

ABSTRACT
Adoptive T-cell therapy (ACT) is an emerging paradigm in which T cells are genetically modified to target cancer-associated antigens and eradicate tumors. However, challenges treating epithelial cancers with ACT reflect antigen targets that are not tumor-specific, permitting immune damage to normal tissues, and preclinical testing in artificial xenogeneic models, preventing prediction of toxicities in patients. In that context, mucosa-restricted antigens expressed by cancers exploit anatomical compartmentalization which shields mucosae from systemic antitumor immunity. This shielding may be amplified with ACT platforms employing antibody-based chimeric antigen receptors (CARs), which mediate MHC-independent recog-nition of antigens. GUCY2C is a cancer mucosa antigen expressed on the luminal surfaces of the intestinal mucosa in mice and humans, and universally overexpressed by colorectal tumors, suggesting its unique utility as an ACT target. T cells expressing CARs directed by a GUCY2C-specific antibody fragment recognized GUCY2C, quantified by expression of activation markers and cytokines. Further, GUCY2C CAR-T cells lysed GUCY2C-expressing, but not GUCY2C-deficient, mouse colorectal cancer cells. Moreover, GUCY2C CAR-T cells reduced tumor number and morbidity and improved survival in mice harboring GUCY2C-expressing colorectal cancer metastases. GUCY2C-directed T cell efficacy reflected CAR affinity and surface expression and was achieved without immune-mediated damage to normal tissues in syngeneic mice. These observations highlight the potential for therapeutic translation of GUCY2C-directed CAR-T cells to treat metastatic tumors, without collateral autoimmunity, in patients with metastatic colorectal cancer.
KEYWORDS: Adoptive immunotherapy, chimeric antigen receptors, colorectal cancer, gene therapy, guanylyl cyclase C
Abbreviations
- ACT
adoptive cell therapy
- CAR
chimeric antigen receptor
- GFP
green fluorescent protein
- GUCY2C
guanylyl cyclase C
- TBI
total body irradiation
Introduction
Colorectal cancer (CRC) is the fourth leading cause of cancer, and the second leading cause of cancer-related death in the United States and world.1 While surgical excision of primary tumors can be curative, particularly at the earliest stages of disease, about 50% of patients with colorectal cancer ultimately die of distant metastases.1 While chemo-, radio-, and targeted therapies extend survival to about 24 mo, less than 15% of patients with metastatic CRC survive beyond 5 y,1 highlighting the unmet need for new therapeutic paradigms for this disease.
Adoptive T-cell therapy (ACT) is an emerging platform2 to treat patients with advanced cancer employing autologous tumor-specific T cells that are expanded ex vivo and transferred back into patients. While initial approaches employed tumor-infiltrating lymphocytes (TILs) to treat melanoma,3 genetic modification of bulk peripheral blood T cells to express antigen-specific receptors theoretically extends this approach to all cancers, with notable success in treating leukemia and neuroblastoma.4-6 However, ACT has had limited utility against epithelial tumors, reflecting unresolved issues surrounding toxicities. Indeed, employing receptors directing genetically modified T cells to target antigens that are shared by tumors and normal tissues can produce severe autoimmune damage and patient death.7,8 Moreover, ACT products examined clinically have been tested in preclinical mouse models devoid of endogenous target antigen, incompletely characterizing the potential for toxicities in normal tissues.9,10 In that context, T cells engineered to express an affinity-enhanced TCR targeting the colorectal tumor antigen carcinoembryonic antigen (CEA), also broadly expressed by intestinal epithelial cells, produced severe colitis in patients.8 Similarly, T cells modified to express an antibody-based chimeric antigen receptor (CAR) targeting the tumor antigen ERBB2 (Her-2) produced lethal pneumonitis in the only patient receiving this therapy, reflecting Her-2 expression in lung.7 These considerations highlight the importance of identifying tumor-selective antigens, immune cell platforms that optimally discriminate tumor and normal tissues, and syngeneic preclinical models to define the biology, efficacy, and safety of new ACT paradigms.11-13
Guanylyl cyclase C (GUCY2C) is a membrane-bound cyclase whose cell-surface expression is confined to the apical surfaces of intestinal epithelial cells and exhibits limited expression in extra-intestinal tissues of humans and mice.14,15 Of significance, GUCY2C is a cancer mucosa antigen,16 universally overexpressed by primary and metastatic human CRCs and is ectopically expressed in esophageal and gastric cancers associated with intestinal dysplasia.14,15,17,18 Moreover, anatomical segregation of GUCY2C on the luminal surface of the intestinal epithelium19-22 limits access to systemically delivered GUCY2C-targeted molecules permitting diagnostic imaging23 and monoclonal antibody-based therapy24,25 of colorectal cancer metastasis without recognition of intestinal epithelium. Further, GUCY2C vaccines induce CD8+ T cell and antibody responses that eliminate metastatic colorectal tumors, without autoimmunity, in syngeneic mouse models26-28 and this platform is currently being tested in humans.29
Beyond vaccines, luminal compartmentalization of GUCY2C offers an intriguing solution to toxicities of current ACT paradigms against metastatic CRC. Moreover, a syngeneic mouse model, in which endogenous target antigen expression in normal tissue and tumors closely models humans, offers a unique opportunity to directly test CAR-T cell therapeutic efficacy and toxicity. The present study examined the ability of CAR-T cells directed to murine GUCY2C to treat established parenchymal CRC metastases without autoimmunity. This study establishes proof-of-principle for safe and effective GUCY2C CAR-T cell therapy, which can be translated to CRC patients.
Results
GUCY2C CAR-T cells
Monoclonal antibodies targeting the GUCY2C extracellular domain (GUCY2CECD) generated from hybridomas (MS7, MS20, and MS24) recognized purified GUYC2C (Fig. 1A), GUCY2C in the colon (Fig. 1B), and small intestine (Fig. S1) of Gucy2c+/+, but not Gucy2c−/−, mice; and GUCY2C-expressing, but not GUCY2C-deficient, CT26 murine colorectal cancer cells (Fig. 1C). Heavy and light chain variable region sequences from each of the GUCY2C-specific hybridomas were used to generate third-generation CARs (Fig. 1D), which were inserted into a retroviral construct used to infect T cells with ˜65% transduction efficiencies (Fig. S2). GUCY2C-binding avidity was quantified by incubating CAR-T cells with increasing concentrations of purified 6xHis-tagged GUCY2CECD, followed by detection with labeled α6xHis antibody and assessment by flow cytometry (Fig. 1D and Fig. S3). GUCY2C binding was detected with constructs derived from the MS7 and MS24, but not MS20, antibodies. CARs derived from MS24 exhibited ˜22-fold greater avidity (Kav 44.3 nM vs. 994.2 nM; p = 0.0567) and ˜5-fold greater surface expression (Bmax 741.5 vs. 144.2; p < 0.0001) compared to MS7-derived CARs (Fig. 1D), similar to the higher avidity of MS24 monoclonal antibody in comparison to MS7 (Fig. S4).
Figure 1.
