

Abstract
Background
Blow flies (Diptera: Calliphoridae) are important medical, veterinary and forensic insects encompassing 8 % of the species diversity observed in the calyptrate insects. Few genomic resources exist to understand the diversity and evolution of this group.
Results
We present the hybrid (short and long reads) draft assemblies of the male and female genomes of the common North American blow fly, Phormia regina (Diptera: Calliphoridae). The 550 and 534 Mb draft assemblies contained 8312 and 9490 predicted genes in the female and male genomes, respectively; including > 93 % conserved eukaryotic genes. Putative X and Y chromosomes (21 and 14 Mb, respectively) were assembled and annotated. The P. regina genomes appear to contain few mobile genetic elements, an almost complete absence of SINEs, and most of the repetitive landscape consists of simple repetitive sequences. Candidate gene approaches were undertaken to annotate insecticide resistance, sex-determining, chemoreceptors, and antimicrobial peptides.
Conclusions
This work yielded a robust, reliable reference calliphorid genome from a species located in the middle of a calliphorid phylogeny. By adding an additional blow fly genome, the ability to tease apart what might be true of general calliphorids vs. what is specific of two distinct lineages now exists. This resource will provide a strong foundation for future studies into the evolution, population structure, behavior, and physiology of all blow flies.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-016-3187-z) contains supplementary material, which is available to authorized users.
Keywords: Genome, Phormia regina, Calliphoridae, Blow fly, Sex determination, X chromosome, Y chromosome
Background
One in ten species on earth are flies (Diptera). They are the most derived group within arthropods, and have experienced an explosive radiation in the last 50 million years [1, 2]. Over the past decade, dipteran draft genomes including the fruit fly (Drosophila melanogaster, [3]), the house fly (Musca domestica, [4]), and the malaria mosquito (Anopheles gambiae, [5]) have been published. Within Diptera, the family Calliphoridae, commonly known as blow flies, comprises ~1500 species [6, 7], and contributes 8 % of species diversity in calyptrate flies [8]. Calliphorids are ubiquitous, distributed world-wide, and are important in the medical [9–12], veterinary/agricultural [13–16] and forensic fields [17, 18]. For example, blow flies are responsible for the transmission of human disease [19–22]. Mihalyi’s danger-index was calculated for seven blow fly species in South America, with consideration for the synanthropic index. Six of the seven species posed a greater sanitary risk than the house fly [23], a known disease vector [24, 25]. Interestingly, other closely related blow fly species have been shown to be medically advantageous as a means of wound debridement, otherwise known as maggot therapy [9, 12, 26, 27]. In these cases, maggots physically debride the wound of decaying tissue while simultaneously excreting antimicrobial compounds [26, 28–36] effective against antibiotic resistant bacteria such as methicillin resistant Staphylococcus aureus (MRSA) [29, 37].
Recently, the first calliphorid genome, from the sheep blow fly (Lucilia cuprina, [38]), was released. The publication of the L. cuprina draft genome brings with it the potential for studying a group of flies that have evolved recently [1, 2, 39], and have adopted many different life histories [6]. For example, the sheep blow fly specimen(s) that was sequenced was from a location in which it is predominantly, if not exclusively, an obligate ectoparasite (infestation of living vertebrate tissue by fly larvae) [38], and has presumably adapted under selective pressures from subsisting on carrion to infesting live animals. Calliphorid genomes will provide the necessary resources needed to understand the basic biological processes of lineage-specific traits in myiasis-causing flies.
The black blow fly, Phormia regina (Meigen), is a Palearctic fly with records throughout North America and Northern Europe, and is the dominant carrion fly for most of Canada and the United States [40]. We chose to examine the genome of this species because 1) it plays an important role in ecosystems via carrion decomposition and nutrient recycling [41], 2) its’ abundance in North America, and 3) because it exhibits no specialized parasitic adaptations or unusual sex determination strategies (i.e. it is not monogenic), though the sex determination strategy for Phormia regina is largely unknown. Like other calliphorid flies, P. regina contains 2n = 12 chromosomes, including heteromorphic sex chromosomes [42, 43]. These characteristics make it a good candidate for comparisons with species that have more specialized life histories. Furthermore, providing an additional reference genome from Calliphoridae will allow for a more complete understanding of the clade and adaptive processes that take place within it. We sequenced the male and female genomes (~40X), allowing us to characterize sex chromosomes and sex determining pathways, as well as evolutionary relationships of chemoreceptors, antimicrobial peptides, and insecticide resistance pathways in relation to other calliphorids and dipterans.
Methods
Genome sequencing – short reads
Genomic DNA was extracted from whole flies using the DNeasy Blood and Tissue DNA Extraction kit (Qiagen Inc., Valencia, CA) and pooled from five female and five male Phormia regina flies housed in a laboratory colony (approximately 4–6 months old). The founders originated from Indianapolis, IN (39.7684° N, 86.1581° W) and were collected during the summer of 2012. The extracted DNA from each individual was quantified using a Qubit fluorometer (ThermoFisher Scientific, Grand Island, NY) and mixed in equal proportions to yield the two pooled extracts, one for each sex. DNA libraries were prepared using TruSeq DNA sample preparation (Illumina, San Diego, CA) and sequenced (2 × 100 bp) using one half lane of an Illumina HiSeq2000 platform at the Purdue University Genomics Core Facility (West Lafayette, IN). Additional 454 reads were obtained as described in [44].
Genome sequencing – long reads
Genomic DNA was extracted from a whole single male P. regina specimen that had been in colony for >15 generations (different colony from above, but same originating location, Indianapolis). DNA library preparation and sequencing was performed according to the manufacturer’s instructions and reflects the P6-C4 sequencing enzyme and chemistry, respectively, at the Icahn School of Medicine at Mount Sinai Genomics Core Facility. 14 % of the input library eluted from the agarose cassette and was available for sequencing. For all cases, this yield was sufficient to proceed to primer annealing and DNA sequencing on the PacBio RSII instrument (Pacific Biosciences, Menlo Park, CA). SMRTcell libraries were placed onto the RSII machine at a sequencing concentration of 150 pM and configured for a 240-min continuous sequencing run. Sequencing was conducted to achieve a 7401 bp subread N50 across a total of 1.5 Gb of data comprised of 268,000 reads on 2 SMRTcells.
Due to the high error rate of reads generated from PacBio sequencing [45], error correction was performed using the Correct PacBio Reads tool of CLC’s Genome Finishing Module plug-in v1.5.1 (Qiagen Inc) on a local workstation. Additional error correction was also performed using the SMRT Analysis PacBioToCA correction module, with the assistance of the high quality Illumina reads, using default settings.
Genome processing and assembly
Genome processing and assemblies were accomplished using CLC Genomics Workbench v6.0.5 (CLC-GWB; Qiagen Inc.). However, in order to evaluate the effectiveness of the CLC genome assembler, preliminary assemblies were generated using de novo genome assemblers: Velvet [46] and SOAPdenovo [47]. The CLC assemblies were accomplished using a desktop computer with enhanced memory (32 Gb RAM), whereas the Velvet and SOAPdenovo assemblies were performed using a large memory supercomputing cluster (Indiana University, Bloomington, IN). The male and female Illumina short reads were initially assembled into two draft genomes with the three assemblers. Each assembly was carried out using a range of kmer values until the ‘best’ assembly was captured as determined by contig number and contig N50 size (the kmer values varied with each assembly). The CLC-GWB de novo assembly using only Illumina reads resulted in the smallest number of contigs and the longest N50 (Table 1). With this data, we therefore decided to rely solely on CLC-GWB for the remainder of our analyses and any additional assemblies produced using additional reads (454 and PacBio).
Table 1.
Comparative assembly statistics of preliminary assemblies generated using CLC-GWB, Velvet and SOAPdenovo
| Assembler | Sex | Optimal kmer (bp) | # contigs | N50 (bp) | % readsa |
|---|---|---|---|---|---|
| CLC-GWB | Female | 60 | 251,115 | 3684 | 96.0 |
| Male | 60 | 306,273 | 2487 | 93.5 | |
| Velvet | Female | 75 | 317,916 | 1095 | 47.0 |
| Male | 75 | 310,045 | 956 | 37.9 | |
| SOAPdenovo | Female | 75 | 1,953,762 | 451 | 68.8 |
| Male | 75 | 2,010,365 | 351 | 58.6 |
For each assembly pipeline, different kmers were used to generate the optimal assembly, and the main metric for determining quality of assembly was the combination of the number of contigs and the N50
a% reads = the number of reads that mapped to each assembled genome
The pipeline for assembly using the Illumina and 454 reads on CLC-GWB was as follows: low quality reads (phred scores < 20) and adaptor sequences were removed, duplicate reads were removed and overlapping pairs were merged (mismatch cost was set to 2 and a gap cost was set to 3). To remove extraneous or contaminating DNA, we used a filtering pipeline that included the mapping and subsequent removal of the above processed reads to 1405 phage (www.phantome.org, downloaded 03/2014) and 595 bacterial genomes (NCBI, www.ncbi.nlm.nih.gov, downloaded 03/2014). Further filtering removed mitochondrial reads, which were assembled separately using methods described below. Approximately 0.38 and 0.49 % of reads were identified as bacteriophage and bacterial contaminants, respectively. Following the removal of these contaminants, 5 % of overlapping paired reads were merged, 1 % of duplicates and low quality reads were removed, leaving a total of 253,233,928 male and 254,306,608 female reads for downstream analyses.
Preliminary assembly iterations involved further optimization of kmer values (ranging from 24 to 60 nucleotides) and bubble sizes (ranging 100–1000 bp). Assemblies were evaluated based on total number of assembled contigs, estimated genome sizes, contig N50 values, and completeness as per CEGMA v2.4.010312 (see below [48]).
Once optimal kmer sizes were determined (45 bp for each individual genome), reads were mapped back to the assemblies using CLC-GWB (mismatch cost of 2, insertion cost of 3, deletion cost of 3, length fraction of 0.5, similarity fraction of 0.8, bubble size 300 bp). PacBio (error-corrected) and 454 reads were combined with the Illumina reads to create a hybrid de novo assembly by using them to scaffold the contigs using CLC’s Join Contigs Tool from the genome finishing module plug-in.
Mitochondrial genome assembly
The mitochondrial genomes of seven calliphorid flies (Cochliomyia hominivorax, NC_002660; Protophormia terraenovae, NC_019636.1; Chrysomya albiceps, NC_019631.1; Chrysomya bezziana, NC_019632.1; Chrysomya rufifacies, NC_019634.1; Chrysomya megacephala, NC_019633.1; Lucilia sericata, NC_009733; Lucilia cuprina, NC_019573.1) were obtained from NCBI (www.ncbi.nlm.nih.gov) and used as local reference genomes for the pooled male and female reads. Reads that mapped to these reference genomes were then extracted and assembled using CLC-GWB v7.0.3 to generate a mitochondrial genome assembly. The mitochondrial genome sequence was 99 % similar to a previously published genome from P. regina (KC005712, 15,513 bp), 93 % to Protophormia terranovae (JX913743.1, 15,170 bp), 92 % to Chrysomya megacephala (AJ426041, 15,831 bp), 91 % to Lucilia sericata (AJ422212.1, 15,945 bp), and 90 % to Cochliomyia hominivorax (AF260826, 16,022 bp), among others. For all calliphorid genomes, sequence similarity was 90 % or greater. All mitochondrial amino acid sequences were 100 % identical to other calliphorid mitochondrial protein sequences.
Genome assembly assessment
Assembly statistics were calculated using CLC-GWB and the genome assessment tool QUAST v3.1 [49]. To evaluate the completeness of the genome assemblies, CEGMA v2.4.010312 [48] was used to detect the number of complete and partial core eukaryotic genes present in the assembled genomes. This analysis was completed using GeneWise v2.4.1, NCBI-BLAST+ v2.2.28 and geneid v1.4.4 to return DNA sequences of each prediction and their associated statistical reports.
Gene prediction & gene ontology
AUGUSTUS [50] was used for ab initio prediction of gene sequences based on reference Drosophila melanogaster sequences as it is an extensively studied and annotated genetic model organism. Predicted protein sequences were annotated by BLASTp v2.2.28+ using a non-redundant protein BLAST database and an E-value cutoff ≤ 1e-5. Comparative analysis of the male and female gene set was performed by using CD-HIT-2D v4.5.6 [51] to compare the two protein datasets (90 % identity, word size (-n) of 5, and length difference cutoff (-s2) of 90 %). The unique protein sequences for the male were blast searched (BLASTp, E-value cutoff ≤ 1e-10) against the complete predicted amino acid sequences of the female to confirm their uniqueness. The same was done for the female protein set against the male protein set. Proteins without hits were assumed to be unique to each sex.
Functional characterization of the predicted gene sequences was performed using default settings in Blast2GO v3.1.3 [52]. Gene ontology (GO) terms were assigned to the annotated sequences. GO slim functionality in Blast2GO was used to simplify the annotation into functional categories and the proteins were categorized at level 2 into the three main GO classifications of biological process, cellular component and molecular function. InterProScan [53] statistics and KEGG [54] map pathways were also extracted from Blast2GO v3.1.3 using default values. GO terms from biological processes for each of the unique gene sets were summarized after the removal of redundant GO terms by the web server REViGO (reduce and visualize gene ontology) to create a visual representative subset of the terms using a clustering algorithm that uses similarity measures [55]. The allowed similarity was set to 0.5 (small) and the database of GO terms selected was from D. melanogaster.
