonic stem cells (ESCs), we were able to identify only a small number of genes depleted to significant levels (Figure 1A). We reasoned that this may be secondary to non-uniform CRISPR-Cas9 efficiency across the large numbers of gRNAs in the library, leading to reduced technical and statistical robustness. To identify factors that affect gRNA efficiency, we first compared nucleotide composition between efficient and inefficient gRNAs in the mouse ESC screen. This analysis revealed strong nucleotide biases between positions 16 and 20 (Figure 1B). These biases also have been observed in human cells (Wang et al., 2014) as well as Caenorhabditis elegans (Farboud and Meyer, 2015), suggesting that they may be an intrinsic feature of the current S. pyogenes CRISPR-Cas9 platform.
Figure 1.

Optimization of CRISPR Dropout Screens and Validation
(A–D) Results of dropout screens in mouse ESCs (A and C) and nucleotide-level biases on gRNA efficiency (B and D) identified with version 1 (v1; A and B) and version 2 (v2; C and D) of the mouse genome-wide CRISPR libraries.
(E–G) Comparisons between gRNA counts (E) or gene-level significance of dropout and gene expression (F and G). An RNA-seq dataset (GSE44067; Zhang et al., 2013) was used and a cutoff of 0.5 FPKM was applied to distinguish expressed and non-expressed genes. The vast majority of gRNAs targeting non-expressed genes (E, left panel) exhibited equal representation between plasmid and day 14 mouse ESCs, indicating that the library complexity was maintained and that off-target effects were negligible. By contrast, a significant number of expressed genes are under- or over-represented in surviving day 14 ESCs. This is also evident at the gene-level analysis (F and G). The Kolmogorov-Smirnov test was used in (G).
To increase CRISPR-Cas9 efficiency, we first tested a gRNA scaffold optimized for CRISPR imaging (Chen et al., 2013) and found that, consistent with the results shown in a recent report (Dang et al., 2015), gRNAs with the improved scaffold exhibited significantly higher knockout efficiency than those with the conventional scaffold (Figures S1A and S1B). In addition, to generate an optimal gRNA library, we re-designed gRNAs for the mouse genome using a new design pipeline (see Supplemental Experimental Procedures) and generated a murine lentiviral gRNA library (version 2 [v2]) composed of 90,230 gRNAs targeting a total of 18,424 genes (Table S1). We then tested the performance of the v2 library, with regard to depletion (dropout) of genes, with the same experimental setting as with our first version (v1). With the optimized platform, many more genes were depleted at statistically significant levels (360 and 1,680 genes depleted at a false discovery rate [FDR] of 0.1 with the v1 and v2 library, respectively; Figure 1C; Data S1). Furthermore, the nucleotide biases observed in v1 were not observed with the v2 library (Figure 1D), indicating that on-target efficiency prediction (Doench et al., 2016, Wang et al., 2015) may not be necessary with the improved gRNA scaffold. The abundances of gRNAs targeting non-expressed genes (fragments per kilobase of transcript per million mapped reads [FPKM] ≤ 0.5) remained the same as the initial pool (plasmid), whereas large numbers of gRNAs with increased or decreased abundance in surviving ESCs were readily observed for expressed genes (FPKM > 0.5) (Figure 1E). At the gene level, the vast majority of depleted genes were expressed at FPKM > 0.5 in mouse ESCs (Figures 1F and 1G). Taken together, these data show that the sensitivity of our optimized CRISPR dropout screens for detecting cell-essential genes is markedly increased, whereas the off-target effects are negligible.
Generation and Validation of a Toolkit for CRISPR Dropout Screens in Human Cells
To perform CRISPR dropout screens in cancer cells, we generated a CRISPR functional screening toolkit composed of (1) lentiviral gRNA expression vectors harboring the improved scaffold (Figures S1C–S1E), (2) Cas9 activity reporters (Figures S1F–S1M), and (3) a human genome-wide CRISPR library (v1) consisting of 90,709 gRNAs targeting a total of 18,010 genes (Table S1). We then generated a pool of Cas9-expressing HT-29 colon cancer cells by lentiviral transduction and analyzed Cas9 activity using our reporter system. We found that a proportion of cells did not show detectable Cas9 activity despite growing under antibiotic selection (Figure S2A). Because the presence of Cas9-inactive cells can have an adverse impact on the efficiency of dropout screens, we sub-cloned Cas9-expressing cells and found that this eliminated Cas9-inactive cells (Figure S2B). We consistently observed the presence of Cas9-inactive cells in every cancer cell line tested thus far and found that these cells harbored mutations in the proviral Cas9 coding sequence with an APOBEC3 mutational signature (Hultquist et al., 2011) (Figure S2C). This Cas9-inactive fraction could be reduced by approximately 70% using a lentiviral construct carrying Cas9 upstream, rather than downstream, of the Blasticidin-resistant gene (Figures S1C, S2D, and S2E).
We proceeded to perform dropout screens in clonal Cas9-expressing HT-29 cells. Cells were harvested every 3 days from days 7 to 25 after transduction, and gRNA sequencing was performed (Data S2). As with the mouse ESC screen, a comparison between the screening results and RNA sequencing (RNA-seq) data revealed that the vast majority of depleted genes were expressed in HT-29 cells (Figures S3A and S3B), indicating that off-target effects were also negligible in our human CRISPR library. We identified approximately 2,000 depleted genes at a cutoff of FDR 20% and found that essential biological processes were enriched among them (Figures S3C–S3E).
Cancer cells often exhibit genomic instability associated with multiple copy number alterations (Beroukhim et al., 2010, Bignell et al., 2010, Zack et al., 2013). To investigate whether copy number affects CRISPR efficiency, we analyzed the distributions of dropout p values for individual genes according to their copy numbers and found no noticeable differences in dropout efficiency for genes with up to five copies (Figure 2A), although genes with three copies showed a modest but statistically significant reduction (adjusted p = 0.0217). By contrast, genes with eight copies, located on the Myc-centered distal region on chromosome 8 displayed a depletion pattern, which was very distinct to that of the surrounding region (Figures 2B and S3F). A similar depletion pattern in a continuous chromosome segment was previously observed in a highly amplified region in K562 cells (Wang et al., 2015). These results indicate that most genomic locations are amenable to dropout even when amplified and that knowledge of genome-wide copy number can help interpretation of genome-wide screens.
Figure 2.

Validation of the Human CRISPR Library in the HT-29 Colon Cancer Cell Line
(A) Effects of copy numbers on dropout efficiency in human colon cancer cell line, HT-29. Genes that were significantly depleted at day 25 (FDR < 10%) were grouped according to their copy number.