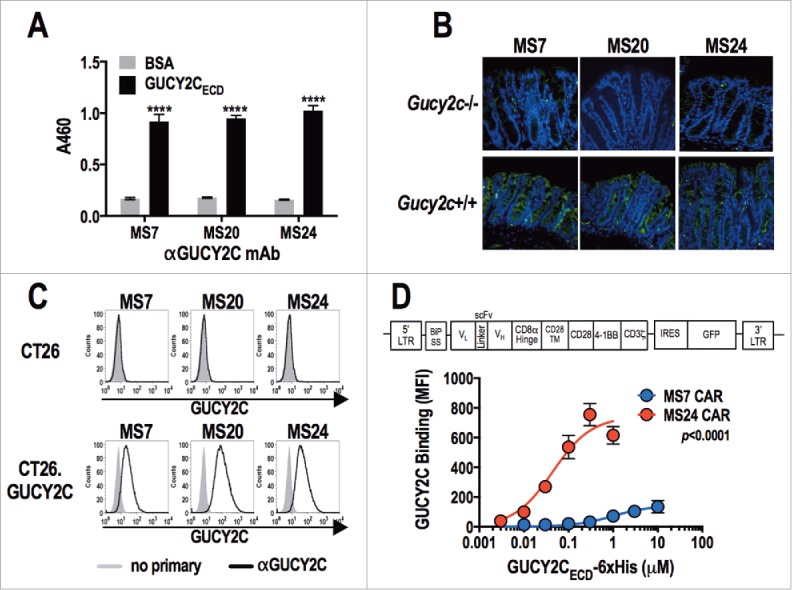
Characterization of GUCY2C-specific antibodies and CAR constructs. (A) Monoclonal antibodies generated against GUCY2C (MS7, MS20, and MS24) were assessed by ELISA for specific binding to GUCY2CECD or negative control bovine serum albumin (BSA) plated at 1 μg/mL, ****p < 0.0001 (Two-way ANOVA of GUCY2C vs. BSA – binding for each mAb). (B) Wild-type (Gucy2c+/+) or GUCY2C-deficient (Gucy2c−/−) mouse colon sections were stained with GUCY2C-specific monoclonal antibodies (green), demonstrating specificity of antibodies for GUCY2C in the intestine. DAPI (blue). Representative of three sections each. (C) Flow cytometry analysis was performed on GUCY2C-deficient (CT26) and GUCY2C-expressing (CT26.GUCY2C) CT26 mouse colorectal cancer cells stained with GUCY2C mAbs. Results are representative of two experiments. (D) A third generation CAR construct was synthesized containing the BiP signal sequence, scFv, the CD8α hinge region, the transmembrane and intracellular domain of CD28, the intracellular domain of 4-1BB (CD137) and the intracellular domain of CD3ζ. The CAR construct was inserted into the MSCV retroviral plasmid pMIG upstream of an IRES-GFP marker. (D) Murine CD8+ T cells transduced with a retrovirus containing a Control CAR or CARs derived from GUCY2C antibodies (MS7 and MS24) were labeled with varying concentrations of purified 6xHis-GUCY2CECD (0–10 μM) detected with α5xHis-Alexa-647 conjugate. Flow plots (Fig. S3) were gated on live CD8+ cells and the mean fluorescence intensity (MFI) indicated 6xHis-GUCY2CECD binding on live CD8+ transduced (GFP+) cells. MS7 and MS24 binding curves were compared by extra-sum-of-squares F test. Data represent mean ± standard deviation of three experiments.
GUCY2C CARs mediate antigen-specific activation of T cells
MS7 and MS24, but not MS20 or control, CAR-T cells upregulated CD25 and CD6930 when stimulated with immobilized GUCY2CECD, but not BSA (Fig. 2). All T cells produced comparable levels of CD25 and CD69 when stimulated with PMA and ionomycin, confirming that transduction with different CARs did not impact T cell activation. MS24 CAR mediated greater activation than MS7 CAR (Fig. 2), consistent with its ˜22-fold higher avidity (Fig. 1D). MS20 CAR was excluded from further consideration reflecting its inability to bind GUCY2CECD (Fig. S3) or mediate GUCY2C-dependent T cell activation (Fig. 2). MS7 and MS24, but not control, CAR-T cells produced effector cytokines IFNγ, TNFα, and MIP-1α when stimulated with immobilized GUCY2CECD, but not BSA (Fig. 3). Importantly, MS24 CAR-T cells exhibited greater polyfunctionality than MS7 CAR-T cells (Fig. 3B) characterized by a reduction in cells producing no cytokines (p < 0.0001, Two-way ANOVA) and an increase in cells producing two or three cytokines (p < 0.05 and p < 0.0001, respectively, Two-way ANOVA) following GUCY2C stimulation. Similarly, MS7 and MS24, but not control, CAR-T cells lysed GUCY2C-expressing, but not GUCY2C-deficient, CT26 mouse colon cancer cells (Fig. 4). As expected, MS24 CAR T cells lysed GUCY2C-expressing CT26 cells more rapidly than MS7 CAR-T cells (half-maximal lysis at 54 min vs. 200 min; p < 0.05; Figs. 4B and D) consistent with its higher avidity (Fig. 1D).
Figure 2.
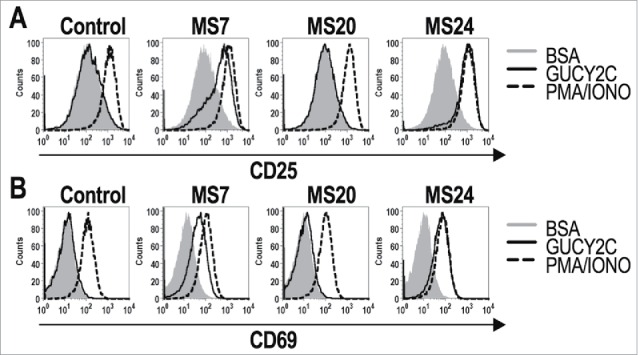
GUCY2C-specific CARs mediate antigen-dependent T cell activation. 1 × 106 CAR-expressing T cells were stimulated for 6 h with plate-coated antigen (BSA or GUCY2C) or PMA and Ionomycin (PMA/IONO). T cell activation markers CD25 (A) and CD69 (B) were quantified by flow cytometry. Histograms are gated on live CD8+GFP+ T cells. Results are representative of three experiments.
Figure 3.
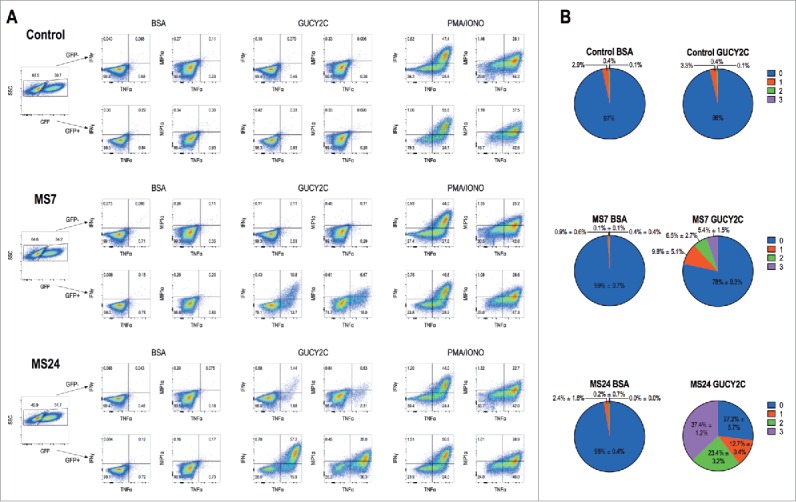
GUCY2C-specific CARs mediate cytokine production. 1 × 106 CAR-expressing T cells were stimulated for 6 h with plate-coated antigen (BSA or GUCY2C) or PMA and Ionomycin (PMA/IONO) in the presence of protein transport inhibitor. Cells were fixed, permeabilized, and stained for the intracellular cytokines IFNγ, TNFα, or MIP-1α and analyzed via flow cytometry. (A) Plots are gated on live GFP- (top) or GFP+ (bottom) CD8+ T cells. (B) Polyfunctional cytokine graphs depict the percentages of CAR-T cells producing 0, 1, 2, or 3 cytokines. Plots in (A) are representative of two experiments, and polyfunctional cytokine analysis (B) represent means ± standard deviation of two experiments.
Figure 4.