Sex chromosome identification
In order to characterize the sex chromosomes, we used the chromosome quotient (CQ) [56] approach which discovers sex chromosome sequences by using a stringent aligning criterion of the male and female reads onto each other’s genomes. The stringent alignment required a whole read to map onto the reference contigs with a zero mismatch in order to reduce the number of false positives [56]. The female to male ratio of the alignments was then used to distinguish contigs from the X or Y chromosome. Male contigs with a CQ of less than the arbitrary 0.05 were grouped as putative Y chromosome contigs and female contigs with a CQ ranging between 1.9 and 2.5 were grouped as putative X chromosome contigs. The sex chromosome contigs were then blast searched against an arthropod database (BLASTn, E-value ≤ 1e-5) to determine any homology with other insects in the database. The contigs were also compared against the well-characterized sex chromosomes of D. melanogaster (tBLASTx, E-value ≤ 1e-5).
Putative gene approaches
All candidate genes were discovered using one or all of three approaches. First, genes of interest (in particular pathways or associated with potential adaptive traits, see below) were initially acquired from Flybase (www.flybase.org, [57]) using queries for specific genes, or using genes associated with specific GO terms. Contigs with hits were identified via local blast (BLASTn) with an E-value cutoff of ≤ 1e-5. Gene sequences were individually annotated for gene structure using the web server version of the AUGUSTUS prediction tool [50], aligned using MUSCLE [58] and viewed with MVIEW [59]. For comparison purposes with other calliphorid or dipteran species, if the gene sequences were available in Genbank, they were acquired and included in our nucleotide (and subsequent predicted amino acid) sequence alignments. If BLASTn approaches of Drosophila sequences failed to produce hits, a second approach to discovering candidate genes was to use keyword searches in our annotated gene dataset. A third approach was to use tBLASTx and homologous sequences from other taxa (such as Lucilia or Musca) on the P. regina genomes.
Sex-determining genes
Putative sex determining genes were isolated and characterized by querying a set of known genes (transformer (tra) – CG16724, transformer2 (tra2) – CG10128, sex lethal (sxl) – CG43770, doublesex (dsx) – CG11094, fruitless (fru) – CG14307, daughterless (da) – CG5102, and maleless (mle) – CG11680) from D. melanogaster against our male and female assemblies. tra and tra2 gene sequences did not result in contig hits using BLASTn, therefore we used the coding sequences from closely related blow fly species (Lucilia cuprina – FJ461621.1 and FJ461620.1, Cochliomyia macellaria – JX315619.1, Cochliomyia hominivorax – JX315618.1 and Lucilia sericata – JX315620.1) as well as sequences from other calliphorid genomes (unpublished) for which we have transcriptomic data using discontinuous megablast.
Chemoreceptors
D. melanogaster gene sequences of odorant binding proteins (OBPs), odorant receptors (ORs), gustatory receptors (GRs) and ionotropic receptors (IRs) were used as query sequences for local blast searches against the P. regina genome (E-value cutoff of ≤ 1e-5) using tBLASTx (Additional file 1: Table S1). Protein sequences of the ionotrophic receptor IR25a were acquired from NCBI from the following species: Lucilia cuprina-KNC28739, Calliphora stygia-AID61273, Stomoxys calcitrans-XP013104244, Musca domestica-NP001273813, Batrocera oleae-XP014086336, Ceratitis capitata-XP004530416, and Drosophila melanogaster-NP001260049.
Antimicrobial peptides
A set of immune-related genes obtained from flybase.org was used to query the Phormia regina genome for antimicrobial peptides (attacins, cecropins, defensins, diptericins, Additional file 2: Table S2). A second approach simply queried our BLASTp results (from the predicted) to identify putative immune-related genes that had homologs to other immune-related genes in insects.
Insecticide resistance genes
Genes associated with the metabolism of foreign material (xenobiotics) are primarily cytochrome P450′s (Additional file 3: Table S3), glutathione S-transferases (GST, Additional file 4: Table S4) and esterases/hydrolases (Additional file 5: Table S5). These genes were identified by manually searching the BLASTp results from the annotation step of the predicted genes; and KEGG pathways for terms that included cytochrome P450s, GSTs and esterases.
Repetitive elements
Repetitive elements in the male and female Phormia regina genomes as well as the putatively identified X and Y chromosomes were identified using RepeatMasker [60] and a library of all known dipteran transposable elements (TEs; RepBase; accessed 14 March 2015). In addition to known transposable elements, RepeatMasker searches were used to identify low-complexity regions including mini and microsatellite sequences. Output from RepeatMasker was used to quantify overall repeat content and an accumulation profile. For the accumulation profile, which reflects relative rates and periods of TEs in a genome, the Kimura 2-parameter [61] distance between a transposable element insertion and the assumed ancestral sequence were calculated using the calcDivergenceFromAlign.pl script packaged with RepeatMasker.
Results and discussion
Genome assembly
Raw reads and genome assemblies have been submitted to GenBank (BioProject ID PRJNA338752, accession numbers MINK00000000.1 and MINJ00000000.1 for the male and female genomes, respectively). Mitochondrial reads (8,378,416) were removed from the main genomic dataset and assembled into a mtDNA genome (15,801 bp, Additional file 6: Figure S1, GenBank accession KX853042).
In order to refine and scaffold our assemblies, we added longer 454 (average read length 344 bp) and error-corrected PacBio reads (average read length 5698 bp). We repeated the de novo assembly process in CLC-GWB using a range of kmer values. Our ‘best’ hybrid assembled genomes contained a combination of smaller numbers of contigs and longer N50s (Table 2). The male (534 Mbp) and female (550 Mbp) genomes had average coverages of 44X with >97 % of the reads mapping back to the genomes.
Table 2.
Final draft genome assembly statistics of the male and female genomes following the addition of 454 and PacBio reads, including a measure of the robustness of the assembly in the number of core eukaryotic genes assembled (CEGMA)
| Parameter | Female | Male |
|---|---|---|
| Optimal kmer (bp) | 60 | 50 |
| No. of contigs | 192,662 | 187,700 |
| N50 (bp) | 7918 | 7177 |
| Avg. contig (bp) | 2859 | 2846 |
| Max. contig (bp) | 116,828 | 691,679 |
| % reads used | 98.03 | 97.38 |
| Avg. coverage | 44X | 44X |
| % AT | 72 % | 72 % |
| Genome Size (Mb) | 550 | 534 |
| No. of contigs (> = 1000 bp) | 96,027 | 98,029 |
| Total length Mb (> = 1000 bp) | 485 | 473 |
| No. of N’s per 100 kb | 1565 | 1800 |
| CEGMA % (complete) | 93.95 | 96.77 |
| CEGMA % (partial) | 99.19 | 99.60 |
These values are larger than the experimentally estimated sizes of 529 Mbp and 517 Mbp for the female and male P. regina, respectively [43]. This is likely due to the presence of repetitive sequences that do not assemble well [62], as well as the presence of allelic variation due to pooled sequencing of five male and five female individuals [63]. The robustness of the protein coding portion of the assembly was assessed using CEGMA [48], where 93.95 and 96.77 % of complete core eukaryotic genes were identified in the female and male genomes (see Additional file 7: Table S6 for completeness report).
Gene prediction and ontology
A total of 9490 and 8312 full length genes were predicted by AUGUSTUS [50] (Table 3) in the male and female genomes, respectively. The genic characteristics of the exonic regions and intronic regions of the predicted genes in both sexes show a total of ~30,000 exons and 20,000 introns (Table 3). The total number of predicted protein-encoding genes in our assembled genomes was small compared to other recently sequenced Dipterans such as Lucilia cuprina (14,544 genes) [38] and Musca domestica (15,345 genes) [4]. This may be due to the high stringency we used for gene prediction where only complete genes (gene sequences with start and stop codons present within individual contigs) were allowed. With a more contiguous version of the genome, and inclusion of transcriptome data in the prediction process, the predicted gene count will probably increase. For comparison, we predicted complete genes in the scaffolded version of the L. cuprina genome (ASM118794v1) and found 10,681 genes (compared to the expected 14,554 genes, data not shown), these results demonstrate that our pipeline for predicting genes is more conservative.
Table 3.
Male and female P. regina gene predictions including the total number of complete genes, the genic structure characteristics (number and length distribution of the intronic and exonic regions), the proportion genes that produced an NCBI result, and the proportion with characterized identifiable protein domains (InterProScan)
| Female | Male | |
|---|---|---|
| No. of predicted protein-encoding genes | 8312 | 9490 |
| Min length (aa) | 66 | 66 |
| Max length (aa) | 14,023 | 12,632 |
| No. of exons | 31,254 | 33,298 |
| Min length (bases) | 3 | 3 |
| Max length (bases) | 14,580 | 14,589 |
| No. of introns | 22,942 | 23,808 |
| Min length (bases) | 42 | 42 |
| Max length (bases) | 55,816 | 47,389 |
| No. of predicted genes with blast results (NCBI) | 7792 (94 %) | 8789 (93 %) |
| InterProScan Statistics | ||
| With InterPro ID’s | 6909 (83 %) | 7964 (84 %) |
In order to compare the genic structures of the predicted genes with other flies, the total counts and average lengths of the exons and introns were compared to recently published genomes of the common housefly M. domestica [4], the blow fly L. cuprina [64] and the fruit fly Drosophila melanogaster (Table 4). The average lengths of gene and intron sequences of L. cuprina and M. domestica are approximately double the size of P. regina’s. However, the average lengths of the exons are similar in size among the three species. The cause in the length disparity is likely due to the scaffolded nature of the Musca and Lucilia genomes, which contain a large number of N’s as placeholders – thus giving rise to seemingly larger introns.
Table 4.
An overview comparing the genic structure and statistics in P. regina (P.reg), L. cuprina (L.cup) and M. domestica (M.dom) genome assemblies
| P.reg (Male) | P.reg (Female) | L.cupa | M.doma | |
|---|---|---|---|---|
| Genes Count | 9490 | 8312 | 14,554 | 15,345 |
| Mean length | 6042 bp | 6869 bp | 12,197 bp | 13,553 bp |
| Exons Count | 33,298 | 31,254 | 65,493 | 67,886 |
| Mean length | 403 bp | 393 bp | 432 bp | 431 bp |
| Introns Count | 23,808 | 22,942 | – | 52,875 |
| Mean length | 871 bp | 936 bp | 2560 bp | 3889 bp |
aStatistics of the genic structure for L. cuprina were obtained from Anstead et al in [38], and for M. domestica from Scott et al. in
A total of 7792 (94 %) and 8789 (93 %) of the predicted genes in the female and male, respectively, had homology to sequences in GenBank with E-values less than 1e-5. Therefore, approximately 6 % of the predicted genes (701 male, 520 female) have no apparent homologs in the arthropod database and could be unique to P. regina. Additionally, annotation by InterProScan classified 83.12 and 83.91 % protein domains in the female and male predicted genes, respectively.
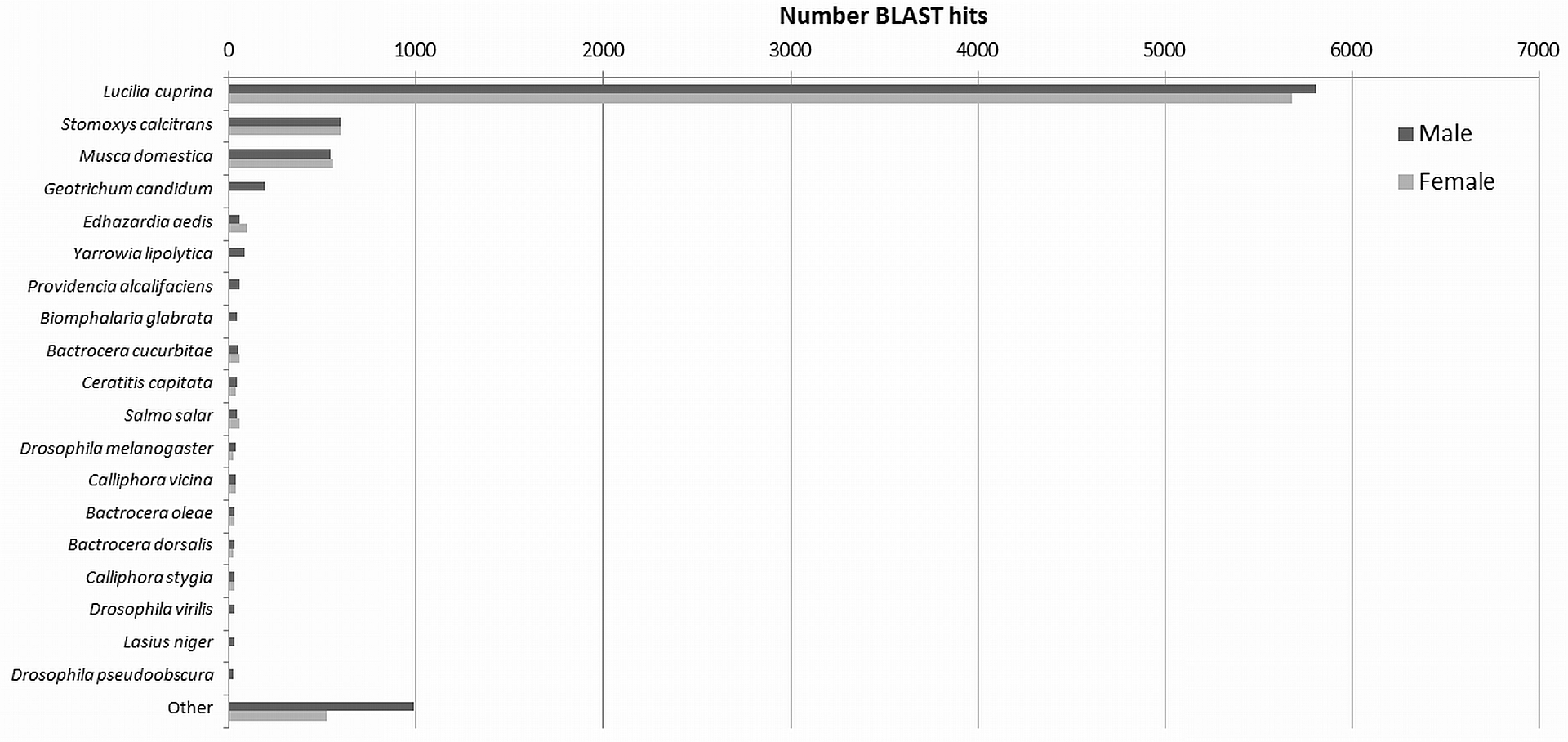
The species that were most represented in the BLASTp results for both sexes came from calyptrate flies, specifically the sheep blow fly (Lucilia cuprina), followed distantly by the stable fly (Stomoxys calcitrans) and the common house fly (Musca domestica) reflecting the phylogenetic relatedness among these species [65]. A total of 5681 (68 %) gene sequences from the female and 5806 (61 %) gene sequences from the male had hits to L. cuprina gene sequences while ~7 % of gene sequences from both sexes had top hits from the stable fly and M. domestica (Additional file 8: Figure S2).
