

Abstract
The initiation and progression of breast cancer from the transformation of the normal epithelium to ductal carcinoma in situ (DCIS) and invasive disease is a complex process involving the acquisition of genetic alterations and changes in gene expression, alongside microenvironmental and recognized histological alterations. Here, we sought to comprehensively characterise the genomic and transcriptomic features of the MCF10 isogenic model of breast cancer progression, and to functionally validate potential driver alterations in three‐dimensional (3D) spheroids that may provide insights into breast cancer progression, and identify targetable alterations in conditions more similar to those encountered in vivo. We performed whole genome, exome and RNA sequencing of the MCF10 progression series to catalogue the copy number and mutational and transcriptomic landscapes associated with progression. We identified a number of predicted driver mutations (including PIK3CA and TP53) that were acquired during transformation of non‐malignant MCF10A cells to their malignant counterparts that are also present in analysed primary breast cancers from The Cancer Genome Atlas (TCGA). Acquisition of genomic alterations identified MYC amplification and previously undescribed RAB3GAP1–HRAS and UBA2–PDCD2L expressed in‐frame fusion genes in malignant cells. Comparison of pathway aberrations associated with progression showed that, when cells are grown as 3D spheroids, they show perturbations of cancer‐relevant pathways. Functional interrogation of the dependency on predicted driver events identified alterations in HRAS, PIK3CA and TP53 that selectively decreased cell growth and were associated with progression from preinvasive to invasive disease only when cells were grown as spheroids. Our results have identified changes in the genomic repertoire in cell lines representative of the stages of breast cancer progression, and demonstrate that genetic dependencies can be uncovered when cells are grown in conditions more like those in vivo. The MCF10 progression series therefore represents a good model with which to dissect potential biomarkers and to evaluate therapeutic targets involved in the progression of breast cancer. © 2016 The Authors. The Journal of Pathology published by John Wiley & Sons Ltd on behalf of Pathological Society of Great Britain and Ireland.
Keywords: breast cancer progression, 3D spheroid assays, next‐generation sequencing
Introduction
The initiation and progression of breast cancer from the transformation of the normal epithelium to carcinoma in situ and invasive disease is a multifaceted process that results in the acquisition of multiple genomic alterations, including changes in genomic copy number, structural rearrangements, acquisition of mutations, altered gene expression, and pathway dysregulation 1, 2, 3, 4. The transition through these states, i.e. non‐invasive to invasive disease, is a well‐defined and staged process, through which breast cancers progress to acquire the capacity to grow, persist, and eventually spread to secondary sites.
High‐throughput molecular profiling of breast cancers and their precursor lesions has revealed that they have distinct genomic and transcriptomic alterations 3, 5, 6, 7, 8; however, matched preinvasive lesions and invasive counterparts from the same patient are remarkably similar 6, 7, 8, 9, 10, suggesting that the extent of genomic heterogeneity is determined early in breast cancer development. There is evidence suggesting that the progression from in situ to invasive disease is not exclusively driven by specific genomic aberrations in the preinvasive cells, but is a result of paracrine interactions of tumour cells with the surrounding stromal environment 3, 11, 12, 13.
The MCF10 progression series is a product of the ‘normal’ mammary epithelial cell line MCF10A that is spontaneously immortalised from the MCF10 mortal cell line (MCF10M), which originated from benign fibrocystic disease 14. As MCF10A cells are non‐tumorigenic, cells were HRAS‐transformed to produce MCF10neoT and MCF10AT1 cells 15, 16 (Figure 1A). MCF10AT1 cells were subsequently serially passaged in vivo to produce carcinoma in situ MCF10DCIS.com 17 and the invasive carcinoma cells MCF10Ca1a, MCF10Ca1d and MCF10Ca1h 18, 19. MCF10Ca1a and MCF10Ca1d are in vitro clones derived from the same in vivo tumour, whereas MCF10Ca1h is derived from a separate tumour (Figure 1A). This series of cell lines therefore represents an isogenic model of disease progression, and provides a useful tool for the investigation of molecular changes during the progression of human breast neoplasia and the generation of tumour heterogeneity on a common genetic background 19.
Figure 1.

Spectrum of acquired alterations in the MCF10 progression series. (A) Diagrammatic representation of the generation of the MCF10 progression series. Non‐invasive cell lines are highlighted in grey, DCIS.com cell are highlighted in green, and invasive cell lines are highlighted in red. Circled numbers represent the number of days for which cell lines were grown in vivo before replantation. (B) Matrix of identified somatic mutations acquired from MCF10A cells that also occur in TCGA breast cancer data. Mutations were classified according to membership of Cancer Gene Census (navy blue) 79, the results of the gene driver prediction algorithm FATHMM (purple) 61 and other prediction algorithms (grey) are shown (see Materials and methods). (C) Heatmap of gains (red) and losses (blue) identified from GISTIC. The genomic position is plotted along the x‐axis, and samples are plotted on the y‐axis. A heatmap of focal (<10 Mb) amplifications and homozygous deletions identified from GISTIC is shown. The colour scale bar depicts homozygous deletions to amplifications (–2 to +2). Note the presence of focal amplification of MYC (8q24.21) acquired from MCF10neoT cells onwards. (D) Chromosome 8 plots of MCF10A, MCF10neoT and MCF10Ca1a cells of log2‐normalized sequencing reads (y‐axis) plotted against base pair position cross chromosome 8. Green represents an amplification log2 ratio of >1.8. (E) Unrooted phylogenetic tree generated by neighbour joining of the MCF10 progression series based on variant data and whole arm chromosomal changes acquired from MCF10A cells onwards. Driver mutations and chromosomal changes acquired are indicated in grey.
Numerous studies have characterised different cell lines from the MCF10 progression series through the use of genomic, transcriptomic and proteomic profiling 20, 21, 22, 23, 24, 25, 26. These have shown that alterations that differ between the cell lines can identify drivers of different stages of breast cancer progression. Indeed, proteomic profiling has identified increased expression of AKT and STAT signalling in the invasive cell lines, events that are also known to occur in primary disease 26. Similar studies also identified secreted biomarkers that are known to be involved in metastasis of non‐invasive and invasive cells 27. The model has also proven useful in dissecting the role of poor prognostic biomarkers, such as BRMS1 and FSP1, in vitro 22, 28, and for the identification and functional assessment of novel biomarkers of progression from ductal carcinoma in situ (DCIS) to invasive disease with both three‐dimensional (3D) culture and in vivo models 21, 23, 29.
Here we sought to: (1) define the genomic characteristics, at base pair resolution, of the MCF10 breast cancer progression series of cell lines that are associated with different stages of progression; (2) determine the enrichment of pathway alterations in the progression from preinvasive to invasive disease; (3) establish an in vitro functional screening tool using cancer cell line spheroids, which more accurately recapitulate in vivo models; and (4) use this platform as biological proof of concept to identify potential driver genetic alterations of breast cancer progression.
Materials and methods
Cell lines
The isogenic MCF10 cell line series includes the initial untransformed normal cell line, MCF10A, the benign proliferation stages (MCF10AT1 and MCF10NeoT), the carcinoma in situ stage (MCF10DCIS.com), and the invasive carcinoma stages (MCF10Ca1a cl1, MCF10Ca1d cl1, and MCF10Ca1h). Cell lines were kindly provided by The Barbara Ann Karmanos Cancer Institute (Detroit, MI, USA), except for MCF10A cells, which were purchased from The American Type Culture Collection (LGC, Teddington, UK), and MCF10DCIS.com cells from Asterand (Royston, UK). Cells were authenticated by short tandem repeat typing with the Geneprint10 Kit (Promega, Southampton, UK), and routinely tested for mycoplasma infection by use of an enzyme‐linked immunosorbent assay‐based test (MycoAlert Mycoplasma detection kit; Lonza, Basel, Switzerland). Cells were grown as described previously 30 and in supplementary materials, Supplementary materials and methods.
Nucleic acid isolation
DNA was isolated with the DNeasy Blood and Tissue kit (Qiagen, Crawley, UK), and RNA was extracted with Trizol (Life Technologies, Paisley, UK), according to the manufacturers' instructions. Nucleic acids were quantified using the Qubit Fluorometer assay (Life Technologies), and RNA integrity was defined using a Bioanalyzer (Agilent, Santa Clara, CA, USA). All samples had an RNA integrity number of >9.