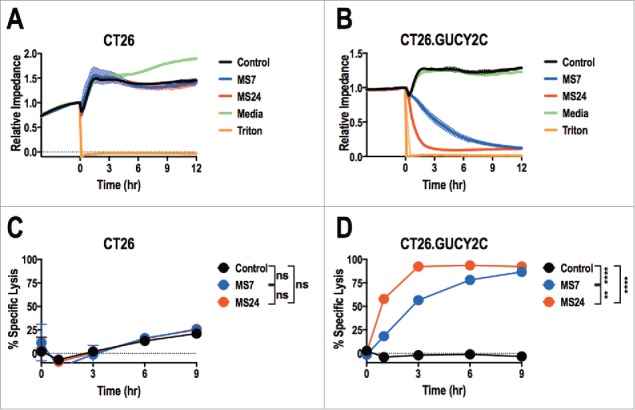
Real-time GUCY2C-specific CAR T cell-mediated cytotoxicity. CT26 (A and C) or CT26.GUCY2C (B and D) mouse colorectal cancer cells were seeded at 10,000 cells/well in an E-Plate and CAR-T cells, media, or Triton-X 100 (Triton) were added to the plate 24 h later (time = 0). The E-Plate was scanned every 15 min to quantify relative electrical impedance (normalized to time = 0). (A and B) Solid lines indicate the mean of duplicate wells and surrounding clouds indicate standard deviation. Results are representative of two experiments. (C and D) % specific lysis values for each CAR-T cell and target cell combination were calculated from the impedance data at the indicated time points. All statistical tests in (C) and (D) compared (1) Control to MS7, (2) Control to MS24, and (3) MS7 to MS24 (**p < 0.01, ****p < 0.0001, Two-way ANOVA). (C and D) show the means ± standard deviation of two experiments.
GUCY2C CAR-T cells oppose metastatic colorectal cancer
Mice received CT26.GUCY2C cells by tail vein to induce lung metastases,31 followed 3 d later by 5 Gy total body irradiation (TBI) and 1 × 107 T cells.32 A non-myeloablative dose of 5 Gy TBI was administered prior to T cell transfer to enhance the efficacy of adoptive T cell therapy by reducing sinks for the homeostatic cytokines IL-7 and IL-15.33,34 The number of tumors in lungs of mice treated with GUCY2C CAR-T cells was significantly reduced compared to mice treated with control CAR-T cells (Figs. 5A and B). Moreover, GUCY2C CAR-T cell-treated mice exhibited reduced morbidity, quantified by cachexia (Fig. 5C), and improved survival (Fig. 5D). As expected, morbidity (Fig. 5C) and survival (Fig. 5D) were significantly better with MS24, compared to MS7, CAR-T cells.
Figure 5.
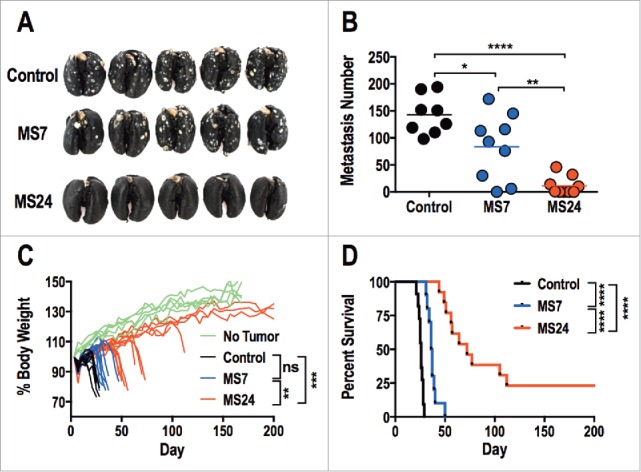
GUCY2C-specific CAR T cells oppose parenchymal colorectal cancer metastases. (A–D) BALB/c mice were injected i.v. with 5 × 105 CT26.GUCY2C cells to establish lung metastases. On day 3, mice received a dose of 5 Gy TBI followed by an i.v. injection of ˜1 × 107 CAR-T cells. Mice were sacrificed on day 24 after tumor inoculation, lungs were stained with India ink, and tumor nodules were counted (A and B), or mice were followed longitudinally for morbidity (C) and survival (D). (A) Representative images of lungs collected and stained on day 24. (B) Quantification of metastases in each mouse. (N = 8–9 mice/group; *p < 0.05, **p < 0.01, and ****p < 0.0001, One-way ANOVA). (C) Body weight curves of mice relative to initial body weight indicating progression of cancer cachexia, where each line represents an individual mouse. No Tumor represents curves of unmanipulated mice. (n = 10–13 mice/group; ns = not significant, ** p< 0.01, *** p< 0.001, One-way ANOVA of total area under the curve). (D) Survival analysis (n = 10–13 mice/group; ****p < 0.0001, Mantel-Cox log-rank test). All statistical tests in (B–D) compared (1) Control to MS7, (2) Control to MS24, and (3) MS7 to MS24.
GUCY2C CAR-T cells do not induce autoimmunity
MS24 CAR-T cells produce the greatest GUCY2C-dependent T cell activation (Fig. 2), cytokine production (Fig. 3), cytolysis (Fig. 4), and antitumor efficacy (Fig. 5) without autoimmunity (Fig. 6). Mice receiving MS24 CAR-T cells were healthy with no signs or symptoms of inflammatory bowel disease including failure to thrive, altered bowel habits, or rectal bleeding. MS24 CAR-T cells accumulated in GUCY2C-expressing CT26 lung metastases (Figs. 6A and B), mediating antitumor immunity (Fig. 5), but were absent from intestines (Figs. 6A and B), producing no T cell-mediated damage quantified by histopathology (Figs. 6C and D). Similarly, MS24 CAR-T cell-treated mice were free of immune-mediated damage in extra-intestinal tissues by histopathology (Fig. 6D) and exhibited normal organ and metabolic functions quantified by serum chemistries (Fig. S5).
Figure 6.
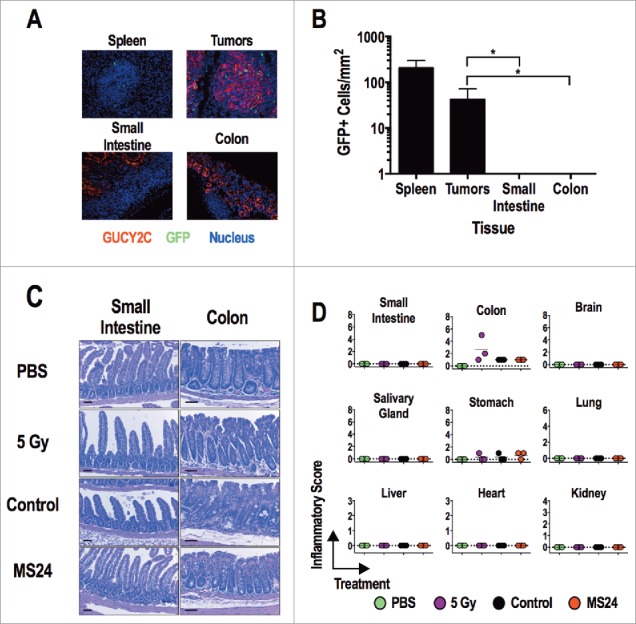
GUCY2C-specific CAR T cells do not induce tissue damage. (A and B) MS24 CAR-T cells were administered to BALB/c mice with CT26.GUCY2C lung metastases established 14 d earlier following 5 Gy TBI. (A) Lungs, spleens, and intestines were collected 2 d later, and tissue sections were stained with anti-GUCY2C (red) and anti-GFP (green) antibodies and counterstained with DAPI. (B) GFP+ MS24 CAR-T cells were quantified in remnant follicles in spleen, tumor metastases in lungs, and Peyer's patches in intestines by immunostaining of GFP. Data represent the mean of four mice. (****p < 0.001, One-way ANOVA compared to accumulation in lung metastases). (C and D) BALB/c mice were treated with PBS, 5 Gy TBI, 5 Gy TBI + 1 × 107 Control or MS24 CAR-T cells. On day 6 post-treatment, mice were sacrificed and tissues collected, fixed in formalin, and paraffin embedded. Slides were stained with H&E and scored for pathology. Scale bars indicate 100 μm. (C) Representative H&E-stained small intestine and colon sections. (D) Inflammatory scoring for all tissues collected. No significant differences between Control and MS24 CAR-T cell treatment groups were detected (One-way ANOVA).