The two most abundant Biological Process GO categories for both sexes were cellular processes (female 72.4 %, male 73.7 %) and metabolic processes (female 60.4 %, male 67.0 %) (Fig. 1). The GO terms that were associated with Molecular Function were mainly assigned to binding (female 41.8 %, male 42.5 %) and catalytic activity (female 32.3 %, male 35.4 %). While the top GO terms associated with Cellular Component were assigned to cell (female 71.3 %, male 70.5 %) and organelle (female 48.9 %, male 46.4 %) (Additional file 9: Table S7). The overall distribution of genes within GO classifications in P. regina was very similar to M. domestica and D. melanogaster [3] (Additional file 10: Table S8).
Fig. 1.
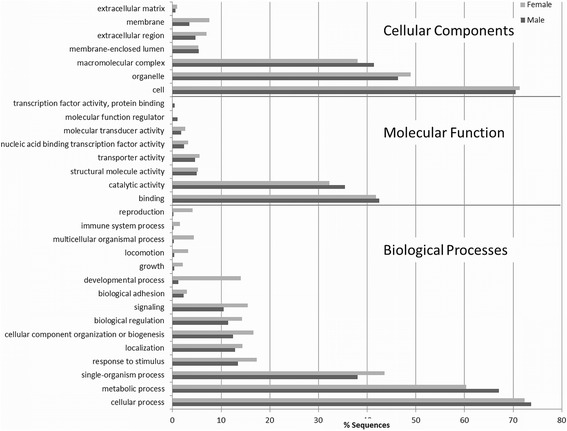
GO term classification of the 3 functional categories (biological processes, molecular function and cellular component) of the predicted genes in the male and female genome assemblies
Categorization of information from molecular-level interactions extracted from the annotated GO terms was performed by the KEGG component in Blast2GO. A total of 111 and 107 KEGG pathways from the GO-slim Blast2GO analysis were identified for the male and female gene sets, respectively. A visual representation of the pathways with greater than 10 sequences is shown in Fig. 2. The top pathways for both sexes were purine metabolism, thiamine metabolism and biosynthesis of antibiotics. A full list of the KEGG pathways can be found in Additional file 11: Table S9.
Fig. 2.
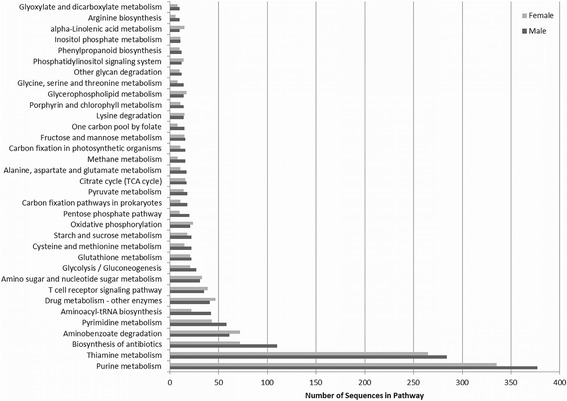
The top 35 KEGG biological pathways of the male and female gene sets extracted from the Blast2GO analysis
Sex chromosomes
Typically, sex determination is carried out through the heteromorphic XX/XY system where Y-linked male determining genes or the presences of a Y-linked male determining factor are proposed to repress female development causing male sexual differentiation thus promoting the male phenotype [66, 67]. This has been observed in the Mediterranean fruit fly (Ceratitis capitata), the olive fruit fly (Bactrocera oleae) and the common house fly (M. domestica) [68].
Putative sex chromosomes for both the male and female genomes were isolated using the chromosome quotient (CQ) approach [49, 56] and 9134 and 10,721 contigs were identified to putatively belong to the X and Y chromosome, respectively. Adding the sizes of each contig, these result in a putative X chromosome size of ~21.2 Mbp and ~14.5 Mbp for the putative Y chromosome, which approximates measured differences between the male and female flies of ~9 Mbp [43]. A direct comparison with the Drosophila X and Y chromosomes yielded 608 (~7 %) female contigs with homology to the X chromosome, and 233 male contigs (~2 %) with homology to the Y chromosome. BLASTn results against the arthropod database (Additional file 12: Table S10) resulted in 47 % (4321 contigs) of the X chromosome contigs and 26 % (2789 contigs) of the Y chromosome contigs identified as having homologous sequences in the database. Most putative Y chromosome contigs did not yield BLAST hits. A reasonable explanation for the limited number of BLAST hits may be the fact that very few model species have characterized and annotated Y chromosomes in the database due to repeat-rich heterochromatic sequences [69, 70].
The majority of the hits from the BLASTn results of the X and Y chromosomes corresponded to repetitive sequences. For example, in the BLAST results from the X chromosome, of the contigs that produced hits, 58 % hit to multiple BAC sequences in Calliphora vicina achaete-scute complex, AS-C (accession numbers LN877230-LN877235). Even though these sequences contain the AS-C complex genes, these blast hits are likely hitting to the repetitive regions present in 20–25 % of these BACs [71]. Furthermore, in Drosophilidae, the AS-C complex is found in the X chromosome, where scute plays an additional role in sex determination acting as an X chromosome signal element [64, 72, 73]. The presence of homologous sequences to the AS-C complex in both the male and female putative sex contigs is an indicator that this complex may also be involved in sex determination pathway in P. regina.
Unique sex genes
A comparative analysis between the male and female predicted genes showed that 1480 genes were unique to the male and 727 predicted genes were unique to the female. These unique sets of genes are a likely combination of sex-biased genes that drive phenotypic differences leading to sex-specific developmental trajectories [74], and/or be present in either male or female assembly because a complete gene was predicted in one sex and not the other. Approximately 73 % of the male and female unique genes had homology to sequences in NCBI’s Arthropoda NR database. Gene ontology analysis of the unique set of genes for each sex in the biological processes category produced a total of 1589 and 1841 GO terms for the male and female, respectively. Comparing the two sets of GO terms indicated that 517 of the GO terms were unique to the male while 769 were unique to the female. The long lists of the unique GO terms for each sex was summarized by clustering GO terms that belong to the same family and removing redundant terms using the web server REViGO [55].
An example of a clustered category in the male is the category flocculation (Additional file 13: Figure S3A) which include the GO terms sperm motility, energy taxis and positive chemotaxis. The genes functionalized with these GO terms may be specific to the males and involved in sperm chemotaxis where sperm from the male fly is guided by a chemoattractant excreted by the female to fertilize an oocyte [75]. One of the categories clustered in the female (Additional file 13: Figure S3B) is response to xenobiotic stimulus. These are genes expressed during an immune response or during exposure to toxic foreign material (xenobiotic) producing enzymes such as cytochrome P450 and acetyl-CoA synthetases. These female specific genes may be involved in the protection of female flies against the diverse array of pathogens it comes across while laying eggs, or from components of male ejaculates after mating [76]. Similar GO categories connected to immune response were detected to be enriched in female D. melanogaster flies as compared to males [77].
Sex determining genes
In most dipterans investigated thus far, sex determination is regulated by a cascade of genes which exhibit a hierarchical interaction during development where a product of one gene controls the sex-specific splicing of the gene immediately downstream [66, 78]. Some of the key players involved in dipteran sex determination pathway include the genes sex lethal (sxl), doublesex (dsx), transformer (tra), transformer 2 (tra2), fruitless (fru), daughterless (da) and maleless (mle) (Additional file 14: Table S11).
The gene tra is one of the main sex determining genes in various insects whose ancestral function is to introduce variation in sex determining mechanisms [79]. We queried both the male and female P. regina genomes for homologs using D. melanogaster sex determining genes but found none. We then searched for P. regina tra and tra2 using homologs from closely related species - L. cuprina, Cochliomyia hominivorax, C. macellaria and L. sericata. This search also failed to produce any homologs to tra or tra2 – rather producing homologs to hypothetical L. cuprina proteins (FF38_00928 and FF38_09888, respectively). The only homology shared between our putative hits and those in the reference databases are due to the presence of zinc finger domains. For tra – Cochliomyia macellaria and C. hominivorax only share 60.8 % sequence identity. Our putative tra gene in Phormia regina shares 48.7 % and 52.3 % sequence identity with Cochliomyia and Chrysomya rufifacies, respectively (data not shown). For tra2 – a query of our genomes yielded hundreds of hits with e-values less than 1e-52 – suggesting not an assembly error, but rather a common domain is detected. Additional approaches are necessary to more fully annotate these genes in P. regina.
The gene doublesex (dsx) is another transcription factor that controls the activity of genes involved in sexual differentiation [66, 80]. Doublesex is differentially spliced, encoding male and female sex specific dsx proteins [66]. Homologous sequences of dsx were detected in both sexes with an E-value less than 1e-42. The sex determining gene daughterless is a member of the basic helix-loop-helix (bHLH) family of DNA binding domains and is a transcription factor [81]. Da is essential for neurogenesis, oogenesis and sex determination [82]. We annotated da in P. regina for both sexes. The length of the predicted da gene was determined to be 4494 bp and 8479 bp long in the male and female, respectively, in comparison to the D. melanogaster (FBgn0267821) da gene (5124 bp) (Fig. 3). The difference between the two appears to be in the 5’ UTR region in which some noncoding gene sequences are predicted in the female, but the male’s 5′ UTR was not completely assembled (the data are missing, Fig. 3).
Fig. 3.
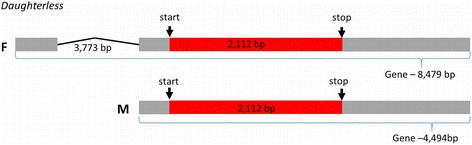
Predicted gene structure of the sex determining gene daughterless for the female (F) and male (M) P. regina. The red boxes represent the exon, the grey boxes inclusive of the red represent the mRNA, and the black line represent the intron. Image is not drawn to scale
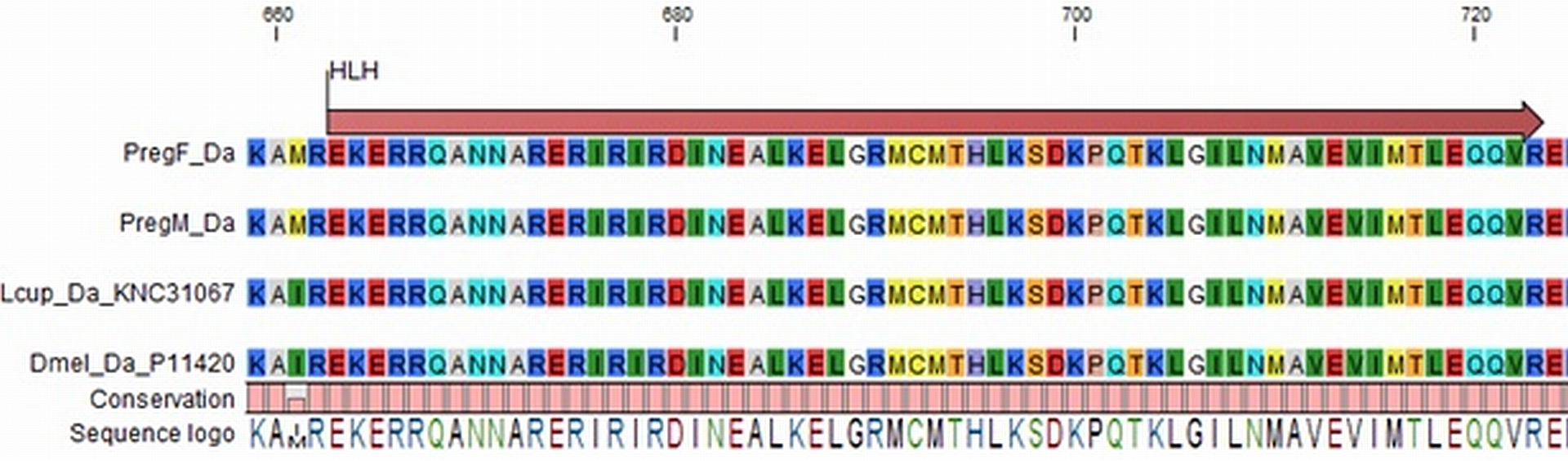
The coding sequences of da for both sexes were nearly identical (99.5 %) and predicted to be 2112 bp long, which is comparable to that of D. melanogaster of 2133 bp (J03148.1) and L. cuprina 2278 bp (JRES01000453.1 – scaffold966, locus tag FF38_09934). A multiple sequence alignment of the coding sequences demonstrates the high degree of variation with only 87.83 % similarity to L. cuprina, and 57.22 % to D. melanogaster (Additional file 15: Figure S4). Protein sequences of da were also compared between the three species. The length of da was 726 amino acid sequences for both sexes in P. regina compared to 758 amino acid sequences in L. cuprina (KNC31067.1) and 710 amino acid sequences in D. melanogaster (P11420). Amino acid sequence alignment shows L. cuprina to be 95 % identical to P. regina, while D. melanogaster is 59 % identical (Additional file 16: Figure S5). A conserved region shared among the three species is the helix-loop-helix domain (Additional file 17: Figure S6).
The maleless (mle) gene is one of the regulatory genes required for dosage compensation of X-linked genes in the X chromosome of male flies [83]. Annotation of the gene maleless (mle) resulted in gene sequences of total length 6375 bp and 6374 bp predicted for the male and female sexes, respectively, approximating the size of mle in D. melanogaster (6016 bp, JQ663522.1). The length of the protein sequence of mle in P. regina for both sexes was 1253 amino acids, comparable to D. melanogaster (1293 aa, AFI26242) with 72.27 % similarity to P. regina (Additional file 18: Figure S7).
Chemoreceptor genes
A fly’s ability to detect and respond to a chemical signal is integral to its survival. In particular, for blow flies, the adults must be able to detect odors associated with decay immediately following death in order to be among the first insects to show up and lay eggs. Following detection of carrion, an insect must be able to determine the quality of the resource and decide if it is suitable by using a variety of gustatory receptors (GRs), ionotropic receptors (IRs), odorant receptors (ORs), and odorant binding proteins (OBPs). These four gene families are the main chemoreceptors that function in the olfactory and gustatory system of insects [84]. To determine the presence of chemoreceptors in the P. regina genomes, we queried D. melanogaster’s chemoreceptor genes using tBLASTx. D. melanogaster has a predicted total of 68 GRs through alternative splicing, 62 ORs [85], 65 genes encoding IRs [86] and 52 genes encoding OBPs [87]. The tBLASTx results of the predicted chemoreceptors (including alternatively spliced sequences) in D. melanogaster resulted in a total of 61 GRs, 40 OBPs, 64 ORs and 63 IR’s with homologous sequences in the female assembled genome. These homologous sequences were detected in 28 contigs (61 GRs), 25 contigs (40 OBPs), 37 contigs (64 ORs) and 41 contigs (63 IRs) (Additional file 1: Table S1).
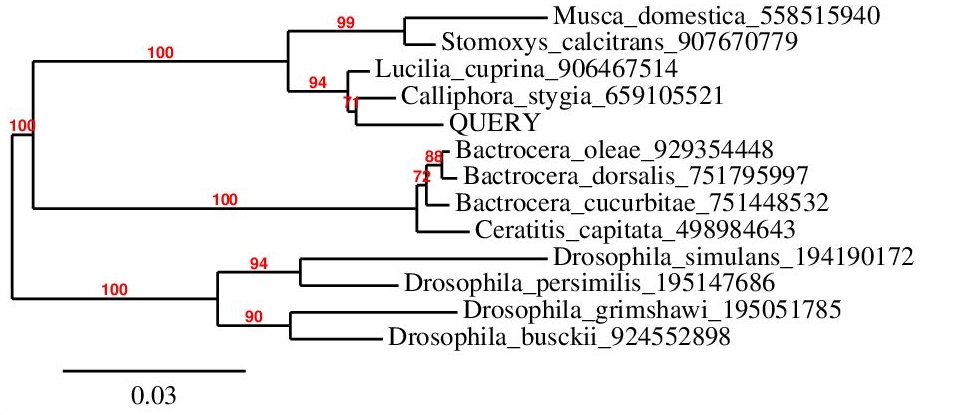
In addition to the reception of chemical stimulus in olfaction, IR’s have recently been found to be involved in thermosensation, and the circadian cycle [88]. In comparison to ORs, the IR family in insects is relatively conserved suggesting that it is an ancient chemosensory receptor family [89]. IR25a is one of the most highly conserved IRs in many species [90]. It acts as a co-receptor with other different odor-sensing IRs. In D. melanogaster, it is expressed in sensory neurons in the antennae and also in the proboscis [91]. We compared IR25a protein sequences from the female P. regina to L. cuprina, M. domestica, Calliphora stygia, Stomoxys calcitrans, D. melanogaster, C. capitata and B. oleae via multiple sequence alignment, and subsequent PhyML for tree building, and TreeDyn for tree drawing [92–97]. Overall there was high similarity, with sequence identity ranging from 86 to 97 %. The predicted P. regina IR25a sequence was most similar to L. cuprina with 97.19 %. The amino acid sequences in regions annotated as putative peptide binding sites were mostly conserved (Additional file 19: Figure S8). The remarkable sequence conservation of IR25a implies the presence of a unique and essential evolutionary conserved role of the IR25a receptors in various insect species (Additional file 20: Figure S9).
Antimicrobial genes
Blow flies develop in an environment of decomposing vertebrate tissue that is overrun with bacteria and as such, they not only compete with bacteria for resources but also need protection from infection. Insects in general have a diverse innate immunity pathway that provides protection from various microbes that can inhibit their survival [4, 98, 99]. As a result, they possess different mechanisms that signal the expression of genes activating an antimicrobial defense system to fight bacterial and fungal infections [100]. Some blow fly species have proven useful in wound debridement therapy as they also excrete many potent antimicrobial compounds [31, 33].
Two approaches were used to discover antimicrobial peptide genes: BLASTn using known D. melanogaster genes and keyword searching for known antimicrobial peptide sequences against annotation information assigned to the predicted protein sequences (Additional file 21: Table S12). Thirty-six Drosophila genes were queried and hits were identified for 25 using BLASTn. Using the BLASTp approach, 115 predicted protein sequences produced BLASTp hits with homology to genes present in four major signaling pathways in insects that are involved with protection from bacterial and fungal infections. These include the Toll, immune deficiency (Imd), Janus kinase signal transducer and activator of transcription (JAK/STAT), and JNK pathways [98, 101]. These pathways recognize different types of microbes and induce the transcription of immune-related genes which degrade pathogens or act as signaling molecules [99]. Both male and female had near identical BLASTp results. We therefore selected the female genome for the annotation of the contigs and genes of interest.
The Toll signaling pathway is activated by the presence of gram-positive bacteria or fungi while the Imd pathway is mainly activated by gram-negative bacteria. Both pathways lead to the production of antimicrobial peptides to fight pathogens that cause infection [99, 102]. A number of genes involved in the Toll signaling pathway were predicted in the P. regina female genome, including Spaetzle, tube, toll, cactus, G protein-coupled receptor kinase 2 [103, 104]. This is consistent with the suggestions that the Toll signaling pathway may be involved in the antimicrobial defense system in blow flies as it is in other insects [101, 103, 104]. Furthermore, protein recognition receptors involved in pathogen recognition were identified in P. regina. These include peptidoglycan-recognition proteins – PGRP-LE, PGRP-LC, PGRP-SC2 and gram negative binding proteins – GNBP1 and GNBP3. The NF-kappa β-like gene, relish, which is involved in inducing the humoral immune response in Drosophila as well as antibacterial and antifungal factors [105] was also identified. Both relish and PGRP-LC are involved in the Imd pathway.
Similar to M. domestica and D. melanogaster, P. regina harbors the four antimicrobial families, attacins, diptericins, cecropins and defensins. We found homology to genes related to antimicrobial humoral responses (par-1, cecA1 and tlk [106, 107]), as well as genes responsible for responses to bacteria (Gprk2 and Relish [108–110]), all possessing kinase or peptidase activity capable of breaking down bacteria cell walls (Additional file 21: Table S12). These are likely produced and excreted in the salivary glands of the developing larvae as they feed on the decaying tissue. A summary of the top hits from BLASTn results after querying our female genome for the homologous sequences from D.melanogaster are listed in Additional file 21: Table S12. The four antimicrobial families were represented by cecropin A1 (cecropin), iconoclast (defensing), Hephaestus (diptericin) and relish (attacin). These results based on homology to immune-related proteins in other insects, imply that the immune signaling pathway in P. regina is similar to other model insects, and may have evolved to work within the harsh sanitary conditions to which P. regina is normally exposed.
Xenobiotic resistance
The enhancement of xenobiotic (foreign compounds) metabolism in insects due to the extensive use of insecticides, has led to the evolution of xenobiotic resistance and tolerance in insects creating a challenge in pest management [111]. The three major groups of genes involved in producing metabolic enzymes to protect insects against plant defense systems (plant allelochemicals) and insecticides are cytochrome P450 monooxygenases, esterases/hydrolases, and glutathione-S-transferases [112, 113]. The presence of these detoxifying enzymes likely helps P. regina to withstand high pathogen loads from decaying carrion. Cytochrome P450 genes are the family of enzymes primarily associated with metabolism of xenobiotics and resistance to most insecticides [114, 115]. They are also involved in the catalysis of a range of chemical reactions important for developmental processes.
A total of 41 and 44 predicted genes (Additional file 3: Table S3) were annotated via the D. melanogaster database as cytochrome P450 genes from the female and male P. regina genes, respectively. This is fewer than in any of the three species we compared; M. domestica has 146 P450 genes, 90 in D. melanogaster, and 72 in Glossina morsitans [116]. However, when we applied our methods to the Lucilia cuprina published predicted gene set, we recovered 57 P450 genes (data not shown). CYP4 and CYP6 were the predominant P450 families in both male and female draft genomes (Additional file 3: Table S3) where they occupied approximately 36 and 30 % of the total P450 genes respectively (in L. cuprina, they occupied 25 and 23 %, respectively). The predominance of CYP4 and CYP6 P450 genes was also observed in the genomes of D. melanogaster [117] and M. domestica where they occupy 50 and >60 % of the total cytochrome P450 genes in their genomes, respectively. The reduction in the number of P450 genes (assumed to be closer to 100 in insect genomes) is likely due to the stringent gene prediction criteria used in our fragmented genomes (only complete genes predicted).
An increase in the expression or activity of the metabolic enzymes that belong to the esterases and hydrolases family has also been linked to insecticide resistance [118] and correlated with resistance to two major insecticide classes: pyrethroids and organophosphates [113]. This is mainly due to the presence of ester bonds in most insecticides which are hydrolyzed by the esterase [119]. A total of 103 and 131 genes with hydrolase/esterase activities were predicted in the female and male draft genomes, respectively. The common ones in both sexes included phosphodiesterase, thioesterase, carboxylesterase and phosphatase (Additional file 5: Table S5).
Glutathione S-transferases (GST) are multifunctional enzymes that are not only involved in detoxification of xenobiotic compounds, but also in other physiological processes in insects including intracellular transport and hormone biosynthesis [120]. In the detoxification process, they function by metabolizing insecticides producing water-soluble metabolites that are easily excreted [121, 122]. A total of 9 and 11 GST genes were predicted in the female and male P. regina genomes, respectively (Additional file 4: Table S4). Unfortunately, insecticide resistance is a growing problem [123] in many insects, including mosquitos [124–126] and blow flies [127–129] has in some cases been attributed to glutathione S-transferase activity [120, 123, 130–132].
Repetitive elements
Repeat identification in the male and female genomes via homology-based searches identified close to 38 and 46 Mbp of repetitive DNA accounting for 7.3 and 8.7 % of the male and female assemblies (Additional file 22: Table S13). Approximately 10.5 (male) and 18 Mbp (female) of repetitive sequences were transposable elements, and the remaining were low complexity sequences including mini- and microsatellites. Several major transposable elements super-families were identified in the P. regina genome, but the vast majority (>70 %) of elements belonged to 5 families or super families: Jockeys, LOAs, Gypsys, Tc-Mariners, and Helitrons. DNA transposons and retrotransposons were present in roughly equal proportions. Low complexity sequences were the dominant type of repetitive sequence making up ~66 % of the male and female genome assemblies.
Dipteran species show significant variability in SINE content but SINE elements appear to be missing from the P. regina genome entirely. For example, SINEs are absent in Drosophila [133] but present in high copy numbers in other dipterans [5]. Only 39 SINE insertions were identified in P. regina, most of which are distantly related, or highly mutated version of SINE-3_QC and SINE-4_QC from Culex quinquefasciatus, the southern house mosquito. In C. quinquefasciatus, SINE-3_QC and SINE-4_QC are present more than ten thousand times [134].
Based on the female (Fig. 4a) and male (Fig. 4b) accumulation profiles, it appears that transposable elements in P. regina tend to be old, with little accumulation in the recent past. In general, transposable element insertions for each family tend to be more numerous in the female genome assembly. Transposable elements present in the female assembly, but absent in the male assembly follow a similar accumulation profile to the genomes as a whole (Fig. 4c) ruling out temporally biased accumulation in either sex. Class II transposons, LINEs, and LTRs accumulated at similar times given that the majority of elements in each group are between 37 and 45 % diverged from their putative ancestral partner. DNA transposons have been accumulating for a slightly longer period than LINEs and LTRs with most element divergences ranging from 23 to 41 %. In all, >97 % of all elements are >10 % divergent from their respective consensus elements. This implies that the minimal transposable elements accumulation has occurred in the recent past, or that newly inserted transposable elements are being actively removed from the genome [135]. However, with high divergences between potentially novel transposable elements in P. regina and transposable elements in RepBase is possible that lineage-specific SINEs are present but unidentifiable using homology based searches [136] and a full TE curation of the genome is necessary.
Fig. 4.