Exome sequencing
Genomic DNA (1 µg) was subjected to DNA capture with the Human All Exome V4 XT kit (Agilent, Santa Clara, CA, USA), and sequenced on 50% of a lane on an Illumina HiSeq2500, resulting in a minimum of × 109 median depth of coverage. Paired‐end reads in FASTQ format were aligned to the reference human genome build GRCh37, by the use of Burrows–Wheeler Aligner (BWA) 31. Variants were identified with the Genome Analysis Toolkit v3 (GATK) 32, and variant annotation was performed according to GATK Best Practices recommendations with Refseq and excluding decoy sequences 33, 34. Exome DNA sequencing was also performed with the Ion Torrent AmpliSeq technology (Life Technologies), according to the manufacturer's instructions (supplementary materials, Supplementary materials and methods), with a median depth of >100 for all samples. The Torrent Suite v4.0.2 pipeline (Life Technologies) was used to align raw reads and identify variants. Calls from the GATK were overlapped with calls from the Torrent Suite v4.0.2 pipeline to identify robust mutations. Candidate somatic mutations were called on the basis of filtering of variants with minor allele frequencies of >1% according to dbSNP build 132 and with <5 supporting reads. Mutations associated with progression in any cell line from MCF10neoT onwards were subsequently annotated on the basis of calls by Strelka 35, with MCF10As as the baseline comparator and manual review with the Integrative Genomics Viewer 36 to rule out the presence of reads supporting a given mutation in the ‘negative’ cell lines. Variants were subsequently annotated with Annovar 37. Mutations were overlaid with annotated data from primary breast cancers from The Cancer Genome Atlas (TCGA) 38 and subjected to functional prediction algorithms (supplementary materials, Supplementary materials and methods). A subset of variants taken forward for functional analysis were validated in all cell lines by Sanger sequencing, as described previously 39.
Whole genome sequencing
Libraries for whole genome sequencing were prepared with the NEBNext Ultra DNA Library Preparation kit (New England Biolabs, Hitchin, UK) from 1 µg of DNA, according to the manufacturer's protocol. Whole genome sequencing FASTQ files were aligned to the human genome (hg19) with BWA 40, and copy number variations were identified with the Patchwork 41 and GISTIC algorithms 42, as described in supplementary materials, Supplementary materials and methods. DNA was also subjected to high‐resolution microarray comparative genomic hybridization (aCGH) as described previously 43 and in supplementary materials, Supplementary materials and methods.
Paired‐end massively parallel RNA sequencing
RNA sequencing was performed with 100 ng of ribosomal‐depleted RNA from cell lines grown on plastic [two‐dimensional (2D) and as spheroids] as described previously 44 (see supplementary materials, Supplementary materials and methods). RNA sequencing FASTQ files were aligned to the human genome (GRCh37.73) with TopHat version 2.0.8b 45. Reads mapping to two or more locations were removed from the analysis. Differential gene expression analysis was performed with DESeq2, with an adjusted p‐value cut‐off of ≤0.01 46. Gene expression of the preinvasive cells (MCF10A, MCF10AT1, and MCF10neoT) was compared with that of the invasive cells (MCF10Ca1a, MCF10Ca1d, and MCF10Ca1h). MCF10DCIS.com cells were omitted from this analysis, given that they form preinvasive lesions in vivo that spontaneously become invasive 47. Fusion genes were identified with Chimerascan 48 and deFuse 49 algorithms. Pathway enrichment was performed with ConsensusPathDB 50.
Reverse transcription‐quantitative PCR (RT‐qPCR) and Sanger sequencing validation
Reverse transcription was performed with Superscript III (Invitrogen), with 500 ng of RNA per reaction, as described previously 51 and in supplementary materials, Supplementary materials and methods. Sequences were visualized by the use of 4Peaks (http://nucleobytes.com/4peaks/). In‐frame fusion genes were quantified in the cell line series with RT‐qPCR, and the abundance of the fusion transcript relative to β‐actin mRNA (ACTB) was calculated with the delta‐delta CT method. Primer sequences are listed in supplementary materials, Table S1.
Small interfering RNA (siRNA) screen
Genes were chosen to be screened with siGENOME smartpool siRNA (GE Healthcare, Little Chalfont, UK) targeting wild‐type genes in 96‐well spheroid assays, based on the presence of either: (1) recurrent amplifications and homozygous deletions, or (2) non‐synonymous coding mutations in the progression series. Alterations were chosen that were also present in primary tumours assessed in METABRIC 52 and aCGH studies 53, 54, 55 for copy number alterations, and from TCGA and other published studies at a frequency >0.5% for somatic mutations 56, 57, 58, 59, 60. For amplifications and homozygous deletions, those that are either known drivers or predicted drivers (for amplifications) as assessed from a significant correlation of amplification with gene expression 55 were selected. For mutations, those that are known to be drivers or predicted to be drivers from the prediction algorithm FATHMM 61 were triaged for functional assessment.
Three‐dimensional spheroid cultures
Five thousand cancer cells per well of a 96‐well low‐attachment plate (Corning, Amsterdam, The Netherlands) were reverse‐transfected with 37.5 nm siGENOME smartpool siRNA (GE Healthcare) or siControls [positive (ubiquitin B) and negative (pool 1)] by the use of Lullaby reagent (Oz Biosciences, Marseille, France) in 180 µl of cold culture medium, as described previously 62. Spheroid area was calculated with Celigo S (Nexcelom, Lawrence, MA, USA), and viability was measured with the CellTiter‐Glo assay (Promega). Relative growth was calculated relative to siControl. Hits were scored as >1.2 for increased spheroid growth and as <0.8 for reduced cell growth (see supplementary materials, Supplementary materials and methods).
Transfections of mammalian cells on plastic
Two thousand five hundred cancer cells per well of a 96‐well plate (Sigma‐Aldrich, Dorset, UK) were reverse‐transfected with 37.5 nm siGENOME siRNA by the use of Lullaby reagent (Oz Biosciences), as described previously 54 and in supplementary materials, Supplementary materials and methods.
Immunohistochemistry of spheroid cultures
Spheroids were grown for 7 days, and fixed in 3.8% formaldehyde for 30 min, washed with phosphate‐buffered saline three times, and stored at 4 °C. Spheroids were then pooled, dehydrated, embedded in paraffin, and sectioned. The spheroid sections were then deparaffinized with xylene, rehydrated, microwaved, and incubated overnight with primary antibodies against Ki67, TP53, and pAKT (see supplementary materials, Supplementary materials and methods).
Statistical analyses
p‐Values of <0.05 (heteroscedastic, two‐sided) were considered to be statistically significant for comparisons of the siRNA screen.
Data availability
Raw whole genome, exome and RNA sequencing data have been deposited in the NCBI Sequence Read Archive under the accession number PRJNA308098.
Results
Genomic alterations associated with breast cancer progression
To better understand the role of genomic and transcriptomic alterations in breast cancer progression, we performed whole exome, low‐depth whole genome and RNA sequencing of the MCF10 progression series (Figure 1A) to comprehensively define the repertoire of mutations, copy number alterations, expressed fusion genes and transcriptional alterations. Whole exome sequencing was performed with both capture and amplicon‐based sequencing at an average depth of × 100. This identified 7275 coding non‐synonymous variations [single‐nucleotide variants (SNVs)] in MCF10A, 7327 in MCF10neoT, 7336 in MCF10AT1, 7364 in MCF10DCIS.com, 7354 in MCF10Ca1a, 7351 in MCF10Ca1d, and 7358 in MCF10Ca1h. Taking MCF10A cells as the baseline, we identified mutations in 196 genes that were acquired in the malignant cell lines (i.e. not present in MCF10A non‐malignant cells; supplementary materials, Table S2), including 64 genes that also occur in TCGA and other published DNA sequencing studies 56, 57, 58, 59, 60 (Figure 1B). These included a PIK3CA hotspot mutation (H1047R) acquired in MCF10DCIS.com cells and maintained in the invasive cell lines MCF10Ca1a and MCF10Ca1h (in agreement with previous reports 63), and novel convergent mutations in TP53 in MCF10Ca1a and MCF10Ca1h cells. We next defined the presence of relevant breast cancer predicted driver gene mutations that were acquired in the malignant cells by annotating the variants with a combination of functional prediction algorithms (see Materials and methods) and the specific cancer driver prediction algorithm FATHMM 61. This analysis revealed 53 mutations that were predicted to disrupt protein function, and seven predicted cancer driver mutations. These encompassed four predicted cancer driver mutations that were acquired during transformation of non‐malignant MCF10A cells to malignant DCIS.com cells (HRAS, EPHA7, MAP3K12, and PCSK5), and three that were acquired during transformation of MCF10DCIS.com cells to invasive cells (MCF10Ca1a and MCF10Ca1h) (PTPRD, TP53, and VSP13A) (Figure 1E; supplementary material, Figures S1 and S2). Furthermore, 57% of all variants were expressed at the RNA level (supplementary material, Table S2).