Discussion
This study provides the first proof-of-principle for GUCY2C CAR-T cells to treat parenchymal colorectal cancer metastases without autoimmunity in a syngeneic mouse model. Immunologic competence of GUCY2C CARs was reflected by the ability of GUCY2C, but not control, CAR-T cells to engage GUCY2C and induce T cell activation (Fig. 2) and effector function (Figs. 3 and 4). Immunologic competence was associated with the ability of GUCY2C, but not control, CAR-T cells to induce GUCY2C-dependent cytokine production (Fig. 3) and in vitro colorectal cancer cell cytolysis (Fig. 4). Further, GUCY2C, but not control, CAR-T cells reduced disease burden, improved morbidity, and produced durable eradication of disease in a mouse model of GUCY2C-expressing colorectal cancer metastases in lung (Fig. 5). Moreover, therapeutic efficacy was achieved without immune-mediated tissue damage, in small and large intestine or other extra-intestinal tissues (Fig. 6 and Fig. S5). Importantly, the superior therapeutic efficacy and safety of GUCY2C CAR-T cells was achieved in a syngeneic mouse model in which GUCY2C was endogenously expressed in normal tissues in a pattern recapitulating that in humans, colorectal tumors expressed an identical antigen, and T cells were directed by CAR receptors to that endogenous antigen. Taken together, these observations suggest that GUCY2C-targeted CAR-T cells could eradicate tumors without autoimmunity in patients with metastatic colorectal cancer.
One key limitation to translating ACT to epithelial tumors generally, and colorectal cancer specifically, is the paucity of tumor-specific antigens to which immune cells can be targeted. In that context, targeting tumor-associated self-antigens risks development of “on-target, off-tumor” toxicities and therapy-limiting autoimmunity.35,36 CEA is an antigen that is normally expressed by intestinal and lung epithelial cells, and overexpressed by colorectal tumors. While T cells directed to CEA exhibit antitumor efficacy, their administration to patients produced treatment-limiting colitis.8 Similarly, Her-2 also is expressed by normal epithelial cells and overexpressed by colorectal tumors, and T cells targeting this antigen produced lethal immune-mediated damage to normal lung in a patient.7 In contrast, GUCY2C may be uniquely suited to direct immune cells to colorectal metastases without autoimmunity due to its sequestered expression on apical surfaces of intestinal epithelia.19-21 Thus, GUCY2C-targeted CAR T cells eradicated GUCY2C-expressing pulmonary metastases without immune damage to intestinal epithelia (Fig. 6). GUCY2C-targeted imaging agents,23 immunotoxins,25 and vaccines26-28 recognize GUCY2C-expressing metastatic colorectal tumors, but not normal intestinal epithelia. Indeed, universal overexpression of GUCY2C on the surface of metastatic tumors,17,18,37 but its absence on basolateral membranes of epithelial cells19-21 may shield normal intestine from systemic GUCY2C-targeted therapies. In that context, T cells directed to CEA, whose epitopes are presented by MHC in apical and basolateral membranes of intestinal cells, were characterized by antigen-dependent intestinal accumulation and colitis in mouse models38,39 and patients.8 In contrast, mice treated with GUCY2C CAR-T cells were free of intestinal accumulation and toxicity (Fig. 6), underscoring the importance of GUCY2C sequestration in apical membranes to prevent CAR-T cell recognition, accumulation, and toxicity. Beyond intestinal mucosa, GUCY2C expression recently was described in neurons of hypothalamus, an immunologically privileged compartment, where it regulates satiety and appetite.40 GUCY2C levels in hypothalamus are 10-fold lower than those in intestine.40 In that context, sufficient quantities of antigen on the surface of cellular targets are necessary to induce CAR T cell activation.41 GUCY2C-targeted CAR T cells or immunotoxins25 did not damage hypothalamic neurons, supporting the suggestion that they are sequestered from systemically targeted agents and/or express GUCY2C at levels below detection by our monoclonal antibodies. Taken together, the narrow pattern of expression in normal tissues, expression in compartments which are anatomically shielded or immunologically privileged, and universal overexpression by metastatic tumors suggest that GUCY2C is uniquely qualified as an antigenic target to which T cells can be directed to treat metastatic CRC without autoimmunity.
Beyond selectivity of expression in normal tissues and tumors, aligning ACT platforms with patterns of antigen expression within tissues can minimize treatment-limiting adverse events. For example, CEA is expressed in apical membranes of intestinal epithelial cells, and is presumably inaccessible to targeted CAR-T cells which recognize native antigen in an MHC-independent fashion.5,42 Indeed, CAR-T cells directed to human CEA did not produce colitis in a transgenic mouse model recapitulating human CEA expression.38 However, T cells directed by recombinant TCRs recognize peptide antigens in the context of MHC.43 In turn, MHC-peptide complexes can be presented on basolateral membranes of intestinal epithelial cells44 or cross-presented by antigen-presenting cells in tissues or lymphoid organs,45 abrogating the anatomical shielding of natively-expressed apical membrane proteins. Indeed, T cells directed by CEA-specific TCRs produce severe colitis in mice and humans.8,39 Thus, aligning GUCY2C as an apical target antigen, with CAR platforms to direct MHC-independent elimination of tumor cells, should maximize the benefit of luminal sequestration in normal tissues, optimizing therapeutic discrimination between tumors and intestine, as observed here.
Utilization of receptors targeting self-antigens to direct engineered T cells, and the associated incomplete discrimination of normal tissues and tumors, creates an imperative to define their safety and therapeutic efficacy in preclinical models that emulate the expression of target antigens in patients. However, the translation of ACT to patient-based trials has been based largely on the safety and efficacy of these paradigms in artificial human tumor xenograft systems. In some cases, the target antigen was not expressed in normal tissues, making it impossible to assess safety.9,10 In other cases, transgenic models attempt to emulate the expression of the human antigen in normal tissues in patients. For example, CEA CAR-T cells exhibited antitumor efficacy without immune-mediated damage to normal tissues in CEA-transgenic mice.38 Nevertheless, safety profiles achieved with artificial xenogeneic preclinical models must be viewed with some caution. Indeed, in other CEA transgenic mouse models, antitumor efficacy of CEA-specific T cells was associated with severe immune-mediated colitis.39 More significantly, a recent clinical trial of CEA-specific T cells induced severe autoimmune colitis, which represented dose-limiting toxicity and required discontinuation of therapy.8 Further, human Her-2 transgenic mice treated with large doses of CAR-T cells derived from the human Her-2 specific antibody 4D5,46 but not the FRP5 antibody,47 caused death within 4 d of treatment. This effect was not achieved with lower doses of 4D5 CAR-T cells; however, a colorectal cancer patient treated with 4D5 CAR-T cells also died following treatment, with potential evidence of antigen-specific toxicities in lung epithelium.7 These conflicting reports highlight the challenges of extrapolating safety profiles of ACT defined in transgenic mouse models, particularly if antigen expression levels do not mimic those in humans.