(B) Depletion p values (top) and copy number (bottom) of genes on chromosomes 8 and 20. Note that an eight-copy region containing Myc shows a clear distinction in the depletion pattern. Copy number data in HT-29 were obtained from the Catalogue of Somatic Mutations in Cancer (COSMIC) cell line database (http://cancer.sanger.ac.uk/cell_lines/).
(C and D) Hierarchical clustering of gene depletion. Genes that were significantly depleted on day 25 (FDR < 10%) were analyzed.
(E) Representative gene sets enriched in early intermediate- and late-depletion groups. The full list can be found in Table S2.
The Kolmogorov-Smirnov test was used in (A). See also Figures S1–S3, Tables S1, S2, and S3, and Data S2.
To investigate the timing of cell-essential gene depletion, we performed a longitudinal dropout analysis using the HT-29 dataset. A quarter of genes that were depleted at day 25 were already depleted by day 7, but the remaining cell-essential genes were depleted during the next 18 days (Figure S3D). An unsupervised cluster analysis of the depletion patterns identified seven clusters (Figure 2C). We further classified these clusters into three groups according to the time point at which depletions reached maximum significance (Figure 2D). Early genes, represented by clusters 1 and 5, were those that reached the highest significance before day 10. The intermediate group (clusters 2, 4, and 6) reached the highest depletion significance on day 13 or 16, whereas the late-depleting group (clusters 3 and 7) showed slow, gradual depletion, which reached maximal significance at later time points. Gene set enrichment analysis (GSEA) revealed dynamic changes in gene signatures over time (Figure 2E; Table S2). Essential biological processes for cell survival were significantly enriched in the early-depletion group, whereas processes involved in proliferation were depleted at early-to-intermediate time points. Genes in the late-depleting group seemed to represent genes whose loss was likely to have a lesser impact on proliferation. For example, this group included genes involved in glycosylphosphatidylinositol anchor biosynthesis, disruption of which leaves cells viable but slower to proliferate (Koike-Yusa et al., 2014). Taken together, CRISPR-Cas9-based dropout screens with our improved lentiviral libraries identified essential genes with high precision and performance in human cancer cells. Our findings establish a technical framework for the performance and interpretation of genome-wide dropout screens using the CRISPR-Cas9 technology.
Identification of Genetic Vulnerabilities in AML
Having optimized our platform, we proceeded to perform genome-wide dropout screens in five AML cell lines (MOLM-13, MV4-11, HL-60, OCI-AML2, and OCI-AML3) and the fibrosarcoma line HT-1080 as a second non-AML reference. Similar to HT-29, bulk Cas9-expressing cells included a fraction of cells without Cas9 activity, but single-cell cloning effectively eliminated this population and showed uniform Cas9 activity (Figures S4A and S4B). The karyotypes of the selected Cas9-expressing clones were analyzed for all AML lines and found to agree closely with the published karyotypes of the parental lines (Figures S4C and S4D). The selected clones were transduced with the human CRISPR library, cultured for 30 days, and harvested to determine their gRNA content (Table S3; Data S2). The genome-wide screens, performed using two biological replicates per line, identified circa 1,000–1,500 depleted genes in each AML cell line (Figure 3A). We first determined that significantly depleted genes were almost exclusively derived from those expressed at FPKM > 0.5 in the corresponding cell line (Figures S5A–S5F), showing that off-target effects were very limited and that gene dropouts were likely to have phenotypic consequences on cellular growth and/or survival. We also compared dropout efficiency of known cell-essential genes according to the number of copies of the chromosomes on which they are located and we found no significant difference (Figures S5G–S5K), indicating that Cas9 disrupted genes equally effectively irrespective of copy number in our AML cell lines.
Figure 3.

Identification of AML-Cell-Line-Specific Essential Genes
(A) Numbers of depleted genes in each of the seven cancer cell lines screened according to FDRs.
(B) Venn diagram depicting AML-cell-line-specific cell-essential genes defined as those depleted in at least one AML cell line and not in HT-1080 or HT-29.
(C) Gene ontology analysis of the 66 genes essential to three or more AML cell lines.
(D) Depletion of five gRNA against MLL according to their location relative to the MLL breakpoint region.
(E) Depletion of the FLT3, NRAS, and NPM1 genes affected by known oncogenic mutations in the specified AML cell lines and of BCL2, which was depleted in all AML cell lines except OCI-AML3, which carries a frameshift mutation in BAX.
See also Figures S4 and S5, Table S3, and Data S2.
To identify AML-specific vulnerabilities, we focused on genes depleted in one or more AML, but not in either of the non-AML cell lines (Table S3). This analysis identified 66–223 essential genes for each cell line (492 genes in total; Figure 3B), including 66 genes essential to three or more and 5 genes essential to all five AML cell lines. Gene ontology analysis of these genes showed particular enrichment in processes pertaining to chromatin modification and organization and transcriptional regulation (Figure 3C), in keeping with the fact that AML is driven by corrupted epigenetic and transcriptional networks.
We also specifically checked for depletion of driver mutations present in the AML cell lines screened. First, we looked at MLL (also known as KMT2A) and found that gRNAs targeting the exons upstream of the MLL breakpoint region, and therefore predicted to disrupt the MLL-AF9 and MLL-AF4 oncogenes, were depleted in both MOLM-13 and MV4-11 (Figure 3D). In addition, gRNAs against FLT3 and NRAS showed specific depletion in cell lines carrying activating mutations in these genes, whereas NPM1 was depleted in four of the five AML lines including OCI-AML3 (Figure 3E). Interestingly, BCL2 was depleted in all AML cell lines except OCI-AML3, which carries a BAX pE41fs∗33 mutation (Figure 3E), suggesting BAX mutations as candidate mediators of resistance to BCL2 inhibitors, a promising therapeutic strategy in AML (Chan et al., 2015, Pan et al., 2014).
Genetic and Pharmacological Validation of the Screening Results
To validate the results of our screen, we first demonstrated genetically the cell-essential nature of the five dropout genes shared by all AML cell lines (Figure S5M). We then selected eight dropout genes and a control non-dropout gene (HDAC6) for targeted inhibition using genetic and pharmacological approaches. We first followed a gene-by-gene knockout approach using the CRISPR-Cas9 system. Two gRNAs (one from our library and one new) were designed per gene, and the relative growth of gRNA-transduced and non-transduced cells were compared in competitive co-culture assays. Results were in close agreement with the findings of our dropout screens (Figures 4A, 4B, and S5N). We then tested the ability of existing clinical compounds to inhibit the growth of the five AML cell lines and again found these to be in consonance with the findings of our genome-wide screens (Figure 4C). MAP2K1 (also known as MEK1) and MAP2K2 (also known as MEK2) are thought to have redundant functions, but OCI-AML2 was sensitive to depletion of either gene. To test MEK1/2 dependency in the other AML cell lines, we devised a lentiviral dual gRNA expression vector (Figures S1C and S1E) and found that HL-60 and OCI-AML3 were sensitive only to double MEK1/2 knockout. This differential sensitivity to MEK1/2 genetic perturbation was mirrored in responses to the dual MEK1/2 inhibitor trametinib.
