

Abstract
Advanced lung cancer has poor prognosis owing to its low sensitivity to current chemotherapy agents. Therefore, discovery of new therapeutic agents is urgently needed. In this study, we investigated the antitumor effects of peperomin E, a secolignan isolated from Peperomia dindygulensis, a frequently used Chinese folk medicine for lung cancer treatment. The results indicate that peperomin E has antiproliferative effects, promoting apoptosis and cell cycle arrest in non‐small‐cell lung cancer (NSCLC) cell lines in a dose‐dependent manner, while showing lower toxicity against normal human lung epidermal cells. Peperomin E inhibited tumor growth in A549 xenograft BALB/c nude mice without significant secondary adverse effects, indicating that it may be safely used to treat NSCLC. Furthermore, the mechanisms underlying the anticancer effects of peperomin E have been investigated. Using an in silico target fishing method, we observed that peperomin E directly interacts with the active domain of DNA methyltransferase 1 (DNMT1), potentially affecting its genome methylation activity. Subsequent experiments verified that peperomin E decreased DNMT1 activity and expression, thereby decreasing global methylation and reactivating the epigenetically silenced tumor suppressor genes including RASSF1A,APC,RUNX3, and p16INK4, which in turn activates their mediated pro‐apoptotic and cell cycle regulatory signaling pathways in lung cancer cells. The observations herein report for the first time that peperomin E is a potential chemotherapeutic agent for NSCLC. The anticancer effects of peperomin E may be partly attributable to its ability to demethylate and reactivate methylation‐silenced tumor suppressor genes through direct inhibition of the activity and expression of DNMT1.
Keywords: Anticancer activity, DNA (cytosine‐5‐)‐methyltransferase 1, non‐small‐cell lung cancer, peperomin E, tumor suppressor genes
Lung cancer is the leading cause of cancer‐related death worldwide.1 Owing to a large smoking population and serious air pollution, the incidence of lung cancer has grown rapidly in China, resulting in a large social and economic burden.2, 3, 4 Diagnosis of lung cancer, especially non‐small‐cell lung cancer (NSCLC), often occurs so late that approximately two‐thirds of patients have lost the opportunity for radical surgery.4 Four chemotherapeutic regimens – cisplatin and gemcitabine, cisplatin and docetaxel, carboplatin and paclitaxel, and cisplatin and paclitaxel – have been used for clinical treatment of NSCLC.5 However, the aforementioned chemotherapy is not very efficient and causes severe side‐effects, including neurotoxicity, renal damage, and bone marrow inhibition.6, 7, 8 Patients usually die within 1–2 years of diagnosis. Therefore, identifying new drug targets and novel therapeutic agents to reduce mortality and improve quality of life for patients with advanced lung cancer remains a major goal for researchers and drug developers.
Peperomia dindygulensis (Piperaceae) is commonly used in southern China as a folk medicine to treat various cancers.9 Chemical analysis of this plant has indicated that it contains secolignans,10, 11, 12, 13 tetrahydrofuran lignans,14 flavonoids,15 and polyketides.16, 17, 18 Secolignans are characteristic of the Peperomia species and have been shown to possess various bioactivities that make them effective treatments against tumors,10, 11, 12, 13, 19 inflammation,20 and HIV infection.21 Among the secolignans that have been isolated, peperomin E (PepE; Fig. 1a), which is characterized by an α‐methylene‐γ‐butyrolactone moiety, has shown the strongest inhibition of cancer cell growth in lung,11 breast,19 leukemia,19 liver,11 and cervical cancer cells.19 However, antitumor activity and safety of PepE have not been investigated in vivo, and the mechanism of its anticancer activity has not been elucidated.
Figure 1.
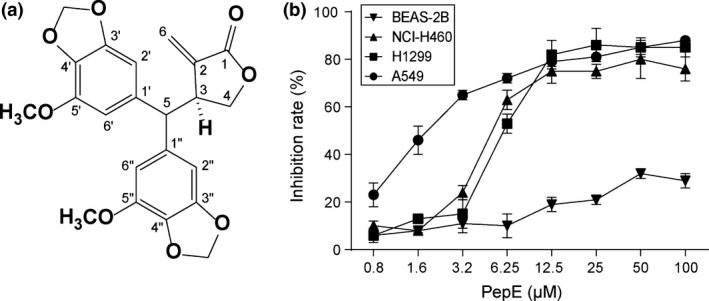
In vitro antitumor activity of peperomin E (PepE) against non‐small‐cell lung cancer cells. (a) Chemical structure of peperomin E (PepE); and (b) growth inhibition rates of normal cell line BEAS‐2B and lung cancer cell lines treated with PepE at the indicated concentrations for 48h. Data shown are means±SD (n = 3).
In this study, we evaluated the safety and efficacy of PepE for treatment of NSCLC. We undertook an in silico target fishing study of PepE, which showed that PepE possessed the highest affinity to the active pocket of the DNA methyltransferase 1 (DNMT1) enzyme. Subsequent experiments investigated PepE activity against DNMT1 activity and expression and evaluated the effect of PepE on the expression of the epigenetically silenced tumor suppressors (i.e., RASSF1A and APC genes in A549 cells, p16INK4 gene in H1299 cells, and RUNX3 gene in NCI‐H460 cells), which may further activate pro‐apoptotic and cell cycle regulatory signaling pathways in these NSCLC cell lines.
Materials and Methods
Plant material
Peperomia dindygulensis (whole plant) was collected from Yunnan Province, China, in February 2014 and identified by Professor She‐ban Pu from the China Pharmaceutical University (Nanjing, China). Voucher specimens (PDg 2014‐2) were deposited at the College of Pharmacy, Nanjing University of Chinese Medicine, (Nanjing, China).
Chemicals and antibodies
Peperomin E was previously isolated from P. dindygulensis in our laboratory by a series of chromatographic procedures. Its structure (Fig. 1) was unequivocally elucidated by spectroscopic methods (i.e., mass spectrometry, proton (1H) nuclear magnetic resonance (NMR), and carbon‐13 (13C) NMR; Fig. S1). Purity of PepE was verified by HPLC peak area normalization and peak purity analysis. Results showed that the purity was >98% and the peak purity angle/peak purity threshold was <1 (Fig. S2).
Peperomin E powder was dissolved in DMSO (Sigma‐Aldrich, St. Louis, MO, USA) to produce a 10−2 M solution, which was stored at −20°C. 5‐Aza‐2‐deoxycytidine (5‐Aza‐dC, CAS No. 2353‐33‐5) was purchased from Sigma‐Aldrich and dissolved in water immediately before use.
Monoclonal rabbit antibodies against DNMT1, Ras association domain family member 1 (RASSF1A), macrophage stimulating (MST)1, MST2, Bax, Bcl‐2, cleaved caspase 3, cleaved caspase 9, poly(ADP‐ribose) polymerase (PARP), and GAPDH were all purchased from Abcam (San Francisco, CA, USA). Cyclin D1, runt related transcription factor 3 (RUNX3), p16INK4, adenomatous polyposis coli (APC), modulator of apoptosis 1 (MOAP1), Connector enhancer of kinase suppressor of ras 1 (CNK1), β‐catenin, and HRP‐conjugated secondary antibodies (goat anti‐rabbit) were all purchased from Santa Cruz Biotechnology (Dallas, TX, USA). Gibco FBS and high‐glucose DMEM media were obtained from Thermo Fisher Scientific (Rockford, IL, USA).
Cell culture
BEAS‐2B, A549, H1229, and NCI‐H460 cells were obtained directly from ATCC (Manassas, VA, USA) and passaged in our laboratory for <6 months after resuscitation. Cells were cultured and maintained in high‐glucose DMEM supplemented with 10% (v/v) FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin and grown at 37°C in a 5% CO2 humidified incubator.
In vitro cytotoxicity assay
The effects of PepE on lung cancer cell viability were investigated using MTT assays. The tumor cells (4 × 104 cells/well in 100 μL medium) were seeded into 96‐well plates for 24 h before drug treatment. We used 5‐Aza‐dC, which exhibits activity against lung cancer cells, as a positive control.22 After treatment with various concentrations of PepE (0–100 μM) and 5‐Aza‐dC for 48 h, the cell plates were treated with MTT solution (20 μL; 5 mg/mL in PBS) for an additional 4 h at 37°C. The formazan crystals in viable cells were solubilized with DMSO (150 μL) and the absorbance was measured on a microplate reader (ELX 800; BioTek Instruments, Winooski, VT, USA) at a wavelength of 490 nm. Cell viability was calculated as the inhibition ratio (I%) using the following equation (optical density at 490 nm [OD490]):
In vivo tumorigenicity assay
Six‐week‐old male BALB/c athymic nude mice were purchased from Cavens animal center (Changzhou, China; No. SCXK 2011‐0003). The mice were kept under specific pathogen‐free conditions, provided with sterilized food and water, and housed in positive pressure isolators with 12:12‐h light:dark cycles. A549 cells (5.0 × 106; 0.2 mL) were injected s.c. into the right armpit. Tumor growth and bodyweight were monitored every other day. When tumor diameter reached 5 mm, mice were randomly divided into treatment and control groups (n = 6/group). Peperomin E was dissolved in soybean oil and given by i.v. injection at a dose of 25 or 50 mg/kg every other day. As a positive control, 5‐Aza‐dC was given by i.v. injection at a dose of 5 mg/kg twice a week. The control group received soybean oil only. Mice were treated for 3 weeks. Tumor volume was calculated using the formula V = 1/2 (A × B 2), where A indicates the length and B indicates the width of the tumor as measured by calipers. At the end of the treatment period, blood samples were collected from the retro‐orbital plexus. Serum was tested for alanine aminotransferase and creatinine levels. Plasma was tested for blood platelet and neutrophilic granulocyte cell levels. Analyses were carried out using a DXC800 Synchron spectrophotometer (Beckman Coulter, Brea, CA, USA). Mice were then killed and the tumor tissue, lung, liver, and kidney were surgically removed from the body, fixed in 10% neutral formalin, and embedded in paraffin for analysis. Experiments were carried out in accordance with the Guidelines for Animal Experimentation and approved by the Animal Ethics Committee at Nanjing University of Chinese Medicine.