Somatic copy number alterations associated with progression
We used low‐depth [on average, ×7 (range of coverage, ×6–9] whole genome sequencing to characterise the repertoire of copy number alterations of cell lines within the progression series. Consistent with previous observations 24, 63, MCF10A cells had high‐level gains of 1q, gains of 5q, 8q, 19q, and 20q, and homozygous deletion of 9p encompassing CDKN2A/B (supplementary material, Figure S3). Indeed, the other cell lines were comparable, with high‐level focal amplification of 8q24.21, encompassing MYC, 10q22.1–q22.2, and 17p11.2 (Figure 1D; supplementary material, Table S3). They did not have gain of 1q, and were therefore probably derived from a clone of the parental line that had normal 1q (Figure 1C). Interestingly, MCF10DCIS.com cells had both the gain of 1q seen in the parental MCF10A cells and the three focal high‐level amplifications. A number of homozygous deletions were acquired during progression, including 8p23.1 (MCF10DCIS.com, MCF10Ca1a, and MCF10Ca1d), 12p13.2 (MCF10AT1, MCF10DCIS.com, and MCF10Ca1a) and 22q12.2 (MCF10neoT, MCF10AT1, MCF10DCIS.com, MCF10Ca1a, and MCF10Ca1d) (Figure 1C; supplementary material, Figures S3 and S4 and Table S3). In addition, acquisition of a focal intragenic homozygous deletion of RUNX1 (MCF10DCIS.com, MCF10Ca1a, and MCF10Ca1d) was identified (in agreement with previous reports 63).
Fusion gene transcripts associated with progression
Previous studies have reported that breast cancers can show extensive large‐scale genomic rearrangements 64, and have documented the presence of expressed fusion genes that drive the malignant phenotype of the cells and present therapeutic opportunities 43, 65. RNA sequencing analysis of the MCF10 progression series identified expressed fusion genes in MCF10DCIS.com (n = 1), MCF10Ca1d (n = 1) and MCF10Ca1h (n = 2) that were identified by both deFUSE 49 and Chimerascan 48 fusion gene detection algorithms (supplementary material, Table S4). These included two fusion transcripts predicted to result in novel functional proteins (i.e. in‐frame) that were not present in MCF10A cells, namely an interchromosomal fusion involving RAB3GAP1 and HRAS, detected in MCF10DCIS.com and MCF10Ca1d cells, and an intrachromosomal fusion on chromosome 19q involving UBA2 and PDCD2L in MCF10Ca1h cells (Figure 2A, B). Validation of the fusion transcripts with RT‐qPCR and Sanger sequencing demonstrated that the in‐frame RAB3GAP1–HRAS fusion was present in all cell lines that had been subjected to HRAS transformation (i.e. from MCF10AT1 onwards), whereas UBA2–PDCD2L was only seen in MCF10Ca1h cells (Figure 2C, D; supplementary material, Table S4). Neither fusion was detected in the control cell line MCF7. Furthermore, the levels of RAB3GAP1–HRAS transcript expression increased from MCF10neoT cells across the progression series, with MCF10Ca1d cells showing the highest expression. This observation mirrored the HRAS gene expression levels detected in the RNA sequencing data (Figure 2E), suggesting that the differences in HRAS expression may be attributable to the presence of the fusion gene. Interestingly, the reciprocal fusion HRAS–RAB3GAP1 was detected in MCF10DCIS.com cells; however, this was not predicted to result in a functional protein. Mining of the TCGA Fusion gene Data Portal (http://54.84.12.177/PanCanFusV2/) 66 and other published fusion datasets in breast cancer 43, 64, 65 identified two additional in‐frame fusion genes involving RAP3GAP1 in breast cancer (RAB3GAP1–ACMSD and RAB3GAP1–MAP4K3). An in‐frame HRAS fusion gene was identified in a head and neck primary tumour (RNH1–HRAS) that leads to increased levels of HRAS expression 66; however, no additional HRAS fusion genes were detected in primary breast cancers. An out‐of‐frame UBA2–PDCD2L fusion was detected in a primary ovarian cancer, but none was observed in breast cancer.
Figure 2.
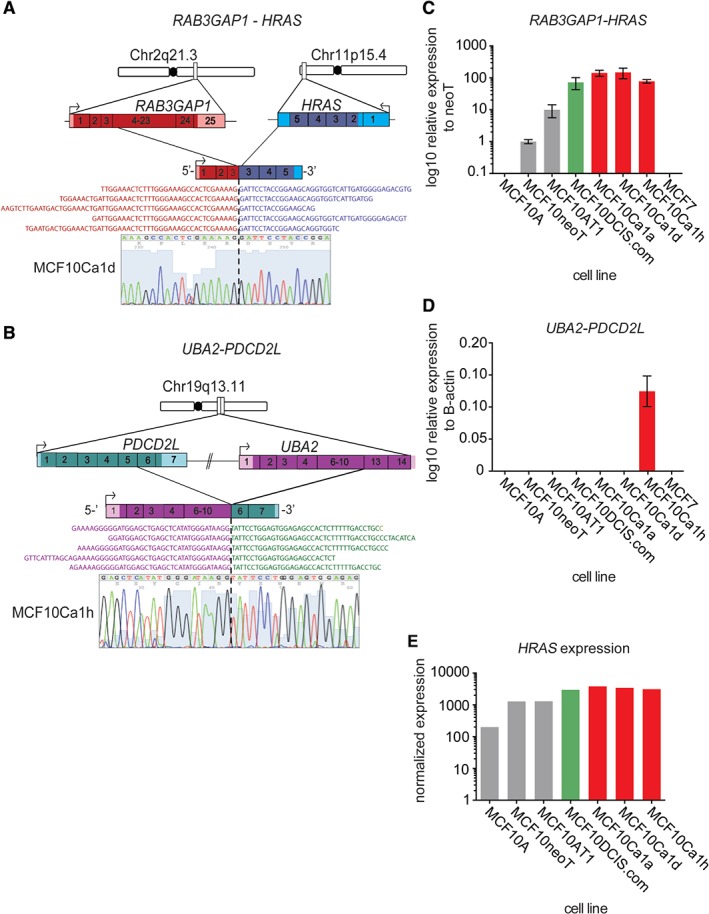
Identification of expressed fusion genes in the MCF10 progression series. (A) Cartoon representation of genomic location, orientation and architecture of the expressed RAB3GAP1–HRAS fusion gene. Representative RNA sequencing reads spanning the fusion are also displayed. The RT‐qPCR product was Sanger‐sequenced; confirming the fusion junction, and a representative chromatogram from MCF10Ca1d cells is shown. (B) Cartoon representation of genomic location, orientation and architecture of the expressed UBA2–PDCD2L fusion gene. Representative RNA sequencing reads spanning the fusion are also displayed. The RT‐qPCR product was Sanger‐sequenced, confirming the fusion junction, and a representative chromatogram from MCF10Ca1h cells is shown. (C) Bar plot showing relative expression of the RAB3GAP1–HRAS fusion gene in the MCF10 progression series detected by RT‐qPCR. (D) Bar plot showing relative expression of the UBA2–PDCD2L fusion gene in the MCF10 progression series detected by RT‐qPCR (E) Bar plot of normalized reads of HRAS in the MCF10 progression series from RNA sequencing.
Pathway alterations associated with breast cancer progression
We next sought to assess the differences in gene expression during progression from preinvasive to invasive disease. Differential gene expression of preinvasive cell lines (MCF10A, MCF10AT1, and MCF10NeoT) and invasive cell lines (MCF10Ca1a, MCF10Ca1d, and MCF10Ca1h) identified 236 significantly differentially expressed genes [false discovery rate (FDR) p‐value of <0.01; DEseq2] (Figure 3A; supplementary material, Table S5). These genes were enriched in pathways involved in platelet amyloid precursor protein processing, senescence, autophagy, and arachidonic acid metabolism (Figure 3B; supplementary material, Table S6). Previous studies have demonstrated that the MCF10 progression series behave differently when grown in 3D culture, and provide a useful model for studying driver alterations associated with oncogenic transformation 67, 68 and disease progression 69, 70. Indeed, cell lines from the progression series formed spheroids, and showed good growth kinetics and positive histological staining of the proproliferative markers Ki67 and phospho‐AKT (supplementary material, Figure S5). To further evaluate functional pathways that may be dysregulated in breast cancer progression, in cells grown in more in vivo‐like conditions 62, we performed RNA sequencing of the series of cell lines grown as 3D spheroids. This analysis identified 1022 genes that were differentially expressed between preinvasive and invasive cell lines (supplementary material, Table S5). Functional annotation of these genes identified significant over‐representation of pathways involved in nuclear receptor signalling, epidermal growth factor receptor (EGFR) signalling, ErbB receptor signalling, fibroblast growth factor receptor (FGFR) signalling, signal transduction, integrin signalling, and extracellular matrix organization (Figure 3A, B; supplementary material, Table S6). These findings indicate that more cancer‐relevant pathways are active when cells are grown in a 3D environment, possibly reflecting the way in which the cells were selected for in vivo when they were generated 19.