These considerations underscore the importance of establishing safety and efficacy profiles of ACT in syngeneic models in which engineered T cells are directed by receptors targeting antigens endogenously expressed in normal tissues.11 Indeed, CAR-T cells targeting murine CD19 eliminate B cell leukemia in mice with concomitant loss of normal B cells,12,48 recapitulating observations in clinical trials.4,5 Separately CAR-T cells targeting stromal FAP and VEGFR-2 also produce significant toxicities in mice.13,49 CAR-T cells targeting FAP cause lethal bone marrow toxicity in mice reflecting FAP expression on multipotent bone marrow stem cells.13 Further, VEGFR-2 CAR-T cells caused toxicity only in tumor-bearing mice, mediated by CAR-expressing CD4+, but not CD8+, T cells.49 In the present study, we demonstrated that CAR-T cells directed to murine GUCY2C eradicated GUCY2C-expressing colorectal tumors metastatic to lungs, without inducing autoimmunity in intestine or other normal tissues in mice endogenously expressing the target antigen. However, CAR-T cell toxicity typically reflects acute cytokine production and tissue damage,50 and the long-term toxicity of GUCY2C CAR-T cell therapy could not be evaluated here. In that context, GUCY2C CAR-T cell persistence was limited (Fig. S6), reflecting their production in the presence of IL-2. Recent analysis of the impact of cytokine milieu during CAR-T cell production and in vivo following their administration, suggest that alternative cytokine combinations may produce CAR-T cells with enhanced persistence and antitumor efficacy.51 Indeed, CAR-T cell production in the presence of IL-2 produced highly differentiated CAR-T cells with the least in vivo persistence and antitumor efficacy, compared to IL-7, IL-15, IL-18, IL-21, or no cytokines. Thus, as cell production methods continue to be refined, toxicity will be re-evaluated to determine if improving GUCY2C CAR-T cell persistence and antitumor efficacy concomitantly increases their associated toxicity targeting antigen-expressing tissues.
Taken together, these observations provide proof-of-principle in a uniquely relevant preclinical syngeneic model that GUCY2C-targeted CAR-T cells could be clinically effective, without inducing autoimmune tissue damage, in patients with metastatic colorectal cancer. This paradigm leverages the unique structural compartmentalization of GUCY2C endogenously expressed in normal tissues,19-22 and the universal overexpression of this antigen by metastatic colorectal tumors.17,18,37 It offers an immunotherapeutic strategy to treat bulky metastatic disease, which is complementary to GUCY2C vaccines as secondary prevention of metastatic disease in CRC patients at risk.26-28 The significance of these observations can best be appreciated by considering that metastatic CRC is nearly always fatal, without curative therapeutic options.1 Moreover, beyond CRC, GUCY2C-targeted ACT offers a unique therapeutic option to treat metastases in patients with gastric, esophageal, and pancreatic cancers which also are universally fatal and which ectopically express GUCY2C after transformation.17 Translation of these observations into patients will require the development of monoclonal antibodies to human GUCY2C, their incorporation into CAR constructs, and testing of their therapeutic efficacy in mice harboring human colorectal cancer metastases.
Materials and methods
Mouse GUCY2C monoclonal antibodies
The monoclonal antibodies (MS7, MS20, and MS24) recognize mouse GUCY2C,40 CT26.GUCY2C cells,28 and 6xHis-tagged GUCY2CECD protein.28 GUCY2C-specific antibodies were tested by ELISA as previously described.28 FACS: CT26 tumor cells were stained with 10 μg/mL GUCY2C-specific antibody follow by detection with anti-mouse conjugated to Alexa-488 and assessed for surface expression using the BD LSR II flow cytometer. Immunohistochemistry: intestinal tissues from wild-type and Gucy2c−/− BALB/c mice were fixed in formalin and paraffin embedded. Tissue sections were stained with 5 μg/mL GUCY2C-specific monoclonal antibody followed by detection with anti-mouse antibody conjugated to Alexa-488 and mounted in ProLong Gold Antifade Reagent with DAPI (Life Technologies). Images were captured using the EVOS FL Auto Cell imaging system.
CAR construction
A third generation codon-optimized CAR was synthesized containing the BiP (GRP-78) signal peptide, and scFv from the murine 4D5 monoclonal antibody specific for human ERBB2,52 CD8α hinge region, CD28 transmembrane and intracellular domains, and 4-1BB (CD137) and CD3ζ intracellular domains in the pMA entry plasmid (GeneArt). CARs derived from the 4D5 antibody served as the negative control CAR in all experiments performed. VL and VH variable regions were cloned from MS7, MS20, and MS24 hybridomas by RT-PCR using degenerate primers and linked with a glycine-serine linker (G4S)4 by overlap extension PCR. Resulting GUCY2C-specific scFv constructs were subcloned into the synthesized CAR construct, replacing 4D5.
Retrovirus production and T cell transduction
CARs were subcloned into the pMSCV-IRES-GFP (pMIG) retroviral vector using XhoI and EcoRI restriction sites. The Phoenix-Eco retroviral packaging cell line (Gary Nolan, Stanford University) was transfected with CAR-pMIG vectors and the pCL-Eco retroviral packaging vector (Imgenex) using the Calcium Phosphate ProfectionR Mammalian Transfection System (Promega). Retrovirus-containing supernatants were collected 48 h later, filtered through 0.45 μM filters, and aliquots were frozen at −80°C. Murine CD8+ T cells were purified from BALB/c splenocytes using the CD8α+ T cell Isolation Kit II (Miltenyi Biotec) and subsequently stimulated with αCD3/αCD28-coated beads (T Cell Activation/Expansion Kit, Miltenyi Biotec) at a 1:1 bead:cell ratio at 1 × 106 cells/mL in cRPMI (RMPI + 10% FBS, 10 μM HEPES, 0.05 μM 2-mercaptoethanol) with 100 U/mL recombinant human IL-2 (NCI Repository). The day following stimulation, ½ of the culture media was carefully replaced with an equal volume of thawed retroviral supernatant in the presence of polybrene (Millipore). Spinoculation was performed at room temperature for 90 min at 2,500 rpm followed by incubation at 37°C for 2.5 h at which point cells were pelleted and resuspended in fresh media containing IL-2. T cells were expanded for 7–10 d by daily dilution to 1 × 106 cells/mL with fresh cRPMI and IL-2 at which point they were used for functional assays. All cell counts employed a Muse Cell Analyzer and Count and Viability Assay (Millipore). Serum lots were not pretested for performance.
CAR surface detection
CAR-transduced T cells were stained with the Live/Dead Fixable Aqua Dead Cell Stain kit (Invitrogen), labeled with varying concentrations of 6xHis-tagged GUCY2CECD for 1 h, stained with the α5xHis Alexa-647 conjugate (Qiagen) and αCD8b-PE (clone H35.17.2, BD Biosciences) for 1 h, fixed with 2% PFA and analyzed using the BD LSR II flow cytometer and FlowJo software (Tree Star). GUCY2C binding was quantified by determining the mean fluorescence intensity of Alexa-647 on live CD8+ GFP+ cells. Non-linear regression analysis (GraphPad Prism v6) was used to determine the Kav and Bmax of GUCY2C-CAR binding.
Surface activation marker and intracellular cytokine staining
CAR-transduced T cells were stimulated for 6 h with antigen coated on tissue culture plates at 1 μg/mL in PBS overnight at 4°C, or with Cell Stimulation Cocktail (PMA/Ionomycin, eBioscience). Incubation included the Protein Transport Inhibitor Cocktail (eBioscience) when assessing intracellular cytokines. Cells were stained with Live/Dead fixable Aqua Dead Cell stain kit (Invitrogen) and subsequently stained for surface markers using the following antibodies: αCD8α-PerCP-Cy5.5 (clone 53.6-7) and αCD69-PE (clone H1.2F3) from BD Biosciences and αCD25-PE (clone PC61.5, eBioscience). Intracellular cytokine staining was performed using the BD Cytofix/Cytoperm Kit (BD Biosciences) and staining with the following antibodies: αGFP-Alexa-488 (Invitrogen), αIFNγ-APC-Cy7 (XMG1.2) and αTNFα-PE-Cy7 (MP6-XT22) from BD Biosciences, and αMIP1α-PE (clone 39624, R&D Systems). Cells were fixed in 2% PFA and analyzed on a BD LSR II flow cytometer. Analyses were performed using FlowJo software (Tree Star).