Apoptosis and cell cycle analysis
Cell apoptosis was determined using an annexin V–FITC apoptosis detection kit (KeyGen Biotechnology, Nanjing, China) according to the manufacturer's instructions. Briefly, A549 cells were treated with PepE (0.5, 2.0, or 4.0 μM) or 5‐Aza‐dC (2.0 μM) for 48 h. Cells were harvested by trypsinization, washed twice with pre‐chilled PBS, and centrifuged at 671 × g for 5 min. The cell pellet was resuspended in binding buffer and stained with annexin V–FITC/propidium iodide. After incubation at 15°C for 15 min in the dark, the samples were quantified by Becton Dickinson FACSCalibur flow cytometry (Franklin Lakes, NJ, USA), and data were analyzed using CellQuest software (Becton Dickinson).
Cell cycle analysis was undertaken using a cell cycle detection kit (KeyGen Biotechnology) according to the manufacturer's instructions. Briefly, A549 cells were treated with PepE (2.0 μM) or 5‐Aza‐dC (2.0 μM) for 48 h. The cells were harvested and fixed with 70% alcohol at 4°C for 12 h, DNA was stained with propidium iodide in the presence of 1% DNase‐free RNase A at 37°C for 30 min before flow cytometry analysis (FACSCalibur; Becton Dickinson). The distribution of cells in distinct cell cycle phases was analyzed using CellQuest software (Becton Dickinson).
In silico target fishing studies
Target fishing and molecular docking studies were carried out with Discovery Studio version 4.0 (Accelrys, San Diego, CA, USA) using the DS Target Fishing program (based on the LibDock and Pipeline Pilot module designed by Neotrident Corp., Beijing, China). To identify potential binding proteins of PepE, the Annotated Database of Druggable Binding Sites from the Protein DataBank (sc‐PDB), (http://bioinfo-pharma.u-strasbg.fr/scPDB/), which contains structural information (e.g., active sites) of more than 3678 known or potential protein drug targets, was used. A protein preparation protocol was used to perform tasks such as inserting missing atoms in incomplete residues, deleting alternate conformations, removing water, and protonating titratable residues. For energy minimization of PepE, an Merck Molecular Force Field (MMFF) was used in the parameterization step. Molecular docking studies were carried out to generate bioactive binding poses of PepE in the active site (or binding pocket) of druggable targets using the LibDock and CDOCKER programs. Peperomin E was docked to each druggable target and ranked by LibDock score with the target fishing program. More details on the target fishing procedures are given in Figure S3.
Determination of genomic DNA methylation levels
A549, NCI‐H460, and H1299 cells were treated with PepE (0.5, 2.0, or 4.0 μM) or 5‐Aza‐dC (2.0 μM) for 48 h. Genomic DNA was isolated and purified from the above cells and tumor tissues extracted from nude mice using the DNeasy tissue kit (Qiagen, Valencia, CA, USA) following the manufacturer's instructions and stored at −80°C. Global methylation levels were determined by the liquid chromatography–electrospray ionization tandem mass spectrometry (MS/MS) method as described previously.23 Briefly, 500 ng genomic DNA was enzymatically hydrolyzed to single nucleotides using the Epiquik one‐step DNA hydrolysis kit (Cat. No. P‐1023; Epigentek, Farmingdale, NY, USA) following the manufacturer's instructions. Digested DNA was separated on a Prominence ultra‐fast liquid chromatography system (Shimadzu, Kyoto, Japan) using an Acquity ultra performance liquid chromatography high strength silica T3 column (100 mm × 2.1 mm, i.d. 1.7 μm; Waters, Dublin, Ireland) with a guard cartridge at 35°C. The mobile phase was composed of acetonitrile and 0.1% formic acid aqueous solution (elution gradient, 0–10 min, 2–15% acetonitrile; flow rate, 0.3 mL/min; injection volume, 2 μL). Detection was carried out on a triple quadrupole linear ion trap mass spectrometer equipped with a TurboIonSpray ion source (5500 Q‐Trap; Applied Biosystems, Foster City, CA, USA). Quantification was undertaken in multiple reaction monitoring mode by monitoring transition pairs of m/z 242.0/126.1 for 5‐methyl‐deoxy‐cytidine (5mdC) and 267.9/152.0 for deoxyguanosine (dG). Genomic DNA methylation was expressed as [5mdC]/[dG]%, which was quantified by using a calibration curve between the peak area ratio of 5mdC to dG (A5mdC/AdG) versus [5mdC]/[dG]%.
In vitro DNA methyltransferase inhibition assay
Nuclear extracts were prepared from A549 cells using the EpiQuik nuclear extraction kit (Cat. No. OP‐0002‐1; Epigentek) following the manufacturer's instructions and stored at −80°C. Nuclear extract quantity was determined using Quick Start Bradford protein assay kit (Bio‐Rad, Hercules, CA, USA). The DNMT activity was determined using an Epiquik DNMT activity/inhibition assay kit (Cat. No. P‐3001; Epigentek) following the manufacturer's instructions. Briefly, 10 μg A549 nuclear extract was added to sample wells coated with cytosine‐rich DNA substrate. Then PepE or 5‐Aza‐dC was added to the inhibition wells; control wells received DMSO only. The substrate reaction was read spectrophotometrically at 450 nm using the 96‐well Multiskan GO microplate reader (Thermo Scientific). Inhibition of DNMT was calculated using the following formula; IC50 was determined from the dose–inhibition curves.
Bisulfite modification and methylation‐specific PCR
DNA was extracted from PepE‐treated and untreated A549, NCI‐H460, and H1229 cells using the DNeasy tissue kit (Qiagen) according to the manufacturer's instructions. Extracted DNA was modified with sodium bisulfite and analyzed according to a modification of the method described by Herman et al.24 Briefly, 1 μg genomic DNA was modified with bisulfite using a CpGenome DNA modification kit (Chemicon, Temecula, CA, USA). The modified DNA was amplified with primers for the methylated and unmethylated gene sequences designed according previously published reports (Table S1).25, 26, 27, 28 The reactions were carried out using the Veriti 96‐well thermal cycler (Applied Biosystems). Polymerase chain reaction conditions are listed in Table S2. The PCR products were then loaded onto 3.5% low‐range agarose gels (Thermo Fisher Scientific), stained with ethidium bromide, and observed under UV illumination using Gel Doc XR (Bio‐Rad). CpGenome Universal methylated DNA and unmethylated DNA (Chemicon) were used as positive controls for DNA methylation and unmethylation.
Quantitative real‐time PCR assays
RNA was extracted from NSCLC cells (including A549, NCI‐H460, and H1299 cells) using the RNeasy RNA isolation kit (Qiagen), and on‐column DNA digestion was done using the RNase‐Free DNase Set (Qiagen). cDNA was synthesized from 2 μg RNA with the iScript cDNA synthesis kit (Bio‐Rad) according to the manufacturer's instructions, and gene expression levels of DNMT1, RASSF1A, and APC in A549 cells, RUNX3 in NCI‐H460 cells, and p16INK4 in H1299 cells were quantitatively measured in triplicate with a real‐time quantitative PCR system (Tianlong TL988‐IV; Tianlong Scientific Technology Corp., Xi'an, China), using SYBR Green I (Applied Biosystems) and corresponding primers (Table S3, designed by the software Primer premier 5.0 and synthesized by Genewiz Corp., Suzhou, China). The PCR conditions are listed in Table S4. The mRNA levels of all tested genes were normalized against GAPDH, results were reported as fold‐change () relative to non‐treated samples. To visually evaluate the mRNA expression of RASSF1A, APC, RUNX3, and p16INK4 genes, the amplified products derived from the Veriti 96‐well thermal cycler (Applied Biosystems) were loaded onto 1.8% agarose gels containing ethidium and observed using the Gel Doc XR system (Bio‐Rad).
Western blot analysis
Protein levels were analyzed by Western blotting. Briefly, A549, NCI‐H460, or H1299 cells were cultured at 2 × 106 cells/mL in a 100 × 20‐mm cell culture dish and treated with different doses of PepE or 5‐Aza‐dC (2.0 μM) for 48 h. Subsequently, the cells were collected and incubated in RIPA buffer (Santa Cruz Biotechnology). Cell lysates were centrifuged and used as protein extracts. The concentration of each extract was determined by Bradford protein assay (Bio‐Rad). Proteins (approximately 25–50 μg) were subjected to 10% SDS‐PAGE (Thermo Fisher Scientific). Proteins were then transferred onto a nitrocellulose transfer membrane (Abcam) and blocked for 1 h using 5% non‐fat dried milk at room temperature. After incubating in TBS/0.1% Tween‐20, primary antibodies against DNMT1, RASSF1A, MST1/2, Bax, Bcl‐2, cleaved caspase 3, cleaved caspase 9, cleaved PARP, GAPDH (Abcam), and cyclin D1, RUNX3, p16INK4, APC, MOAP1, CNK1, and β‐catenin (Santa Cruz Biotechnology) were added (1:1000 v/v). After overnight incubation, the primary antibodies were washed away and the appropriate secondary antibodies (diluted 1:5000) were added. Then, protein bands were visualized by chemiluminescence using an ECL Plus chemiluminescence kit (Applygen Technologies, Beijing, China) on X‐ray film. Gray levels were analyzed using Gel‐Pro 32 software (Media Cybernetics, Rockville, MD, USA) with normalization to GAPDH or β‐actin. Experiments were repeated three times.