Figure 3.
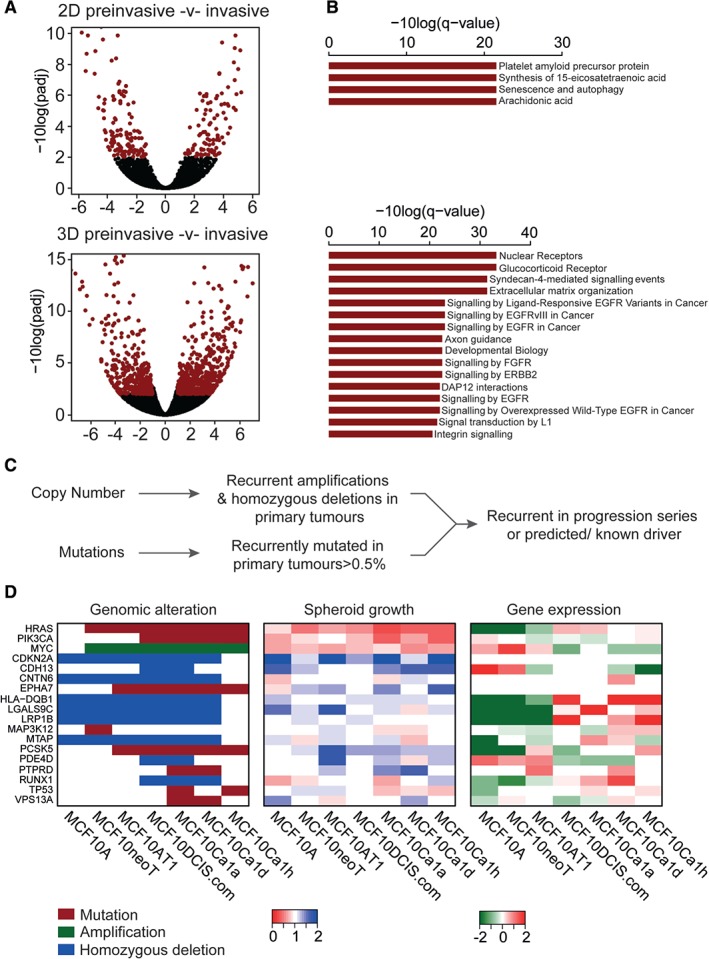
Evaluation of pathways and driver alterations in spheroid cultures. (A) Volcano plots showing the differentially expressed transcripts between preinvasive and invasive cells from the MCF10 progression series cultured under both 2D and 3D conditions. Red: FDR p‐values of <0.01. (B) Bar plot showing the significantly over‐represented pathways (ConsensusDb q‐value of <0.01) from (A). (C) Schematic of gene selection for the siRNA screen. (D) Matched heatmaps of genomic status (mutation, amplification, and homozygous deletion); results of spheroid growth after siRNA‐mediated silencing and gene expression. Relative spheroid growth was measured according to the survival fraction of treated cells relative to siControl. Hits were triaged as a relative survival fraction as compared with non‐targeting control of <0.8 or >1.2. Gene expression is the log2 median centred normalized reads from the RNA sequencing data.
Functional characterisation of driver alterations upon progression
Given our observations that, when grown in spheroid cultures, the MCF10 cell line series show enrichment of cancer‐relevant pathways associated with progression to invasive disease, we sought to functionally test which genomic alterations (amplifications, homozygous deletions, and mutations) that are also seen in primary breast cancers (Figure 3C; see Materials and methods) would be driving the growth of these cells. Cell lines were optimized for siRNA‐mediated gene depletion, whereby ablation of the genes encoding the tumour suppressor phosphatase and tensin homolog and ubiquitin B resulted in increased and decreased spheroid growth relative to control siRNA, respectively (supplementary material, Figure S5). A siRNA‐based screen of 18 genes identified three that constitute potential driver events, namely PIK3CA (p = 0.0485, t‐test), HRAS, and TP53 (p < 0.0001, t‐test), and that, when silenced, decreased spheroid growth and were associated with genomic status (Figure 3D; supplementary material, Table S7). Deconvolution of the siRNA oligonucleotide pools showed that all of these genes were oncogenic drivers, resulting in decreased spheroid growth when silenced (supplementary material, Figure S6). These included PIK3CA, whereby cells with an H1047R activating mutation showed selective dependency on PIK3CA silencing (Figure 4A). In addition, PIK3CA mutant cells were also selectively dependent on AKT1 silencing (p = 0.046, t‐test), perhaps reflective of the subsequent increased AKT1 activation, (Figure 4A; supplementary material, Figures S5 and S6); however, this appeared to be an effect specific to cells grown as spheroids, and was not observed in traditional 2D culture (Figure 4B; supplementary material, Table S7). Furthermore, the oestrogen receptor‐negative breast cancer cell line BT20, which harbours an H1047R PIK3CA mutation, showed a similar effect (Figure 4B). Moreover, breast cancer cell line spheroids showed dependency on PIK3CA according to their PIK3CA status, with mutant MCF7 and T47D cells (harbouring E575K and H1047R PIK3CA mutations, respectively) being sensitive to PIK3CA silencing, and MDA‐MB‐231 cells (wild type) showing no change in viability after PIK3CA silencing. It is tempting to posit that this is due to the maintenance of AKT activity under unfavourable conditions imparted by the spheroid architecture, as spatial AKT activity was observed in the preinvasive cell lines, whereas stable and high phospho‐AKT staining was observed in the invasive cell line spheroids that harboured the activating mutation in PIK3CA (Figure 4E).
Figure 4.
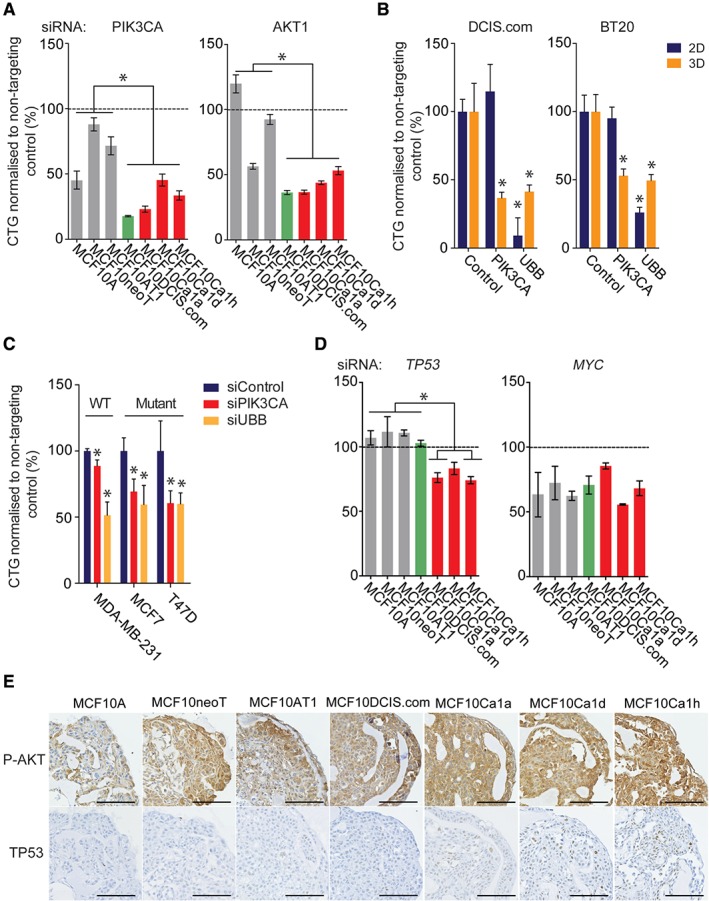
Functional validation of dependency on driver events in the MCF10 progression series. (A) Progression series cell lines were reverse‐transfected with siRNAs against PIK3CA and AKT1, and with a non‐targeting control. Spheroids were formed after 24 h in low‐attachment plates, and the medium was topped up every 3 days. After 7 days, spheroid viability was determined with Cell Titre Glo. (B) DCIS.com and BT20 cell lines were reverse‐transfected with siRNAs targeting PIK3CA and UBB, and with a non‐targeting control, under both 2D and 3D conditions. The medium was topped up every 3 days. Viability was determined with Cell Titre Glo. Statistical comparisons were performed with Student's t‐test (*p ≤ 0.05). (C) MDA‐MB‐231, MCF7 and T47D cell lines were reverse‐transfected with siRNAs targeting PIK3CA and UBB, and with a non‐targeting control, under 3D conditions. The medium was topped up every 3 days, and spheroid viability was determined with Cell Titre Glo. Statistical comparisons were performed with Student's t‐test (*p ≤ 0.05). (D) Progression series cell lines were reverse‐transfected with siRNAs against TP53 and MYC, and with a non‐targeting control. Spheroids were formed after 24 h in low‐attachment plates, and the medium was topped up every 3 days. After 7 days, spheroid viability was determined with Cell Titre Glo. (E) The progression series was grown for 7 days. The medium was topped up every 3 days. After 7 days, spheroids were fixed with formaldehyde, embedded, sectioned, and stained for phospho‐AKT (P‐AKT) (473) and total TP53. Representative images are shown. Scale bar: 100 µm.
