

Abstract
Medicinally relevant spirocyclic indolenines, carbazoles and quinolines can each be directly synthesised selectively from common indolyl ynone starting materials by catalyst variation. The high yielding, divergent reactions all proceed by an initial dearomatising spirocyclisation reaction to generate an intermediate vinyl–metal species, which then rearranges selectively by careful choice of catalyst and reaction conditions.
Keywords: carbazoles, catalysis, diversity, quinolines, spirocycles
The synthesis of structurally diverse compounds is central to the discovery of pharmaceutical lead compounds.1 However, the formation of distinct compound sets usually requires multiple synthetic routes, which is time‐consuming and labour‐intensive; therefore, strategies capable of selectively forming multiple products from common starting materials are of high value. The concept underpinning our approach is the formation of a common reactive intermediate (from a simple, inexpensive starting material), which depending on the catalyst used can rearrange into different scaffolds (e.g., spirocycles, aromatics and heterocycles/carbocycles; Figure 1). This approach has the potential to significantly streamline existing synthetic methods, and lead to a broader understanding of catalysis and reaction mechanisms. Although there have been numerous examples of catalyst variation leading to different products in recent years,2, 3 such methods have mainly focused on the formation of products with similar frameworks (e.g., redox isomers, regioisomers or stereoisomers). In this work, our aim was to develop a series of divergent processes capable of selectively delivering multiple products with the level of scaffold diversity outlined in Figure 1.
Figure 1.
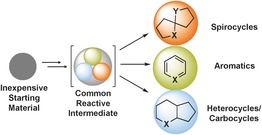
Catalyst‐driven scaffold diversity.
To demonstrate the synthetic potential of our scaffold‐diversity approach, we chose to explore the formation and subsequent reaction of spirocyclic vinyl–metal intermediates of the form 2 (Scheme 1). Previous work in our research group has demonstrated that the dearomatising spirocyclisation4 of ynones 1 into spirocyclic indolenines 3 can be catalysed by AgOTf, with vinyl–silver species 2 ([M]=Ag) as likely intermediates.5 A key design feature of our strategy was the idea that varying the catalyst would alter the nature and reactivity of the vinyl–metal intermediate 2 in a programmable way, such that alternative products could be formed by different rearrangement reactions. Herein, we report the successful realisation of this approach. Notably, by judicious choice of catalyst, simple, inexpensive ynone starting materials 1 can be converted into spirocyclic indolenines6 3 using AgI, carbazoles 5 using AuI and quinolines 7 using AgI/AlIII in high yield, each by a simple, catalytic and atom‐economical process. Furthermore, in suitable cases, tetracyclic scaffolds 8 can be formed with complete diastereoselectivity, by a telescoped spirocyclisation/nucleophilic addition sequence, which was performed using a chiral AgI salt to furnish an enantiopure product.
Scheme 1.
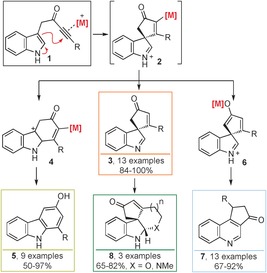
Divergent synthesis of spirocycles 3, carbazoles 5, quinolines 7 and tetracyclic scaffolds 8 from indolyl ynones 1.
The spirocyclisation of 1 a using AgOTf formed indolenine 3 a in quantitative yield (Scheme 2);5 the mild reaction conditions are believed to play a key role in this process, stabilising the spirocycle with respect to further reactions. However, in the proposed scaffold diversity approach, in which the synthesis of carbazole 5 a was an initial goal, the challenge was to deliberately promote 1,2‐migration7 in a controlled manner.8 A Ph3PAuNTf2 catalyst was chosen based on the prediction that the π‐acidic gold(I) catalyst would effectively promote the initial spirocyclisation reaction and that the intermediate vinyl–gold species (2 a‐Au) would be prone to 1,2‐migration, based on known reactivity of related vinyl–gold and gold–carbenoid species.9 This idea was validated (94 % yield of 5 a) with a likely reaction mechanism depicted in Scheme 3; the ring enlargement is believed to proceed either via cyclopropane intermediate 9 a, or by a direct 1,2‐migration reaction (2 a‐Au→10 a) based on related precedent.7, 9 The importance of vinyl–gold intermediate 2 a‐Au in the 1,2‐migration is evidenced by the fact that no reaction takes place when spirocycle 3 a is treated with Ph3PAuNTf2 under the same conditions.
Scheme 2.
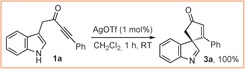
Formation of spirocyclic indolenine 3 a.
Scheme 3.