Figure 4.

Genetic and Pharmacological Validation of Screen Hits
(A) Significance levels for cell essentiality of selected genes in AML cell lines from our dropout screens.
(B) Validation of the findings of the screen using a 12-day competitive co-culture assay. Cells were transduced with lentivirus expressing one of two gRNAs per gene, and the BFP-positive fraction was compared with the non-transduced population. Results were normalized to day 4 for each gRNA. Data are shown as mean ± SD (n = 2). The full dataset can be found in Figure S5N.
(C) Effects of selected clinical inhibitors on cell growth. The results were normalized to DMSO-treated cells from each cell line cultured in parallel. Data are shown as mean ± SD (n = 3).
(D) Drug Gene Interaction database (DGIdb) (Griffith et al., 2013) categorization of AML-specific cell-essential genes into “druggable” categories defined by the DGIdb. Three categories are depicted. Full categorization can be found in Table S4. In the druggable set, representative genes in each of the three categories are listed.
Reassured by the concordance between the results of our screening and validation experiments, we searched the “druggability” of the 492 genes specifically depleted in our AML cell lines using the Drug Gene Interaction database (DGIdb) (Griffith et al., 2013) and found that 227 (46%) of the genes are in druggable categories (Figure 4D; Table S4). Among these were 33 genes, for which “clinically actionable” compounds are available, which overlap only partially with the “Kinase” and “histone modification” categories. However, the majority of genes in the druggable categories were not previously considered potential therapeutic targets (Figure 4D). Of note, at least 12 dropout genes, including BRD4, that were essential to at least three AML cell lines, as well as to HT-29 and HT-1080, are targets of clinical inhibitors (Table S4), indicating that “pan-essential” genes should not be dismissed as potential therapeutic targets.
Selection of Rational Therapeutic Development Targets
Our approach thus far has enabled us to define a set of genes that are essential to AML, but not to either of two solid cancer cell lines. However, it is probable that some of these AML-essential genes are also essential to normal blood cells including hemopoietic stem cells (HSCs), and such genes may not represent plausible therapeutic targets. Because currently no methods are available for systematic identification of essential genes in normal HSCs, we took an alternative strategy to identify therapeutic targets. In particular, we hypothesized that genes displaying cell line or oncogene specificity were less likely to cause toxicity to normal HSCs but could still be relevant to multiple AML genotypes. To do this, we compared the cell-essential genes of the MOLM-13 and MV4-11 cell lines. These both carry an internal tandem duplication in the FLT3 gene (FLT3-ITD) and multiple copies of chromosome 8, and exhibit comparable response to DOT1L and BRD4 inhibitors, but harbor the distinct, but related, fusion genes, MLL-AF9 (MOLM-13) and MLL-AF4 (MV4-11), known to directly establish leukemogenic transcriptional programs. Looking at the depleted genes (FDR < 0.2), we noted that MOLM-13 and MV4-11 showed significant overlap, but also many differences (Figure 5A; Table S3). Among these differentially essential genes, we selected two druggable genes for further study: the histone acetyltransferase gene KAT2A (also known as GCN5) and the spliceosome kinase gene SRPK1. We also chose CHEK1, a known therapeutic target (Daud et al., 2015, Zabludoff et al., 2008), as a control gene with a similar depletion pattern in both. In addition, we chose AURKB and HDAC3 as control essential genes to both and HDAC6 as essential to neither cell line (Figure 5A).
Figure 5.

Differential Vulnerabilities between MLL-AF4- and MLL-AF9-Driven Leukemias
(A) Comparison of dropout p values between MOLM-13 and MV4-11. AURKB and HDAC3 were significantly depleted in both lines, but HDAC6 was not in either line. In contrast, CHEK1, KAT2A, and SRPK1 were depleted only in MOLM-13. Genes that are specifically depleted in either cell line (FDR < 0.1) but not in either non-AML cell line are highlighted in pale red.
(B) Schematic of CRISPR-based validation of genotype-specific essentialities using ex vivo mouse leukemia model.
(C and D) Normal percentages of LK (Lin−/Kit+) and LSK (Lin−/Sca1+/Kit+) hemopoietic stem-progenitor cells were identified in the bone marrow of Rosa26Cas9/+ mice. Data are shown as mean ± SD (n = 3).
(E) Colony-forming assays of bone marrow cells derived from WT and Rosa26Cas9/Cas9 mice, showing no differences in replating ability of Rosa26Cas9/Cas9 cells compared with WT.
(F) Validation of Cas9 activity in Lin− or LK/LSK cells from Rosa26Cas9/+ mice using the Cas9 activity reporter.
(G) Growth kinetics of primary Lin− cells from Flt3ITD/+;Rosa26Cas9/+ mice transformed with a retrovirus expressing MLL-AF4 or MLL-AF9. Data are shown as mean ± SD (n = 4).
(H) Competitive co-culture assay showing oncogene-specific vulnerabilities in the ex vivo leukemia model. As a normal cell control, non-leukemic HPC-7 mouse hematopoietic cells were used. Results were normalized to day 4 for each gRNA. Data are shown as mean ± SD (n = 3).
The Student’s t test was performed in (D) and (E). Two-way ANOVA was performed in (G). See also Figure S6 and Table S6.