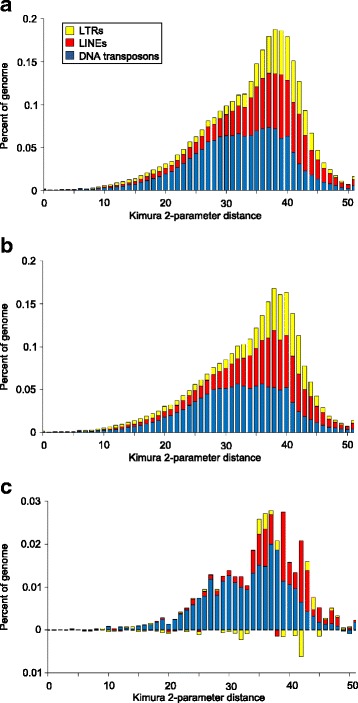
Transposable element accumulation in the female (a) and male (b) Phormia regina genome assemblies. Kimura 2-parameter distances were calculated between transposable element insertions in the genome and the homologous element in the dipteran Repbase library. Larger divergences indicate elements with larger mutation loads, and by extension, were deposited in the genome in the more distant past. Less than 40 SINE elements are present in either the female or male assemblies and are not shown here. Transposable elements are slightly more abundant in the female genome assembly. The accumulation of female specific repeats (c) follows that of the whole genome in general
Close to 1.7 Mb of the X chromosome (8.1 % of the total X chromosomes) was derived from repeats, compared to ~390 Kb of the Y chromosome (2.7 % of the total Y chromosome). For both chromosomes, more than half the repetitive sequences are in the form of simple repeats (60 and 55 % for the X and Y chromosomes, respectively). The increased amount of repeats on the X chromosome is likely due to its larger size, however, even though the X chromosome is proportionally more repetitive than the Y, it is not large enough to be significant.
Conclusions
Although all impacts are impossible to foresee, we anticipate four important fields will benefit from this data: (1) insecticide resistance and/or sensitivity; (2) adaptive evolution in a rapidly evolving clade (Calliphoridae); (3) sex chromosome evolution; and (4) genotype-phenotype correlations of development rate variation.
Insecticide resistance
There are four main calliphorids that are serious agricultural pests causing myiasis (infestation by fly larvae): Chrysomya bezziana (Old World screwworm), Cochliomyia hominivorax (New World screwworm), Lucilia sericata (green bottle fly) and L. cuprina (sheep blow fly) [14, 137, 138], although other species have been implicated in secondary myiasis, including P. regina ([139–143]. Two of these, Ch. bezziana and Co. hominivorax are obligate parasites, while the remaining two Lucilia species are either obligate, or facultative ectoparasites, depending on where they reside [144]. The genome of the obligate ectoparasite L. cuprina (in Australia/New Zealand, this species does not appear to ever develop on carrion) was recently sequenced in an effort to determine how insecticide resistance has persisted and to develop novel pest management targets that would not harm beneficial insects. In order to better understand these processes and assess target efficacy, it is important to understand how these genes are structured in other closely related flies. The P. regina reference genome presented here will provide the opportunity to extract candidate genes, determine the structure-function relationship, and produce new ligands with the potential to slow or eradicate these pest species.
Adaptive evolution
Calliphoridae includes approximately 1500 different species and accounts for ~8 % of calyptrate species diversity [8]. Many lineages within Calliphoridae have evolved specialized adaptations. As the above example demonstrates, ectoparasitism has evolved at least twice within Calliphoridae, perhaps in response to selective pressures of the usually ephemeral carrion resource – if the insect can arrive at a resource before it becomes available to all its competitors, its genes have a greater probability of being maintained. P. regina does not have these features or life-strategies, suggesting the retention of the ancestral life history, and can thus serve as a robust reference genome from which more derived features can be understood.
Sex chromosome evolution
Many calyptrate flies utilize XY sex determination. In many other species sharing this system, the X chromosome is relatively stable, and the Y chromosome has experienced loss of function due to the absence of recombination over time and the presence of repetitive DNA. Therefore, the structure of the Y chromosome represents its approximate evolutionary age. With individual sex genomes, and putative chromosomes sequenced and assembled herein, we can compare sex chromosome structure within the Calliphoridae, where some species have vastly different sex chromosome sizes (i.e. Lucilia cuprina male and female flies differ by nearly 100 Mbp, [43]), or within the monogenic fly Chrysomya rufifacies with no difference in genome sizes of the male and female flies [43]. Additionally, this reference genome will be useful in studying sex determining pathways in Calliphoridae, which could be useful as a mechanism to target for pest control [145].
Development rate variation
As many of these flies are primary colonizers of vertebrate carrion [17], they have forensic uses in cases of decomposition when the time of death cannot be determined using traditional physiological approaches. The collected larvae serve as a clock to the minimum time since the individual died, as they will only colonize a body following death (excluding species known to cause myiasis). Then, the age of the larvae is estimated for a minimum postmortem interval [146, 147]. The age, however, is estimated from a reference data set in which laboratory conditions permit multiple temperatures and conditions [148–150]. These models of development assume little to no population-level variation even though variation has been clearly demonstrated [151–153]. Thus, it is important to understand the fundamental developmental processes in blow flies if they are to be used for postmortem interval estimates, and to be capable to predicting the possible variation based on genotypes of the individual larvae collected. Once again, the availability of a reference genome, differentiating male and female genomes [154], will be invaluable to understanding the molecular basis of development, and its associated variation.
This first draft of the P. regina genome represents a critical step in calliphorid genomics. The accessibility of the P. regina genome will quicken the pace of the exploration and comparisons in the evolutionary relationships and developmental analysis among blow fly species and also with other dipteran species. In time, these findings could have significant agricultural, medical and forensics fields.
Acknowledgements
We are grateful to Phillip San Miguel (Director of the Purdue Genomics Core facility at Purdue University West Lafayette), and Gintaras Deikus & Robert Sebra (Director of Technology Development & Assistant Professor, Icahn School of Medicine at Mount Sinai, Institute for Genomics and Multiscale Biology) for library construction and DNA sequencing of our samples (Illumina and Pacific Biosciences, respectively). We also would like to thank Carrie Ganote and Le-Shin Wu of NCGAS at IU Bloomington for technical support and Scott Orr (IUPUI) for his expertise in data storage. We also appreciate the comments and suggestions during the critical review of this manuscript by two anonymous reviewers.
Funding
Financial support has come from the School of Science (IUPUI) for providing start-up funds (CJP). This research is based upon work supported by the National Science Foundation under Grants DBI-1458641 and ABI-1062432 to Indiana University (NCGAS) and CNS-0521433 (Data Capacitor II); Lilly Endowment, Inc., through its support for the Indiana University Pervasive Technology Institute and in part by the Indiana METACyt Initiative (Karst); and Shared University Research grants from IBM, Inc., to Indiana University (Scholarly Data Archive). Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Science Foundation, the National Center for Genome Analysis Support, or Indiana University.
Availability of data and materials
This Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under the accession MINK00000000 and MINJ00000000 for the male and female genomes, respectively. The Phormia regina genomes and raw sequence reads are available from NCBI under BioProject PRJNA338752 and BioSamples SAMN05567885 and SAMN05567884.
Authors’ contributions
AAA carried out the de novo assembly, analyzed and interpreted the data, and drafted the manuscript. RNP performed the repetitive element analysis, contributed to writing and editing the manuscript. DAR participated in analysis and interpretation of data, and critically revised the draft manuscript. CJP conceived and designed the study, participated in coordination and contributed to draft manuscript writing and revisions. All authors read and approved the final manuscript.
Competing interests
The authors declare they have no competing interests.
Consent for publication
Not applicable.
Ethics approval
Not applicable.
Abbreviations
- aa
Amino acid
- AS-C
Achaete-scute complex
- BAC
Bacterial artificial chromosome
- bHLH
Basic helix-loop-helix
- bp
Base pair
- CEGMA
Core eukaryotic genes mapping approach
- CoA
Coenzyme A
- CQ
Chromosome quotient
- D. melanogaster
Drosophila melanogaster
- da
daughterless
- dsx
doublesex
- fru
fruitless
- Gb
Gigabase
- GNBP
Gram negative binding proteins
- GO
Gene ontology
- GRs
Gustatory receptors
- GSTs
Glutathione S-transferases
- Imd
Immune deficiency
- IRs
Ionotropic receptors
- JAK/STAT
Janus kinase signal transducer and activator of transcription
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- L. cuprina
Lucilia cuprina
- LINEs
Long interspersed nuclear elements
- LTRs
Long terminal repeats
- Mb
Megabases
- Mbp
Million base pair
- mle
Maleless
- MRSA
Methicillin resistant Staphylococcus aureus
- OBPs
Odorant binding proteins
- ORs
Odorant receptors
- P. regina
Phormia regina
- PGRP
Peptidoglycan-recognition proteins
- REViGO
Reduce and visualize gene ontology
- SINEs
Short interspersed nuclear elements
- sxl
Sex lethal
- tra
Transformer
- tra2
Transformer2
- UTR
Untranslated region
Additional files
tBLASTx results of contigs with D. melanogaster’s homologus gene sequences of gustatory receptors, odorant binding proteins, odorant receptors and ionotropic receptors. (XLSX 22 kb)
BLASTn (27 hits) and BLASTp (115 hits) results for antimicrobial peptide queries using D. melanogaster’s homologous gene and protein sequences as queries. (XLSX 19 kb)
A list showing the distribution of cytochrome P450 monooxygenases families in the male and female P. regina genomes. (XLSX 18 kb)
A list showing the distribution of glutathione S-transferases in the male and female P. regina genomes. (XLSX 9 kb)
A list of metabolic enzymes that belong to the esterases and hydrolases family that are predicted to be linked with insecticide resistance. (XLSX 22 kb)
{kind=link}
The mitochondrial DNA (mtDNA) genome of Phormia regina, annotated using the mtDNA genome of Cochliomyia hominivorax mtDNA genome. The AT-rich region is colored in purple, tRNA and rRNA genes in red, and protein coding genes are in yellow. (JPG 102 kb)
CEGMA Completeness report of the female and male P. regina genomes. Number of proteins equals the number of proteins found from the 248 ultra-conserved CEGs present in the genome with % completeness representing the percentage. (DOC 48 kb)
{kind=link}
Top-hit species distribution from Blast2GO for the male and female P. regina. The top hit species is the blow fly L. cuprina. The species listed are those with >25 hits. Those with less than the threshold are summed and grouped in the ‘Other’ category. (JPG 551 kb)
Comparative gene ontology terms of the male and the female Phormia regina predicted genes. (DOC 69 kb)
A comparative gene ontology showing the ranking of different functional categories in each species among P. regina (both sexes), M. domestica and D. melanogaster with the rank of 1 indicating the most abundant GO term. (DOC 62 kb)
List of biological KEGG pathways extracted from the annotations of the male and female predicted protein-encoding genes. Each tab represents biological KEGG pathways extracted from the male and female genomes. (XLSX 66 kb)
BLASTn results of the putative sex chromosome contigs (chromosome X and Y) queried against the arthropoda database in GenBank. (XLSX 729 kb)
{kind=link}
A visual representation showing a summary of GO terms categorized in the biological processes that are unique to the male (A) and female (B). (JPG 1742 kb)
Top hits of sex determining genes homologous to D. melanogaster in the male and female P. regina assembled genomes. (DOC 34 kb)
{kind=link}
Multiple sequence alignment of the coding sequences of the sex determining gene daughterless of male and female P. regina, L. cuprina (scaffold 966) and D. melanogaster (J03148). Sequence similarities between the male and female P. regina is 99.95 %, between P. regina (F) and L. cuprina is 87.83 %, and between P. regina (F) and D. melanogaster is 57.22 %. (JPG 1974 kb)
{kind=link}
Multiple sequence alignment of protein sequences of the sex determining gene daughterless of P. regina, L. cuprina (KNC31067) and D. melanogaster (P11420). Sequence similarity of P. regina to L. cuprina is 95 % and to D. melanogaster is 59 %. (JPG 1589 kb)
{kind=link}
A section of a multiple sequence alignment of the sex determining gene, daughterless, of the conserved region of 60 amino acid sequences of the helix-loop-helix domain of the da protein. (JPG 593 kb)
{kind=link}
Multiple sequence alignment of protein sequences of the sex determining gene maleless of P. regina and D. melanogaster (P24785). Sequence similarities between the male and female P. regina is 100 %, and the similarity to D. melanogaster is 72.27 %. (JPG 1574 kb)
{kind=link}
Multiple amino acid sequence alignment of the predicted ionotropic receptor IR25a in Phormia regina (g4045.t1), Lucilia cuprina (KNC28739, sequence similarity 97.19 %), Stomoxys calcitrans (XP_013104244, sequence similarity 94.76 %), Bactrocera oleae (XP_014086336, sequence similarity 88.12 %), Ceratitis capitata (XP_004530416, sequence similarity 87.85 %). Also included are protein sequences generated from sequenced RNA from Calliphora stygia (AID61273, sequence similarity 97.06 %), Musca domestica (NP_001273813, 93.36 %), and Drosophila melanogaster (NP_001260049, sequence similarity 86.08 %). The P. regina amino acid sequence is incomplete at the amino terminus, however, the conservation in this protein is demonstrated by the similarities between the wide taxonomic groups represented here. (JPG 1574 kb)
{kind=link}
Maximum likelihood phylogenetic tree of the amino acid sequences of IR25a based on alignment generated for Additional file 19: Figure S8. Scale bar represents evolutionary distance (number of amino acid substitutions). (JPG 148 kb)
A summary of the top BLASTn results of antimicrobial related genes homologous to Drosophila. The four antimicrobial families are represented by cecropin A1 (cecropin), iconoclast (defensin), hephaestus (diptericin) and relish (attacin). (DOC 37 kb)
A summary of copy number, number of bases, and percent of the genome derived from repetitive sequences in the female and male Phormia regina genome. Repeats were identified based on homology to known dipteran repetitive sequences in RepBase. (DOC 72 kb)
Contributor Information
Anne A. Andere, Email: aaandere@iupui.edu
Roy N. Platt, II, Email: neal.platt@gmail.com.