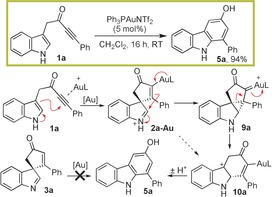
Formation of carbazole 5 a; [Au]=Ph3PAuNTf2, L=ligand.
We next examined whether we could initiate an alternative rearrangement commencing from ynone 1 a, by seeking to promote cyclopropanation of an enolate from the less substituted branch of the cyclopentenone; more oxophilic catalysts were chosen for this task, as it was thought that they would better promote the necessary enolate formation. We were unable to uncover a catalyst that could successfully initiate spirocyclisation and subsequent rearrangement on its own. However, first performing the spirocyclisation using 2 mol % of AgOTf as catalyst in isopropanol, followed by the addition of 5 mol % of AlCl3⋅6H2O and subsequent heating in a microwave gave quinoline 7 a in high yield (Scheme 4).10 Following AgI‐mediated spirocyclisation, it is thought that the AlIII catalyst promotes enolate formation and subsequent cyclopropanation to form 12 a, which can then fragment to form 13 a and aromatise to give quinoline 7 a (either by simple proton shuttling, or by a series of 1,5‐sigmatropic H‐transfer reactions).
Scheme 4.
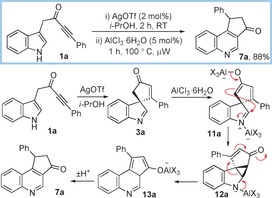
Formation of quinoline 7 a; X=Cl or iPrO.
Supporting evidence for this unprecedented rearrangement was obtained: treatment of spirocycle 3 a with LHMDS in THF (i.e. conditions which almost certainly would result in enolate formation) also led to the formation of quinoline 7 a, in 81 % yield. Furthermore, the importance of the carbonyl group was shown by the fact that treatment of known cyclopentenol 14 11 with AlCl3⋅6H2O did not result in quinoline formation. Instead, 1,2‐migration of the alkenyl group took place, furnishing carbazole 15 following tautomerisation and dehydration (Scheme 5).
Scheme 5.
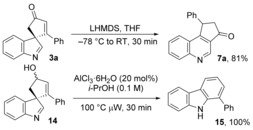
Base‐mediated formation of quinoline 7 a and the contrasting reactivity of spirocyclic cyclopentenol 14.
To probe the scope of all three reaction manifolds, various functionalised indole‐tethered ynones 1 a–1 m were prepared, substituted in several positions with electron‐rich and ‐poor aromatics, alkyl substituents, O‐ and N‐protected alkyl groups and PhS.12 First, using the AgOTf‐mediated spirocyclisation methodology, substrates 1 a–1 m were cleanly converted into the corresponding spirocyclic indolenines 3 a–3 m, all in excellent yields (Table 1, conditions A). The Ph3PAuNTf2‐mediated carbazole‐forming reaction was similarly broad in scope (conditions B); some reactions were less efficient than the analogous spirocycle formations, and ynone 1 d did not produce any of the desired product (instead stalling at the formation 3 d), but the majority of the carbazole products 5 a–j were isolated in very good yields.13 Finally, the quinoline‐forming reaction sequence was also found to be very general (conditions C). For ynones 1 a–1 e,1 g,1 k–1 l, the sequential AgOTf spirocyclisation and AlCl3⋅6 H2O mediated rearrangement steps could both be performed in iPrOH in one‐pot as described, whereas for ynones with more sensitive functional groups (1 f, 1 h, 1 i, 1 j, 1 m), the process benefited from a solvent swap, with the spirocyclisation first being performed in CH2Cl2 before concentration and addition of iPrOH prior to the AlCl3⋅6 H2O step. The AlCl3⋅6 H2O reactions were typically performed under microwave irradiation at 100 °C, but they were also shown to proceed well on a gram scale with conventional heating, albeit with a longer reaction time being required.14 The structure of quinoline 7 f was confirmed by X‐ray crystallography.15
Table 1.
Reaction scope for the formation of spirocyclic indolenines, carbazoles and quinolones.