Pathologic and immunohistochemical observation and TUNEL analysis
The tumor, lung, liver, and kidney were excised from the mice, fixed in 10% neutral formalin, embedded in paraffin, and cut into 4‐μm‐thick sections. At least 10 randomly selected tissue sections from each group were stained with HE for histopathological analysis. The tumor tissue sections were incubated in 3% hydrogen peroxide for 15 min and then in normal goat serum for 20 min to block endogenous peroxidase activity and unspecific binding sites, respectively. Sections were then incubated for 2 h with primary rabbit antibodies against DNMT1, RASSF1A, APC, MST1/2, MOAP1, CNK1, cyclin D1, β‐catenin, and TUNEL (1:100 dilution) (KeyGen Biotechnology). Sections were then treated with biotinylated secondary antibodies (1:200 dilution), and antibody‐binding sites were visualized by 3,3′‐diaminobenzidine staining (Dako, Carpinteria, CA, USA). Sections were observed under an inverted phase contrast microscope (IX51; Olympus, Tokyo, Japan). Photographs were taken and analyzed using Image‐Pro Plus software (Media Cybernetics, Inc, Rockville, MD, USA).
Statistical analysis
Statistical analyses were undertaken using GraphPad Prism 6.0 software (Graphpad software Inc., La Jolla, CA, USA). One‐way anova followed by Student's t‐test was used for multiple comparisons. P < 0.05 was considered significant and P < 0.01 was considered more significant.
Results
Peperomin E inhibits cell viability, promotes apoptosis, and induces G1/S phase cell cycle arrest of lung cancer cells in vitro and in vivo
Human lung cancer cell lines A549, NCI‐H460, and H1229 were treated with PepE at various concentrations (0–100 μM) for 48 h. The inhibitory effect of PepE on these cell lines was dose‐dependent (Fig. 1b). The PepE IC50 values were as follows: A549, 2.177 μM; H1229, 6.862 μM; and NCI‐H460, 6.485 μM. Selectivity for tumor versus normal cells was determined by comparing the efficacy of PepE on the above tumor cells to its efficacy on the normal human lung epithelial cell line BEAS‐2B. Inhibition of PepE on BEAS‐2B cells was far less potent than on the gastric tumor cell lines (Fig. 1b), indicating that PepE is specific for cancer cells.
Peperomin E given at 25 or 50 mg/kg every other day slowed the growth of A549 tumors in BALB/c mice (Fig. 2a). Mice treated with PepE showed significantly lower tumor volume on day 21 compared to negative control mice (P < 0.01, Fig. 2a). As indicated in Figure 2(a), the proliferation inhibition efficacy of PepE at 50 mg/kg every day was lower than that of 5‐Aza‐dC (positive control) at 10 mg/kg twice a week (57.35% and 67.11% inhibition, respectively). However, lower toxicity was observed in PepE‐ than in 5‐Aza‐dC‐treated mice based on parallel monitoring of body weight. As shown in Figure 2(b), 5‐Aza‐dC treatment led to significant weight loss in mice with xenograft tumors compared with PepE treatment. As shown in Figure 2(c), no significant alterations of blood biochemical parameters were observed after PepE treatment. Additionally, no significant histological or pathologic alterations were found in the lung, liver, or kidney after PepE treatment (Fig. S4). However, 5‐Aza‐dC treatment led to a significant decrease in platelet and neutrophilic granulocyte cell levels in xenograft mice compared with negative control mice (Fig. 2c). These results indicated that PepE can effectively protect against tumor growth in vivo with fewer adverse effects than 5‐Aza‐dC.
Figure 2.
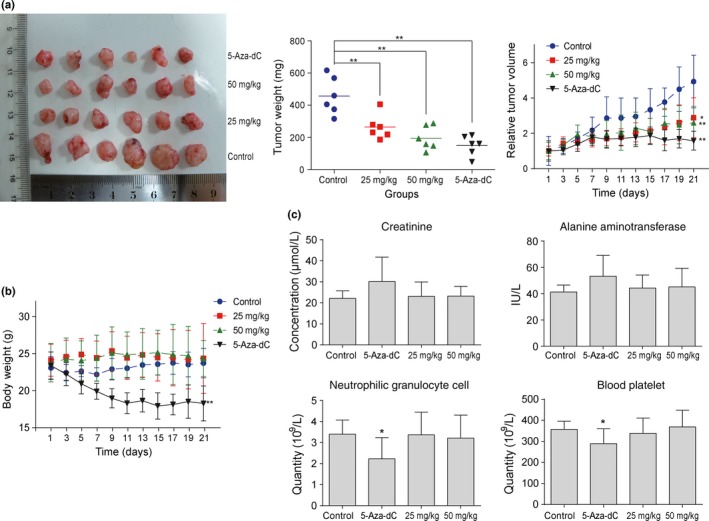
In vivo antitumor efficacy and safety evaluation of peperomin E against non‐small‐cell lung cancer. (a) Illustration shows the tumor excised, the mean tumor weight, and the tumor growth curve for mice with subcutaneous tumors when treated with peperomin E and 5‐Aza‐dC; (b) The change in body weight during the treatment; and (c) Measurement of serum or plasma parameters of the xenograft mice. Data represents the mean ± SD (n = 6). *P < 0.05; **P < 0.01. 5‐Aza‐dC, 5‐Aza‐2‐deoxycytidine.
Typical characteristics of apoptosis, such as cell shrinkage and pyknosis, were observed in PepE‐treated lung cancer cells, indicating that PepE may cause lung cancer cell death by inducing cell apoptosis. Therefore, flow cytometry using FITC–annexin V/propidium iodide staining coupling were used to detect cancer cell apoptosis and Western blot analysis was used to detect apoptosis‐related proteins after PepE treatment. Following 48 h of treatment, the proportion of apoptotic cells was significantly higher in cells exposed to 0.5 μM PepE compared with vehicle (P < 0.01; Fig. 3a). A dose‐dependent increase was also observed in the percentage of apoptotic cells (17.5%, 24.9%, and 39.2% following treatment with 0.5, 2.0, and 4.0 μM PepE, respectively) (Fig. 3a). Meanwhile, a concentration‐dependent increase of the pro‐death protein Bax was observed following PepE treatment, whereas Bcl‐2 levels decreased with increasing PepE concentration (Fig. 3b). Decrease in the Bcl‐2/Bax ratio led to the cleavage of caspase 9 and caspase 3 (Fig. 3b), and cleaved PARP, a hallmark of apoptosis, significantly increased after PepE treatment (Fig. 3b). The above results indicated that PepE may cause lung cancer cell death by inducing cell apoptosis. The extent of apoptosis in tumor tissues was further examined using the TUNEL assay. The PepE (50 mg/kg) and 5‐Aza‐dC groups showed more TUNEL‐positive cells than the control group (Fig. 3c), indicating that PepE can also induce lung cancer cell apoptosis in vivo.
Figure 3.
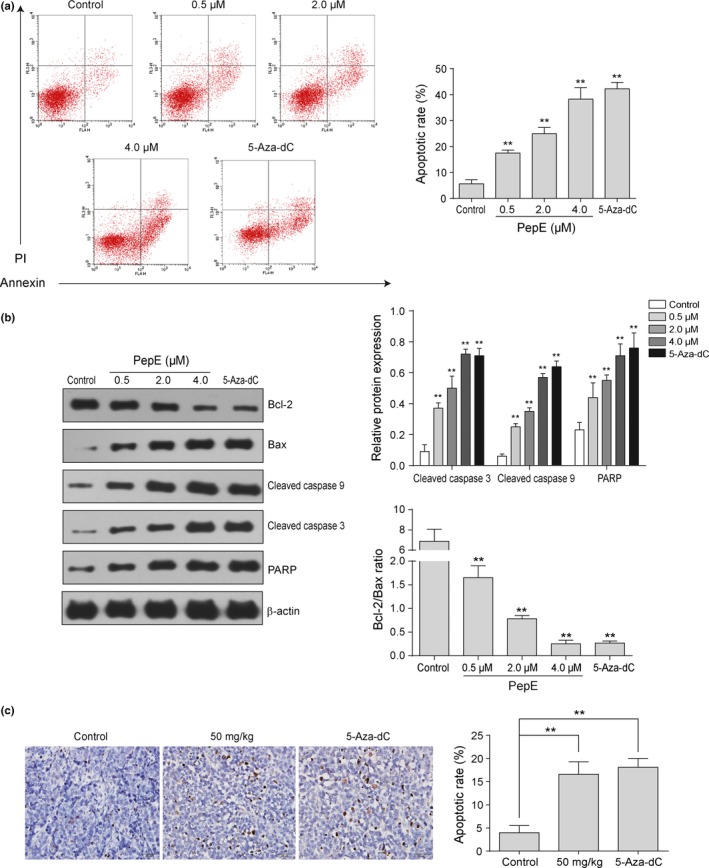
Effects of peperomin E (PepE) on the changes of apoptotic rate and apoptosis‐related proteins of A549 non‐small‐cell lung cancer cells. (a) The Annexin V/PI double‐staining assay of A549 cells, cell populations in the lower right (Annexin+/PI−) represents early apoptotic cells, upper right (Annexin+/PI+) represents late apoptotic cells; (b) Expression of Bcl‐2, Bax, cleaved caspase 9, cleaved caspase 3 and PARP proteins in A549 cells, the intensity of the bands was quantified by optical density (OD) and normalized to the OD of β‐actin; and (c) Representative TUNEL staining of tumors harvested 21 days after subcutaneous inoculation is shown for control group, 50 mg/kg PepE and 5‐Aza‐dC treatment group. Data are presented as means ± SD (n = 3 for a and b; n=6 for c). **P < 0.01. 5‐Aza‐dC, 5‐Aza‐2‐deoxycytidine; PI, propidium iodide.