Real time cell-mediated cytotoxicity assay
The xCELLigence system (Acea Biosciences Inc.) was utilized for assessment of T cell-mediated cytotoxicity.53 Briefly 1 × 104 CT26 or CT26.GUCY2C targets were plated in 150 μL of DMEM 10% FBS in each well of an E-Plate 16 and grown overnight, quantifying electrical impedance every 15 min, using the RTCA DP Analyzer system (Acea Biosciences Inc.). Approximately 24 h later, 50 μL of CAR-T cells was added at an effector-to-target (E:T) ratio of 5:1 or 50 μL of media or 10% Triton-X 100 were added as negative and positive controls, respectively. Cell-mediated killing was quantified over the next 16 h reading electrical impedance every 15 min. Percent-specific lysis values were calculated using GraphPad Prism Software v6 for each replicate at each time point, using impedance values following the addition of media and Triton for normalization (0% and 100% specific lysis, respectively).
Metastatic tumor model
BALB/c mice were obtained from the NCI Animal Production Program (Frederick, MD). Animal protocols were approved by the Thomas Jefferson University Institutional Animal Care and Use Committee. Male BALB/c mice were challenged with 5 × 105 CT26.GUCY2C cells by tail vein injection to establish lung metastases. On day 3 following tumor injections, mice received a non-myeloablative dose of 5 Gy total body irradiation in a PanTak, 310 kVe x-ray machine. Mice received ˜1 × 107 fresh CAR-T cells in 100 μL PBS following irradiation by tail vein injection. Mice were followed for cachexia (twice weekly body weights) and survival, or sacrificed at day 23 after tumor cell injection and lungs were stained with India ink and fixed in Fekete's solution for tumor enumeration.28
Toxicity
For T cell accumulation, 1 × 107 MS24 CAR-T cells were administered to BALB/c mice with CT26.GUCY2C lung metastases established 14 d earlier following 5 Gy TBI. Lungs, spleens, and intestines were collected 2 d later, and tissue sections were stained with anti-GUCY2C (MS20) and anti-GFP antibodies and counterstained with DAPI. GFP+ MS24 CAR-T cells were quantified in remnant follicles in spleen, tumor metastases in lungs, and Peyer's patches in intestines and normalized to area (mm2). A cell was considered to be GFP+ if fluorescence intensity was above the background level (set using corresponding GFP-negative control tissues) associated with a DAPI-stained nucleus in the same plane. For histopathology, tissues and serum were collected from mice 6 d after treatment with PBS, 5 Gy TBI, or 5 Gy TBI and CAR-T cells. Tissues were fixed in formalin and embedded in paraffin. Sections were stained with hematoxylin and eosin and scored for toxicity by a blinded pathologist (P.L.). Scoring criteria are listed in Table S1. Serum chemistries were commercially determined (Charles River Laboratories).
Statistical analyses
Statistical analyses were conducted using GraphPad Prism Software v6. All results are representative of at least three experiments unless otherwise indicated.
Supplementary Material
Disclosure of potential conflicts of interest
SAW was the Chair of the Data Safety Monitoring Board for the CHART-1 Trial™ sponsored by Cardio3 Biosciences, and the Chair (uncompensated) of the Scientific Advisory Board to Targeted Diagnostics and Therapeutics, Inc. which provided research funding that, in part, supported this work and has a license to commercialize inventions related to this work.
Funding
Funding was provided by NIH (R01 CA75123, R01 CA95026, RC1 CA146033, P30 CA56036, R01 CA170533 to SAW; F31 CA171672 to MM); Targeted Diagnostic and Therapeutics Inc. (to SAW); PhRMA Foundation (to AES); and Margaret Q. Landenberger Research Foundation (to AES). SAW is the Samuel M.V. Hamilton Professor of Thomas Jefferson University. This project was funded, in part, by grants from the Pennsylvania Department of Health (SAP #4100059197, SAP #4100051723). The Department specifically disclaims responsibility for any analyses, interpretations or conclusions. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Howlader N, Noone AM, Krapcho M, Garshell J, Miller D, Altekruse SF, Kosary CL, Yu M, Ruhl J, Tatalovich Z,Mariotto A, Lewis DR, Chen HS, Feuer EJ, Cronin KA (eds). SEER Cancer Statistics Review, 1975-2012, National Cancer Institute. Bethesda, MD, http://seer.cancer.gov/csr/1975_2012/, based on November 2014 SEER data submission, posted to the SEER web site, April 2015. [Google Scholar]
- 2.Couzin-Frankel J. Breakthrough of the year 2013. Cancer immunotherapy. Science 2013; 342:1432-3; PMID:24357284; http://dx.doi.org/ 10.1126/science.342.6165.1432 [DOI] [PubMed] [Google Scholar]
- 3.Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, Topalian SL, Sherry R, Restifo NP, Hubicki AM et al.. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science 2002; 298:850-4; PMID:12242449; http://dx.doi.org/ 10.1126/science.1076514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, Teachey DT, Chew A, Hauck B, Wright JF et al.. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med 2013; 368:1509-18; PMID:23527958; http://dx.doi.org/ 10.1056/NEJMoa12-15134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med 2011; 365:725-33; PMID:21830940; http://dx.doi.org/ 10.1056/NEJMoa1103849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, Rossig C, Russell HV, Diouf O, Liu E et al.. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 2011; 118:6050-6; PMID:21984804; http://dx.doi.org/ 10.1182/blood-2011-05-354449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther 2010; 18:843-51; PMID:20179677; http://dx.doi.org/ 10.1038/mt.2010.24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan DA, Feldman SA, Davis JL, Morgan RA, Merino MJ, Sherry RM et al.. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther 2011; 19:620-6; PMID:21157437; http://dx.doi.org/ 10.1038/mt.2010.272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao Y, Wang QJ, Yang S, Kochenderfer JN, Zheng Z, Zhong X, Sadelain M, Eshhar Z, Rosenberg SA, Morgan RA. A herceptin-based chimeric antigen receptor with modified signaling domains leads to enhanced survival of transduced T lymphocytes and antitumor activity. J Immunol 2009; 183:5563-74; PMID:19843940; http://dx.doi.org/ 10.4049/jimmunol.0900447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parkhurst MR, Joo J, Riley JP, Yu Z, Li Y, Robbins PF, Rosenberg SA. Characterization of genetically modified T-cell receptors that recognize the CEA:691-699 peptide in the context of HLA-A2.1 on human colorectal cancer cells. Clin Cancer Res 2009; 15:169-80; PMID:19118044; http://dx.doi.org/ 10.1158/1078-0432.CCR-08-1638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sampson JH, Choi BD, Sanchez-Perez L, Suryadevara CM, Snyder DJ, Flores