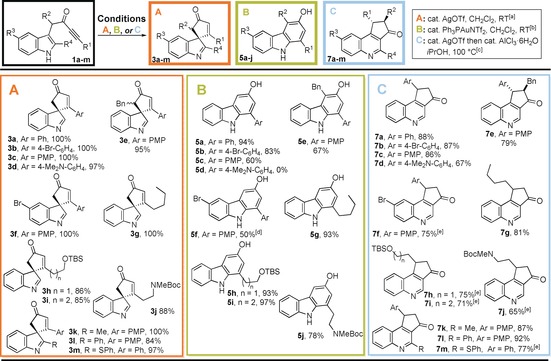
|
[a] AgOTf (1 mol %) in CH2Cl2 (0.1 m) at RT for 0.1–3.5 h. [b] Ph3PAuNTf2 (2–5 mol %) in CH2Cl2 (0.1 m) at RT for 7–18 h. [c] AgOTf (1 mol %) in iPrOH (0.1 m) at RT for 1–3 h, then AlCl3⋅6 H2O (5–10 mol %) at 100 °C μW for 1–2 h. [d] Reaction performed in toluene. [e] AgOTf (1 mol %) in CH2Cl2 (0.1 m) at RT for 1–3 h, then solvent swap for iPrOH (0.1 m) then AlCl3⋅6 H2O (5–10 mol %) at 100 °C μW for 1–2 h. PMP=para‐methoxyphenyl.
Another strand of scaffold diversity starting from more functionalised ynones 1 h–1 j was briefly explored. Tetracyclic scaffolds 8 h–j, equipped with additional complexity, were easily obtained following reaction of ynones 1 h–1 j with AgOTf and subsequent acid‐mediated protecting group cleavage in one pot (Scheme 6, and see the Supporting Information for details).16 The tetracycles were formed as the single diastereoisomers shown, and in addition, (S)‐8 h was prepared in enantioenriched form (89:11 e.r.) by utilising (R)‐CPA silver(I) salt 16 in place of AgOTf.17 The e.r. of (S)‐8 h could be increased to ≈100:0 by recrystallisation from ethanol, and its structure was confirmed by X‐ray crystallography (see the Supporting Information).15
Scheme 6.
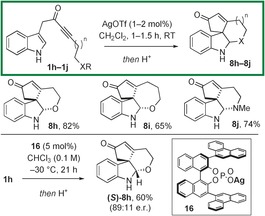
One‐pot spirocyclisation/trapping to form tetracycles 8 h–8 j.
In summary, readily available indolyl ynones have been shown to be versatile starting materials for the synthesis of spirocyclic indolenines 3 a–m, carbazoles 5 a–j, quinolines 7 a–m and tetracyclic compounds 8 h–j using a catalyst‐driven scaffold diversity approach. The reactions are typically high yielding, work on a wide range of indolyl ynone substrates, are operationally simple and can all be performed with no effort to exclude air or moisture. All of the procedures are thought to proceed by an initial dearomatising spirocyclisation to form a key vinyl–metal intermediate before diverging at this point depending on the nature of the catalyst used. The synthetic methods are expected to be of value both in target synthesis projects18 and to enable the rapid generation of compound libraries for biological screening.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors wish to thank the EPSRC (J.T.R.L. EP/M018601/01) and the University of York (M.J.J., A.K.C. and W.P.U.) for funding and Dr A. C. Whitwood (University of York) for X‐ray crystallography.
J. T. R. Liddon, M. J. James, A. K. Clarke, P. O'Brien, R. J. K. Taylor, W. P. Unsworth, Chem. Eur. J. 2016, 22, 8777.
Contributor Information
Prof. Richard J. K. Taylor, Email: richard.taylor@york.ac.uk
Dr. William P. Unsworth, Email: william.unsworth@york.ac.uk
References
- 1.