To test whether the observed essentialities for KAT2A and SRPK1 are indeed attributable to the different MLL oncogenic fusions rather than other differences between MOLM-13 and MV4-11, we developed a genetically defined experimental model (Figure 5B). First, we generated mice expressing Cas9 constitutively under the control of the ubiquitous EF1a promoter from the Rosa26 locus (Figure S6). Rosa26Cas9/+ mice reproduced in the expected Mendelian ratios exhibited normal long-term survival and had normal hemopoietic stem and progenitor cell numbers, and normal proportions of blood cell subtypes (Figures 5C, 5D, S6G, and S6H). Cas9-expressing stem-progenitor cells exhibited comparable colony-forming and serial replating activity to wild-type (WT) cells and displayed highly efficient Cas9 function (Figures 5E and 5F). These results indicate that Cas9 expression has no detectable effect on the hematopoietic system and that any phenotype observed in gRNA-expressing cells is likely caused by genetic perturbation of a target gene. Rosa26Cas9/+ mice were crossed to Flt3ITD/+ mice (Lee et al., 2007), and lineage-negative hemopoietic progenitors from RosaCas9/+;Flt3ITD/+ double transgenic mice were transduced with retroviral vectors expressing MLL-AF4 (Montes et al., 2011) or MLL-AF9 (Dawson et al., 2011) and cultured in vitro for 10–12 days, displaying similar exponential growth rates and a myeloid phenotype (Figure 5G). The cells were then independently transduced with individual lentiviruses carrying one of two gRNAs against the Kat2a, Srpk1, Aurkb, Chek1, Hdac3, and Hdac6 genes. In keeping with the results of our screen, this revealed significant differences in cell growth between MLL-AF4- and MLL-AF9-driven cells transduced with Chek1, Kat2a, and Srpk1 gRNAs, whereas Aurkb and Hdac3 gRNAs were equally effective against both cell types (Figure 5H). In addition, we tested the essentiality of each gene to the non-leukemic mouse multipotent HPC-7 cells, which represent an early blood stem-progenitor cell and are capable of generating functional hematopoietic cells in vivo (Wilson et al., 2016). As shown in Figure 5H, all three of the MLL-AF9-specific essential genes had no effects on proliferation of HPC-7 cells. Taken together, these results support our strategy to use oncogene-specific essentialities for candidate prioritization and provide genetic evidence that KAT2A and SRPK1 are attractive drug targets. We chose to investigate KAT2A further because this gene is essential to three of the five AML cell lines studied (MOLM-13, OCI-AML2, and OCI-AML3), and as such may be relevant to a wider group of AML patients. Of note, the OCI-AML3 line carries a mutation in NPM1, which is also found in 25%–35% of primary AMLs.
Mechanistic Insights into the Effects of KAT2A Inhibition in AML
First, using two separate gRNAs, we confirmed that genetic disruption of KAT2A reduced the growth of MOLM-13, OCI-AML2, and OCI-AML3, but not MV4-11 and HL-60 (Figures 6A and S5N). We confirmed that targeting with KAT2A-specific gRNA was associated with significantly reduced levels of KAT2A protein (Figure 6B). We then tested the effects of the KAT2A inhibitor MB-3 (Biel et al., 2004) on the growth of these lines and found that drug response mirrored the genetic validation studies (Figures 6C and 6D). To obtain mechanistic insights into the molecular effects of pharmacological KAT2A inhibition, we performed RNA-seq analysis on the sensitive MOLM-13 line after a 48-hr exposure to MB-3. We identified significant changes in gene expression including downregulation of genes associated with the MLL-AF9 leukemogenic program such as HOXA9, HOXA10, MEIS1, and MYC, and concomitant upregulation of genes associated with myeloid differentiation including ANPEP (CD13), ITGB2 (CD18), ITGAM (CD11b), and IL17RA (CD23) (Figures 6E and 6F). In keeping with these effects being the results of reduced KAT2A function, we confirmed that using chromatin immunoprecipitation (ChIP)-qPCR MB-3 led to reduction of the acetylation level at lysine-9 and lysine-27 of histone H3 at the downregulated gene loci: HOXA9, HOXA10, MEIS1, and MYC (Figure 6G). Microscopic and flow cytometry analyses of MOLM-13 cells after a 48 hr exposure to MB-3 confirmed monocytic-macrophage differentiation (Figure 6H) and increased CD13 surface expression (Figure 6I), whereas neither was observed in the MB-3-insensitive MV4-11 cells. Furthermore, prolonged incubation with MB-3 caused a marked increase in apoptosis of MOLM-13 and OCI-AML3, but not MV4-11 (Figure 6J). Taken together, these results indicated that KAT2A inhibition suppresses AML cell proliferation through inhibition of leukemogenic transcriptional programs and induction of differentiation leading to cell death by apoptosis.
Figure 6.

KAT2A Suppression Induces Myeloid Differentiation and Apoptosis
(A) CRISPR-based validation of KAT2A depletion in the five AML cell lines. Full results can be found in Figure S5N.
(B) Western blot analysis of KAT2A expression in MOLM-13 targeted by KAT2A-specific gRNA.
(C and D) Drug response (C) and 50% inhibitory concentration (IC50) values (D) of the five AML cell lines treated with the KAT2A inhibitor MB-3.
(E) Differentially expressed genes in MB-3-treated MOLM-13. AML program genes (downregulated) and myeloid marker genes (upregulated) are highlighted.
(F) Gene set enrichment analysis (GSEA) showing significant enrichment for the AML program and myeloid differentiation.
(G) Histone H3 acetylation status of genes downregulated by MB-3 treatment using ChIP-qPCR assay.
(H and I) Microscopic (H) and flow cytrometric (I) analyses of myeloid differentiation after 24-hr treatment with 100 μM MB-3. No changes were observed in MB-3-insensitive MV4-11 cells. Scale bar, 10 μm.
(J) Increased apoptosis after treatment with 100 μM MB-3.
Data are shown as mean ± SD (n = 3 in C, D, and J; n = 2 in G). The Student’s t test was performed in (D) and (G). ∗p < 0.05; ∗∗p < 0.01. See also Figure S5 and Table S6.
Clinical Potential of KAT2A Inhibition in AML Therapy
We next investigated whether KAT2A inhibition reduces cell proliferation in vivo. We first introduced the luciferase gene into MOLM-13-Cas9 cells and then transduced the cells with either an empty gRNA scaffold or a gRNA targeting the KAT2A gene. After 3 days of puromycin selection, transduced cells were transplanted into immunocompromised Rag2−/−;Il2rg−/− mice, which were then imaged for bioluminescence until death. We found that KAT2A disruption was associated with a significant reduction in AML cell expansion (Figures 7A and 7B) and prolongation of mouse survival (Figure 7C), indicating that KAT2A inhibition suppresses AML cell proliferation in vivo. Encouraged by these results, we proceeded to test the effects of MB-3 on primary human AML cells. Treatment of 10 primary AMLs of diverse genotypes (Table S5) with MB-3 led to significant reduction of colony formation in methylcellulose media at both 100 and 200 μM concentration (Figures 7D and 7E). By contrast, the colony-forming cell (CFC) efficiency of CD34+ human cord blood cells was not significantly affected by 100 or 200 μM MB-3 (Figure 7F). Taken together, our results show that KAT2A inhibition does not exhibit adverse effects on hematopoietic stem-progenitor cells and offers itself as a potential anti-AML therapeutic strategy for future studies.