Interestingly we observed two independent SNVs in MCF10Ca1a and MCF10Ca1h cells in the DNA‐binding domain of TP53, suggestive of convergent evolution. TP53 silencing in the progression series significantly correlated with smaller spheroid size in mutant cells, suggesting that these mutations act in an oncogenic manner (p = 0.0051, t‐test). Moreover, there was a significant dependency on TP53 associated with increased progression (preinvasive versus invasive cells; p = 0.0021, t‐test), regardless of mutation status, that appeared to correlate with increased nuclear accumulation of TP53 protein in these cells (Figure 4D, E). It is of note that all cell lines showed sensitivity to MYC silencing, suggesting that cells are dependent on MYC transcriptional activity independently of amplification status (Figure 4D).
Given that MCF10A cells underwent HRAS transformation to produce subsequent cell lines, we tested whether cells would still be dependent on oncogenic RAS signalling for their survival further along the course of progression for cell survival. Indeed, silencing of HRAS reduced spheroid growth of all cells subsequent to MCF10A (Figure 5A); however, this association appeared to be significantly correlated with expression of the RAP3GAP1–HRAS fusion gene (r = –0.7857, p = 0.0480, Spearman rank correlation) rather than on total HRAS expression (r = –0.5714, p = 0.2, Spearman rank correlation), and, in a similar manner to PIK3CA and AKT1, seemed to be more specific to cells grown as spheroids (supplementary material, Figure S5). Specific silencing of the RAB3GAP1–HRAS fusion gene, however, had no effect on spheroid growth (Figure 5A, B), perhaps indicative of the subclonal nature (as evidenced by the low percentage of the transcript involved in the fusion, i.e. isoform fraction) of the cells in which the fusion was detected with RNA sequencing (supplementary material, Table S4).
Figure 5.
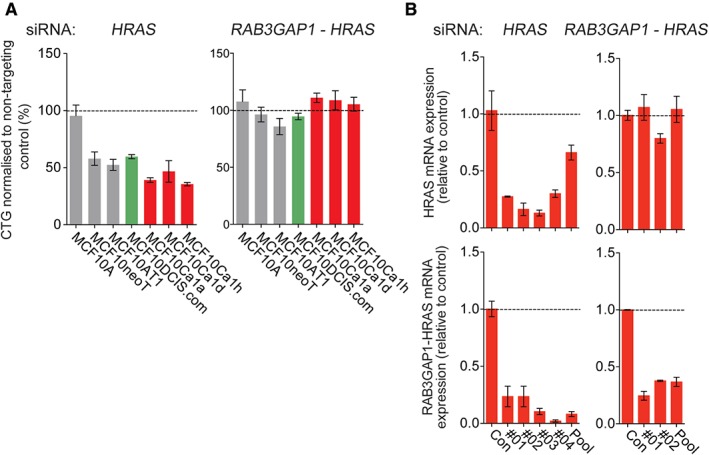
Functional validation of dependency on TP53 and HRAS in the MCF10 progression series. (A) Progression series cell lines were reverse‐transfected with siRNAs against HRAS and RAB3GAP1–HRAS, and with a non‐targeting control. Spheroids were formed after 24 h in low‐attachment plates, and the medium was topped up every 3 days. Spheroid viability was determined with Cell Titer Glo. (B) MCF10Ca1a cells were reverse‐transfected with single and pooled siRNAs targeting HRAS and RAB3GAP1–HRAS, and with a non‐targeting control, for 72 h. HRAS expression and RAB3GAP1–HRAS expression were determined with RT‐qPCR. Expression was normalized to loading controls B2M and β‐actin (ACTB).
Discussion
Here, we have performed a comprehensive analysis of both the genomes (at base pair resolution) and transcriptomes of the MCF10 cell line series that represent different stages of breast cancer progression when grown in vivo, and have demonstrated that these cell lines harbour relevant driver alterations seen in primary breast cancers, and represent a good model for studying breast cancer progression with in vitro spheroid models.
Overall, the patterns of genomic copy number alterations are similar between the cell lines; however, there are key differences, suggestive of subclonal selection from the parental MCF10A cells. In particular, at base pair resolution, the number of mutations varied across cell lines, with a number of key driver mutations being selected for at different stages of progression. These included a PIK3CA hotspot mutation in MCF10DCIS.com cells that leads to increased AKT signalling, and TP53 mutations in the more aggressive invasive cell lines MCF10Ca1a and MCF10Ca1h 71, as shown by increased nuclear accumulation of TP53 protein in these cells. We identified a number of mutations that were clonally selected through progression for which the allele fraction differed in the different cell lines. It may be the case that such alterations are merely passengers (i.e. do not confer a selective advantage to the cells); however, they may provide a growth advantage at different stages of progression, which is in agreement with recent studies on triple‐negative breast cancers, where genuine driver alterations have been shown to be subclonal and heterogeneously distributed 59. At the copy number level, focal high‐level MYC amplification was acquired in the malignant cell lines, as previously reported 63; however, it was not seen in MCF10A cells, which harboured gain of the entire arm of 8q. This finding is in agreement with other reports showing gain of MYC to be an initiating event in this cell line panel, rather than focal amplification 72. Indeed, MYC amplification has been associated with a poor prognosis 73, 74, and is often acquired in metastatic disease 75, 76. However, all cells in the progression series appeared to be sensitive to MYC silencing. Although the majority of the cell lines appeared to be derived from a clone lacking 1q gain, the presence of 1q gain in MCF10DCIS.com cells may alternatively be a result of isochromosome 1q being lost in culture.
Consistent with previous observations that members of the MCF10 progression series behave differently when grown in 3D culture 69, 70, we found a number of distinct differentially regulated pathways associated with progression when cells were grown in spheroids as compared with traditional 2D culture, perhaps reflecting the nature of the nutrient and oxygen gradients in these models 62. Indeed, functional assessment of recurrent alterations identified oncogenic dependencies that were only observed when assessed in spheroid models, including a selective dependency on PIK3CA signalling in cells harbouring an H1047R mutation that was corroborated in additional breast cancer cells harbouring the H1047R mutation. Indeed, the H1047R mutation has been shown to promote metabolic adaption by increasing de novo lipogenesis 77, a feature observed in aggressive cancers.
Through exome sequencing, we identified independent non‐synonymous coding mutations in TP53 in the MCF10Ca1a and MCF10Ca1h cell lines. Consistent with observations that TP53 mutations can be late events in breast cancer progression 59 and are associated with a poor prognosis 78, it has been shown that these sublines can spontaneously metastasise when grown in vivo 19. Interestingly, in all the invasive cells (MCF10Ca1a‐1 h), increased nuclear TP53 protein accumulation was observed, which correlated with sensitivity to TP53 silencing, suggesting that TP53 dysregulation is associated with increased cellular invasiveness and an oncogenic dependency in this model.