The cell cycle progression of A549 cells was also evaluated. As shown in Figure 4(a), the percentage of cells in G1 was significantly increased after treatment with 2 μM PepE and 5‐Aza‐dC for 48 h than with the negative control (PepE, 77.3% vs 57.4%; 5‐Aza‐dC, 84.7% vs 57.4%; P < 0.01), suggesting that 2 μM PepE arrested A549 cells in the G1 phase. The proportion of sub‐G1 phase cells was significantly increased in A549 cells exposed to 2 μM PepE and 5‐Aza‐dC compared with negative control (PepE, 23.4% vs 2.3%; 5‐Aza‐dC, 37.1% vs 2.3%; P < 0.01), suggesting that 2 μM PepE led to a notable level of cell apoptosis (Fig. 4a). The expression of cell cycle‐related protein cyclin D1 in A549 cells was then evaluated following PepE treatment. A concentration‐dependent decrease of cyclin D1, which can lead to G1/S‐phase cell cycle arrest in cancer cells, was observed in vitro after PepE treatment (Fig. 4b).
Figure 4.
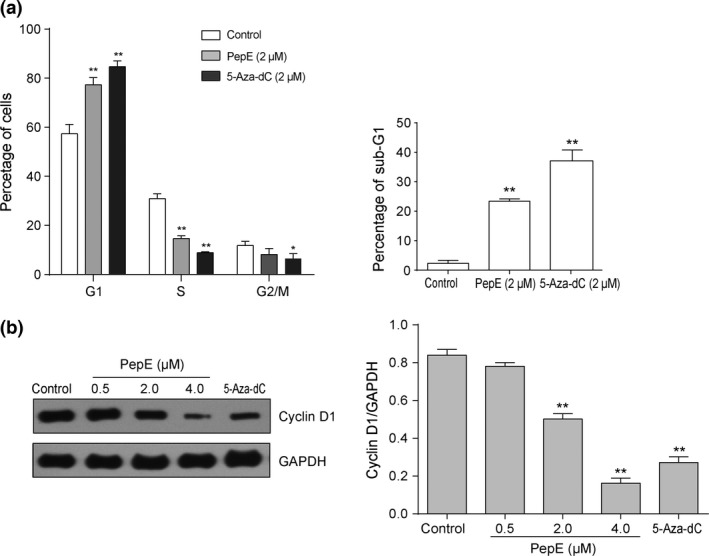
Induction of G1 cell cycle arrest and cyclin D1 downregulation in non‐small‐cell lung cancer cells in response to peperomin E (PepE) treatment. (a) The cell cycle distributions (including G1, S, G2/M and Sub‐G1 phases) of A549 cells after treatment with PepE and 5‐Aza‐dC for 48h are presented as a histogram graph. (b) Effects of PepE and 5‐Aza‐dC on changes of Cyclin D1 protein for 48h.All data are presented as means ± SD (n = 3). *P < 0.05; **P < 0.01. 5‐Aza‐dC, 5‐Aza‐2‐deoxycytidine.
The above results indicate that the antiproliferation effect of PepE was mediated primarily by promoting apoptosis and suppressing the cell cycle in vitro and in vivo.
Identification of potential PepE targets by computational target fishing
The top 10 tumor‐related targets, ranked by LibDock score in descending order, are shown in Table 1. Human DNMT1 (PDB ID: 3SWR) and mouse DNMT1 (PDB ID: 3AV6) ranked first and second, respectively. Peperomin E docking on DNMT1 (PDB ID: 3SWR) active sites (Fig. 5a) showed three hydrogen bond formations with glycine (GLY1147), glutamic acid (GLU1168), and phenylalanine (PHE1145). Similarly, molecular binding of 5‐Aza‐dC, a standard inhibitor of DNMT1, showed five hydrogen bonds with glycine (GLY1147), glutamic acid (GLU1168, GLU1266), asparagine (ASN1578), and phenylalanine (PHE1145). Moreover, as shown in Figure 5(b) and Table 2, superimposition of co‐crystallized sinefungin and docked compounds PepE and 5‐Aza‐dC on DNMT1 showed similar binding affinity. Peperomin E bound well within the DNMT1 active pocket (Fig. 5b), but showed slightly lower binding affinity than 5‐Aza‐dC and sinefungin. The molecular docking 2‐D diagram for PepE, sinefugin, 5‐Aza‐dC, and S‐adenosylmethionine (Fig. 5c–f) shows interaction of DNMT1 hydrophobic amino acid residues with bond ligands.
Table 1.
Top 10 potential tumor‐related targets of peperomin E predicted by LibDock of Discovery Studio 4.0
| Rank | PDB ID. | Libdock score | Targets |
|---|---|---|---|
| 1 | 3SWR | 155.7 | DNA (cytosine‐5)‐methyltransferase 1 (human) |
| 2 | 3AV6 | 139.5 | DNA (cytosine‐5)‐methyltransferase 1 (mouse) |
| 3 | 3TLM | 136.9 | Sarcoplasmic/endoplasmic reticulum calcium ATPase 1 (cattle) |
| 4 | 4EKL | 134.8 | RAC‐alpha serine/threonine‐protein kinase (human) |
| 5 | 1T5T | 132.1 | Sarcoplasmic/endoplasmic reticulum calcium ATPase 1 (rabbit) |
| 6 | 4FJY | 130.4 | Phosphatidylinositol 4,5‐bisphosphate 3‐kinase catalytic subunit gamma isoform (human) |
| 7 | 3QKK | 130.1 | RAC‐alpha serine/threonine‐protein kinase (human) |
| 8 | 3ZK6 | 129.1 | Bcl‐2 like protein 1 (human) |
| 9 | 3ML8 | 124.0 | Phosphatidylinositol‐4,5‐bisphosphate 3‐kinase catalytic subunit gamma isoform (human) |
| 10 | 3OW4 | 123.8 | RAC‐alpha serine/threonine‐protein kinase (human) |
Annotated Database of Druggable Binding Sites from the Protein DataBank (PDB) was used to identify potential binding proteins of peperomin E.
Figure 5.
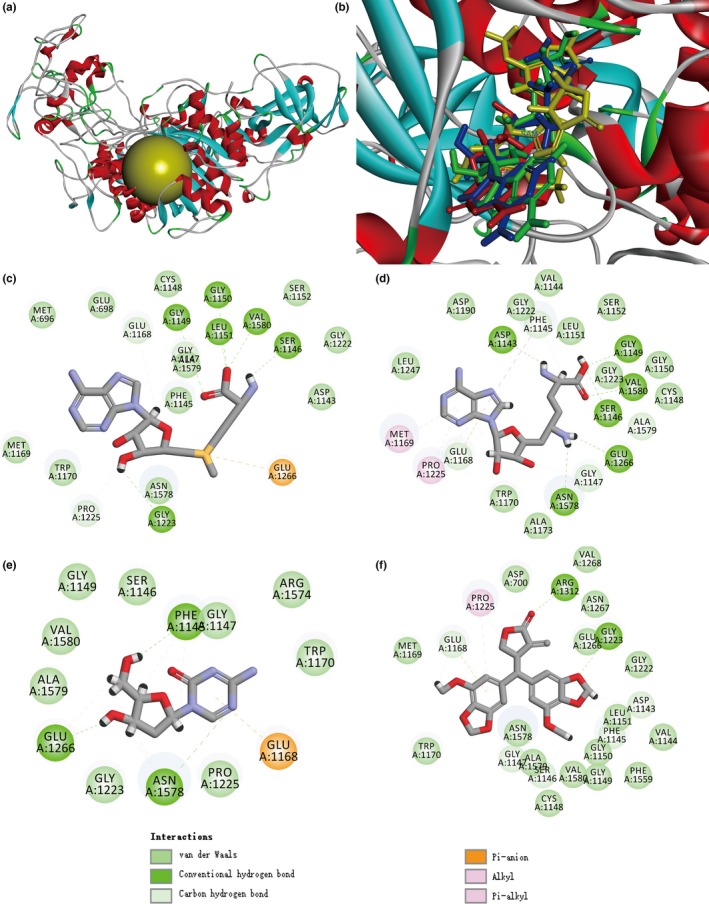
Molecular interactions between peperomin E and DNA methyltransferase 1. (a) Structure model of DNMT1 (PDB: 3SWR) with co‐crystallized ligand Sinefugin binding site (yellow color). (b) Superimposition of the best conformation of 5‐Aza‐dC (in red), Sinefugin (in green), S‐adenosylmethionine (SAM, in blue) and PepE (in yellow) docked in the active site pocket of DNMT1 by Discovery Studio 4.0 Libdock protocol. 2‐D diagrams illustrating protein‐ligand interactions of (c) SAM, (d) Sinefugin, (e) 5‐Aza‐dC and (f) PepE.
Table 2.