Figure 7.

KAT2A Inhibition Shows Suppression of Leukemic Cell Growth In Vivo and Human Primary AML Cells
(A) Bioluminescence imaging of mice transplanted luciferase-labeled gRNA-transduced MOLM-13 cells at indicated time points.
(B) Quantification of luminescence. ∗∗∗∗p < 0.0001.
(C) Kaplan-Meier plot showing survival of mice transplanted with MOLM-13 expressing the indicated gRNA. Log rank test was performed.
(D and E) Colony-forming cell (CFC) assay of 10 primary AMLs of diverse genotypes with 100 and 200 μM MB-3. Detailed information can be found in Table S5. Mean values of 10 samples are shown in (E). Error bars represent SD. ∗p < 0.05.
(F) CFC efficiency of CD34+ human cord blood cells (n = 4).
The Student’s t test was performed in (B), (E), and (F). See also Table S5. N, normal karyotype; ND, not determined.
Discussion
Despite important advances in understanding their genomic and molecular pathogenesis, many cancers including AML continue to represent unmet clinical challenges (Cancer Genome Atlas Research Network., 2013, Döhner et al., 2015). It is therefore crucial to develop additional therapeutic strategies by identifying vulnerabilities in cancer cells. This can be achieved by either hypothesis-driven mechanistic studies or hypothesis-free unbiased genetic screening. In AML, recent detailed mechanistic studies have identified DOT1L as a vulnerability of MLL-rearranged leukemia (Bernt et al., 2011), and both a mechanistic and an RNAi-based epigenetics-focused screen identified BRD4 as a therapeutic target against AMLs of different genotypes (Dawson et al., 2011, Zuber et al., 2011). Drug development against these targets has rapidly progressed and their therapeutic efficacy is now being tested in clinical trials. Nevertheless, despite these successes, AML remains a lethal disease for most patients, and a complete set of genetic vulnerabilities for this and other cancers remains unknown, leaving many candidates with a therapeutic potential undiscovered.
To this end, we optimized and validated a robust CRISPR-Cas9 platform for the performance of genome-wide essentiality screens and applied this to catalog genetic vulnerabilities in AML. Our results have not only confirmed known therapeutic targets but also revealed a large number of genetic vulnerabilities in the AML cell lines studied, many of which represent plausible direct or indirect targets for drug development. Importantly, the unbiased nature of genome-wide screens such as ours makes them a powerful instrument for the identification of such targets, which is both orthogonal and complementary to mechanistic studies of disease pathogenesis and also able to reveal both intuitive and non-intuitive vulnerabilities.
Nevertheless, not all genetic vulnerabilities represent viable therapeutic targets. An important hurdle in selecting these is the real possibility that any genes essential to AML cells may also be essential to normal hemopoietic and/or non-hemopoietic cells, making their pharmacological inhibition harmful. To select targets that are likely to exhibit minimal adverse effects and thus have a higher likelihood of success in drug development, we applied a differential essentiality filter to our screen dataset and identified and characterized a potential AML therapeutic target, namely KAT2A. Genetic or pharmacological suppression of KAT2A did not show detectable adverse effects in either mouse HPC-7 hematopoietic precursor cell line or human cord blood CD34+ cells, further supporting that our approach was valid. It would be important to identify any toxic effects on hemopoietic stem-progenitor cells using a potent and bioavailable KAT2A inhibitor. Of course, in the absence of comprehensive datasets from other normal cell types, we cannot rule out the possibility that KAT2A suppression can cause side effects, and the generation of such datasets would significantly enhance our ability to predict clinical toxicity and identify the most promising therapies. It is, however, noteworthy that at least a dozen targets that have been in clinical use already were essential to cell types other than AML, suggesting that valuable targets can be found even among genes within this category and may potentially have a broad spectrum of antitumor activity.
Notwithstanding limitations in predicting clinical toxicity, our results demonstrate that KAT2A inhibition induces cellular differentiation and apoptosis of AML cells. Although the precise molecular basis of these effects will need to be investigated in future studies, the transcriptional changes associated with KAT2A inhibition suggest that the effects may be secondary to inhibition of leukemogenic transcriptional programs, in a manner reminiscent of BRD4 and DOT1L inhibition (Bernt et al., 2011, Dawson et al., 2011). KAT2A encodes a histone lysine acetyltransferase that functions within the multi-protein transcriptional co-activator complexes SAGA (Spt-Ada-Gcn5-acetyltransferase) or ATAC (Ada2a-containing); the former predominantly localizes at a subset of active promoters, whereas the latter localizes at distinct active promoters and enhancers (Krebs et al., 2011). As such, KAT2A influences diverse transcriptional programs and participates in multiple developmental and cellular processes (Wang and Dent, 2014). It has been shown that leukemia induction by MLL-AF9 requires the Myb-p300 interaction, which is thought to be responsible for the methylation-to-acetylation switch at the lysine-27 residue of histone H3 upon MLL-AF9 expression in HSCs (Pasini et al., 2010). One hypothesis is that a KAT2A-containing complex serves as a transcriptional coactivator that is also recruited to the target sites by MLL-AF9 and activates and/or maintains the leukemic transcriptional program. Alternatively, KAT2A might maintain the leukemic program through acetylation of non-histone proteins as exemplified by direct acetylation of the RUNX1/MDS1/EVI1 (Senyuk et al., 2003) and E2A-PBX1 (Holmlund et al., 2013) fusion oncoproteins by KAT2A and its homolog KAT2B (also known as PCAF). Further work is required to investigate the molecular function of KAT2A and determine the full therapeutic potential of this finding.
Our work demonstrates the power of unbiased genome-wide screens to catalog a comprehensive set of genetic vulnerabilities in cancer cells. Such catalogs enable not only the rapid identification of new targets and development of therapeutic strategies, but also generate hypotheses pertinent to the study of molecular mechanisms underlying tumorigenesis.
Experimental Procedures
All reagents and detailed methods are described in the Supplemental Information.