In addition to the identification of mutations associated with progression, we also identified two in‐frame fusion genes in the cell line series, included a HRAS fusion that was acquired in MCF10neoT cells, which were transformed by oncogenic HRAS, and was maintained in all subsequent cell lines. This fusion gene joins the promoter of RAB3GAP1, which encodes a GTPase‐activating protein, to exon 3 of HRAS. Although it is interesting to speculate that this fusion gene would also lead to selective HRAS dependency, given an observed correlation of HRAS oncogenic dependency and expression of the fusion gene, we observed no effect on spheroid growth with selective inhibition of the fusion gene. This may be because the fraction of fusion transcript as compared with that wild‐type transcript represents ∼1% of all HRAS reads. As no additional HRAS fusion genes were observed on analysis of published RNA sequencing data, this fusion most likely represents a consequence of HRAS v12 transformation.
Our study, although comprehensive, is not without limitations. HRAS mutation is not a common genetic alteration in human breast cancer, so the cell line series model might not accurately reflect the tumorigenic process in human breast cancers. This is exemplified by the identification of a fusion gene involving HRAS in the model and the lack of such fusion genes in primary breast cancer. Although exome sequencing identified the acquisition of mutations in the malignant cells, we cannot rule out the possibility that these are present at very low subclonal populations in the parental MCF10A cells, given that we did not perform high‐depth targeted resequencing. Nevertheless, through exome sequencing with an average depth of × 100, we observed clonal selection of alterations across different cell lines, suggesting that MCF10A cells are oligoclonal. Although our copy number data support this, the low depth of coverage may also limit the detection of subclonal events. Our triage of genomic alterations for functional assessment mainly identified mutational events and homozygous deletions rather than amplifications that were representative of primary breast cancers. This may be due to our triage strategy; however, it is known that, in general, triple‐negative breast cancers lack many recurrent amplification events 59. Moreover, genomic alterations tested that did not cause a difference in spheroid growth may score in additional assays, and may warrant further testing. In addition, functional assessment of differentially expressed genes may provide further insights into the drivers of progression in this model.
In conclusion, comprehensive characterisation of the MCF10 isogenic progression series has identified a number of driver alterations that are associated with progression from preinvasive to invasive cellular phenotypes that model the genomic alterations seen in primary breast cancer. Moreover, more accurate modelling of the in vivo tumour environment with 3D culture methods allows the validation of founder (HRAS transformation) and acquired (PIK3CA and TP53 mutations) events that would not have been appreciated with traditional techniques. The MCF10 progression series therefore represents a good model with which to dissect potential biomarkers and evaluate therapeutic targets involved in the progression of breast cancer.
Author contributions statement
The authors contributed in the following way: RN: conceived the study; KB, GF: provided materials; BP, PTW, SV, FD, RN carried out the experiments: SLM, JC, HB, AG: performed the bioinformatics analysis; SLM, BP, PH, CJL, RN: discussed and interpreted the results; SLM, BP, CJL, RN: wrote the first draft. All authors read and approved the final manuscript.
SUPPLEMENTARY MATERIAL ONLINE.
Supplementary materials and methods
Supplementary figure legends
Figure S1. Variant allele frequency plots of acquired mutations.
Figure S2. IGV screenshots of acquired mutations in the MCF10 progression series.
Figure S3. Copy number alterations in the MCF10 progression series.
Figure S4. Focal amplifications in the MCF10 progression series.
Figure S5. RNAi modulation of the isogenic MCF10 model in cancer cell line spheroids.
Figure S6. Oligo deconvolution in MCF10Ca1a cells in spheroids and on plastic.
Table S1. Details of primer sequences, siRNA sequences and qPCR assays.
Table S2. Mutations identified in the MCF10 progression series.
Table S3. Gains and losses and focal amplifications and deletions from GISTIC.
Table S4. Expressed fusion genes identified from RNA‐sequencing of cell lines grown as spheroids identified by both DeFuse and Chimerascan algorithms.
Table S5. Differentially expressed genes between pre‐invasive and invasive cells grown on plastic and as spheroids.
Table S6. Pathway analysis of differentially expressed genes between pre‐invasive and invasive cell lines grown on plastic and as spheroids.
Table S7. Summary of siRNA screen of cells grown as spheroids and on plastic.
Supporting information
Supplementary materials and methods
Supplementary figure legends
Figure S1: Variant allele frequency plots of acquired mutations Variant allele frequency (VAF) plots of predicted driver mutations acquired in malignant cells, depicting the cell line (x‐axis) and VAF (y‐axis). The size of the dots corresponds to the depth of coverage.
Figure S2: IGV screenshots of acquired mutations in the MCF10 progression series Screenshots from Integrative Genomics Viewer (IGV) of driver genes acquired from in malignant cells, showing the presence or absence of mutant reads in the cell lines. Reads are sorted according to mutant reads.
Figure S3: Copy number alterations in the MCF10 progression series Genome plots showing normalised coverage of reads (y‐axis) from whole genome sequencing are plotted according to the genomic position in the genome (x‐axis).
Figure S4: Focal amplifications in the MCF10 progression series (A) Chromosome 10 and (B) Chromosome 17 plots showing focal amplifications acquired during progression. Normalised coverage (y‐axis) is plotted according to genomic position (x‐axis)
Figure S5: RNAi modulation of the isogenic MCF10 model in cancer cell line spheroids (A) Micrograph images of MCF10DCIS.com cells subsequent to transfection of siRNA. Images were taken at day 7. (B) Heatmap of spheroid growth after siRNA mediated silencing and gene expression on plastic (2D). Relative spheroid growth is measured by the survival fraction of treated cells relative to siControl. (C) Micrograph images of spheroids at day 1, 4 and 7 of the MCF10 progression series, demonstrating good growth kinetics. (D) Images of spheroids stained with H&E and antibodies against Ki67, phosphor‐AKT, and TP53.
Figure S6: Oligo deconvolution in MCF10Ca1a cells in spheroids and on plastic. (A) Barplots showing relative mRNA levels after gene silencing. Statistically significant alterations in mRNA expression were calculated using Student's t test (*p ≤ 0.05) (B) Barplots of relative cell growth in 2D and as 3D spheroids with individual oligos. Relative spheroid growth is measured by the survival fraction of treated cells relative to siControl. Statistical comparisons were performed using Student's t‐test (*p ≤ 0.05)
Table S1 Details of primer sequences, siRNA sequences and RT‐qPCR assays.
Table S2 Mutations identified in the MCF10 progression series.
Table S3 Gains and losses and focal amplifications and deletions from GISTIC.
Table S4 Expressed fusion genes identified from RNA‐sequencing of cell lines grown as spheroids identified by both DeFuse and Chimerascan algorithms.
Table S5 Differentially expressed genes between pre‐invasive and invasive cells grown on plastic and as spheroids.
Table S6 Pathway analysis of differentially expressed genes between pre‐invasive and invasive cell lines grown on plastic and as spheroids.
Table S7 Summary of siRNA screen of cells grown as spheroids and on plastic.
Acknowledgements
This study was funded by Breast Cancer Now. RN is the recipient of a Breast Cancer Now Career Development Fellowship (2011MaySF01).
No conflicts of interest were declared.