David A. Ray, Email: david.a.ray@ttu.edu
Christine J. Picard, Phone: (317) 278-1050, Email: cpicard@iupui.edu
References
- 1.Misof B, Liu S, Meusemann K, Peters RS, Donath A, Mayer C, Frandsen PB, Ware J, Flouri T, Beutel RG, et al. Phylogenomics resolves the timing and pattern of insect evolution. Science. 2014;346:763–767. doi: 10.1126/science.1257570. [DOI] [PubMed] [Google Scholar]
- 2.Wiegmann BM, Trautwein MD, Winkler IS, Barr NB, Kim JW, Lambkin C, Bertone MA, Cassel BK, Bayless KM, Heimberg AM, et al. Episodic radiations in the fly tree of life. Proc Natl Acad Sci U S A. 2011;108:5690–5695. doi: 10.1073/pnas.1012675108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, Amanatides PG, Scherer SE, Li PW, Hoskins RA, Galle RF, et al. The genome sequence of Drosophila melanogaster. Science. 2000;287:2185–2195. doi: 10.1126/science.287.5461.2185. [DOI] [PubMed] [Google Scholar]
- 4.Scott JG, Warren WC, Beukeboom LW, Bopp D, Clark AG, Giers SD, Hediger M, Jones AK, Kasai S, Leichter CA, et al. Genome of the house fly, Musca domestica L., a global vector of diseases with adaptations to a septic environment. Genome Biol. 2014;15:466. [DOI] [PMC free article] [PubMed]
- 5.Holt RA, Subramanian GM, Halpern A, Sutton GG, Charlab R, Nusskern DR, Wincker P, Clark AG, Ribeiro JM, Wides R, et al. The genome sequence of the malaria mosquito Anopheles gambiae. Science. 2002;298:129–149. doi: 10.1126/science.1076181. [DOI] [PubMed] [Google Scholar]
- 6.Norris KB. The bionomics of blow flies. Ann Rev Entomol. 1965;10:47–68. doi: 10.1146/annurev.en.10.010165.000403. [DOI] [Google Scholar]
- 7.Pape T, Blagoderov V, Mostovski MB. Order diptera linnaeus in animal biodiversity: an outline of higher-level classification and survey of taxonomic richness. Zootaxa. 2011;2148:222–229. [Google Scholar]
- 8.Singh B, Wells JD. Molecular systematics of the Calliphoridae (Diptera: Oestroidea): evidence from one mitochondrial and three nuclear genes. J Med Entomol. 2013;50:15–23. doi: 10.1603/ME11288. [DOI] [PubMed] [Google Scholar]
- 9.Chleir F. Maggot therapy for wound debridement. A randomized multicenter trial. Phlebologie Ann Vasc. 2012;65:75. doi: 10.1001/archdermatol.2011.1895. [DOI] [PubMed] [Google Scholar]
- 10.Kirinoki-Ichikawa S, Miyamoto M, Tezuka A, Kubota Y, Takagi G, Iimura T, Takami Y, Matsuda N, Mizuno K. Limb salvage achievement by maggot debridement therapy (Mdt); the analysis of 164 chronic wound patients. Wound Repair Regen. 2014;22:A13. [Google Scholar]
- 11.Sherman RA, Pechter EA. Maggot therapy: a review of the therapeutic applications of fly larvae in human medicine, especially for treating osteomyelitis. Med Vet Entomol. 1988;2:225–230. doi: 10.1111/j.1365-2915.1988.tb00188.x. [DOI] [PubMed] [Google Scholar]
- 12.Steenvoorde P, Jacob CE, Van Doorn L, Oskam J. Maggot debridement therapy of infected ulcers: patient and wound factors influencing outcome - a study on 101 patients with 117 wounds. Ann R Coll Surg Engl. 2007;89:598–604. doi: 10.1308/003588407X205404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bisdorff B, Wall R. Sheep blowfly strike risk and management in Great Britain: a survey of current practice. Med Vet Entomol. 2008;22:303–308. doi: 10.1111/j.1365-2915.2008.00756.x. [DOI] [PubMed] [Google Scholar]
- 14.Colwell DD, Dorchies P, Scholl PJ, Losson B, Boulard C, Chaudhury MF, Graf JF, Jacquiet P, Barillet E, Carta A, et al. Management of myiasis: current status and future prospects. Vet Parasitol. 2004;125:93–104. doi: 10.1016/j.vetpar.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 15.Hall M, Wall R. Myiasis of humans and domestic animals. Adv Parasitol. 1995;35:257–334. doi: 10.1016/S0065-308X(08)60073-1. [DOI] [PubMed] [Google Scholar]
- 16.Knipling EF, Rainwater HT. Species and incidence of dipterous larvae concerned in wound myiasis. J Parasitol. 1937;23:451–455. doi: 10.2307/3272391. [DOI] [Google Scholar]
- 17.Byrd JH, Castner JL. Forensic entomology: the utility of arthropods in legal investigations. 2. Boca Raton: CRC Press; 2010. [Google Scholar]
- 18.Amendt J, Zehner R, Johnson DG, Wells J. Future Trends in Forensic Entomology. Curr Concepts Forensic Entomol. 2010:353-68.
- 19.Greenberg B. Flies and disease. Princeton: Princeton University Press; 1971. [Google Scholar]
- 20.Hubalek Z, Halouzka J. Persistence of clostridium botulinum type C toxin in blow fly (Calliphoridae) larvae as a possible cause of avian botulism in spring. J Wildl Dis. 1991;27:81–85. doi: 10.7589/0090-3558-27.1.81. [DOI] [PubMed] [Google Scholar]
- 21.Fischer OA, Matlova L, Dvorska L, Svastova P, Bartl J, Weston RT, Pavlik I. Blowflies Calliphora vicina and Lucilia sericata as passive vectors of Mycobacterium avium subsp. avium, M. a. paratuberculosis and M. a. hominissuis. Med Vet Entomol. 2004;18:116–122. doi: 10.1111/j.0269-283X.2004.00477.x. [DOI] [PubMed] [Google Scholar]
- 22.Asgari S, Hardy JR, Sinclair RG, Cooke BD. Field evidence for mechanical transmission of rabbit haemorrhagic disease virus (RHDV) by flies (Diptera:Calliphoridae) among wild rabbits in Australia. Virus Res. 1998;54:123–132. doi: 10.1016/S0168-1702(98)00017-3. [DOI] [PubMed] [Google Scholar]
- 23.Maldonado MA, Centeno N. Quantifying the potential pathogens transmission of the blowflies (Diptera: Calliphoridae) Mem Inst Oswaldo Cruz. 2003;98:213–216. doi: 10.1590/S0074-02762003000200008. [DOI] [PubMed] [Google Scholar]
- 24.Brown CJ. House flies and Helicobacter pylori. Can Med Assoc J. 1997;157:130. [Google Scholar]
- 25.Graczyk TK, Cranfield MR, Fayer R, Bixler H. House flies (Musca domestica) as transport hosts of Cryptosporidium parvum. Am J Trop Med Hyg. 1999;61:500–504. doi: 10.4269/ajtmh.1999.61.500. [DOI] [PubMed] [Google Scholar]
- 26.Barnes KM, Dixon RA, Gennard DE. The antibacterial potency of the medicinal maggot, Lucilia sericata (Meigen): Variation in laboratory evaluation. J Microbiol Methods. 2010;82:234–237. doi: 10.1016/j.mimet.2010.06.005. [DOI] [PubMed] [Google Scholar]
- 27.Tantawi TI, Willams KA, Villet MH. An accidental but safe and effective use of lucilia cuprina (Diptera: Calliphoridae) in maggot debridement therapy in Alexandria, Egypt. J Med Entomol. 2010;47:491–494. doi: 10.1603/ME09183. [DOI] [PubMed] [Google Scholar]
- 28.Bexfield A, Bond AE, Roberts EC, Dudley E, Nigam Y, Thomas S, Newton RP, Ratcliffe NA. The antibacterial activity against MRSA strains and other bacteria of a < 500 Da fraction from maggot excretions/secretions of Lucilia sericata (Diptera : Calliphoridae) Microbes Infect. 2008;10:325–333. doi: 10.1016/j.micinf.2007.12.011. [DOI] [PubMed] [Google Scholar]
- 29.Bexfield A, Nigam Y, Thomas S, Ratcliffe NA. Detection and partial characterisation of two antibacterial factors from the excretions/secretions of the medicinal maggot Lucilia sericata and their activity against methicillin-resistant Staphylococcus aureus (MRSA) Microbes Infect. 2004;6:1297–1304. doi: 10.1016/j.micinf.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 30.Cerovsky V, Zdarek J, Fucik V, Monincova L, Voburka Z, Bem R. Lucifensin, the long-sought antimicrobial factor of medicinal maggots of the blowfly Lucilia sericata. Cell Mol Life Sci. 2010;67:455–466. doi: 10.1007/s00018-009-0194-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jaklic D, Lapanje A, Zupancic K, Smrke D, Gunde-Cimerman N. Selective antimicrobial activity of maggots against pathogenic bacteria. J Med Microbiol. 2008;57:617–625. doi: 10.1099/jmm.0.47515-0. [DOI] [PubMed] [Google Scholar]
- 32.Kruglikova AA. Antimicrobial components of haemolymph and exosecretion of Larvae Lucilia sericata (Meigen) (Diptera, Calliphoridae) J Evol Biochem Physiol. 2011;47:534–542. doi: 10.1134/S0022093011060044. [DOI] [PubMed] [Google Scholar]
- 33.Margolin L, Gialanella P. Assessment of the antimicrobial properties of maggots. Int Wound J. 2010;7:202–204. doi: 10.1111/j.1742-481X.2010.00234.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van der Plas MJA, Dambrot C, Dogterom-Ballering HCM, Kruithof S, van Dissel JT, Nibbering PH. Combinations of maggot excretions/secretions and antibiotics are effective against Staphylococcus aureus biofilms and the bacteria derived therefrom. J Antimicrob Chemother. 2010;65:917–923. doi: 10.1093/jac/dkq042. [DOI] [PubMed] [Google Scholar]
- 35.Dogandemir G, Koru O, Bedir O, Kilic A, Araz RE, Tanyuksel M, Basustaoglu AC. Evaluation of in vitro antimicrobial activity of whole body extracts of Lucilia Sericata Maggots. Am J Trop Med Hyg. 2010;83:62. [Google Scholar]
- 36.Barnes KM, Gennard DE, Dixon RA. An assessment of the antibacterial activity in larval excretion/secretion of four species of insects recorded in association with corpses, using Lucilia sericata Meigen as the marker species. Bull Entomol Res. 2010;100:635–640. doi: 10.1017/S000748530999071X. [DOI] [PubMed] [Google Scholar]
- 37.Huberman L, Gollop N, Mumcuoglu KY, Black C, Galun R. Antimicrobial properties of whole body extracts and haemolymph of Lucilia sericata maggots. J Wound Care. 2007;16:123–7. doi: 10.12968/jowc.2007.16.3.27011. [DOI] [PubMed] [Google Scholar]
- 38.Anstead CA, Korhonen PK, Young ND, Hall RS, Jex AR, Murali SC, Hughes DS, Lee SF, Perry T, Stroehlein AJ, et al. Lucilia cuprina genome unlocks parasitic fly biology to underpin future interventions. Nat Commun. 2015;6:7344. doi: 10.1038/ncomms8344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bertone MA, Courtney GW, Wiegmann BM. Phylogenetics and temporal diversification of the earliest true flies (Insecta: Diptera) based on multiple nuclear genes. Syst Entomol. 2008;33:668–687. doi: 10.1111/j.1365-3113.2008.00437.x. [DOI] [Google Scholar]
- 40.Whitworth T. Keys to the genera and species of blow flies (Diptera : Calliphoridae) of America North of Mexico. Proc Entomol Soc Wash. 2006;108:689–725. [Google Scholar]
- 41.Putnam RJ. The role of carrion-frequenting arthropods in the decay process. Ecol Entomol. 1978;3:133–139. doi: 10.1111/j.1365-2311.1978.tb00911.x. [DOI] [Google Scholar]
- 42.Ullerich FH, Schottke M. Karyotypes, constitutive heterochromatin, and genomic DNA values in the blowfly genera Chrysomya, Lucilia, and Protophormia (Diptera: Calliphoridae) Genome. 2006;49:584–597. doi: 10.1139/G06-013. [DOI] [PubMed] [Google Scholar]
- 43.Picard CJ, Johnston JS, Tarone AM. Genome sizes of forensically relevant Diptera. J Med Entomol. 2012;49:192–197. doi: 10.1603/ME11075. [DOI] [PubMed] [Google Scholar]
- 44.Ramakodi MP, Singh B, Wells JD, Guerrero F, Ray DA. A 454 sequencing approach to dipteran mitochondrial genome research. Genomics. 2015;105:53–60. doi: 10.1016/j.ygeno.2014.10.014. [DOI] [PubMed] [Google Scholar]
- 45.Koren S, Schatz MC, Walenz BP, Martin J, Howard JT, Ganapathy G, Wang Z, Rasko DA, McCombie WR, Jarvis ED, Adam MP. Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nat Biotechnol. 2012;30:693–700. doi: 10.1038/nbt.2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zerbino DR. Using the Velvet de novo assembler for short-read sequencing technologies. Curr Protoc Bioinformatics. 2010;Chapter 11:Unit 11 15. doi: 10.1002/0471250953.bi1105s31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, He G, Chen Y, Pan Q, Liu Y, et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience. 2012;1:18. doi: 10.1186/2047-217X-1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Parra G, Bradnam K, Korf I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics. 2007;23:1061–1067. doi: 10.1093/bioinformatics/btm071. [DOI] [PubMed] [Google Scholar]
- 49.Gurevich A, Saveliev V, Vyahhi N, Tesler G. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 2013;29:1072–1075. doi: 10.1093/bioinformatics/btt086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stanke M, Morgenstern B. AUGUSTUS: a web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 2005;33:W465–467. doi: 10.1093/nar/gki458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 52.Conesa A, Gotz S. Blast2GO: a comprehensive suite for functional analysis in plant genomics. Int J Plant Genomics. 2008;2008:619832. doi: 10.1155/2008/619832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jones P, Binns D, Chang HY, Fraser M, Li W, McAnulla C, McWilliam H, Maslen J, Mitchell A, Nuka G, et al. InterProScan 5: genome-scale protein function classification. Bioinformatics. 2014;30:1236–1240. doi: 10.1093/bioinformatics/btu031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Supek F, Bosnjak M, Skunca N, Smuc T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One. 2011;6:e21800. doi: 10.1371/journal.pone.0021800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hall AB, Qi Y, Timoshevskiy V, Sharakhova MV, Sharakhov IV, Tu Z. Six novel Y chromosome genes in Anopheles mosquitoes discovered by independently sequencing males and females. BMC Genomics. 2013;14:273. doi: 10.1186/1471-2164-14-273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Attrill H, Falls K, Goodman JL, Millburn GH, Antonazzo G, Rey AJ, Marygold SJ, Consortium F. FlyBase: establishing a Gene Group resource for Drosophila melanogaster. Nucleic Acids Res. 2016;44:D786–D792. doi: 10.1093/nar/gkv1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brown NP, Leroy C, Sander C. MView: a web-compatible database search or multiple alignment viewer. Bioinformatics. 1998;14:380–381. doi: 10.1093/bioinformatics/14.4.380. [DOI] [PubMed] [Google Scholar]
- 60.Smit AFA, Hubley R, Green P. RepeatMasker Open-4.0. http://www.repeatmasker.org; 2013-2015.
- 61.Kimura M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol. 1980;16:111–120. doi: 10.1007/BF01731581. [DOI] [PubMed] [Google Scholar]
- 62.Treangen TJ, Salzberg SL. Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat Rev Genet. 2012;13:36–46. doi: 10.1038/nrg3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vinson JP, Jaffe DB, O’Neill K, Karlsson EK, Stange-Thomann N, Anderson S, Mesirov JP, Satoh N, Satou Y, Nusbaum C, et al. Assembly of polymorphic genomes: algorithms and application to Ciona savignyi. Genome Res. 2005;15:1127–1135. doi: 10.1101/gr.3722605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Younger-Shephard S, Vaessin H, Bier E, Jan LY, Jan YN. Deadpan, an essential pan-neural gene encoding an HLH protein, acts as a denominator in Drosophila sex determination. Cell. 1992;70:911–922. doi: 10.1016/0092-8674(92)90242-5. [DOI] [PubMed] [Google Scholar]
- 65.McAlpine JF. Manual of Nearctic Diptera. Ottawa: Research Branch, Agriculture Canada; 1981. [Google Scholar]
- 66.Sanchez L. Sex-determining mechanisms in insects. Int J Dev Biol. 2008;52:837–856. doi: 10.1387/ijdb.072396ls. [DOI] [PubMed] [Google Scholar]
- 67.Concha C, Scott MJ. Sexual Development in Lucilia cuprina (Diptera, Calliphoridae) Is Controlled by the Transformer Gene. Genetics. 2009;182:785–798. doi: 10.1534/genetics.109.100982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dubendorfer A, Hediger M, Burghardt G, Bopp D. Musca domestica, a window on the evolution of sex-determining mechanisms in insects. Int J Dev Biol. 2002;46:75–79. [PubMed] [Google Scholar]
- 69.Carvalho AB. Origin and evolution of the Drosophila Y chromosome. Curr Opin Genet Dev. 2002;12:664–668. doi: 10.1016/S0959-437X(02)00356-8. [DOI] [PubMed] [Google Scholar]
- 70.Carvalho AB, Vibranovski MD, Carlson JW, Celniker SE, Hoskins RA, Rubin GM, Sutton GG, Adams MD, Myers EW, Clark AG. Y chromosome and other heterochromatic sequences of the Drosophila melanogaster genome: how far can we go? Genetica. 2003;117:227–237. doi: 10.1023/A:1022900313650. [DOI] [PubMed] [Google Scholar]
- 71.Negre B, Simpson P. The achaete-scute complex in Diptera: patterns of noncoding sequence evolution. J Evol Biol. 2015;28:1770–1781. doi: 10.1111/jeb.12687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wrischnik LA, Timmer JR, Megna LA, Cline TW. Recruitment of the proneural gene scute to the Drosophila sex-determination pathway. Genetics. 2003;165:2007–2027. doi: 10.1093/genetics/165.4.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Erickson JW, Cline TW. A bZIP protein, sisterless-a, collaborates with bHLH transcription factors early in Drosophila development to determine sex. Genes Dev. 1993;7:1688–1702. doi: 10.1101/gad.7.9.1688. [DOI] [PubMed] [Google Scholar]
- 74.Ingleby FC, Flis I, Morrow EH. Sex-biased gene expression and sexual conflict throughout development. Cold Spring Harb Perspect Biol. 2015;7:a017632. doi: 10.1101/cshperspect.a017632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yang Y, Lu X. Drosophila sperm motility in the reproductive tract. Biol Reprod. 2011;84:1005–1015. doi: 10.1095/biolreprod.110.088773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Innocenti P, Morrow EH. Immunogenic males: a genome-wide analysis of reproduction and the cost of mating in Drosophila melanogaster females. J Evol Biol. 2009;22:964–973. doi: 10.1111/j.1420-9101.2009.01708.x. [DOI] [PubMed] [Google Scholar]
- 77.Innocenti P, Morrow EH. The sexually antagonistic genes of Drosophila melanogaster. PLoS Biol. 2010;8:e1000335. doi: 10.1371/journal.pbio.1000335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Eirin-Lopez JM, Sanchez L. The comparative study of five sex-determining proteins across insects unveils high rates of evolution at basal components of the sex determination cascade. Dev Genes Evol. 2015;225:23–30. doi: 10.1007/s00427-015-0491-6. [DOI] [PubMed] [Google Scholar]
- 79.Verhulst EC, van de Zande L, Beukeboom LW. Insect sex determination: it all evolves around transformer. Curr Opin Genet Dev. 2010;20:376–383. doi: 10.1016/j.gde.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 80.Price DC, Egizi A, Fonseca DM. The ubiquity and ancestry of insect doublesex. Sci Rep. 2015;5:13068. doi: 10.1038/srep13068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cronmiller C, Cummings CA. The daughterless gene product in Drosophila is a nuclear protein that is broadly expressed throughout the organism during development. Mech Dev. 1993;42:159–169. doi: 10.1016/0925-4773(93)90005-I. [DOI] [PubMed] [Google Scholar]
- 82.Caudy M, Vassin H, Brand M, Tuma R, Jan LY, Jan YN. Daughterless, a Drosophila gene essential for both neurogenesis and sex determination, has sequence similarities to myc and the achaete-scute complex. Cell. 1988;55:1061–1067. doi: 10.1016/0092-8674(88)90250-4. [DOI] [PubMed] [Google Scholar]
- 83.Kuroda MI, Kernan MJ, Kreber R, Ganetzky B, Baker BS. The maleless protein associates with the X chromosome to regulate dosage compensation in Drosophila. Cell. 1991;66:935–947. doi: 10.1016/0092-8674(91)90439-6. [DOI] [PubMed] [Google Scholar]
- 84.Dahanukar A, Hallem EA, Carlson JR. Insect chemoreception. Curr Opin Neurobiol. 2005;15:423–430. doi: 10.1016/j.conb.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 85.Robertson HM, Warr CG, Carlson JR. Molecular evolution of the insect chemoreceptor gene superfamily in Drosophila melanogaster. Proc Natl Acad Sci U S A. 2003;100(Suppl 2):14537–14542. doi: 10.1073/pnas.2335847100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rytz R, Croset V, Benton R. Ionotropic receptors (IRs): chemosensory ionotropic glutamate receptors in Drosophila and beyond. Insect Biochem Mol Biol. 2013;43:888–897. doi: 10.1016/j.ibmb.2013.02.007. [DOI] [PubMed] [Google Scholar]
- 87.Vieira FG, Rozas J. Comparative genomics of the odorant-binding and chemosensory protein gene families across the Arthropoda: origin and evolutionary history of the chemosensory system. Genome Biol Evol. 2011;3:476–490. doi: 10.1093/gbe/evr033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chen C, Buhl E, Xu M, Croset V, Rees JS, Lilley KS, Benton R, Hodge JJ, Stanewsky R. Drosophila Ionotropic Receptor 25a mediates circadian clock resetting by temperature. Nature. 2015;527:516–520. doi: 10.1038/nature16148. [DOI] [PubMed] [Google Scholar]
- 89.Cao D, Liu Y, Walker WB, Li J, Wang G. Molecular characterization of the Aphis gossypii olfactory receptor gene families. PLoS One. 2014;9:e101187. doi: 10.1371/journal.pone.0101187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Croset V, Rytz R, Cummins SF, Budd A, Brawand D, Kaessmann H, Gibson TJ, Benton R. Ancient protostome origin of chemosensory ionotropic glutamate receptors and the evolution of insect taste and olfaction. PLoS Genet. 2010;6:e1001064. doi: 10.1371/journal.pgen.1001064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Benton R, Vannice KS, Gomez-Diaz C, Vosshall LB. Variant ionotropic glutamate receptors as chemosensory receptors in Drosophila. Cell. 2009;136:149–162. doi: 10.1016/j.cell.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Dereeper A, Audic S, Claverie JM, Blanc G. BLAST-EXPLORER helps you building datasets for phylogenetic analysis. BMC Evol Biol. 2010;10:8. doi: 10.1186/1471-2148-10-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dereeper A, Guignon V, Blanc G, Audic S, Buffet S, Chevenet F, Dufayard JF, Guindon S, Lefort V, Lescot M, et al. Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008;36:W465–469. doi: 10.1093/nar/gkn180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17:540–552. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- 95.Anisimova M, Gascuel O. Approximate likelihood-ratio test for branches: A fast, accurate, and powerful alternative. Syst Biol. 2006;55:539–552. doi: 10.1080/10635150600755453. [DOI] [PubMed] [Google Scholar]
- 96.Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- 97.Chevenet F, Brun C, Banuls AL, Jacq B, Christen R. TreeDyn: towards dynamic graphics and annotations for analyses of trees. Bmc Bioinformatics. 2006;7:439. doi: 10.1186/1471-2105-7-439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Imler JL. Overview of Drosophila immunity: a historical perspective. Dev Comp Immunol. 2014;42:3–15. doi: 10.1016/j.dci.2013.08.018. [DOI] [PubMed] [Google Scholar]
- 99.Stokes BA, Yadav S, Shokal U, Smith LC, Eleftherianos I. Bacterial and fungal pattern recognition receptors in homologous innate signaling pathways of insects and mammals. Front Microbiol. 2015;6:19. doi: 10.3389/fmicb.2015.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ferrandon D, Imler JL, Hetru C, Hoffmann JA. The Drosophila systemic immune response: sensing and signalling during bacterial and fungal infections. Nat Rev Immunol. 2007;7:862–874. doi: 10.1038/nri2194. [DOI] [PubMed] [Google Scholar]
- 101.Lemaitre B, Hoffmann J. The host defense of Drosophila melanogaster. Annu Rev Immunol. 2007;25:697–743. doi: 10.1146/annurev.immunol.25.022106.141615. [DOI] [PubMed] [Google Scholar]
- 102.Valanne S, Wang JH, Ramet M. The Drosophila Toll signaling pathway. J Immunol. 2011;186:649–656. doi: 10.4049/jimmunol.1002302. [DOI] [PubMed] [Google Scholar]
- 103.Weber AN, Tauszig-Delamasure S, Hoffmann JA, Lelievre E, Gascan H, Ray KP, Morse MA, Imler JL, Gay NJ. Binding of the Drosophila cytokine Spatzle to Toll is direct and establishes signaling. Nat Immunol. 2003;4:794–800. doi: 10.1038/ni955. [DOI] [PubMed] [Google Scholar]
- 104.Lemaitre B, Nicolas E, Michaut L, Reichhart JM, Hoffmann JA. The dorsoventral regulatory gene cassette spatzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell. 1996;86:973–983. doi: 10.1016/S0092-8674(00)80172-5. [DOI] [PubMed] [Google Scholar]
- 105.Hedengren M, Asling B, Dushay MS, Ando I, Ekengren S, Wihlborg M, Hultmark D. Relish, a central factor in the control of humoral but not cellular immunity in Drosophila. Mol Cell. 1999;4:827–837. doi: 10.1016/S1097-2765(00)80392-5. [DOI] [PubMed] [Google Scholar]
- 106.Kleino A, Valanne S, Ulvila J, Kallio J, Myllymaki H, Enwald H, Stoven S, Poidevin M, Ueda R, Hultmark D, et al. Inhibitor of apoptosis 2 and TAK1-binding protein are components of the Drosophila Imd pathway. EMBO J. 2005;24:3423–3434. doi: 10.1038/sj.emboj.7600807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Samakovlis C, Kimbrell DA, Kylsten P, Engstrom A, Hultmark D. The immune response in Drosophila: pattern of cecropin expression and biological activity. Embo Journal. 1990;9:2969–2976. doi: 10.1002/j.1460-2075.1990.tb07489.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Valanne S, Myllymaki H, Kallio J, Schmid MR, Kleino A, Murumagi A, Airaksinen L, Kotipelto T, Kaustio M, Ulvila J, et al. Genome-wide RNA interference in Drosophila cells identifies G protein-coupled receptor kinase 2 as a conserved regulator of NF-kappaB signaling. J Immunol. 2010;184:6188–6198. doi: 10.4049/jimmunol.1000261. [DOI] [PubMed] [Google Scholar]
- 109.Holzl H, Kapelari B, Kellermann J, Seemuller E, Sumegi M, Udvardy A, Medalia O, Sperling J, Muller SA, Engel A, Baumeister W. The regulatory complex of Drosophila melanogaster 26S proteasomes. Subunit composition and localization of a deubiquitylating enzyme. J Cell Biol. 2000;150:119–130. doi: 10.1083/jcb.150.1.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Meinander A, Runchel C, Tenev T, Chen L, Kim CH, Ribeiro PS, Broemer M, Leulier F, Zvelebil M, Silverman N, Meier P. Ubiquitylation of the initiator caspase DREDD is required for innate immune signalling. Embo Journal. 2012;31:2770–2783. doi: 10.1038/emboj.2012.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hemingway J, Ranson H. Insecticide resistance in insect vectors of human disease. Annu Rev Entomol. 2000;45:371–391. doi: 10.1146/annurev.ento.45.1.371. [DOI] [PubMed] [Google Scholar]
- 112.Scott JG. Insect cytochrome P450s:Thinking beyond detoxification. Recent Adv Insect Physiol Toxicol Mol Biol. 2008:117–24.
- 113.Li X, Schuler MA, Berenbaum MR. Molecular mechanisms of metabolic resistance to synthetic and natural xenobiotics. Annu Rev Entomol. 2007;52:231–253. doi: 10.1146/annurev.ento.51.110104.151104. [DOI] [PubMed] [Google Scholar]
- 114.David JP, Ismail HM, Chandor-Proust A, Paine MJ. Role of cytochrome P450s in insecticide resistance: impact on the control of mosquito-borne diseases and use of insecticides on Earth. Philos Trans R Soc Lond B Biol Sci. 2013;368:20120429. doi: 10.1098/rstb.2012.0429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Scott JG. Cytochromes P450 and insecticide resistance. Insect Biochem Mol Biol. 1999;29:757–777. doi: 10.1016/S0965-1748(99)00038-7. [DOI] [PubMed] [Google Scholar]
- 116.International Glossina Genome I Genome sequence of the tsetse fly (Glossina morsitans): vector of African trypanosomiasis. Science. 2014;344:380–386. doi: 10.1126/science.1249656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Tijet N, Helvig C, Feyereisen R. The cytochrome P450 gene superfamily in Drosophila melanogaster: annotation, intron-exon organization and phylogeny. Gene. 2001;262:189–198. doi: 10.1016/S0378-1119(00)00533-3. [DOI] [PubMed] [Google Scholar]
- 118.Montella IR, Schama R, Valle D. The classification of esterases: an important gene family involved in insecticide resistance--a review. Mem Inst Oswaldo Cruz. 2012;107:437–449. doi: 10.1590/S0074-02762012000400001. [DOI] [PubMed] [Google Scholar]
- 119.Sogorb MA, Vilanova E. Enzymes involved in the detoxification of organophosphorus, carbamate and pyrethroid insecticides through hydrolysis. Toxicol Lett. 2002;128:215–228. doi: 10.1016/S0378-4274(01)00543-4. [DOI] [PubMed] [Google Scholar]
- 120.Enayati AA, Ranson H, Hemingway J. Insect glutathione transferases and insecticide resistance. Insect Mol Biol. 2005;14:3–8. doi: 10.1111/j.1365-2583.2004.00529.x. [DOI] [PubMed] [Google Scholar]
- 121.Ranson H, Hemingway J. Mosquito glutathione transferases. Methods Enzymol. 2005;401:226–41. doi: 10.1016/S0076-6879(05)01014-1. [DOI] [PubMed] [Google Scholar]
- 122.Fang S. Insect glutathione S-transferase: a review of comparative genomic studies and response to xenobiotics. Bull Insectol. 2012;65:265–271. [Google Scholar]
- 123.Ffrench-Constant RH, Daborn PJ, Le Goff G. The genetics and genomics of insecticide resistance. Trends Genet. 2004;20:163–170. doi: 10.1016/j.tig.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 124.Weill M. Overview of 40 years of insecticide resistance genes evolution in the mosquito culex pipiens. Pathogens Global Health. 2013;107:433–434. [Google Scholar]
- 125.Liu N. Insecticide resistance in mosquitoes: Impact, mechanisms, and research directions. Ann Rev Entomol. 2015;60:537–559. doi: 10.1146/annurev-ento-010814-020828. [DOI] [PubMed] [Google Scholar]
- 126.Hoppe MA. The importance of insecticide resistance management in the control of the mosquito vectors of malaria. Am J Trop Med Hyg. 2010;83:178. [Google Scholar]
- 127.Roxburgh NA, Shanahan GJ. Carbamate resistance in the sheep blowfly, Lucilia cuprina (Wied) Vet Rec. 1973;93:467. doi: 10.1136/vr.93.17.467-a. [DOI] [PubMed] [Google Scholar]
- 128.Hughes PB, Levot GW. Toxicity of 3 avermectins to insecticide susceptible and resistant larvae of lucilia-cuprina (Wiedemann) (Diptera, Calliphoridae) J Aust Entomol Soc. 1990;29:109–111. doi: 10.1111/j.1440-6055.1990.tb00329.x. [DOI] [Google Scholar]
- 129.Sales N, Levot GW, Hughes PB. Monitoring and selection of resistance to pyrethroids in the Australian sheep blowfly, Lucilia-Cuprina. Med Vet Entomol. 1989;3:287–291. doi: 10.1111/j.1365-2915.1989.tb00230.x. [DOI] [PubMed] [Google Scholar]
- 130.Carvalho RA, Azeredo-Espin AML, Torres TT. Deep sequencing of New World screw-worm transcripts to discover genes involved in insecticide resistance. Bmc Genomics. 2010;11:695. doi: 10.1186/1471-2164-11-695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Che-Mendoza A, Penilla RP, Rodriguez DA. Insecticide resistance and glutathione S-transferases in mosquitoes: a review. Afr J Biotechnol. 2009;8:1386–1397. [Google Scholar]
- 132.Cochrane BJ, Hargis M, Debelligny PC, Holtsberg F, Coronella J. Evolution of glutathione S-Transferases associated with insecticide resistance in drosophila. ACS Symp Ser. 1992;505:53–70. doi: 10.1021/bk-1992-0505.ch005. [DOI] [Google Scholar]
- 133.Kramerov DA, Vassetzky NS. Origin and evolution of SINEs in eukaryotic genomes. Heredity (Edinb) 2011;107:487–495. doi: 10.1038/hdy.2011.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Jurka J. Non-LTR retrotransposons from the southern house mosquito. 2011;11:595.
- 135.Lavoie CA, Platt RN, 2nd, Novick PA, Counterman BA, Ray DA. Transposable element evolution in Heliconius suggests genome diversity within Lepidoptera. Mob DNA. 2013;4:21. doi: 10.1186/1759-8753-4-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Platt RN, 2nd, Blanco-Berdugo L, Ray DA. Accurate transposable element annotation is vital when analyzing new genome assemblies. Genome Biol Evol. 2016;8:403–410. doi: 10.1093/gbe/evw009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Erzinclioglu YZ. The larvae of some blowflies of medical and veterinary importance. Med Vet Entomol. 1987;1:121–125. doi: 10.1111/j.1365-2915.1987.tb00332.x. [DOI] [PubMed] [Google Scholar]
- 138.Zumpt F. Myiasis in man and animals in the Old World. London: Butterworths; 1965. [Google Scholar]
- 139.Ali-Khan FE, Ali-Khan Z. A case of traumatic dermal myiasis in Quebec caused by Phormia regina (Meigen) (Diptera: Calliphoridae) Can J Zool. 1975;53:1472–1476. doi: 10.1139/z75-178. [DOI] [PubMed] [Google Scholar]
- 140.Bolek MG, Coggins JR. Observations on myiasis by the calliphorid, Bufolucilia silvarum, in the eastern American toad (Bufo americanus americanus) from southeastern Wisconsin. J Wildl Dis. 2002;38:598–603. doi: 10.7589/0090-3558-38.3.598. [DOI] [PubMed] [Google Scholar]
- 141.Chodosh J, Clarridge JE, Matoba A. Nosocomial conjunctival ophthalmomyiasis with cochliomyia macellaria. Am J Ophthalmol. 1991;111:520–521. doi: 10.1016/S0002-9394(14)72399-2. [DOI] [PubMed] [Google Scholar]
- 142.Ferraz ACP, Proenca B, Gadelha BQ, Faria LM, Barbalho MGM, Aguiar-Coelho VM, Lessa CSS. First record of human myiasis caused by association of the species chrysomya megacephala (Diptera: Calliphoridae), Sarcophaga (Liopygia) ruficornis (Diptera: Sarcophagidae), and Musca domestica (Diptera: Muscidae) J Med Entomol. 2010;47:487–490. doi: 10.1603/ME09143. [DOI] [PubMed] [Google Scholar]
- 143.Hall RD, Anderson PC, Clark DP. A case of human myiasis caused by Phormia regina (Diptera: Calliphoridae) in Missouri, USA. J Med Entomol. 1986;23:578–579. doi: 10.1093/jmedent/23.5.578. [DOI] [PubMed] [Google Scholar]
- 144.Stevens J, Wall R. Species, sub-species and hybrid populations of the blowflies Lucilia cuprina and Lucilia sericata (Diptera:Calliphoridae) Proc Biol Sci. 1996;263:1335–1341. doi: 10.1098/rspb.1996.0196. [DOI] [PubMed] [Google Scholar]
- 145.Concha C, Belikoff EJ, Carey BL, Li F, Schiemann AH, Scott MJ. Efficient germ-line transformation of the economically important pest species Lucilia cuprina and Lucilia sericata (Diptera, Calliphoridae) Insect Biochem Mol Biol. 2011;41:70–75. doi: 10.1016/j.ibmb.2010.09.006. [DOI] [PubMed] [Google Scholar]
- 146.VanLaerhoven SL. Blind validation of postmortem interval estimates using developmental rates of blow flies. Forensic Sci Int. 2008;180:76–80. doi: 10.1016/j.forsciint.2008.07.002. [DOI] [PubMed] [Google Scholar]
- 147.Wells JD, LaMotte LR. Estimating the postmortem interval. In: Byrd JH, Castner JL, editors. Forensic Entomology: Utility of Arthropods in Legal Investigations. CRC Press (Boca Raton, FL); 2001: 263–85.
- 148.Byrd JH, Allen JC. The development of the black blow fly, Phormia regina (Meigen) Forensic Sci Int. 2001;120:79–88. doi: 10.1016/S0379-0738(01)00431-5. [DOI] [PubMed] [Google Scholar]
- 149.Byrd JH, Butler JF. Effects of temperature on Cochliomyia macellaria (Diptera: Calliphoridae) development. J Med Entomol. 1996;33:901–905. doi: 10.1093/jmedent/33.6.901. [DOI] [PubMed] [Google Scholar]
- 150.Byrd JH, Butler JF. Effects of temperature on Chrysomya rufifacies (Diptera: Calliphoridae) development. J Med Entomol. 1997;34:353–358. doi: 10.1093/jmedent/34.3.353. [DOI] [PubMed] [Google Scholar]
- 151.Tarone AM, Picard CJ, Spiegelman C, Foran DR. Population and Temperature Effects on Lucilia sericata (Diptera: Calliphoridae) Body Size and Minimum Development Time. J Med Entomol. 2011;48:1062–1068. doi: 10.1603/ME11004. [DOI] [PubMed] [Google Scholar]
- 152.Gallagher MB, Sandhu S, Kimsey R. Variation in developmental time for geographically distinct populations of the common green bottle fly, Lucilia sericata (Meigen) J Forensic Sci. 2010;55:438–442. doi: 10.1111/j.1556-4029.2009.01285.x. [DOI] [PubMed] [Google Scholar]
- 153.Owings CG, Spiegelman C, Tarone AM, Tomberlin JK. Developmental variation among Cochliomyia macellaria Fabricius (Diptera: Calliphoridae) populations from three ecoregions of Texas, USA. Int J Legal Med. 2014;128:709–717. doi: 10.1007/s00414-014-1014-0. [DOI] [PubMed] [Google Scholar]
- 154.Picard CJ, DeBlois K, Tovar F, Bradley JL, Johnston JS, Tarone AM. Increasing precision in development-based PMI estimates: What’s sex got to do with it? J Med Entomol. 2013;5:425–431. doi: 10.1603/ME12051. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under the accession MINK00000000 and MINJ00000000 for the male and female genomes, respectively. The Phormia regina genomes and raw sequence reads are available from NCBI under BioProject PRJNA338752 and BioSamples SAMN05567885 and SAMN05567884.