Plasmids, Cell Lines, Mouse Lines, and Reagents
Guide RNA expression vectors with the improved scaffold, pKLV2-U6gRNA5(BbsI)-PKGpuro2ABFP-W and pKLV2.2-h7SKgRNA5(SapI)-U6gRNA5(BbsI)-PGKpruo2ABFP-W, for a single and dual gRNA expression, respectively, were generated in this study and have been deposited with Addgene. The optimized human and murine CRISPR libraries were also available through Addgene. Guide RNA sequences used in a gene-by-gene approach are listed in Table S6. All AML cell lines (MOLM-13, MV4-11, HL-60, OCI-AML2, and OCI-AML3), colon cancer cell line HT-29, and fibrosarcoma cell line HT-1080 were obtained from the Sanger Institute Cancer Cell Line Panel and were mycoplasma free. Cas9-expressing cell lines were generated by lentiviral transduction using pKLV2-EF1aBsd2ACas9-W, and Cas9 activity in individual subclones was tested using a lentiviral reporter pKLV2-U6gRNA(gGFP)-PGKBFP2AGFP-W. A Cas9-expressing mouse line was generated by inserting the human EF1a promoter-driven Cas9 expression cassette into the Rosa26 locus in mouse ESC line JM8 (Pettitt et al., 2009) and is kept in the C57BL/6N background. See also Supplemental Information. All animal studies were carried out in accordance with the Animals (Scientific Procedures) Act 1986 (UK) and approved by the Ethics Committee at the Sanger Institute.
Generation of Genome-wide Mutant Libraries and Screening
A total of 3.0 × 107 cells were transduced with a predetermined volume of the genome-wide gRNA lentiviral supernatant. Two days after transduction, the cells were selected with puromycin for 4 days and further cultured. For HT-29, approximately 1 × 108 cells were harvested every 3 days between day 7 and day 25 post-transduction. The AML cell lines and HT-1080 were harvested on day 25 post-transduction. See also Supplemental Information.
gRNA Competitive Proliferation Assay
Cas9-expressing cells were transduced with a lentivirus expressing a gene-specific gRNA, and the percentage of blue fluorescent protein (BFP)-positive cells was measured between days 4 and 12 post-transduction and normalized to the percentage of BFP-positive cells at day 4. See also Supplemental Information.
Drug and Proliferation Assays
A total of 3 × 104 human or primary mouse cells were plated onto 96-well plates with vehicle or the indicated concentrations of compounds. Plates were measured 72 hr post-treatment using CellTiter 96 AQueous Non-Radioactive Cell Proliferation Assay (Promega). See also Supplemental Information.
Adult Primary Leukemia and Cord Blood Sample Analysis
All human AML and cord blood samples were obtained with informed consent under local ethical approval (REC 07-MRE05-44). Primary AML cells or cord-blood-derived CD34+ cells were tested for colony-forming efficiency in H4435 semi-solid medium (Stem Cell Technologies) in the presence of the indicated concentration of MB3 or DMSO. Colonies were counted by microscopy 10–11 days (AML cells) or 12–14 days (CD34+ cells) after plating. See also Supplemental Information.
Statistical Analysis
Statistical analyses performed and the numbers of replicates were mentioned in the associated figure legends. Differences were considered significant for p < 0.05.
Author Contributions
G.S.V., K.T., and K.Y. conceived the study and designed the experiments. Y.L. and K.Y. designed the mouse and human gRNA libraries. H.K-Y., S.O., and K.Y. generated the CRISPR toolkit and the Cas9 transgenic mice, and performed CRISPR screens in mouse ESCs and HT-29. K.T. performed CRISPR screens in the AML cell lines and conducted the validation and drug sensitivity assays with help from E.D.B., E.M., and K.Y. K.Y. performed the large-scale data analyses with help from K.T., F.I., E.D.B., H.P., and G.S.V. E.M., A.M., M.M., M.G., O.M.D., T.M., M.P., and J.C. performed cell culture and mouse experiments. O.M.D. and B.C. performed CRISPR screens for HT-1080. V.G. and U.M. generated and analyzed Cas9-expressing HT-29. M.L. performed analysis of RNA-seq data. J.S.-R. and B.J.P.H. contributed to study strategy, technical and analytical aspects. C.P. conceived and designed analysis of KAT2A inhibition in AML cell lines and in primary patient and cord blood samples, and performed the experiments with help from K.T. and A.F.D. S.T. supported RNA-seq analysis of MB3-treated MOLM-13 cells. P.G. and B.J.P.H. contributed with AML patient samples. K.T., E.D.B., G.S.V., and K.Y. wrote the paper with input from all authors.
Conflict of Interest
G.S.V. is a consultant for Kymab and received an educational grant from Celgene.
Acknowledgments
This work was funded by the Kay Kendall Leukaemia Fund (KKLF) and the Wellcome Trust (WT098051). G.S.V. is funded by a Wellcome Trust Senior Fellowship in Clinical Science (WT095663MA) and work in his laboratory is funded by Bloodwise. C.P. is funded by a Kay Kendall Leukaemia Fund Intermediate Fellowship (KKL888). We thank Gary Gilliland and Eric So for providing Flt3-ITD mice and the MSCV-MLL-AF4 plasmid, respectively. We thank Fengtang Yang and Beyuan Fu for M-FISH and cytogenetic analysis; Bee Ling Ng, Jennifer Graham, and Christopher Hall for help with flow cytometry; George Giotopoulos, Faisal Basheer, and the Cambridge Blood and Stem Cell Biobank for human AML sample processing; the Mouse Genetics Program for blood cell subtype analyses; and the Sanger Institute Core Sequencing facility for sequencing. We thank Mike Stubbington for help with RNA-seq analysis. We thank Roland Rad for helpful discussions and Keisuke Kaji for providing the validated gRNA sequence targeting CD54. We gratefully acknowledge the contribution of the recently deceased Professor Marc De Braekeleer to insightful discussion pertaining to this manuscript.
Published: October 18, 2016
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, six figures, six tables, and two datasets and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2016.09.079.
Contributor Information
Cristina Pina, Email: cp533@medschl.cam.ac.uk.
George S. Vassiliou, Email: gsv20@sanger.ac.uk.
Kosuke Yusa, Email: ky1@sanger.ac.uk.
Accession Numbers
The accession numbers for the CRISPR data reported in this paper are European Nucleotide Archive (http://www.ebi.ac.uk/ena): ERP006734 (mouse ESCs), ERP005600 (HT-29), and ERP008475 (AML and HT-1080). The accession numbers for the RNA-seq data reported in this paper are European Nucleotide Archive: ERP006662 and ERP003933. The CRISPR toolkit and the CRISPR libraries are available from Addgene.
Supplemental Information
The statistical result on gene depletion in all human cancer cell lines used in this study is shown. Depletion p values, depletion FDR, and RNA-seq counts (log10-transformed FPKM) are shown for each gene in each cell line. Note that genes whose RNA-seq count is ≤0.001 FPKM are all given a value of −3.5 as a log10-transformed value. Summaries of depleted genes in each human cancer cell line at FDR 20% or 10% are also shown in separate spreadsheets.