References
- 1. Cowell CF, Weigelt B, Sakr RA, et al. Progression from ductal carcinoma in situ to invasive breast cancer: revisited. Mol Oncol 2013; 7 : 859–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lopez‐Garcia MA, Geyer FC, Lacroix‐Triki M, et al. Breast cancer precursors revisited: molecular features and progression pathways. Histopathology 2010; 57 : 171–192. [DOI] [PubMed] [Google Scholar]
- 3. Ma XJ, Dahiya S, Richardson E, et al. Gene expression profiling of the tumor microenvironment during breast cancer progression. Breast Cancer Res 2009; 11 : R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Simpson PT, Reis‐Filho JS, Gale T, et al. Molecular evolution of breast cancer. J Pathol 2005; 205 : 248–254. [DOI] [PubMed] [Google Scholar]
- 5. Vargas AC, McCart Reed AE, Waddell N, et al. Gene expression profiling of tumour epithelial and stromal compartments during breast cancer progression. Breast Cancer Res Treat 2012; 135 : 153–165. [DOI] [PubMed] [Google Scholar]
- 6. Simpson PT, Gale T, Reis‐Filho JS, et al. Columnar cell lesions of the breast: the missing link in breast cancer progression? A morphological and molecular analysis. Am J Surg Pathol 2005; 29 : 734–746. [DOI] [PubMed] [Google Scholar]
- 7. Fleischer T, Frigessi A, Johnson KC, et al. Genome‐wide DNA methylation profiles in progression to in situ and invasive carcinoma of the breast with impact on gene transcription and prognosis. Genome Biol 2014; 15 : 435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Vincent‐Salomon A, Lucchesi C, Gruel N, et al. Integrated genomic and transcriptomic analysis of ductal carcinoma in situ of the breast. Clin Cancer Res 2008; 14 : 1956–1965. [DOI] [PubMed] [Google Scholar]
- 9. Hernandez L, Wilkerson PM, Lambros MB, et al. Genomic and mutational profiling of ductal carcinomas in situ and matched adjacent invasive breast cancers reveals intra‐tumour genetic heterogeneity and clonal selection. J Pathol 2012; 227 : 42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Heselmeyer‐Haddad K, Berroa Garcia LY, Bradley A, et al. Single‐cell genetic analysis of ductal carcinoma in situ and invasive breast cancer reveals enormous tumor heterogeneity yet conserved genomic imbalances and gain of MYC during progression. Am J Pathol 2012; 181 : 1807–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lodillinsky C, Infante E, Guichard A, et al. p63/MT1‐MMP axis is required for in situ to invasive transition in basal‐like breast cancer. Oncogene 2016; 35: 344–357. [DOI] [PubMed] [Google Scholar]
- 12. Osuala KO, Sameni M, Shah S, et al. IL‐6 signaling between ductal carcinoma in situ cells and carcinoma‐associated fibroblasts mediates tumor cell growth and migration. BMC Cancer 2015; 15 : 584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Allen MD, Marshall JF, Jones JL. alphavbeta6 Expression in myoepithelial cells: a novel marker for predicting DCIS progression with therapeutic potential. Cancer Res 2014; 74 : 5942–5947. [DOI] [PubMed] [Google Scholar]
- 14. Soule HD, Maloney TM, Wolman SR, et al. Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF‐10. Cancer Res 1990; 50 : 6075–6086. [PubMed] [Google Scholar]
- 15. Miller FR, Soule HD, Tait L, et al. Xenograft model of progressive human proliferative breast disease. J Natl Cancer Inst 1993; 85 : 1725–1732. [DOI] [PubMed] [Google Scholar]
- 16. Dawson PJ, Wolman SR, Tait L, et al. MCF10AT: a model for the evolution of cancer from proliferative breast disease. Am J Pathol 1996; 148 : 313–319. [PMC free article] [PubMed] [Google Scholar]
- 17. Miller FR, Santner SJ, Tait L, et al. MCF10DCIS.com xenograft model of human comedo ductal carcinoma in situ. J Natl Cancer Inst 2000; 92 : 1185–1186. [DOI] [PubMed] [Google Scholar]
- 18. Strickland LB, Dawson PJ, Santner SJ, et al. Progression of premalignant MCF10AT generates heterogeneous malignant variants with characteristic histologic types and immunohistochemical markers. Breast Cancer Res Treat 2000; 64 : 235–240. [DOI] [PubMed] [Google Scholar]
- 19. Santner SJ, Dawson PJ, Tait L, et al. Malignant MCF10CA1 cell lines derived from premalignant human breast epithelial MCF10AT cells. Breast Cancer Res Treat 2001; 65 : 101–110. [DOI] [PubMed] [Google Scholar]
- 20. Buchanan NS, Zhao J, Zhu K, et al. Differential expression of acidic proteins with progression in the MCF10 model of human breast disease. Int J Oncol 2007; 31 : 941–949. [PubMed] [Google Scholar]
- 21. Elsarraj HS, Hong Y, Valdez KE, et al. Expression profiling of in vivo ductal carcinoma in situ progression models identified B cell lymphoma‐9 as a molecular driver of breast cancer invasion. Breast Cancer Res 2015; 17 : 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hurst DR, Xie Y, Edmonds MD, et al. Multiple forms of BRMS1 are differentially expressed in the MCF10 isogenic breast cancer progression model. Clin Exp Metast 2009; 26 : 89–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kaur H, Mao S, Li Q, et al. RNA‐Seq of human breast ductal carcinoma in situ models reveals aldehyde dehydrogenase isoform 5A1 as a novel potential target. PLoS One 2012; 7 : e50249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Marella NV, Malyavantham KS, Wang J, et al. Cytogenetic and cDNA microarray expression analysis of MCF10 human breast cancer progression cell lines. Cancer Res 2009; 69 : 5946–5953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rhee DK, Park SH, Jang YK. Molecular signatures associated with transformation and progression to breast cancer in the isogenic MCF10 model. Genomics 2008; 92 : 419–428. [DOI] [PubMed] [Google Scholar]
- 26. So JY, Lee HJ, Kramata P, et al. Differential expression of key signaling proteins in MCF10 cell lines, a human breast cancer progression model. Mol Cell Pharmacol 2012; 4 : 31–40. [PMC free article] [PubMed] [Google Scholar]
- 27. Mbeunkui F, Metge BJ, Shevde LA, et al. Identification of differentially secreted biomarkers using LC‐MS/MS in isogenic cell lines representing a progression of breast cancer. J Proteome Res 2007; 6 : 2993–3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Andersen K, Mori H, Fata J, et al. The metastasis‐promoting protein S100A4 regulates mammary branching morphogenesis. Dev Biol 2011; 352 : 181–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Casbas‐Hernandez P, D'Arcy M, Roman‐Perez E, et al. Role of HGF in epithelial–stromal cell interactions during progression from benign breast disease to ductal carcinoma in situ. Breast Cancer Res 2013; 15 : R82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Miller FR. Models of progression spanning preneoplasia and metastasis: the human MCF10AneoT.TGn series and a panel of mouse mammary tumor subpopulations. Cancer Treat Res 1996; 83 : 243–263. [DOI] [PubMed] [Google Scholar]
- 31. Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009; 25 : 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Res 2010; 20 : 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next‐generation DNA sequencing data. Nat Genet 2011; 43 : 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Van der Auwera GA, Carneiro MO, Hartl C, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics 2013; 11 : 11.10.11–11.10.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Saunders CT, Wong WS, Swamy S, et al. Strelka: accurate somatic small‐variant calling from sequenced tumor‐normal sample pairs. Bioinformatics 2012; 28 : 1811–1817. [DOI] [PubMed] [Google Scholar]
- 36. Robinson JT, Thorvaldsdottir H, Winckler W, et al. Integrative genomics viewer. Nat Biotechnol 2011; 29 : 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high‐throughput sequencing data. Nucleic Acids Res 2010; 38 : e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Network T. Comprehensive molecular portraits of human breast tumours. Nature 2012; 490 : 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Natrajan R, Mackay A, Lambros MB, et al. A whole‐genome massively parallel sequencing analysis of BRCA1 mutant oestrogen receptor‐negative and ‐positive breast cancers. J Pathol 2012; 227 : 29–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li H, Durbin R. Fast and accurate long‐read alignment with Burrows–Wheeler transform. Bioinformatics 2010; 26 : 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mayrhofer M, DiLorenzo S, Isaksson A. Patchwork: allele‐specific copy number analysis of whole‐genome sequenced tumor tissue. Genome Biol 2013; 14 : R24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mermel CH, Schumacher SE, Hill B, et al. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy‐number alteration in human cancers. Genome Biol 2011; 12 : R41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Natrajan R, Wilkerson PM, Marchio C, et al. Characterization of the genomic features and expressed fusion genes in micropapillary carcinomas of the breast. J Pathol 2014; 232 : 553–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Maguire SL, Leonidou A, Wai P, et al. SF3B1 mutations constitute a novel therapeutic target in breast cancer. J Pathol 2015; 235 : 571–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Trapnell C, Roberts A, Goff L, et al. Differential gene and transcript expression analysis of RNA‐seq experiments with TopHat and Cufflinks. Nat Protoc 2012; 7 : 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol 2010; 11 : R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Valdez KE, Fan F, Smith W, et al. Human primary ductal carcinoma in situ (DCIS) subtype‐specific pathology is preserved in a mouse intraductal (MIND) xenograft model. J Pathol 2011; 225 : 565–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Iyer MK, Chinnaiyan AM, Maher CA. ChimeraScan: a tool for identifying chimeric transcription in sequencing data. Bioinformatics 2011; 27 : 2903–2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. McPherson A, Hormozdiari F, Zayed A, et al. deFuse: an algorithm for gene fusion discovery in tumor RNA‐Seq data. PLoS Comput Biol 2011; 7 : e1001138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kamburov A, Pentchev K, Galicka H, et al. ConsensusPathDB: toward a more complete picture of cell biology. Nucleic Acids Res 2011; 39 : D712–D717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Natrajan R, Wilkerson PM, Marchio C, et al. Characterization of the genomic features and expressed fusion genes in micropapillary carcinomas of the breast. J Pathol 2014; 232: 553–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Curtis C, Shah SP, Chin SF, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012; 486 : 346–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Natrajan R, Lambros MB, Rodriguez‐Pinilla SM, et al. Tiling path genomic profiling of grade 3 invasive ductal breast cancers. Clin Cancer Res 2009; 15 : 2711–2722. [DOI] [PubMed] [Google Scholar]