- 1a. Burke M. D., Schreiber S. L., Angew. Chem. Int. Ed. 2004, 43, 46–58; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 48–60; [Google Scholar]
- 1b. Lovering F., Bikker J., Humblet C., J. Med. Chem. 2009, 52, 6752–6756; [DOI] [PubMed] [Google Scholar]
- 1c. Hung A. W., Ramek A., Wang Y., Kaya T., Wilson J. A., Clemons P. A., Young D. W., Proc. Natl. Acad. Sci. USA 2011, 108, 6799–6804; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1d. Aldeghi M., Malhotra S., Selwood D. L., Chan A. W. E., Chem. Biol. Drug Des. 2014, 83, 450–461; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1e. Karawajczyk A., Giordanetto F., Benningshof J., Hamza D., Kalliokoski T., Pouwer K., Morgentin R., Nelson A., Muller G., Pierchot A., Tzalis D., Drug Discovery Today 2015, 20, 1310–1316. [DOI] [PubMed] [Google Scholar]
- 2.For a review on ‘Catalytic Selective Synthesis’, see: Mahatthananchai J., Dumas A. M., Bode J. W., Angew. Chem. Int. Ed. 2012, 51, 10954–10990; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 11114–11152. [Google Scholar]
- 3.For more recent examples, see:
- 3a. Dooley J. D., Reddy Chidipudi S., Lam H. W., J. Am. Chem. Soc. 2013, 135, 10829–10836; [DOI] [PubMed] [Google Scholar]
- 3b. Daniels D. S. B., Jones A. S., Thompson A. L., Paton R. S., Anderson E. A., Angew. Chem. Int. Ed. 2014, 53, 1915–1920; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 1946–1951; [Google Scholar]
- 3c. Donets P. A., Cramer N., Angew. Chem. Int. Ed. 2015, 54, 633–637; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 643–647; [Google Scholar]
- 3d. Zhang Y.-S., Tang X.-Y., Shi M., Org. Chem. Front. 2015, 2, 1516–1520; [Google Scholar]
- 3e. Xu L., Li H., Liao Z., Lou K., Xie H., Li H., Wang W., Org. Lett. 2015, 17, 3434–3437; [DOI] [PubMed] [Google Scholar]
- 3f. Liao J.-Y., Shao P.-L., Zhao Y., J. Am. Chem. Soc. 2015, 137, 628–631; [DOI] [PubMed] [Google Scholar]
- 3g. Galván A., Calleja J., González-Pérez A. B., Álvarez R., de Lera A. R., Fañanás F. J., Rodríguez F., Chem. Eur. J. 2015, 21, 16769–16774; [DOI] [PubMed] [Google Scholar]
- 3h. Li D. Y., Chen H. J., Liu P. N., Angew. Chem. Int. Ed. 2016, 55, 373–377; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 381–385; [Google Scholar]
- 3i. Cheng Q.-Q., Yedoyan J., Arman H., Doyle M. P., J. Am. Chem. Soc. 2016, 138, 44–47. [DOI] [PubMed] [Google Scholar]
- 4.For reviews on dearomatising spirocyclisation reactions, see:
- 4a. Zhuo C.-X., Zhang W., You S.-L., Angew. Chem. Int. Ed. 2012, 51, 12662; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 12834; [Google Scholar]
- 4b. Roche S. P., Tendoung J.-J. Y., Tréguier T., Tetrahedron 2015, 71, 3549. [Google Scholar]
- 5. James M. J., Cuthbertson J. D., O'Brien P., Taylor R. J. K., Unsworth W. P., Angew. Chem. Int. Ed. 2015, 54, 7640–7643; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 7750–7753. [Google Scholar]
- 6.For a review on spirocyclic indolenine synthesis, see: James M. J., O'Brien P., Taylor R. J. K., Unsworth W. P., Chem. Eur. J. 2016, 22, 2856–2881. [DOI] [PubMed] [Google Scholar]
- 7.For examples of related 1,2-migration reactions, see:
- 7a. Wu Q. F., Zheng C., You S.-L., Angew. Chem. Int. Ed. 2012, 51, 1680–1683; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 1712–1715; [Google Scholar]
- 7b. Peshkov V. A., Pereshivko O. P., Van der Eycken E. V., Adv. Synth. Catal. 2012, 354, 2841–2848; [Google Scholar]
- 7c. Zheng C., Wu Q.-F., You S.-L., J. Org. Chem. 2013, 78, 4357–4365; [DOI] [PubMed] [Google Scholar]
- 7d. Heffernan S. J., Tellam J. P., Queru M. E., Silvanus A. C., Benito D., Mahon M. F., Hennessy A. J., Andrews B. I., Carbery D. R., Adv. Synth. Catal. 2013, 355, 1149–1159. [Google Scholar]
- 8.For other syntheses of carbazoles involving alkyne activation, see:
- 8a. Wang L., Li G., Liu Y., Org. Lett. 2011, 13, 3786–3789; [DOI] [PubMed] [Google Scholar]
- 8b. Hashmi A. S. K., Yang W., Rominger F., Chem. Eur. J. 2012, 18, 6576–6580; [DOI] [PubMed] [Google Scholar]