References
- Bernt K.M., Zhu N., Sinha A.U., Vempati S., Faber J., Krivtsov A.V., Feng Z., Punt N., Daigle A., Bullinger L. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell. 2011;20:66–78. doi: 10.1016/j.ccr.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beroukhim R., Mermel C.H., Porter D., Wei G., Raychaudhuri S., Donovan J., Barretina J., Boehm J.S., Dobson J., Urashima M. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biel M., Kretsovali A., Karatzali E., Papamatheakis J., Giannis A. Design, synthesis, and biological evaluation of a small-molecule inhibitor of the histone acetyltransferase Gcn5. Angew. Chem. Int. Ed. Engl. 2004;43:3974–3976. doi: 10.1002/anie.200453879. [DOI] [PubMed] [Google Scholar]
- Bignell G.R., Greenman C.D., Davies H., Butler A.P., Edkins S., Andrews J.M., Buck G., Chen L., Beare D., Latimer C. Signatures of mutation and selection in the cancer genome. Nature. 2010;463:893–898. doi: 10.1038/nature08768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutros M., Ahringer J. The art and design of genetic screens: RNA interference. Nat. Rev. Genet. 2008;9:554–566. doi: 10.1038/nrg2364. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013;368:2059–2074. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan S.M., Thomas D., Corces-Zimmerman M.R., Xavy S., Rastogi S., Hong W.J., Zhao F., Medeiros B.C., Tyvoll D.A., Majeti R. Isocitrate dehydrogenase 1 and 2 mutations induce BCL-2 dependence in acute myeloid leukemia. Nat. Med. 2015;21:178–184. doi: 10.1038/nm.3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B., Gilbert L.A., Cimini B.A., Schnitzbauer J., Zhang W., Li G.W., Park J., Blackburn E.H., Weissman J.S., Qi L.S., Huang B. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell. 2013;155:1479–1491. doi: 10.1016/j.cell.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho S.W., Kim S., Kim J.M., Kim J.S. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 2013;31:230–232. doi: 10.1038/nbt.2507. [DOI] [PubMed] [Google Scholar]
- Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A., Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang Y., Jia G., Choi J., Ma H., Anaya E., Ye C., Shankar P., Wu H. Optimizing sgRNA structure to improve CRISPR-Cas9 knockout efficiency. Genome Biol. 2015;16:280. doi: 10.1186/s13059-015-0846-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daud A.I., Ashworth M.T., Strosberg J., Goldman J.W., Mendelson D., Springett G., Venook A.P., Loechner S., Rosen L.S., Shanahan F. Phase I dose-escalation trial of checkpoint kinase 1 inhibitor MK-8776 as monotherapy and in combination with gemcitabine in patients with advanced solid tumors. J. Clin. Oncol. 2015;33:1060–1066. doi: 10.1200/JCO.2014.57.5027. [DOI] [PubMed] [Google Scholar]
- Dawson M.A., Prinjha R.K., Dittmann A., Giotopoulos G., Bantscheff M., Chan W.I., Robson S.C., Chung C.W., Hopf C., Savitski M.M. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. 2011;478:529–533. doi: 10.1038/nature10509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench J.G., Fusi N., Sullender M., Hegde M., Vaimberg E.W., Donovan K.F., Smith I., Tothova Z., Wilen C., Orchard R. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016;34:184–191. doi: 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Döhner H., Weisdorf D.J., Bloomfield C.D. Acute myeloid leukemia. N. Engl. J. Med. 2015;373:1136–1152. doi: 10.1056/NEJMra1406184. [DOI] [PubMed] [Google Scholar]
- Evans J.S., Musser E.A., Mengel G.D., Forsblad K.R., Hunter J.H. Antitumor activity of 1-beta-D-arainofuranosylcytosine hydrochloride. Proc. Soc. Exp. Biol. Med. 1961;106:350–353. [PubMed] [Google Scholar]
- Farboud B., Meyer B.J. Dramatic enhancement of genome editing by CRISPR/Cas9 through improved guide RNA design. Genetics. 2015;199:959–971. doi: 10.1534/genetics.115.175166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrara F., Schiffer C.A. Acute myeloid leukaemia in adults. Lancet. 2013;381:484–495. doi: 10.1016/S0140-6736(12)61727-9. [DOI] [PubMed] [Google Scholar]
- Griffith M., Griffith O.L., Coffman A.C., Weible J.V., McMichael J.F., Spies N.C., Koval J., Das I., Callaway M.B., Eldred J.M. DGIdb: mining the druggable genome. Nat. Methods. 2013;10:1209–1210. doi: 10.1038/nmeth.2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart T., Chandrashekhar M., Aregger M., Steinhart Z., Brown K.R., MacLeod G., Mis M., Zimmermann M., Fradet-Turcotte A., Sun S. High-resolution CRISPR screens reveal fitness genes and genotype-specific cancer liabilities. Cell. 2015;163:1515–1526. doi: 10.1016/j.cell.2015.11.015. [DOI] [PubMed] [Google Scholar]
- Holmlund T., Lindberg M.J., Grander D., Wallberg A.E. GCN5 acetylates and regulates the stability of the oncoprotein E2A-PBX1 in acute lymphoblastic leukemia. Leukemia. 2013;27:578–585. doi: 10.1038/leu.2012.265. [DOI] [PubMed] [Google Scholar]
- Hultquist J.F., Lengyel J.A., Refsland E.W., LaRue R.S., Lackey L., Brown W.L., Harris R.S. Human and rhesus APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H demonstrate a conserved capacity to restrict Vif-deficient HIV-1. J. Virol. 2011;85:11220–11234. doi: 10.1128/JVI.05238-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike-Yusa H., Li Y., Tan E.P., Velasco-Herrera Mdel.C., Yusa K. Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nat. Biotechnol. 2014;32:267–273. doi: 10.1038/nbt.2800. [DOI] [PubMed] [Google Scholar]