Details of LibDock score, CDOCKER energy, CDOCKER interaction energy, active site pocket residues, and hydrogen bonds revealed through molecular docking of S‐adenosylmethionine (SAM), 5‐aza‐2‐deoxycytidine (5‐Aza‐dC), peperomin E (PepE), and sinefugin on DNA methyltransferase 1 (DNMT1) (Protein DataBank ID: 3SWR) of Homo sapiens
| Target | Ligand | LibDock score | CDOCKER energy | CDOCKER interaction energy | Residues of sinefugin binding | Residues involved in H‐bond | No. of H‐bonds |
|---|---|---|---|---|---|---|---|
| Human DNMT1 | Sinefugin | 160.33 | 27.25 | 55.87 | GLY1147, GLY1149, GLY1150, LEU1151, ILE1167, LEU1247, PHE1145, MET1169, ASP1190, VAL1580, ALA1579, GLU1266, GLU1168, PRO1225, ASN1578, CYS1148 | GLY1149, GLY1150, LEU1151, VAL1580, GLU1168, GLU1266 | 6 |
| SAM | 161.44 | 22.05 | 50.26 | ALA1579, MET1169, GLU1168, PHE1145, PRO1225, GLY1147, GLY1149, LEU1151, GLY1150, VAL1580, CYS1148, ALA1579, ASN1578, GLU1266 | GLY1147, GLY1149, GLY1150, GLU1168, LEU1151, VAL1580, PRO1225 | 7 | |
| 5‐Aza‐dC | 160.36 | 4.92 | 38.2 | VAL1580, LEU1151, GLY1149, GLY1147, PHE1145, GLU1168, GLU1266, ALA1579, ASN1578 | GLY1147, GLU1266, GLU1168, ASN1578, PHE1145 | 5 | |
| PepE | 155.7 | –14.81 | 53.19 | PRO1225, GLY1147, GLY1150, LEU1151, PHE1145, GLU1266, VAL1580, GLU1168, ASN1578, MET1169, CYS1148 | GLY1147, PHE1145, GLU1168 | 3 |
The LibDock and CDOCKER programs are provided by Discovery studio version 4.0 software (Accelrys, San Diego, CA, USA).
DNA methyltransferase 1 is responsible for duplicating the pattern of DNA methylation during replication and is essential for mammalian development. Targeting DNMT1 can strongly inhibit the formation of tumors by reverting the hypermethylation of tumor suppressor genes and suppressing cancer‐specific cellular phenotypes.29 Typical DNMT1 inhibitor such as 5‐Aza‐dC can inhibit DNMT1 activity by covalent binding to the active site of the enzyme then blocking the entry of key nucleotide cytosine into such site, thus, preventing DNA methylation of cancer cells.30, 31, 32, 33 The in silico modeling results suggested that PepE might have the same inhibitory effect on DNMT1 by showing similar binding affinity to the active site of this enzyme.
Peperomin E inhibits DNA methyltransferase enzyme activity and expression, thereby decreasing global DNA methylation in NSCLC cells
To validate the results of in silico virtual screening, we determined DNMT activity in the nuclear extract of A549 cells after treatment with various concentrations of PepE (10−4–106 nM) or 5‐Aza‐dC (positive control). We found that both PepE and 5‐Aza‐dC inhibited DNMT activity in the nuclear extracts in a dose‐dependent manner (Fig. 6a), with IC50 values of 0.205 and 2.15 μM, respectively.
Figure 6.
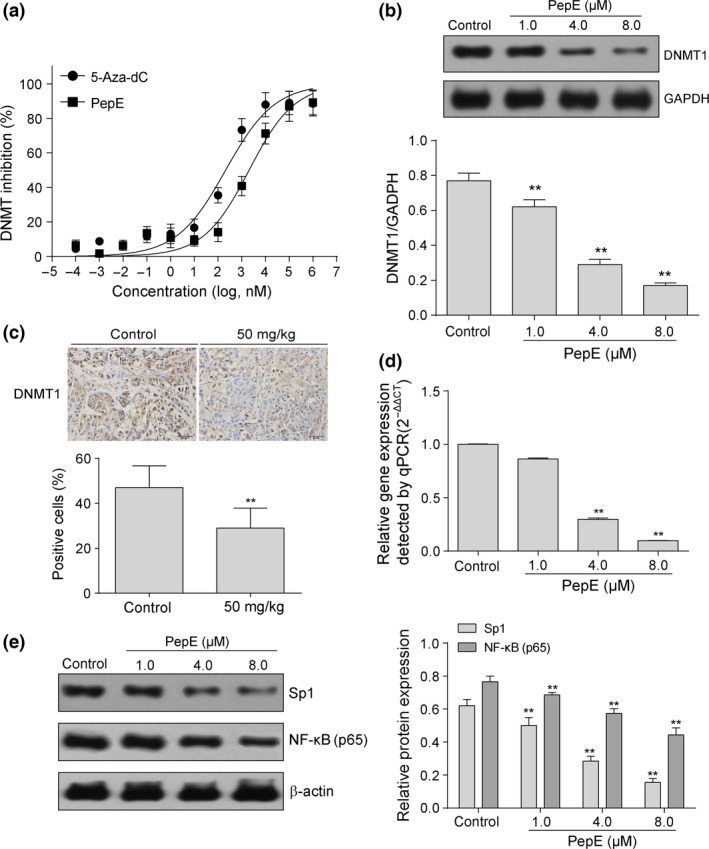
Effects of peperomin E (PepE) on changes of activity and expression of DNA methyltransferase 1 (DNMT1). (a) Dose‐response plots of PepE and 5‐Aza‐dC against DNMT. The IC50 concentrations were determined by biochemical DNMT assays under identical conditions; (b) Western‐blot bands for DNMT1 protein expression in A549 cells before and after PepE treatment. The intensity of the bands was quantified by optical density (OD) and normalized to the OD of GAPDH; (c) Representative immunostaining for DNMT1 in the paraffin section (original magnification ×200). 3 slides per mouse were reviewed. The positive rate was quantified by IOD values and normalized to the IOD values of control group; and (d) Western‐blot bands for Sp1 and NF‐kB (p65) proteins expression in A549 cells before and after PepE treatment. The intensity of the bands was quantified by optical density (OD) and normalized to the OD of β‐actin. All data are presented as means ± SD (n = 3 for a, b, d and e; n = 6 for c). **P < 0.01. NF‐κB, nuclear factor κB; qPCR, quantitative PCR.
To examine whether PepE induces degradation of DNMT1, we carried out Western blot analysis with extracts from A549 cells treated with PepE at different concentrations (1.0, 4.0, and 8.0 μM) and found that PepE treatment was associated with a statistically significant dose‐dependent depletion of DNMT1 in 4.0 and 8.0 μM doses (P < 0.01, Fig. 6b). Next, we measured DNMT1 mRNA levels by quantitative RT‐PCR (RT‐qPCR) following treatment with PepE. As shown in Figure 6(c), DNMT1 mRNA levels decreased in a dose‐dependent manner, indicating that, unlike 5‐Aza‐dC, which induces degradation of DNMT1 through the proteasomal pathway,34 PepE mediates depletion of DNMT1 by altering its mRNA expression. Previous studies indicated that the Sp1/nuclear factor‐κB (NF‐κB) pathway positively modulates the gene expression of DNMT1.35, 36 Therefore, the changing of Sp1 and NF‐κB (p65) proteins in A549 cells after PepE treatment was further investigated. As shown in Figure 6(d), protein levels of Sp1 and NF‐κB (p65) were both significantly downregulated in a dose‐dependent manner after PepE treatment (P < 0.01). This result indicated that the transcriptional repression activity of PepE against DNMT1 might be owing to its ability to reduce the expression of NF‐κB (p65) and Sp1, which correlated with a reduction in binding of these transcription factors to the DNMT1 promoter. This conjecture requires further experimental proof in following studies.
Because DNMT catalyzes the process of DNA methylation, we assessed whether PepE alters genomic DNA methylation levels in NSCLC cells or xenograft tumor tissues. Typical ultra‐fast liquid chromatography tandem mass spectrometry multiple reaction monitoring ion chromatograms for dG and 5‐mdC are shown in Figure S5(A,B). Calibration curves of 5‐mdC and dG (0–10%) are shown in Figure S5(C) (R 2 = 0.9995). DNA methylation levels in NSCLC cells treated with 2.0 and 4.0 μM PepE (i.e., 2.86% and 2.61% methylation in A549 cells, 3.61% and 3.30% methylation in NCI‐H460 cells, and 3.47% and 3.08% methylation in H1299 cells, respectively) were significantly lower than those treated with the negative control (i.e., 3.84%, 4.77%, and 4.20% methylation in untreated A549, NCI‐H460, and H1299 cells, respectively) (Fig. 7a). Similarly, Figure 7(b) shows that genomic methylation levels were significantly reduced in xenograft tumor tissues collected from mice treated with 25 and 50 mg/kg PepE (i.e., 3.76% and 3.12% methylation, respectively) compared to the negative control (4.33% methylation). Treatment of A549 cells and xenograft tumors with 5‐Aza‐dC also significantly decreased global DNA methylation (Fig. 7).
Figure 7.
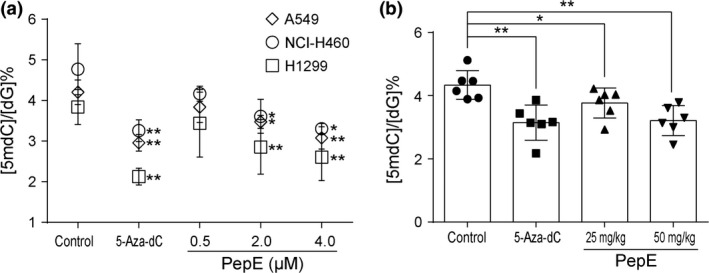
Effect of peperomin E (PepE) on global DNA methylation in non‐small‐cell lung cancer cells in vitro and in vivo. (a) Effects of PepE and 5‐Aza‐dC on global DNA methylation levels in NSCLC cells. DMSO served as control; and (b) Effects of PepE and 5‐Aza‐dC on the global DNA methylation levels in A549 tumor tissues extracted from nude mice. Data are presented as means ± SD (n = 3 for a; n = 6 for b). *P < 0.05; **P < 0.01. 5‐Aza‐dC, 5‐Aza‐2‐deoxycytidine; dG, deoxyguanosine; 5mdC, 5‐methyl‐deoxy‐cytidine.