- 54. Natrajan R, Mackay A, Wilkerson PM, et al. Functional characterization of the 19q12 amplicon in grade III breast cancers. Breast Cancer Res 2012; 14 : R53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Natrajan R, Weigelt B, Mackay A, et al. An integrative genomic and transcriptomic analysis reveals molecular pathways and networks regulated by copy number aberrations in basal‐like, HER2 and luminal cancers. Breast Cancer Res Treat 2010; 121 : 575–589. [DOI] [PubMed] [Google Scholar]
- 56. Ellis MJ, Ding L, Shen D, et al. Whole‐genome analysis informs breast cancer response to aromatase inhibition. Nature 2012; 486 : 353–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Nik‐Zainal S, Alexandrov LB, Wedge DC, et al. Mutational processes molding the genomes of 21 breast cancers. Cell 2012; 149 : 979–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Nik‐Zainal S, Van Loo P, Wedge DC, et al. The life history of 21 breast cancers. Cell 2012; 149 : 994–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Shah SP, Roth A, Goya R, et al. The clonal and mutational evolution spectrum of primary triple‐negative breast cancers. Nature 2012; 486 : 395–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Stephens PJ, Tarpey PS, Davies H, et al. The landscape of cancer genes and mutational processes in breast cancer. Nature 2012; 486 : 400–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Shihab HA, Gough J, Cooper DN, et al. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum Mutat 2013; 34 : 57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Schug ZT, Peck B, Jones DT, et al. Acetyl‐CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell 2015; 27 : 57–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kadota M, Yang HH, Gomez B, et al. Delineating genetic alterations for tumor progression in the MCF10A series of breast cancer cell lines. PLoS One 2010; 5 : e9201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Stephens PJ, McBride DJ, Lin ML, et al. Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature 2009; 462 : 1005–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Robinson DR, Kalyana‐Sundaram S, Wu YM, et al. Functionally recurrent rearrangements of the MAST kinase and Notch gene families in breast cancer. Nat Med 2011; 17 : 1646–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Yoshihara K, Wang Q, Torres‐Garcia W, et al. The landscape and therapeutic relevance of cancer‐associated transcript fusions. Oncogene 2015; 34: 3845–4854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Debnath J, Muthuswamy SK, Brugge JS. Morphogenesis and oncogenesis of MCF‐10A mammary epithelial acini grown in three‐dimensional basement membrane cultures. Methods 2003; 30 : 256–268. [DOI] [PubMed] [Google Scholar]
- 68. Debnath J, Mills KR, Collins NL, et al. The role of apoptosis in creating and maintaining luminal space within normal and oncogene‐expressing mammary acini. Cell 2002; 111 : 29–40. [DOI] [PubMed] [Google Scholar]
- 69. Mullins SR, Sameni M, Blum G, et al. Three‐dimensional cultures modeling premalignant progression of human breast epithelial cells: role of cysteine cathepsins. Biol Chem 2012; 393 : 1405–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Naber HP, Wiercinska E, Ten Dijke P, et al. Spheroid assay to measure TGF‐beta‐induced invasion. J Vis Exp 2011; 57: e3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Peng X, Yun D, Christov K. Breast cancer progression in MCF10A series of cell lines is associated with alterations in retinoic acid and retinoid X receptors and with differential response to retinoids. Int J Oncol 2004; 25 : 961–971. [PubMed] [Google Scholar]
- 72. Worsham MJ, Pals G, Schouten JP, et al. High‐resolution mapping of molecular events associated with immortalization, transformation, and progression to breast cancer in the MCF10 model. Breast Cancer Res Treat 2006; 96 : 177–186. [DOI] [PubMed] [Google Scholar]
- 73. Pereira CB, Leal MF, de Souza CR, et al. Prognostic and predictive significance of MYC and KRAS alterations in breast cancer from women treated with neoadjuvant chemotherapy. PLoS One 2013; 8 : e60576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Rodriguez‐Pinilla SM, Jones RL, Lambros MB, et al. MYC amplification in breast cancer: a chromogenic in situ hybridisation study. J Clin Pathol 2007; 60 : 1017–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Singhi AD, Cimino‐Mathews A, Jenkins RB, et al. MYC gene amplification is often acquired in lethal distant breast cancer metastases of unamplified primary tumors. Mod Pathol 2012; 25 : 378–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wade MA, Sunter NJ, Fordham SE, et al. c‐MYC is a radiosensitive locus in human breast cells. Oncogene 2015; 34 : 4985–4994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Ricoult SJ, Yecies JL, Ben‐Sahra I, et al. Oncogenic PI3K and K‐Ras stimulate de novo lipid synthesis through mTORC1 and SREBP. Oncogene 2016; 35: 1250–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Silwal‐Pandit L, Vollan HK, Chin SF, et al. TP53 mutation spectrum in breast cancer is subtype specific and has distinct prognostic relevance. Clin Cancer Res 2014; 20 : 3569–3580. [DOI] [PubMed] [Google Scholar]
- 79. Futreal PA, Coin L, Marshall M, et al. A census of human cancer genes. Nat Rev Cancer 2004; 4 : 177–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary materials and methods
Supplementary figure legends
Figure S1: Variant allele frequency plots of acquired mutations Variant allele frequency (VAF) plots of predicted driver mutations acquired in malignant cells, depicting the cell line (x‐axis) and VAF (y‐axis). The size of the dots corresponds to the depth of coverage.
Figure S2: IGV screenshots of acquired mutations in the MCF10 progression series Screenshots from Integrative Genomics Viewer (IGV) of driver genes acquired from in malignant cells, showing the presence or absence of mutant reads in the cell lines. Reads are sorted according to mutant reads.
Figure S3: Copy number alterations in the MCF10 progression series Genome plots showing normalised coverage of reads (y‐axis) from whole genome sequencing are plotted according to the genomic position in the genome (x‐axis).
Figure S4: Focal amplifications in the MCF10 progression series (A) Chromosome 10 and (B) Chromosome 17 plots showing focal amplifications acquired during progression. Normalised coverage (y‐axis) is plotted according to genomic position (x‐axis)
Figure S5: RNAi modulation of the isogenic MCF10 model in cancer cell line spheroids (A) Micrograph images of MCF10DCIS.com cells subsequent to transfection of siRNA. Images were taken at day 7. (B) Heatmap of spheroid growth after siRNA mediated silencing and gene expression on plastic (2D). Relative spheroid growth is measured by the survival fraction of treated cells relative to siControl. (C) Micrograph images of spheroids at day 1, 4 and 7 of the MCF10 progression series, demonstrating good growth kinetics. (D) Images of spheroids stained with H&E and antibodies against Ki67, phosphor‐AKT, and TP53.
Figure S6: Oligo deconvolution in MCF10Ca1a cells in spheroids and on plastic. (A) Barplots showing relative mRNA levels after gene silencing. Statistically significant alterations in mRNA expression were calculated using Student's t test (*p ≤ 0.05) (B) Barplots of relative cell growth in 2D and as 3D spheroids with individual oligos. Relative spheroid growth is measured by the survival fraction of treated cells relative to siControl. Statistical comparisons were performed using Student's t‐test (*p ≤ 0.05)
Table S1 Details of primer sequences, siRNA sequences and RT‐qPCR assays.
Table S2 Mutations identified in the MCF10 progression series.
Table S3 Gains and losses and focal amplifications and deletions from GISTIC.
Table S4 Expressed fusion genes identified from RNA‐sequencing of cell lines grown as spheroids identified by both DeFuse and Chimerascan algorithms.
Table S5 Differentially expressed genes between pre‐invasive and invasive cells grown on plastic and as spheroids.
Table S6 Pathway analysis of differentially expressed genes between pre‐invasive and invasive cell lines grown on plastic and as spheroids.
Table S7 Summary of siRNA screen of cells grown as spheroids and on plastic.
Data Availability Statement
Raw whole genome, exome and RNA sequencing data have been deposited in the NCBI Sequence Read Archive under the accession number PRJNA308098.