- Krebs A.R., Karmodiya K., Lindahl-Allen M., Struhl K., Tora L. SAGA and ATAC histone acetyl transferase complexes regulate distinct sets of genes and ATAC defines a class of p300-independent enhancers. Mol. Cell. 2011;44:410–423. doi: 10.1016/j.molcel.2011.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B.H., Tothova Z., Levine R.L., Anderson K., Buza-Vidas N., Cullen D.E., McDowell E.P., Adelsperger J., Fröhling S., Huntly B.J. FLT3 mutations confer enhanced proliferation and survival properties to multipotent progenitors in a murine model of chronic myelomonocytic leukemia. Cancer Cell. 2007;12:367–380. doi: 10.1016/j.ccr.2007.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Xu H., Xiao T., Cong L., Love M.I., Zhang F., Irizarry R.A., Liu J.S., Brown M., Liu X.S. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 2014;15:554. doi: 10.1186/s13059-014-0554-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J., Emanuele M.J., Li D., Creighton C.J., Schlabach M.R., Westbrook T.F., Wong K.K., Elledge S.J. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell. 2009;137:835–848. doi: 10.1016/j.cell.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P., Yang L., Esvelt K.M., Aach J., Guell M., DiCarlo J.E., Norville J.E., Church G.M. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montes R., Ayllón V., Gutierrez-Aranda I., Prat I., Hernández-Lamas M.C., Ponce L., Bresolin S., Te Kronnie G., Greaves M., Bueno C., Menendez P. Enforced expression of MLL-AF4 fusion in cord blood CD34+ cells enhances the hematopoietic repopulating cell function and clonogenic potential but is not sufficient to initiate leukemia. Blood. 2011;117:4746–4758. doi: 10.1182/blood-2010-12-322230. [DOI] [PubMed] [Google Scholar]
- Pan R., Hogdal L.J., Benito J.M., Bucci D., Han L., Borthakur G., Cortes J., DeAngelo D.J., Debose L., Mu H. Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid leukemia. Cancer Discov. 2014;4:362–375. doi: 10.1158/2159-8290.CD-13-0609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasini D., Malatesta M., Jung H.R., Walfridsson J., Willer A., Olsson L., Skotte J., Wutz A., Porse B., Jensen O.N., Helin K. Characterization of an antagonistic switch between histone H3 lysine 27 methylation and acetylation in the transcriptional regulation of Polycomb group target genes. Nucleic Acids Res. 2010;38:4958–4969. doi: 10.1093/nar/gkq244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettitt S.J., Liang Q., Rairdan X.Y., Moran J.L., Prosser H.M., Beier D.R., Lloyd K.C., Bradley A., Skarnes W.C. Agouti C57BL/6N embryonic stem cells for mouse genetic resources. Nat. Methods. 2009;6:493–495. doi: 10.1038/nmeth.1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlabach M.R., Luo J., Solimini N.L., Hu G., Xu Q., Li M.Z., Zhao Z., Smogorzewska A., Sowa M.E., Ang X.L. Cancer proliferation gene discovery through functional genomics. Science. 2008;319:620–624. doi: 10.1126/science.1149200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senyuk V., Sinha K.K., Chakraborty S., Buonamici S., Nucifora G. P/CAF and GCN5 acetylate the AML1/MDS1/EVI1 fusion oncoprotein. Biochem. Biophys. Res. Commun. 2003;307:980–986. doi: 10.1016/s0006-291x(03)01288-9. [DOI] [PubMed] [Google Scholar]
- Shalem O., Sanjana N.E., Hartenian E., Shi X., Scott D.A., Mikkelsen T.S., Heckl D., Ebert B.L., Root D.E., Doench J.G., Zhang F. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J., Wang E., Milazzo J.P., Wang Z., Kinney J.B., Vakoc C.R. Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains. Nat. Biotechnol. 2015;33:661–667. doi: 10.1038/nbt.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva J.M., Marran K., Parker J.S., Silva J., Golding M., Schlabach M.R., Elledge S.J., Hannon G.J., Chang K. Profiling essential genes in human mammary cells by multiplex RNAi screening. Science. 2008;319:617–620. doi: 10.1126/science.1149185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Dent S.Y. Functions of SAGA in development and disease. Epigenomics. 2014;6:329–339. doi: 10.2217/epi.14.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T., Wei J.J., Sabatini D.M., Lander E.S. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014;343:80–84. doi: 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T., Birsoy K., Hughes N.W., Krupczak K.M., Post Y., Wei J.J., Lander E.S., Sabatini D.M. Identification and characterization of essential genes in the human genome. Science. 2015;350:1096–1101. doi: 10.1126/science.aac7041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch J.S., Ley T.J., Link D.C., Miller C.A., Larson D.E., Koboldt D.C., Wartman L.D., Lamprecht T.L., Liu F., Xia J. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150:264–278. doi: 10.1016/j.cell.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson N.K., Schoenfelder S., Hannah R., Sánchez Castillo M., Schütte J., Ladopoulos V., Mitchelmore J., Goode D.K., Calero-Nieto F.J., Moignard V. Integrated genome-scale analysis of the transcriptional regulatory landscape in a blood stem/progenitor cell model. Blood. 2016;127:e12–e23. doi: 10.1182/blood-2015-10-677393. [DOI] [PubMed] [Google Scholar]
- Zabludoff S.D., Deng C., Grondine M.R., Sheehy A.M., Ashwell S., Caleb B.L., Green S., Haye H.R., Horn C.L., Janetka J.W. AZD7762, a novel checkpoint kinase inhibitor, drives checkpoint abrogation and potentiates DNA-targeted therapies. Mol. Cancer Ther. 2008;7:2955–2966. doi: 10.1158/1535-7163.MCT-08-0492. [DOI] [PubMed] [Google Scholar]
- Zack T.I., Schumacher S.E., Carter S.L., Cherniack A.D., Saksena G., Tabak B., Lawrence M.S., Zhsng C.Z., Wala J., Mermel C.H. Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 2013;45:1134–1140. doi: 10.1038/ng.2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Wong C.H., Birnbaum R.Y., Li G., Favaro R., Ngan C.Y., Lim J., Tai E., Poh H.M., Wong E. Chromatin connectivity maps reveal dynamic promoter-enhancer long-range associations. Nature. 2013;504:306–310. doi: 10.1038/nature12716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Zhu S., Cai C., Yuan P., Li C., Huang Y., Wei W. High-throughput screening of a CRISPR/Cas9 library for functional genomics in human cells. Nature. 2014;509:487–491. doi: 10.1038/nature13166. [DOI] [PubMed] [Google Scholar]
- Zuber J., Shi J., Wang E., Rappaport A.R., Herrmann H., Sison E.A., Magoon D., Qi J., Blatt K., Wunderlich M. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478:524–528. doi: 10.1038/nature10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The statistical result on gene depletion in all human cancer cell lines used in this study is shown. Depletion p values, depletion FDR, and RNA-seq counts (log10-transformed FPKM) are shown for each gene in each cell line. Note that genes whose RNA-seq count is ≤0.001 FPKM are all given a value of −3.5 as a log10-transformed value. Summaries of depleted genes in each human cancer cell line at FDR 20% or 10% are also shown in separate spreadsheets.