CT, Schmittling RJ, Nair SK, Reap EA, Norberg PK et al.. EGFRvIII mCAR-modified T-Cell therapy cures mice with established intracerebral glioma and generates host immunity against Tumor-Antigen loss. Clin Cancer Res 2014; 20:972-84; PMID:24352643; http://dx.doi.org/ 10.1158/1078-0432.CCR-13-0709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kochenderfer JN, Yu Z, Frasheri D, Restifo NP, Rosenberg SA. Adoptive transfer of syngeneic T cells transduced with a chimeric antigen receptor that recognizes murine CD19 can eradicate lymphoma and normal B cells. Blood 2010; 116:3875-86; PMID:20631379; http://dx.doi.org/ 10.1182/blood-2010-01-265041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tran E, Chinnasamy D, Yu Z, Morgan RA, Lee CC, Restifo NP, Rosenberg SA. Immune targeting of fibroblast activation protein triggers recognition of multipotent bone marrow stromal cells and cachexia. J Exp Med 2013; 210:1125-35; PMID:23712432; http://dx.doi.org/ 10.1084/jem.20130110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frick GS, Pitari GM, Weinberg DS, Hyslop T, Schulz S, Waldman SA. Guanylyl cyclase C: a molecular marker for staging and postoperative surveillance of patients with colorectal cancer. Expert Rev Mol Diagn 2005; 5:701-13; PMID:16149873; http://dx.doi.org/ 10.1586/14737159.5.5.701 [DOI] [PubMed] [Google Scholar]
- 15.Carrithers SL, Barber MT, Biswas S, Parkinson SJ, Park PK, Goldstein SD, Waldman SA. Guanylyl cyclase C is a selective marker for metastatic colorectal tumors in human extraintestinal tissues. Proc Natl Acad Sci U S A 1996; 93:14827-32; PMID:8962140; http://dx.doi.org/ 10.1073/pnas.93.25.14827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Snook AE, Eisenlohr LC, Rothstein JL, Waldman SA. Cancer mucosa antigens as a novel immunotherapeutic class of tumor-associated antigen. Clin Pharmacol Ther 2007; 82:734-9; PMID:17898707; http://dx.doi.org/ 10.1038/sj.clpt.6100369 [DOI] [PubMed] [Google Scholar]
- 17.Birbe R, Palazzo JP, Walters R, Weinberg D, Schulz S, Waldman SA. Guanylyl cyclase C is a marker of intestinal metaplasia, dysplasia, and adenocarcinoma of the gastrointestinal tract. Hum Pathol 2005; 36:170-9; PMID:15754294; http://dx.doi.org/ 10.1016/j.humpath.2004.12.002 [DOI] [PubMed] [Google Scholar]
- 18.Schulz S, Hyslop T, Haaf J, Bonaccorso C, Nielsen K, Witek ME, Birbe R, Palazzo J, Weinberg D, Waldman SA. A validated quantitative assay to detect occult micrometastases by reverse transcriptase-polymerase chain reaction of guanylyl cyclase C in patients with colorectal cancer. Clin Cancer Res 2006; 12:4545-52; PMID:16899600; http://dx.doi.org/ 10.1158/1078-0432.CCR-06-0865 [DOI] [PubMed] [Google Scholar]
- 19.Charney AN, Egnor RW, Alexander-Chacko JT, Zaharia V, Mann EA, Giannella RA. Effect of E. coli heat-stable enterotoxin on colonic transport in guanylyl cyclase C receptor-deficient mice. Am J Physiol Gastrointest Liver Physiol 2001; 280:G216-21; PMID:11208543 [DOI] [PubMed] [Google Scholar]
- 20.Kuhn M, Adermann K, Jahne J, Forssmann WG, Rechkemmer G. Segmental differences in the effects of guanylin and Escherichia coli heat-stable enterotoxin on Cl− secretion in human gut. J Physiol 1994; 479(Pt 3):433-40; PMID:7837099; http://dx.doi.org/ 10.1113/jphysiol.1994.sp02-0307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guarino A, Cohen MB, Overmann G, Thompson MR, Giannella RA. Binding of E. coli heat-stable enterotoxin to rat intestinal brush borders and to basolateral membranes. Dig Dis Sci 1987; 32:1017-26; PMID:3304888; http://dx.doi.org/ 10.1007/BF01297193 [DOI] [PubMed] [Google Scholar]
- 22.Hodson CA, Ambrogi IG, Scott RO, Mohler PJ, Milgram SL. Polarized apical sorting of guanylyl cyclase C is specified by a cytosolic signal. Traffic 2006; 7:456-64; PMID:16536743; http://dx.doi.org/ 10.1111/j.1600-0854.2006.00398.x [DOI] [PubMed] [Google Scholar]
- 23.Wolfe HR, Mendizabal M, Lleong E, Cuthbertson A, Desai V, Pullan S, Fujii DK, Morrison M, Pither R, Waldman SA. In vivo imaging of human colon cancer xenografts in immunodeficient mice using a guanylyl cyclase C-specific ligand. J Nucl Med 2002; 43:392-9; PMID:11884500 [PubMed] [Google Scholar]
- 24.Almhanna K, Kalebic T, Cruz C, Faris JE, Ryan DP, Jung JA, Wyant T, Fasanmade A, Messersmith WA, Rodon J. Phase I study of the investigational anti-guanylyl cyclase antibody-drug conjugate TAK-264 (MLN0264) in adult patients with advanced gastrointestinal malignancies. Clin Cancer Res 2016; PMID:27178743; http://dx.doi.org/ 10.1158/1078-0432.ccr-15-2474 [DOI] [PubMed] [Google Scholar]
- 25.Marszalowicz GP, Snook AE, Magee MS, Merlino D, Bertman-Booty LD, Waldman SA. GUCY2C lysosomotropic endocytosis delivers immunotoxin therapy to metastatic colorectal cancer. OncoTarget 2014; 5:9460-71; PMID:25294806; http://dx.doi.org/24771148 10.18632/oncotarget.2455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Snook AE, Magee MS, Schulz S, Waldman SA. Selective antigen-specific CD4(+) T-cell, but not CD8(+) T- or B-cell, tolerance corrupts cancer immunotherapy. Eur J Immunol 2014; 44:1956-66; PMID:24771148; http://dx.doi.org/ 10.1002/eji.201444539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Snook AE, Li P, Stafford BJ, Faul EJ, Huang L, Birbe RC, Bombonati A, Schulz S, Schnell MJ, Eisenlohr LC et al.. Lineage-specific T-cell responses to cancer mucosa antigen oppose systemic metastases without mucosal inflammatory disease. Cancer Res 2009; 69:3537-44; PMID:19351847; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-3386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Snook AE, Stafford BJ, Li P, Tan G, Huang L, Birbe R, Schulz S, Schnell MJ, Thakur M, Rothstein JL et al.. Guanylyl cyclase C-induced immunotherapeutic responses opposing tumor metastases without autoimmunity. J Natl Cancer Inst 2008; 100:950-61; PMID:18577748; http://dx.doi.org/ 10.1093/jnci/djn178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robinson E, Bartal A, Mekori T. Results of postoperative treatment of colorectal cancer by radiotherapy, chemotherapy and immunotherapy. Recent Results Cancer Res 1980; 75:80-7; PMID:7232842; http://dx.doi.org/ 10.1007/978-3-642-81491-4_13 [DOI] [PubMed] [Google Scholar]
- 30.Caruso A, Licenziati S, Corulli M, Canaris AD, De Francesco MA, Fiorentini S, Peroni L, Fallacara F, Dima F, Balsari A et al.. Flow cytometric analysis of activation markers on stimulated T cells and their correlation with cell proliferation. Cytometry 1997; 27:71-6; PMID:9000587; http://dx.doi.org/ 10.1002/(SICI)1097-0320(19970101)27:1%3c71::AID-CYT-O9%3e3.0.CO;2-O [DOI] [PubMed] [Google Scholar]
- 31.Mule JJ, Shu S, Schwarz SL, Rosenberg SA. Adoptive immunotherapy of established pulmonary metastases with LAK cells and recombinant interleukin-2. Science 1984; 225:1487-9; PMID:6332379; http://dx.doi.org/ 10.1126/science.6332379 [DOI] [PubMed] [Google Scholar]
- 32.Chamberlain RS, Carroll MW, Bronte V, Hwu P, Warren S, Yang JC, Nishimura M, Moss B, Rosenberg SA, Restifo NP. Costimulation enhances the active immunotherapy effect of recombinant anticancer vaccines. Cancer Res 1996; 56:2832-6; PMID:8665522 [PMC free article] [PubMed] [Google Scholar]
- 33.Gattinoni L, Finkelstein SE, Klebanoff CA, Antony PA, Palmer DC, Spiess PJ, Hwang LN, Yu Z, Wrzesinski C, Heimann DM et al.. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J Exp Med 2005; 202:907-12; PMID:16203864; http://dx.doi.org/ 10.1084/jem.20050732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, Citrin DE, Restifo NP, Robbins PF, Wunderlich JR et al.. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res 2011; 17:4550-7; PMID:21498393; http://dx.doi.org/ 10.1158/1078-0432.CCR-11-0116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hinrichs CS, Restifo NP. Reassessing target antigens for adoptive T-cell therapy. Nat Biotechnol 2013; 31:999-1008; PMID:24142051; http://dx.doi.org/ 10.1038/nbt.2725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stauss HJ, Morris EC. Immunotherapy with gene-modified T cells: limiting side effects provides new challenges. Gene Ther 2013; 20:1029-32; PMID:23804078; http://dx.doi.org/ 10.1038/gt.2013.34 [DOI] [PubMed] [Google Scholar]
- 37.Witek ME, Nielsen K, Walters R, Hyslop T, Palazzo J, Schulz S, Waldman SA. The putative tumor suppressor Cdx2 is overexpressed by human colorectal adenocarcinomas. Clin Cancer Res 2005; 11:8549-56; PMID:16361536; http://dx.doi.org/ 10.1158/1078-0432.CCR-05-1624 [DOI] [PubMed] [Google Scholar]
- 38.Chmielewski M, Hahn O, Rappl G, Nowak M, Schmidt-Wolf IH, Hombach AA, Abken H. T cells that target carcinoembryonic antigen eradicate orthotopic pancreatic carcinomas without inducing autoimmune colitis in mice. Gastroenterology 2012; 143:1095-107 e2; PMID:22750462; http://dx.doi.org/ 10.1053/j.gastro.2012.06.037 [DOI] [PubMed] [Google Scholar]
- 39.Bos R, van Duikeren S, Morreau H, Franken K, Schumacher TN, Haanen JB, van der Burg SH, Melief CJ, Offringa R. Balancing between antitumor efficacy and autoimmune pathology in T-cell-mediated targeting of carcinoembryonic antigen. Cancer Res 2008; 68:8446-55; PMID:18922918; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-1864 [DOI] [PubMed] [Google Scholar]
- 40.Valentino MA, Lin JE, Snook AE, Li P, Kim GW, Marszalowicz G, Magee MS, Hyslop T, Schulz S, Waldman SA. A uroguanylin-GUCY2C endocrine axis regulates feeding in mice. J Clin Invest 2011; 121:3578-88; PMID:21865642; http://dx.doi.org/ 10.1172/JCI57925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chmielewski M, Hombach A, Heuser C, Adams GP, Abken H. T cell activation by antibody-like immunoreceptors: increase in affinity of the single-chain fragment domain above threshold does not increase T cell activation against antigen-positive target cells but decreases selectivity. J Immunol 2004; 173:7647-53; PMID:15585893; http://dx.doi.org/ 10.4049/jimmunol.173.12.7647 [DOI] [PubMed] [Google Scholar]
- 42.Dai H, Wang Y, Lu X, Han W. Chimeric antigen receptors modified T-Cells for cancer therapy. J Natl Cancer Inst 2016; 108; PMID:26819347; https://dx.doi.org/ 10.1093/jnci/djv439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Babbitt BP, Allen PM, Matsueda G, Haber E, Unanue ER. Binding of immunogenic peptides to Ia histocompatibility molecules. Nature 1985; 317:359-61; PMID:3876513; http://dx.doi.org/ 10.1038/317359a0 [DOI] [PubMed] [Google Scholar]
- 44.Bar F, Sina C, Hundorfean G, Pagel R, Lehnert H, Fellermann K, Büning J. Inflammatory bowel diseases influence major histocompatibility complex class I (MHC I) and II compartments in intestinal epithelial cells. Clin Exp Immunol 2013; 172:280-9; PMID:23574324; http://dx.doi.org/ 10.1111/cei.12047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yu P, Spiotto MT, Lee Y, Schreiber H, Fu YX. Complementary role of CD4+ T cells and secondary lymphoid tissues for cross-presentation of tumor antigen to CD8+ T cells. J Exp Med 2003; 197:985-95; PMID:12695490; http://dx.doi.org/ 10.1084/jem.20021804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Globerson-Levin A, Waks T, Eshhar Z. Elimination of progressive mammary cancer by repeated administrations of chimeric antigen receptor-modified T cells. Mol Ther 2014; 22:1029-38; PMID:24572294; http://dx.doi.org/ 10.1038/mt.2014.28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang LX, Westwood JA, Moeller M, Duong CP, Wei WZ, Malaterre J, Trapani JA, Neeson P, Smyth MJ, Kershaw MH et al.. Tumor ablation by gene-modified T cells in the absence of autoimmunity. Cancer Res 2010; 70:9591-8; PMID:21098715; http://dx.doi.org/ 10.1158/0008-5472.CAN-10-2884 [DOI] [PubMed] [Google Scholar]
- 48.Cheadle EJ, Hawkins RE, Batha H, O'Neill AL, Dovedi SJ, Gilham DE. Natural expression of the CD19 antigen impacts the long-term engraftment but not antitumor activity of CD19-specific engineered T cells. J Immunol 2010; 184:1885-96; PMID:20089697; http://dx.doi.org/ 10.4049/jimmunol.0901440 [DOI] [PubMed] [Google Scholar]
- 49.Chinnasamy D, Yu Z, Theoret MR, Zhao Y, Shrimali RK, Morgan RA, Feldman SA, Restifo NP, Rosenberg SA. Gene therapy using genetically modified lymphocytes targeting VEGFR-2 inhibits the growth of vascularized syngenic tumors in mice. J Clin Invest 2010; 120:3953-68; PMID:20978347; http://dx.doi.org/ 10.1172/JCI43490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stauss HJ, Morris EC. Immunotherapy with gene-modified T cells: limiting side effects provides new challenges. Gene Ther 2013; 20(11):1029-32; PMID:23804078; https://dx.doi.org/ 10.1038/gt.2013.34 [DOI] [PubMed] [Google Scholar]
- 51.Xu XJ, Song DG, Poussin M, Ye Q, Sharma P, Rodriguez-Garcia A, Tang YM, Powell DJ. Multiparameter comparative analysis reveals differential impacts of various cytokines on CART cell phenotype and function ex vivo and in vivo. Oncotarget 2016; https://dx.doi.org/ 10.18632/oncotarget.10510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carter P, Presta L, Gorman CM, Ridgway JB, Henner D, Wong WL, Rowland AM, Kotts C, Carver ME, Shepard HM. Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc Natl Acad Sci U S A 1992; 89:4285-9; PMID:1350088; http://dx.doi.org/ 10.1073/pnas.89.10.4285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Peper JK, Schuster H, Loffler MW, Schmid-Horch B, Rammensee HG, Stevanovic S. An impedance-based cytotoxicity assay for real-time and label-free assessment of T-cell-mediated killing of adherent cells. J Immunol Methods 2014; 405:192-8; PMID:24486140; http://dx.doi.org/ 10.1016/j.jim.2014.01.012 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.