The above results indicated that PepE can act as both chemical inhibitor and transcriptional modulator of DNMT1. Such activities might be attributable to its ability to block the active site and downregulate the positive transcriptional mediators (Sp1/NF‐κB pathway) of the DNMT1 enzyme.
Peperomin E demethylates tumor suppressor genes and reactivates their expressions in NSCLC cells
One of the primary biological outcomes of DNA demethylation in cancer cells is transcriptional reactivation of silenced tumor suppressor genes (TSGs).37, 38, 39 The RASSF1A, APC, p16INK4 (in male patients), and RUNX3 genes were discovered more frequently methylated in lung tissue from NSCLC patients than non‐cancerous tissue.40, 41, 42, 43, 44, 45 RASSF1A and APC genes were found fully hypermethylated in A549 cells,46 the RUNX3 gene was found silenced in NCI‐H460 cells by promoter methylation,47 and the promoter of p16INK4 gene was found completely methylated in H1299 cells.40 Demethylation of these TSGs and restoration of their expression in cancer cell lines impairs tumorigenicity. Thus, factors that restore the expression of these TSGs have immense potential in inhibiting tumor growth. As PepE inhibits DNMT1 activity and expression, we speculated that PepE might be involved in demethylation and re‐expression of TSGs in NSCLC cells. We therefore investigated methylation status of RASSF1A and APC genes in A549 cells, the p16INK4 gene in H1299 cells, and the RUNX3 gene in NCI‐H460 cells using methylation‐specific PCR before and after PepE treatment. As expected, RASSF1A and APC genes were fully methylated in untreated A549 cells, confirming that they are complete silenced in A549 cells (Fig. 8a). Following 0.5 μM PepE treatment, there was a noticeable increase in the levels of unmethylated RASSF1A or APC compared to the control; and after 1.0 or 2.0 μM treatment, this effect was even more significant (P < 0.01, Fig. 8a). Quantitative real‐time PCR analysis revealed that PepE and 5‐Aza‐dC each reactivated silenced RASSF1A and APC mRNA expression in a concentration‐dependent manner in A549 cells (i.e., ≥74‐fold for RASSF1A and ≥19 fold for APC at 2 μM dose of PepE, Fig. 8b). The findings are much the same concerning p16INK4 and RUNX3 genes. As shown in Figure 8(a), these two genes were entirely methylated in NCI‐H460 and H1299 cells. After treatment with PepE, the levels of unmethylated p16INK4 and RUNX3 genes were significantly increased, whereas the methylated levels of these two genes were significantly reduced (P < 0.01). The RT‐qPCR results proved that the mRNA expression of p16INK4 and RUNX3 were significantly restored in a dose‐dependent manner following PepE treatment (i.e. ≥30‐fold for p16INK4 and ≥87‐fold for RUNX3 at 4 μM dose of PepE; Fig. 8b).
Figure 8.
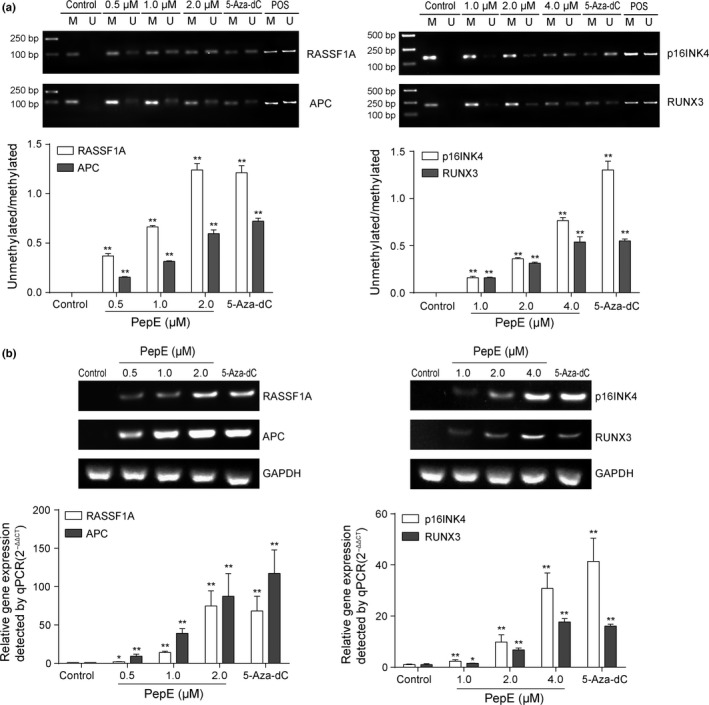
Effects of peperomin E (PepE) on the change of methylation status and gene expressions of tumor suppressor genes in non‐small‐cell lung cancer cells. (a) Methylation‐specific PCR detected RASSF1A and APC methylation status in A549 cells, RUNX3 status in NCI‐H460 cells and p16INK4 status in H1299 cells before and after adding to different doses of PepE and 5‐Aza‐dC (2 μM); and (b) RASSF1A, APC, p16INK4 and RUNX3 genes' expression detected by RT‐qPCR before and after PepE and 5‐Aza‐dC (2 μM) treatment. Data are presented as means ± SD (n = 3). *P < 0.05; **P < 0.01. 5‐Aza‐dC, 5‐Aza‐2‐deoxycytidine; M, methylated; POS, positive control for the methylated or unmethylated products; qPCR, quantitative PCR; U, unmethylated.
In vitro protein reactivation levels were determined using Western blot analysis. As shown in Figure 9(a), PepE and 5‐Aza‐dC each significantly reactivated silenced RASSF1A and APC protein levels in A549 cells in a concentration‐dependent manner (P < 0.01). The protein levels of p16INK4 and RUNX3 were also significantly restored in NCI‐H460 cells in a dose‐dependent manner (P < 0.01). Immunohistochemical analysis showed that there was a significant increase in RASSF1A‐ and APC‐positive cells in the tumor tissues of the 50 mg/kg PepE‐treated group when compared to the control group (Fig. 9b).
Figure 9.
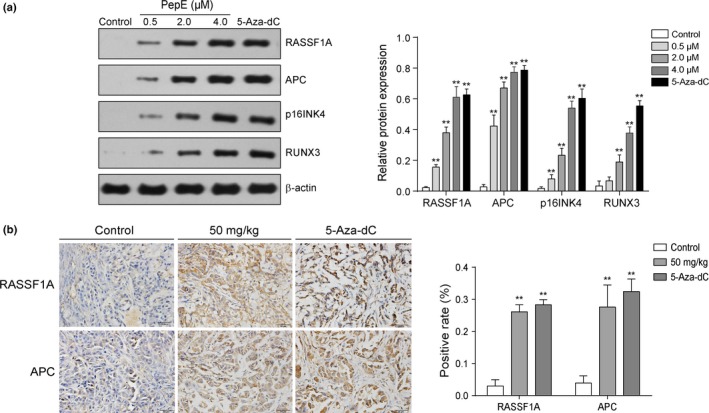
Effect of peperomin E (PepE) on the change of protein expression of epigenetically silenced tumor suppressor in non‐small‐cell lung cancer cells in vitro and in vivo. (a) Western‐blot bands for RASSF1A and APC proteins in A549 cells, p16INK4 and RUNX3 proteins in NCI‐H460 cells. The intensity of the bands was quantified by optical density (OD) and normalized to the OD of β‐actin; and (b) Representative immunostaining for RASSF1A and APC in the paraffin section (original magnification ×200). 3 slides per mouse were reviewed. The positive rate was quantified by IOD values and normalized to the IOD values of control group. Data are presented as means ± SD (n = 3 for a; n = 6 for b). **P < 0.01. 5‐Aza‐dC, 5‐Aza‐2‐deoxycytidine.
According to previous studies, RASSF1A, APC, RUNX3, and p16INK4 tumor suppressors can scaffold multiple pro‐apoptotic pathways and regulate cell cycle progressions in lung cancer. RASSF1A directly activates pro‐apoptotic and cell cycle regulation factors in lung cancer cells including MST1/2 (pro‐apoptotic serine/threonine kinases), CNK1 (a pro‐apoptotic adapter protein), MOAP‐1 (a Bax binding protein), and cyclin D1.48, 49 APC can directly destroy β‐catenin (a crucial regulating protein of Wnt signaling pathway), thus promoting apoptosis and cell cycle arrest of NSCLC cells.50, 51 RUNX3 can induce NSCLC cell apoptosis by blocking the transforming growth factor‐β superfamily signaling pathway.52, 53 p16INK4 mediates apoptosis and cell cycles in NSCLC cells by direct downregulating Rb and indirectly downregulating the anti‐apoptotic protein Bcl‐2.54, 55 In this study, the protein levels of some of the aforementioned pro‐apoptotic and cell cycle regulation factors (i.e., RASSF1A‐mediated MST1/2, CNK1, and MOAP‐1, and APC‐mediated β‐catenin) were also investigated to further elucidate the mechanisms involved in PepE‐induced apoptosis and cell cycle arrest in A549 cells. As shown in Figure S6, after PepE treatment, the protein levels of MST1, MST2, CNK1, and MOAP‐1 were significantly increased, whereas β‐catenin was significantly decreased in a concentration‐dependent manner (P < 0.01). Immunohistochemical analysis showed that there was a significant increase in MST1‐, MST2‐, CNK1‐, and MOAP‐1‐positive cells (P < 0.01) but a decrease in β‐catenin‐positive cells (P < 0.01) in the tumor tissues of the PepE‐treated (50 mg/kg) group (Fig. S7).
Discussion
Both genetic and epigenetic events are implicated in the tumorigenesis of lung cancer. Epigenetic changes, which are reversible, are attractive targets for chemotherapeutic intervention. One of the major epigenetic alterations in lung cancer is aberrant DNA methylation in the promoter region of TSGs.42 Tumor suppressor gene silencing can promote tumorigenesis by increasing cell proliferation and suppressing apoptosis and DNA repair.37 Therefore, therapeutic strategies that target global epigenetic inactivation of TSGs have been pioneered for cancer treatment.
It has been well documented that DNMT1, together with other DNA methyltransferases, regulate and maintain DNA methylation patterns in mammalian cells. Inhibition of DNMT1 has been presented as a possible pathway to demethylation of DNA at the 5‐position of cytosine and reactivation of TSGs silenced by promoter methylation.29 In this study, we first showed that PepE, a naturally derived secolignan, not only inhibits DNMT1 enzymatic activity, but also decreases its transcription levels by blocking the active site and inhibiting the positive transcription regulators (Sp1/NF‐κB pathway) of this enzyme. That is, PepE may serve dual functions: acting as both chemical inhibitor and transcriptional modulator of DNMT1. Both of these activities drive promoter demethylation, resulting in re‐expression of certain silenced TSGs in NSCLC cells. It was also found that, in response to PepE treatment, NSCLC cells underwent apoptosis and became arrested at the G1 phase of the cell cycle. All the aforementioned events occur together and subsequently contribute to PepE's anticancer effects against NSCLC, as evidenced by our in vitro and in vivo studies.
Up to now, the extensively studied DNMT inhibitors were nucleoside analogs including 5‐azacytidine and5‐Aza‐dC, which have been approved by the US FDA for the treatment of myelodysplastic syndrome and cutaneous T‐cell lymphoma.33 Although no DNMT inhibitors are currently approved for treatment of lung cancer, a recently completed clinical trial combining 5‐azacytidine and entinostat (a histone deacetylase inhibitor) for the treatment of advanced NSCLC demonstrated promising results.56 However, there are a number of additional limitations for the use of these nucleoside analogs. In particular, these drugs can cause severe side‐effects, including hematopoietic toxicity and neutropenia. Furthermore, these drugs are unstable, and can be degraded by hydrolytic cleavage and deamination by cytidine deaminase.33, 57 In contrast, PepE, derived from nature, may be devoid of several of these limitations. According to our research data, although the demethylating activity is lower than that of the currently used agent 5‐Aza‐dC, PepE appears to be considerably safer in animals. Therefore, this agent might further serve as a complementary therapy, especially when used in conjunction with nucleoside analogs or in the setting of nucleoside resistance, during treatment of cancer.
In summary, we identified a new DNMT1 inhibitor, PepE, that induced DNA demethylation and reversed the silencing of certain TSGs in NSCLC cells and induced NSCLC cell apoptosis and cell cycle arrest. These findings are of importance for understanding the mechanisms of PepE against lung cancer and provides a promising epigenetically targeting agent for cancer treatment. Further mechanism of action and structural modification studies to increase the efficacy of this natural product are ongoing.
Disclosure Statement
The authors have no conflict of interest.
Supporting information
Fig. S1. Proton nuclear magnetic resonance (a) and carbon‐13 nuclear magnetic resonance (b) spectrum of peperomin E.
Fig. S2. High‐performance liquid chromatography peak purity analysis of peperomin E.
Fig. S3. In silico target fishing protocol using the LibDock module in Discovery Studio 4.0.
Fig. S4. Histopathological analysis of liver, kidney, and lung tissue structures in xenograft mice.
Fig. S5. Chromatogram using ultra‐fast liquid chromatography tandem mass spectrometry of 5‐methyl‐deoxy‐cytidine (5mdC) and deoxyguanosine (dG) in DNA of hydrolyzed A549 cells.
Fig. S6. Effects of peperomin E on changes of Ras association domain family member 1 (RASSF1A)‐modulated pro‐apoptotic and cell cycle arrest proteins.
Fig. S7. Effects of peperomin E on changes of adenomatous polyposis coli (APC)‐modulated pro‐apoptotic protein β‐catenin.
Table S1. Primer list for methylation‐specific PCR.
Table S2. Polymerase chain reaction amplification protocol in methylation‐specific PCR test.
Table S3. Primer list for quantitative RT‐PCR.
Table S4. Polymerase chain reaction amplification protocol in quantitative RT‐PCR test.
Acknowledgments
This work was supported by grants from the Natural Science Foundation of China (Grant No. 81402812) and the Natural Science Foundation of Jiangsu Province (Grant No. BK20130954).
Cancer Sci 107 (2016) 1506–1619
Funding Information
Natural Science Foundation of China (81402812); Natural Science Foundation of Jiangsu Province (BK20130954).
References
- 1. Youlden DR, Cramb SM, Baade PD. The international epidemiology of lung cancer: geographical distribution and secular trends. J Thorac Oncol 2008; 3: 819–31. [DOI] [PubMed] [Google Scholar]
- 2. She J, Yang P, Hong Q, Bai C. Lung cancer in China: challenges and interventions. Chest 2013; 143: 1117–26. [DOI] [PubMed] [Google Scholar]
- 3. Chen W, Zheng R, Zeng H et al Epidemiology of lung cancer in China. Thorac Cancer 2015; 6: 209–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hong QY, Wu GM, Qian GS et al Prevention and management of lung cancer in China. Cancer 2015; 121: 3080–8. [DOI] [PubMed] [Google Scholar]
- 5. Schiller JH, Harrington D, Belani CP et al Comparison of four chemotherapy regimens for advanced non‐small‐cell lung cancer. N Engl J Med 2002; 346: 92–8. [DOI] [PubMed] [Google Scholar]
- 6. Florea AM, Busselberg D. Cisplatin as an anti‐tumor drug: cellular mechanisms of activity, drug resistance and induced side effects. Cancers 2011; 3: 1351–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rajeswaran A, Trojan A, Burnand B et al Efficacy and side effects of cisplatin‐ and carboplatin‐based doublet chemotherapeutic regimens versus non‐platinum‐based doublet chemotherapeutic regimens as first line treatment of metastatic non‐small cell lung carcinoma: a systematic review of randomized controlled trials. Lung Cancer 2008; 59: 1–11. [DOI] [PubMed] [Google Scholar]
- 8. Rowinsky EK, Eisenhauer EA, Chaudhry V et al Clinical toxicities encountered with paclitaxel (Taxol). Semin Oncol 1993; 20: 1–15. [PubMed] [Google Scholar]
- 9. Nanjing New Medical School . Dictionary of Chinese Herbal Drugs. Shanghai: Shanghai Science and Technology Press, 1978; 622. [Google Scholar]
- 10. Govindachari TR, Krishna Kumari GN, Partho PD. Two secolignans from Peperomia dindygulensis . Phytochemistry 1998; 49: 2129–31. [Google Scholar]
- 11. Wu JL, Li N, Hasegawa T et al Bioactive secolignans from Peperomia dindygulensis . J Nat Prod 2006; 69: 790–4. [DOI] [PubMed] [Google Scholar]
- 12. Chen L, Yu Y, Dong JX. Chemical constituents of Peperomia dindygulensis . Chin Tradit Herb Drugs 2007; 38: 491–3. [Google Scholar]
- 13. Lin MG, Yu DH, Wang QW et al Secolignans with antiangiogenic activities from Peperomia dindygulensis . Chem Biodivers 2011; 8: 862–71. [DOI] [PubMed] [Google Scholar]
- 14. Wu JL, Li N, Hasegawa T et al Bioactive tetrahydrofuran lignans from Peperomia dindygulensis . J Nat Prod 2005; 68: 1656–60. [DOI] [PubMed] [Google Scholar]
- 15. Chen L, Zhou Y, Dong JX. Three new flavonoid glycosides from Peperomia dindygulensis . Acta Pharm Sin 2007; 42: 183–6. [PubMed] [Google Scholar]
- 16. Wang QW, Du DH, Lin MG et al Antiangiogenic polyketides from Peperomia dindygulensis Miq. Molecules 2012; 17: 4474–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang XZ, Qu W, Liang JY. New long‐chain aliphatic compounds from Peperomia dindygulensis . Nat Prod Res 2013; 27: 796–803. [DOI] [PubMed] [Google Scholar]
- 18. Wang XZ, Qu W, Liang JY. Two N‐containing polyketide derivatives from Peperomia dindygulensis . Chem Nat Comp 2013; 48: 1027–30. [Google Scholar]
- 19. Xu S, Li N, Ning MM et al Bioactive compounds from Peperomia pellucida . J Nat Prod 2006; 69: 247–50. [DOI] [PubMed] [Google Scholar]
- 20. Tsutsui C, Yamada Y, Ando M et al Peperomins as anti‐inflammatory agents that inhibit the NF‐kappa B signaling pathway. Bioorg Med Chem Lett 2009; 19: 4084–7. [DOI] [PubMed] [Google Scholar]
- 21. Zhang XJ, Yang GY, Wang RR et al 7,8‐Secolignans from Schisandra wilsoniana and their anti‐HIV‐1 activities. Chem Biodivers 2011; 7: 2962–71. [DOI] [PubMed] [Google Scholar]
- 22. Momparle RL, Ayoub J. Potential of 5‐aza‐2′‐deoxycytidine (Decitabine) a potent inhibitor of DNA methylation for therapy of advanced non‐small cell lung cancer. Lung Cancer 2001; 34: 111–5. [DOI] [PubMed] [Google Scholar]
- 23. Song LG, James SR, Kazim L et al Specific method for the determination of genomic DNA methylation by liquid chromatography‐electrospray ionization tandem mass spectrometry. Anal Chem 2005; 77: 504–10. [DOI] [PubMed] [Google Scholar]
- 24. Herman JG, Graff JR, Myohanen S et al Methyation‐specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA 1996; 93: 9821–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kawaguchi K, Oda Y, Saito T et al DNA hypermethylation status of multiple genes in soft tissue sarcomas. Mod Pathol 2006; 19: 106–14. [DOI] [PubMed] [Google Scholar]
- 26. Scarpa M, Scarpa M, Castagliuolo I et al Aberrant gene methylation in non‐neoplastic mucosa as a predictive marker of ulcerative colitis‐associated CRC. Oncotarget 2016; 7: 10322–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chim CS, Pang R, Fung TK et al Epigenetic dysregulation of Wnt signaling pathway in multiple myeloma. Leukemia 2007; 21: 2527–36. [DOI] [PubMed] [Google Scholar]
- 28. Sushma PS, Jamil K, Kumar PU et al PTEN and p16 genes as epigenetic biomarkers in oral squamous cell carcinoma (OSCC): a study on south Indian population. Tumour Biol 2016; 37: 7625–32. [DOI] [PubMed] [Google Scholar]
- 29. Robert RF, Morin S, Beaulieu N et al DNMT1 is required to maintain CpG methylation and aberrant gene silencing in human cancer cells. Nat Genet 2002; 33: 61–5. [DOI] [PubMed] [Google Scholar]
- 30. Erdmann A, Halby L, Fahy J et al Targeting DNA methylation with small molecules: what's next? J Med Chem 2015; 58: 2569–83. [DOI] [PubMed] [Google Scholar]
- 31. Fahy J, Jeltsch A, Arimondo PB. DNA methyltransferase inhibitors in cancer: a chemical and therapeutic patent overview and selected clinical studies. Expert Opin Ther Pat 2012; 22: 1427–42. [DOI] [PubMed] [Google Scholar]
- 32. Stresemann C, Brueckner B, Musch T et al Functional diversity of DNA methyltransferase inhibitors in human cancer cell lines. Cancer Res 2006; 66: 2794–800. [DOI] [PubMed] [Google Scholar]
- 33. Christman JK. 5‐Azacytidine and 5‐aza‐2′‐deoxycytidine as inhibitors of DNA methylation: mechanistic studies and their implications for cancer therapy. Oncogene 2002; 21: 5483–95. [DOI] [PubMed] [Google Scholar]
- 34. Ghoshal K, Datta J, Majumder S et al 5‐Aza‐deoxycytidine induces selective degradation of DNA methyltransferase 1 by a proteasomal pathway that requires the KEN box, bromo‐adjacent homology domain and nuclear localization signal. Mol Cell Biol 2005; 25: 4727–41. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 35. Yu J, Peng Y, Wu LC et al Curcumin down‐regulates DNA methyltransferase 1 and plays an anti‐leukemic role in acute myeloid leukemia. PLoS One 2013; 8: e55934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liu SJ, Liu ZF, Xie ZJ et al Bortezomib induces DNA hypomethylation and silenced gene transcription by interfering with Sp1/NF‐κB‐dependent DNA methyltransferase activity in acute myeloid leukemia. Blood 2008; 111: 2364–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Baylin SB. DNA methylation and gene silencing in cancer. Nat Clin Pract Oncol 2005; 2: 4–11. [DOI] [PubMed] [Google Scholar]
- 38. Jones PA, Takai D. The role of DNA methylation in mammalian epigenetics. Science 2001; 293: 1068–70. [DOI] [PubMed] [Google Scholar]
- 39. Fang MA, Wang YM, Ai N et al Tea polyphenol (−)‐epigallocatechin‐3‐gallate inhibits DNA methyltransferase and reactivates methylation‐silenced genes in cancer cell lines. Cancer Res 2003; 63: 7653. [PubMed] [Google Scholar]
- 40. Shames DS, Girard L, Gao BN et al A genome‐wide screen for promoter methylation in lung cancer identifies novel methylation markers for multiple malignancies. PLoS Med 2006; 3: e486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Drilon A, Sugita H, Sima CS et al A prospective study of tumor suppressor gene methylation as a prognostic biomarker in surgically resected stage I to IIIA non‐small‐cell lung cancers. J Thorac Oncol 2014; 9: 1272–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Langevin SM, Kratzke RA, Kelsey KT et al Epigenetics of lung cancer. Trans Res 2015; 165: 74–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yanagawa N, Tamura G, Oizumi H et al Promoter hypermethylation of RASSF1A and Runx3 genes as an independent prognostic prediction marker in surgically resected non‐small cell lung cancers. Lung Cancer 2007; 58: 131–8. [DOI] [PubMed] [Google Scholar]
- 44. Zhang YW, Wang R, Song HZ et al Methylation of multiple genes as a candidate biomarker in non‐small cell lung cancer. Cancer Lett 2011; 303: 21–8. [DOI] [PubMed] [Google Scholar]
- 45. Yanagawa N, Tamura G, Oizumi H et al Promoter hypermethylation of tumor suppressor and tumor‐related genes in non‐small cell lung cancers. Cancer Sci 2003; 94: 589–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lin Q, Geng JF, Ma KL et al RASSF1A, APC, ESR1, ABCB1 and HOXC9, but not p16INK4A, DAPK1, PTEN and MT1G genes were frequently methylated in the stage I non‐small cell lung cancer in China. J Cancer Res Clin Oncol 2009; 135: 1675–84. [DOI] [PubMed] [Google Scholar]
- 47. Li QL, Kim HR, Kim WJ et al Transcriptional silencing of the Runx3 gene by CpG hypermethylation is associated with lung cancer. Biochem Biophs Res Commun 2004; 314: 223–8. [DOI] [PubMed] [Google Scholar]
- 48. Richter AM, Pfeifer GP, Dammann RH. The RASSF proteins in cancer; from epigenetic silencing to functional characterization. Biochim Biophys Acta 2009; 1796: 114–28. [DOI] [PubMed] [Google Scholar]
- 49. Donninger H, Vos DM, Clark GJ. The RASSF1A tumor suppressor. J Cell Sci 2007; 120: 3163–72. [DOI] [PubMed] [Google Scholar]
- 50. Su LK, Vogelstein B, Kinzler KW et al Association of the APC tumor suppressor protein with catenins. Science 1993; 262: 1734. [DOI] [PubMed] [Google Scholar]
- 51. Stewart DJ. Wnt signaling pathway in non–small cell lung cancer. J Natl Cancer Inst 2014; 106: 1–11. [DOI] [PubMed] [Google Scholar]
- 52. Bae SC, Choi JK. Tumor suppressor activity of Runx3. Oncogene 2004; 23: 4336–40. [DOI] [PubMed] [Google Scholar]
- 53. Ito Y, Miyazono K. Runx transcription factors as key targets of TGF‐β superfamily signaling. Curr Opin Genet Dev 2003; 13: 43–7. [DOI] [PubMed] [Google Scholar]
- 54. Shapiro GI, Edwards CD, Kobzik L et al Reciprocal Rb inactivation and p16INK4 expression in primary lung cancers and cell lines. Cancer Res 1995; 55: 505–9. [PubMed] [Google Scholar]
- 55. Kataoka M, Wiehle S, Schumacher F et al Down‐regulation of bcl‐2 is associated with p16INK4‐mediated apoptosis in non‐small cell lung cancer cells. Oncogene 2000; 12: 1589–95. [DOI] [PubMed] [Google Scholar]
- 56. Juergens RA, Wrangler J, Vendetti FP et al Combination epigenetic therapy has efficacy in patients with refractory advanced non–small cell lung cancer. Cancer Discov 2011; 1: 598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Jean‐Pierre I. Decitabine. Curr Opin Oncol 2003; 15: 446–51. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Proton nuclear magnetic resonance (a) and carbon‐13 nuclear magnetic resonance (b) spectrum of peperomin E.
Fig. S2. High‐performance liquid chromatography peak purity analysis of peperomin E.
Fig. S3. In silico target fishing protocol using the LibDock module in Discovery Studio 4.0.
Fig. S4. Histopathological analysis of liver, kidney, and lung tissue structures in xenograft mice.
Fig. S5. Chromatogram using ultra‐fast liquid chromatography tandem mass spectrometry of 5‐methyl‐deoxy‐cytidine (5mdC) and deoxyguanosine (dG) in DNA of hydrolyzed A549 cells.
Fig. S6. Effects of peperomin E on changes of Ras association domain family member 1 (RASSF1A)‐modulated pro‐apoptotic and cell cycle arrest proteins.
Fig. S7. Effects of peperomin E on changes of adenomatous polyposis coli (APC)‐modulated pro‐apoptotic protein β‐catenin.
Table S1. Primer list for methylation‐specific PCR.
Table S2. Polymerase chain reaction amplification protocol in methylation‐specific PCR test.
Table S3. Primer list for quantitative RT‐PCR.
Table S4. Polymerase chain reaction amplification protocol in quantitative RT‐PCR test.