- 8c. Qiu Y., Fu C., Zhang X., Ma S., Chem. Eur. J. 2014, 20, 10314–10322. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Nieto-Oberhuber C., Muñoz M. P., Buñuel E., Nevado C., Cárdenas D. J., Echavarren A. M., Angew. Chem. Int. Ed. 2004, 43, 2402–2406; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 2456–2460; [Google Scholar]
- 9b. Nieto-Oberhuber C., López S., Muñoz M. P., Jiménez-Núñez E., Buñuel E., Cárdenas D. J., Echavarren A. M., Chem. Eur. J. 2006, 12, 1694–1702; [DOI] [PubMed] [Google Scholar]
- 9c. López-Carrillo V., Huguet N., Mosquera Á., Echavarren A. M., Chem. Eur. J. 2011, 17, 10972–10978; [DOI] [PubMed] [Google Scholar]
- 9d. Wittstein K., Kumar K., Waldmann H., Angew. Chem. Int. Ed. 2011, 50, 9076–9080; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 9242–9246; [Google Scholar]
- 9e. Meiss R., Kumar K., Waldmann H., Chem. Eur. J. 2015, 21, 13526–13530; [DOI] [PubMed] [Google Scholar]
- 9f. Zhdanko A., Maier M. E., ACS Catal. 2015, 5, 5994–6004; [Google Scholar]
- 9g. Dorel R., Echavarren A. M., Chem. Rev. 2015, 115, 9028–9072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.These reaction conditions were originally developed for the cleavage of silyl protecting groups, see: González-Calderón D., Benitez-Puebla L. J., Gonzalez-Gonzalez C. A., Garcia-Eleno M. A., Fuentes-Benitez A., Cuevas-Yañez E., Corona-Becerril D., González-Romero C., Synth. Commun. 2014, 44, 1258–1265. [Google Scholar]
- 11. James M. J., Clubley R. E., Palate K. Y., Procter T. J., Wyton A. C., O'Brien P., Taylor R. J. K., Unsworth W. P., Org. Lett. 2015, 17, 4372. [DOI] [PubMed] [Google Scholar]
- 12.Ynones 1 a–g and 1 k–l were prepared as described in reference [5], while ynones 1 h–j and 1 m were synthesised for the first time in this work (see Supporting Information for preparative details).
- 13.For related carbazole syntheses, see: Wang J., Zhu H.-T., Qiu Y.-F., Niu Y., Chen S., Li Y.-X., Liu X.-Y., Liang Y.-M., Org. Lett. 2015, 17, 3186–3189, and references [8a–c]. [DOI] [PubMed] [Google Scholar]
- 14.Ynone 1 a (3.8 mmol) was converted into quinoline 7 a in 82 % yield upon conventional heating with AlCl3⋅6 H2O at reflux under an air atmosphere for 8 h (see the Supporting Information for full details).
- 15. CCDC 1453490 (7 f) and 1453491 ((S)-8 h) contain the supplementary crystallographic data for this paper. These data are provided free of charge by The Cambridge Crystallographic Data Centre.
- 16.For a related reaction involving in situ indolenine formation and intramolecular trapping, see: Modha S. G., Kumar A., Vachhani D. D., Jacobs J., Sharma S. K., Parmar V. S., van Meervelt L., van der Eycken E. V., Angew. Chem. Int. Ed. 2012, 51, 9572–9575; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 9710–9713. [Google Scholar]
- 17.
- 17a. Hamilton G. L., Kang E. J., Mba M., Toste F. D., Science 2007, 317, 496–499; [DOI] [PubMed] [Google Scholar]
- 17b. Wang Y., Zheng K., Hong R., J. Am. Chem. Soc. 2012, 134, 4096–4099; [DOI] [PubMed] [Google Scholar]
- 17c. Terada M., Li F., Toda Y., Angew. Chem. Int. Ed. 2014, 53, 235–239; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 239–243. [Google Scholar]
- 18.For details of potential natural product targets, see:
- 18a. Magnus P., Mugrage B., Deluca M. R., Cain G. A., J. Am. Chem. Soc. 1990, 112, 5220–5230; [Google Scholar]
- 18b. Kam T. S., Sim K. M., Lim T. M., Tetrahedron Lett. 2001, 42, 4721–4723; [Google Scholar]
- 18c. Mori M., Nakanishi M., Kajishima D., Sato Y., J. Am. Chem. Soc. 2003, 125, 9801–9807; [DOI] [PubMed] [Google Scholar]
- 18d. Crich D., Rumthao S., Tetrahedron 2004, 60, 1513–1516; [Google Scholar]
- 18e. Knölker H.-J., Fröhner W., Heinrich R., Synlett 2004, 2705–2708; [Google Scholar]
- 18f. Yang H., Feng J., Tang Y., Chem. Commun. 2013, 49, 6442–6444; [DOI] [PubMed] [Google Scholar]
- 18g. Unsworth W. P., Cuthbertson J. D., Taylor R. J. K., Org. Lett. 2013, 15, 3306–3309; [DOI] [PubMed] [Google Scholar]
- 18h. Ramkumar N., Nagarajan R., J. Org. Chem. 2013, 78, 2802–2807; [DOI] [PubMed] [Google Scholar]
- 18i. Hieda Y., Choshi T., Fujioka H., Hibino S., Eur. J. Org. Chem. 2013, 7391–7401. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary