

ABSTRACT
We report here the successful vectorization of a hamster monoclonal IgG (namely J43) recognizing the murine Programmed cell death-1 (mPD-1) in Western Reserve (WR) oncolytic vaccinia virus. Three forms of mPD-1 binders have been inserted into the virus: whole antibody (mAb), Fragment antigen-binding (Fab) or single-chain variable fragment (scFv). MAb, Fab and scFv were produced and assembled with the expected patterns in supernatants of cells infected by the recombinant viruses. The three purified mPD-1 binders were able to block the binding of mPD-1 ligand to mPD-1 in vitro. Moreover, mAb was detected in tumor and in serum of C57BL/6 mice when the recombinant WR-mAb was injected intratumorally (IT) in B16F10 and MCA 205 tumors. The concentration of circulating mAb detected after IT injection was up to 1,900-fold higher than the level obtained after a subcutaneous (SC) injection (i.e., without tumor) confirming the virus tropism for tumoral cells and/or microenvironment. Moreover, the overall tumoral accumulation of the mAb was higher and lasted longer after IT injection of WR-mAb1, than after IT administration of 10 µg of J43. The IT injection of viruses induced a massive infiltration of immune cells including activated lymphocytes (CD8+ and CD4+). Interestingly, in the MCA 205 tumor model, WR-mAb1 and WR-scFv induced a therapeutic control of tumor growth similar to unarmed WR combined to systemically administered J43 and superior to that obtained with an unarmed WR. These results pave the way for next generation of oncolytic vaccinia armed with immunomodulatory therapeutic proteins such as mAbs.
KEYWORDS: Monoclonal antibody, oncolytic virotherapy, PD-1, vaccinia virus, vectorization
Abbreviations
- ADCC
antibody-dependent cellular cytotoxicity
- APC
antigen presenting cells
- CDC
complement directed cytotoxicity
- CEF
chicken embryo fibroblast
- CTLA-4
cytotoxic T-lymphocyte-associated protein 4
- DC
dendritic cells
- ECL
enhanced chemiluminescence
- ELISA
Enzyme-linked immunosorbent assay
- Fab
Fragment antigen-binding
- GM-CSF
granulocyte/macrophage-colony stimulating factor
- HC
heavy chain
- HRP
horseradish peroxidase
- ICI
immune checkpoint inhibitors
- IgG
immunoglobulin G
- IT
intratumorally
- LC
light chain
- mAb
monoclonal antibody
- MOI
multiplicity of infection
- MVA
modified vaccinia Ankara virus
- PD-1
programmed cell death- 1
- PD-L1
programmed death-ligand-1
- PFU
plaque forming unit
- Q-PCR
quantitative polymerase chain reaction
- RR
ribonucleotide reductase
- SC
subcutaneously
- scFv
single-chain variable Fragment
- SEC
size exclusion chromatography
- TAA
tumor-associated antigen
- TK
thymidine kinase
- TME
tumor microenvironment
- VEGF
vascular endothelial growth factor
- VGF
virus growth factor
- VH
variable domain of heavy chain
- VL
variable domain of light chain
- WB
Western blot
- WR
Western Reserve (strain of Vaccinia virus)
Introduction
Oncolytic viruses belong to different groups of viruses that share the properties to preferentially target and destroy tumoral cells. Some of those oncolytic viruses are currently evaluated for their safety and efficacy to treat several cancers in different clinical trials.1 The leading product: Imlygic™ (a modified herpes simplex virus 1 expressing human granulocyte-macrophage colony-stimulating factor, GM-CSF) has been recently approved by the FDA for treatment of unresectable cutaneous, subcutaneous and nodal lesions in patients with melanoma recurrent after initial surgery.2 Vaccinia viruses also have provided several promising oncolytic candidates such as JX-594 (SillaJen/Transgene), GL-ONC1 (Genelux), TG6002 (Transgene) and vvDD-CDSR (University of Pittsburgh). These oncolytic vaccinia viruses originate from different strains and carry different genomic modifications (i.e., deletions with or without insertion of transgenes).
Some deletions of viral genes are necessary to improve the safety profile of the virus, by restricting its amplification into actively dividing cells only, including tumor cells.3 Oncolytic vaccinia viruses can also be modified to express a transgene (armed virus) that either increases their safety profile or enhances their oncolytic efficiency.4 For example, TG6002 is a double deleted thymidine kinase (TK-), ribonucleotide reductase (RR-) Copenhagen vaccinia virus strain encoding an enzyme (FCU1) that transform the anti-fungal prodrug 5-fluorocytosine (5-FC) into the cytotoxic 5-Fluorouracil (5-FU).5 The double deletion restricts the replication of the virus to cells containing a high pool of nucleotides (dividing cells). Therefore, TG6002 is unable to replicate in resting cells contrary to tumor cells that are permissive and destroyed by the virus.6 Another example of oncolytic vaccinia virus is JX-594 (Pexa-Vec) that is currently evaluated in several clinical trials for treatment of different solid tumors.4 JX-594 is a TK- Wyeth vaccinia virus that expresses both human GM-CSF and bacterial β-galactosidase. GM-CSF is produced in the tumor where it stimulates the immune system and β-galactosidase is used as marker to monitor the viral replication.
In immuno-competent pre-clinical models, treatment of a tumor by an oncolytic vaccinia virus leads from partial to total regression depending of the host, the nature of the tumor, the dose and route of administration, the strain and the modifications of the virus and the associated treatments. These anti-tumoral effects of oncolytic vaccinia virus are mainly due to a combination of at least three recognized activities: (i) direct lysis or triggered apoptosis of infected tumor cells; (ii) disruption of tumor-associated vasculature by destruction of peri-tumoral endothelial cells and (iii) elicitation of an immune response against tumor cells.6,7,8,9 Concerning the latter point, virus replication stimulates the innate immune system by inducing an immunogenic cell death that is recognized by, and activates, neighboring professional antigen presenting cells (APC) such as dendritic cells (DC).10 The presentation of tumor-associated antigen (TAA) by these activated APC leads to an enhanced adaptive immune response against tumor cells that in turn participates in tumor destruction.11
Moreover, oncolytic vaccinia virus has also been combined with successes in pre-clinical experiments with standard therapeutic treatment of cancer such as chemotherapy, radiotherapy, thermotherapy and immunotherapy.4 Immunotherapies are particularly interesting because of the potential additive or synergistic activities between an oncolytic virus that primes an immune response against the tumor cells, and immunomodulation molecules (such as mAbs) that sustain and/or amplify this response.
Accordingly, John et al. 12 reported the combination of a vaccinia virus WR TK-, virus growth factor (VGF)- with an agonist mAb recognizing the T cell co-stimulation molecule 4-1BB (CD137). Crosslinking of CD137 with an agonist mAb induces proliferation, survival and activation of both CD8+ and CD4+ T cells. In a murine model of breast carcinoma, intratumoral (IT) injections of vaccinia virus combined with systemic administrations of agonist anti-CD137 had a better antitumoral effect than any of the treatment alone. Moreover, Rojas et al. 13 have demonstrated recently in different murine tumor models that combination of a vaccinia virus WR (TK-, B18R-) with an antagonist mAb recognizing the cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), provided a better antitumoral response than either of the treatment alone. Antibodies against the checkpoint molecules PD-1 (Programmed cell death 1), CTLA-4 or PD-L1 (PD-1 ligand 1) are the most documented immune checkpoint inhibitors (ICI) both in pre-clinical and clinical studies.14 PD-1 or CTLA-4 ligations, by their respective ligands, on the surface of the activated T cells inhibit their activation and proliferation in lymphoid organ and in tumor microenvironment. Therefore, mAbs that block the interaction of PD-1 or CTLA-4 with their ligands stimulate proliferation of T cells and then enhance the immune response.15
These results paved the way to test more combinations of vaccinia oncolytic virus with other immunomodulatory antibodies or equivalent molecules (e.g., either antagonists of ICI, or agonists of co-stimulatory molecules). Even if these combinations prove to be pre-clinically advantageous, their medical implementations could be hampered by the development cost associated with the two drugs and would be limited to antibodies or molecules that are already on the market.
One alternative to combination therapy could be the vectorization of mAbs (or equivalent molecules) into oncolytic vaccinia viruses. Vectorization consists in the insertion of sequences coding for the two chains of a monoclonal antibody (or equivalent molecule) into the virus genome under the control of vaccinia promoters. Therefore, the production of “binders” (i.e., mAb, Fab, scFv, ligand…) would occur concomitantly with virus replication and mainly in the tumor. However, to validate this approach, several key questions remain to be addressed such as (i) the kind of molecule that can be vectorized; (ii) the functionality of produced binders; (iii) the duration and the amount of vectorized binder that can be produced in vivo in an immuno-competent host; and (iv) the putative competitive therapeutic advantage of this armed virus in comparison to its parental counterpart.
We present here experimental results providing answers to the above questions. This article focuses on the vectorization, of mAb, Fab and scFv forms of an anti mPD-1 antibody in a vaccinia virus. These three forms of binders have been chosen as they offer different properties that could have an impact on the expected antitumoral effect. Mab are bivalent and therefore bind to target with an increased apparent affinity (avidity effect), whereas scFv and Fab are mainly monovalent. Mab have an Fc that is responsible for high circulating half-life but also for the engagement of complement and recruitment of killer cells (phenomenon known as, Complement directed cytotoxicity, CDC and Antibody-dependent cellular cytotoxicity, ADCC, respectively). Mab are much bigger than scFv or Fab (150 vs. 25 or 50 kDa) and therefore their diffusion into the tumor could be limited by their size. Mab have also complex heterotetrameric structure that may impair their level of expression compared to scFv that are monomeric and Fab that are dimeric.
This article presents the vectorization in vaccinia virus of mAb, Fab, and scFv recognizing mPD-1. MAb, Fab, and scFv have been produced in vitro upon infection of permissive cells by the corresponding recombinant viruses. These molecules have been purified and characterized as functional (i.e., inhibit the PD-L1/PD-1 interaction). The kinetic of expression of the mAb in mice after IT injection of vaccinia virus carrying the sequences coding for the anti-PD-1 heavy and light chains was also investigated. Finally, in an immunocompetent murine model, the antitumoral efficacy of the unarmed virus, combined or not, with an anti-mPD-1 was compared with that of armed vaccinia viruses encoding for either mAb or scFv against mPD1. In this model, armed viruses were found as efficient as the combination of unarmed virus with anti-mPD-1 mAb, in term of effect on tumor growth and survival.
Results
Recombinant mAb, Fab and scFv, vectorized in WR vaccinia virus, are secreted and correctly assembled
J43 mAb DNA sequence was designed using the publically available partially disclosed sequences of heavy and light chain (patent US 7,858,746 B2). The partial sequences were completed by the constant heavy chain of anti-CD79b mAb and the signal sequence of the light chain of anti-CD79b mAb.
Five WR recombinant vaccinia viruses were constructed by insertion at the TK locus of either the light and heavy chains (mAb and Fab) or the corresponding scFv (Fig. 1). In the case of mAb and Fab, two versions were constructed with the heavy and the light chain under the control of either pH5R or p7.5K promoters (i.e., WR-mAb1, WR-mAb2, WR-Fab1 and WR-Fab2). The WR strain was chosen for its ability to better propagate in murine cells in comparison to other vaccinia virus strains. All the WR virus presented in this article were also deleted of the ribonucleotide reductase gene (RR-).
Figure 1.
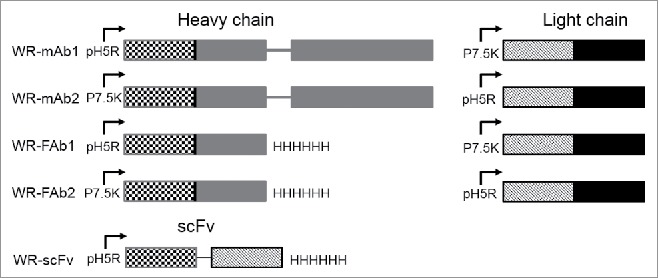
Schematic representation of the expression cassettes inserted in TK locus of the five WR vaccinia virus candidates constructed for this study. The insertion of cassettes disrupted the TK gene. The RR gene (not shown here) was also deleted in all the virus used in this study. For mAb and Fab each chain (heavy and light) is under the control of a different and independent promoter (namely p7.5K or pH5R) with their own strengths (i.e., level of protein expression). Mab corresponds to the whole molecule with two heavy and two light chains assembled to form a bivalent molecule 2 × (Light + Heavy). Fab corresponds to one light chain assembled with one heavy chain lacking their dimerization domains (i.e., hinge and Fc). Fab is a monovalent molecule. ScFv corresponds to the genetic fusion of VH to VL via a poly-GS linker. ScFv is monovalent molecule but a fraction of it can dimerize to form a divalent molecule. The variable and the constant domains of the light and heavy chains are represented with hatched and plain patterns, respectively.
In order to select the combination chain/promoter that allows the best expression of the mAb and Fab, with a correct assembly, Chicken Embryo Fibroblasts (CEF) were infected with the two versions of recombinant virus and cell supernatants analyzed by immunoblot, with a polyclonal anti-hamster IgG. Gel electrophoresis was performed in non-reducing conditions to preserve the assembly of light and heavy chains and to allow an optimal detection (i.e., the polyclonal antibody used for detection did not recognized reduced IgG and Fab chains).
Fig. 2A demonstrates that WR-mAb1 and WR-mAb2 were equally able (in roughly same quantities) to generate a molecule with an assembly pattern comparable to that of commercial J43 (i.e., an apparent size corresponding to two heavy and two light chains linked together). However, in case of WR-mAb2, an extra band between 43 and 55 kDa was clearly visible (lane 3). A band, in the same position, with the same intensity, was also clearly visible in case of WR-Fab2 (lane 4). WR-mAb2 and WR-Fab2 viruses have in common the same light chain under the same strong promoter (pH5R). Moreover, it is well known that antibody light chain can assemble in homodimers when overexpressed.16 This extra band, migrating between 43 and 55 kDa, could correspond to the light chain disulfide cross-linked homodimers with a theoretical mass of 47 kDa.
Figure 2.
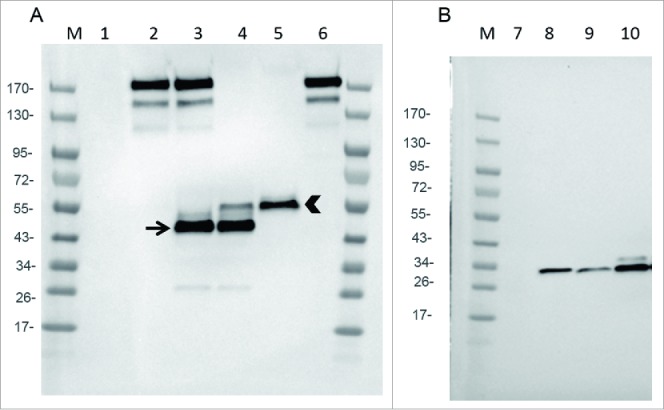
Expression of mAb, Fab and scFv by infected CEF. CEF in six wells plate were infected at MOI 0.2 by either WR (TK- RR-: negative Control: lanes 1 and 7), WR-mAb1 (lane 2), WR-mAb2 (lane 3), WR-Fab2 (lanes 4 and 9), WR-Fab1 (lanes 5 and 10) and WR-scFv (lane 8). After 24 h of infection the culture supernatants were collected and loaded on SDS-PAGE in non-reducing (A) or reducing conditions (B). Commercially available J43 was also loaded (lane 6) as a reference. After transfer onto PVDF membrane, mAb, Fab and scFv were detected using either an anti-hamster IgG (A) or an anti-Histidine tag (B). M: molecular markers. Arrow: putative dimeric light chain. Arrow head: correctly assembled Fab.
Fig. 2A also demonstrates that the WR-Fab1 produced the highest amount of correctly assembled Fab without any detectable misassembled by-product (lane 5).
In the case of WR-Fab1 & 2 and WR-scFv, cell supernatants were analyzed by immunoblot (gel electrophoresis in reducing conditions) with an anti-His tag to detect either the tagged scFv or the tagged heavy chain fragment of the Fab. Fig. 2B demonstrates that the scFv was expressed at the expected size (i.e 27.5 kDa) and that infection by WR-Fab1 generated a larger amount of heavy chain fragment than infection by WR-Fab2. These expression tests were also performed using two mammalian cell lines (BHK-21 and A549) and provided similar results (data not shown). All together, these results showed that infected cells secreted mAb, Fab and scFv at detectable levels and, for some constructions, with the expected light and heavy chain assembly. WR-mAb1 and WR-Fab1 were selected for further experiments, as the expression pattern of their transgenes was closer to the expected ones than those of WR-mAb2 and WR-Fab2.
The replicative and oncolytic abilities of WR-mAb1, WR-Fab1 and WR-scFv were compared to those of the parental virus (WR) to assess the impact of the different transgenes on the virus properties. Three cells lines were used: BHK-21 as permissive and production cell line, B16F10 and MCA 205 as murine tumor cell lines (melanoma and fibrosarcoma respectively). Figs. 3A–C demonstrates that on the three cell lines tested, none of the transgenes had a significant impact on the viral replication. It is noteworthy that the replication of WR viruses was similar in BHK-21 and MCA 205 but significantly slower in B16F10 (even if at 72H post-infection the same plateau of virus titer is reached for the three cell lines. Fig. 3D). The oncolytic activity of the different viruses was evaluated in the three cell lines at two MOI. Because of the intrinsic variability of the virus titration assay, two oncolytic activities are considered different when at least one log difference is observed between them (i.e., same cell viability observed for the two viruses but at MOI different by at least 10-fold). According to this standard, neither the different transgenes nor the addition of J43 monoclonal antibody in culture medium had a significant impact on the oncolytic activity of the WR vaccinia virus tested (Figs. 3E–G; Fig. S1). Moreover, oncolytic sensitivity of the three tested cell lines follows the same pattern as the replication results, with BHK-21 and MCA 205 cell lines being the most sensitive to oncolysis and B16F10 the most resistant. The relative resistance of B16F10 compared to MCA 205 and BHK-21 did not result in a reduced amount of produced mAb1, indicating that WR-mAb1 is clearly infecting and replicating in B16F10 (Fig. 3H).
Figure 3.

In vitro replication and oncolytic activities of the different viruses. Replication of WR-mAb1, WR-Fab1, WR-scFv and WR, and their effects on cell viability, have been assessed on MCA 205, B16F10 and BHK-21 cell lines. The virus replication was monitored over time by q-PCR after an initial infection at MOI 10−2 on BHK-21 (A), B16F10 (B) and MCA 205 (C). The replication of WR-mAb1 on the three cell lines was compared and shown in panel D. Cell viability of BHK-21 (E), B16F10 (F) and MCA 205 (G) was measured using trypan blue exclusion assay after 5 d post-infection with the different viruses and at two MOI (10−2 and 10−3). MAb1 concentration in supernatants collected 5 d post-infection was determined using a quantitative hamster IgG ELISA (H). Represented values are the mean (+/− standard deviation) of at least three measurements (see material and methods for details).
Purification and characterization of recombinant mAb1, Fab1 and scFv expressed by WR-infected cells
Recombinant mAb1, Fab1 and scFv from pooled supernatants of WR-infected CEF were successfully purified to homogeneity by affinity chromatography followed by size exclusion chromatography (SEC). This expression/purification process was repeated once for mAb1 and Fab1 (batches 1 and 2). None of the three purified proteins contained significant amount of aggregated material. ScFv eluted in two separated peaks on gel filtration (Fig. 4A). The peak that eluted first (peak 1) had a surface 7-fold lower than the second one (peak 2). Peak 1 and peak 2 could correspond, respectively, to dimeric and monomeric scFv. Purified mAb and scFv loaded, on SDS-PAGE, displayed the expected band pattern in both non-reducing and reducing conditions without any visible contaminant (Fig. 4B). Recovered quantities and concentrations for each purification are summarized in Table 1. The purified molecules were used as standard on SDS-PAGE to quantify by fluorescence the corresponding protein in the supernatants of infected CEF (i.e., starting material of the purifications). Results are summarized in Fig. 4C that shows that the scFv was expressed at the highest amount (∼2 and 9-fold in mass or ∼4 and 54-fold in mole compared, respectively, with Fab and mAb).
Figure 4.

Characterization and quantification of mAb, Fab and scFv purified from supernatants of infected CEF. Size exclusion chromatography profile of scFv after the affinity chromatography step (A). ScFv eluting from the Ni affinity column was pooled and loaded of Superdex 75 10/300 equilibrated in PBS. Absorbance at 280 nm and elution volume were recorded. Area of peak 2 was about 7-fold area of peak 1. V0 is the void volume of the column. SDS-PAGE profiles of purified recombinant mAb1 and scFv in reducing and non-reducing conditions (B). 1 µg of purified recombinant mAb1 and scFv were loaded on SDS-PAGE in reducing (R) and non-reducing (NR) conditions. J43 (BioXcell) was loaded as reference in the case of mAb1. Quantification of mAb, Fab and scFv in supernatants of the infected cells (C). Supernatants of infected CEF were recovered 48 h after infection and loaded on stain-free SDS-PAGE together with corresponding purified and quantified molecules as standards. Fluorescence intensity of the bands of interest was measured for each supernatant. Quantity of produced protein was determined using the fluorescence of standards as reference. Represented values are the mean (+/− standard deviation) of three measurements.
Table 1.
Quantities and concentrations of purified recombinant mAb1, Fab1 and scFv recovered from supernatants of WR-infected CEF.
| Virus/batch | Conc (µg/mL) | Total quantity (µg) |
|---|---|---|
| WR-scFv peak 1 | 22 | 28 |
| WR-scFv peak 2 | 143 | 287 |
| WR-Fab1 batch1 | 51 | 30 |
| WR-Fab1 batch2 | 33 | 56 |
| WR-mAb1 batch1 | 33 | 13 |
| WR-mAb1 batch2 | 34 | 32 |
The glycosylation of the Fc part of a mAb plays an important role in its ADCC and CDC potencies. Therefore, glycosylation of the purified mAb1, J43 and Rituximab (as reference) was analyzed by LC-MS/MS after trypsin digestion of the antibodies (mass of the glycosylated peptides). The results show that the glycosylation profiles of recombinant mAb1, commercial J43 and Rituximab were comparable (Fig. S2). This result indicates that in these conditions (i.e., hamster IgG vectorized in WR vaccinia virus and expressed by infected CEF) the glycosylation of Fc was similar to the one observed for a human IgG expressed by a stably transfected Chinese Hamster Ovary (CHO) cell line (i.e., Rituximab).17
Recombinant mAb, Fab and scFv are functional
In order to verify that the purified recombinant mAb1, Fab1 and scFv were able to bind to mPD-1, these molecules were incubated with T lymphoma-derived EL4 cells that spontaneously display mPD-1 at their surface. The binding of anti-PD-1 molecules to cells was analyzed by flow cytometry. Fig. 5A shows that recombinant mAb1, Fab1 and scFv efficiently label the murine T lymphocyte cell line.
Figure 5.

Binding of the purified recombinant mAb1, Fab1, scFv to mPD-1. Binding of purified mAb1, Fab1 and scFv to mPD-1-positive EL4 cells (A). Murine T lymphoma EL4 cells were incubated with commercially available J43 (positive control), hamster IgG (negative control), Fab1, monomeric scFv, mCD80-hFc-6xHis (His-tagged positive control, CD80 binds to PD-L1 expressed by EL4 cells) or hErbB2-hFc-6xHis (His-tagged negative control). Binding of mAbs and 6xHis-tagged proteins was detected by flow cytometry using either FITC-conjugated mouse anti-hamster IgG antibody or PE-conjugated mouse anti-His tag antibody. Competition between purified recombinant mAb1, Fab1, scFv (monomeric and dimeric fractions), J43 and mPD-L1 (B and C). Binding of biotinylated mPD-L1-hFc to immobilized mPD-1, or binding of unlabeled mPD-L1-hFc to EL4 cells, in presence of increasing concentrations of competitors (J43, mAb1, Fab1, scFv) or negative control (Hamster IgG) was measured in ELISA (B) or flow cytometry (C) assays. PD-L1 was detected using either streptavidin-HRP or anti-human-Fc-PE. The signal obtained with the lowest concentration of hamster IgG was set as 100%. Represented values are the mean of two normalized measurements.
J43 has been selected for vectorization because it inhibits the interaction of mPD-1 with mPD-L1.18 Therefore, two competitive assays were designed to determine if the recombinant mAb1, Fab1 and scFv, expressed by recombinant WR vaccinia viruses infected CEF, were functional. The first assay was an ELISA in which the binding of labeled mPD-L1 to immobilized mPD-1-Fc was monitored. The second assay is a flow cytometry assay in which the binding of unlabeled mPD-L1 to EL4 cells was monitored. In both assays, the three forms of blocker tested were able to inhibit the interaction mPD-1/PD-L1 in a dose-dependent manner (Figs. 5B and C). Dimeric formats (either mAb1 or scFv peak1) were more potent competitor than their monomeric counterpart formats (Fab1 or scFv peak2) indicating the importance of avidity in the interaction. Interestingly, the purified recombinant mAb1 appeared in both assays to be more potent than the commercial mAb used as positive control indicating that the design of mAb1 from partial J43 sequence and anti-CD79b hamster IgG was correct. Note that Fab does not have any advantage over scFv (either in produced quantity or affinity for mPD-1). Therefore, the characterization of WR-Fab1 was not pursued.
All together these data demonstrate that the vectorization in WR vaccinia virus of the three forms of anti-mPD-1 has yielded to functional molecules.
Vectorization of mAb1 in WR allows its intratumoral expression
In order to determine if the vectorized mAb could be produced in vivo, mice with and without subcutaneous B16F10 or MCA 205 tumors were injected with WR-mAb1 (vehicle and WR were also injected as negative controls for B16F10 and MCA 205 models, respectively). Viruses were injected either SC (without tumor) or IT. In the case of B16F10 model, the virus was injected once whereas in the MCA 205 model it was administered twice (3 d apart). As a benchmark, 10 µg of commercial J43 was also injected IT. Virus intratumoral replication was assessed at different time points, by q-PCR on a dedicated experiment in the MCA 205 model. Concentration of mAb was measured at different time points, both in serum and in tumor after a gentle homogenization to preserve cells integrity (interstitial fluid). All the negative control samples (vehicle in case of B16F10 or unarmed vector in case of MCA 205) did not yield any detectable hamster IgG (i.e., concentration below LOD). Fig. 6 shows that when WR-mAb1 was injected IT, mAb1 was detected in tumor from day 1 to day 11 with the highest concentrations reached at D3 (for MCA 205, before the second injection) or D5 (for B16F10). The concentrations of mAb1 in serum followed the same trend as in tumor with peaks of accumulation at D3 or D5 depending on the model. However, in contrast to the situation in tumor, significant amounts of mAb1 in serum were no more detected after day 5. Interestingly, when the recombinant virus was injected by IT route, the concentration of mAb1 in serum was up to 68 (MCA 205) or 1,900 (for B16F10) times higher than the concentration measured when WR-mAb1 was injected SC (i.e., without tumor). These results strongly support the concept of preferential virus replication in tumor. The maximum concentration of mAb1 either in serum or in tumor was higher in the case of the B16F10 model after a single injection than in the case of MCA 205 model with two injections. This result could reflect the difference of antibody productivity observed in vitro with the two cell lines (Fig. 3H, MOI 10−2), but could be also explained by a difference of tumor mass, and structure, at the first injection (∼600 vs. ∼170 mg for, respectively, B16F10 and MCA 205).
Figure 6.

In vivo expression of mAb1 after injection of WR-mAb1. C57BL/6 mice were implanted SC with either 3 105 B16F10 (A, C) or 8 105 MCA 205 cells (B, D). When tumors reached 100–200 mm2 (B16F10) or 40–60 mm2 (MCA 205), 107 pfu of WR-mAb1 or WR (negative control) or J43 (BioXcell, 10 µg) were injected IT. For mice without tumor, viruses were injected S.C. at the same time points. For MCA 205 tumors only, a second injection of the virus was performed 3 d after the first one. Blood, and tumors of three mice were collected at each time point i.e.: Days 1, 3 (MCA 205 only), 5, 7 (MCA 205 only) and 11 after virus or antibody injections. Concentrations of recombinant mAb or J43 were measured in tumor homogenates (A, B) or in sera (C, D) by sandwich ELISA using anti-hamster IgG antibodies and J43 as standard. The limit of quantification (LOQ = 2-fold the mean of blanks) of the ELISA was 2 ng/mL. The negative controls had no detectable hamster IgG (i.e., concentrations below the LOQ). The mean and the standard deviation of three measurements are represented.
The pharmacokinetics and biodistribution of the antibody after a single IT injection of either J43 or WR-mAb1 are very different. In the case of the IT injection of 10 µg of J43 the maximum concentration is logically measured in plasma and in tumor the day following the injection (D1 for B16F10 or D1 and D5 for MCA 205). In contrast, in the case of the IT injection of WR-mAb1, these maximum concentrations are reached at D3 or D5, and mirror the virus replication (Fig. S3). Moreover, at all time points except D1, mAb1 concentrations in tumor after virus treatment were much higher than the concentration of J43 after IT injection of 10 µg. Finally, the mAb distribution in tumor was assessed by the ratio: [mAb in tumor]/[mAb in serum] at all time points. These tumor accumulation ratios (Table 2) were much higher, for both models and for all time points, after WR-mAb1 than after J43 IT injection illustrating a continuous in situ production in case of treatment with the recombinant virus.
Table 2.
Tumor/Serum ratio of concentrations of hamster IgG after I.T. injection of either J43 (BioXcell) or WR-mAb1. If one of the concentrations was below the LOQ, LOQ was used to calculate the ratio which appears in the table labeled with *. (NA: not applicable, ND: not detected, i.e., both concentrations in tumor and serum were below LOQ).
| Day post-injection | ||||||
|---|---|---|---|---|---|---|
| Models |
treatments |
1 |
3 |
5 |
7 |
11 |
| B16F10 | J43 | 0.6 | NA | 0.2 | NA | 3* |
| WR-mAb1 | 31 | NA | 4 | NA | 605* | |
| MCA 205 | J43 | 0.2 | 0.1 | 0.1 | 4* | ND |
| WR-mAb1 | 8 | 13 | 51 | 84* | 132* | |
These data together demonstrate that vectorization of mAb1 in WR allowed a production of significant amount of the antibody mainly in tumor. The IT accumulation of mAb1, after administration of WR-mAb1, was much higher and lasted longer than after injection of 10 µg of the equivalent protein.
Vectorization of mAb1 and scFv improved the antitumoral efficacy of WR vaccinia virus in the MCA 205 model
In order to determine whether the IT expression of vectorized mAb1, or scFv, could improve the antitumoral efficacy of WR vaccinia virus, the two candidates were tested in two syngeneic tumor models (melanoma: B16F10, fibrosarcoma: MCA 205), where WR oncolytic treatments have effects both on tumor growth and survival. For both models, the tumor growth was measured and compared to those of groups of animals treated with an unarmed WR or a combination of an unarmed WR with J43 (MCA 205 model only). As expected for both models, the IT injections of parental WR inhibited the growth of the tumor (Fig. 7A and Fig. S4A) and improved the survival of the treated animals in case of the B16F10 (Fig. 7B; Fig. S4B). But, in the case of B16F10, neither WR-mAb1 nor WR-scFv, provided an improved antitumoral activity or survival compared to the parental unarmed WR (Fig. S4A and B). However it is worth to note that in B16F10, the systemic administration of anti-mPD-1 in combination with IT injections of WR did not improve the tumor-growth inhibition of the virus treatment alone (data not shown) indicating that this model is not suitable for the evaluation of the WR candidates encoding different forms of anti-mPD-1 molecules. In contrast, MCA 205 model has shown to respond to both WR and J43 with at least an additive effect in case of combination (Fig. 7A). Interestingly, in this permissive model, WR-mAb1 and WR-scFv had an antitumoral efficacy comparable to the combination WR/monoclonal J43 indicating that the levels of intratumoral expression of mAb1 or scFv were sufficient to get the efficacy obtained with large dose of systemic administration (3 × 250 µg) of J43. Moreover, inhibition of MCA 205 tumor growth translated into a significantly enhanced survival in case of WR-mAb1 treatment compared to either WR or J43 alone (Fig. 7B). This gain of survival (vs. WR or J43 alone) achieved with WR-mAb1 was comparable to the one reached with the combination WR/J43.
Figure 7.
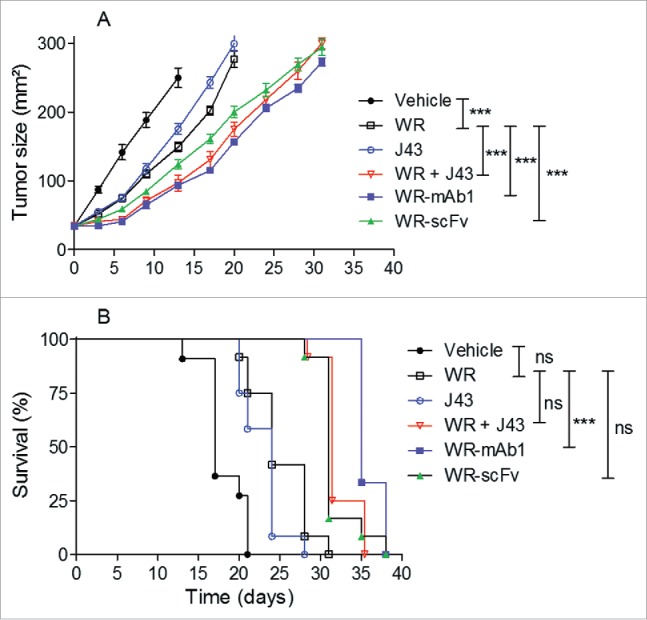
WR-mAb1 and WR-scFv have an improved tumor-growth inhibition activity compared to WR parental virus. MCA 205 tumors were implanted in C57BL/6 (n = 12) and treated as described in Fig. 6. Tumor growth was monitored by measuring length and width of the tumor over time. Mice were euthanized when tumor surface reached 300 mm2. Results are represented as the mean tumor size (A) or as survival percentage (B). Data from two combined experiments are shown. Statistical analysis were performed using a log-rank test to compare the effects of different viruses, pairwise comparison were adjusted with Tukey's correction and a mixed model was used to evaluate the impact of the viruses on the evolution over time of the tumor size. Hochberg's multiple tests correction was used. ***p < 0.001, ns non-significant.
These results together demonstrated that vectorization of an anti-PD-1 monoclonal or scFv in an oncolytic WR vaccinia virus is as efficient as the combination of an “unarmed” WR with systemic administrations of anti-PD-1 antibody.
IT injection of WR oncolytic virus induced a massive infiltration of immune cells
Intratumoral injection into established MCA 205 tumors in C57BL/6 mice of both empty WR and WR-mAb1 significantly altered lymphocyte populations within the tumor microenvironment (TME) 7 d post-initiation of WR treatment. WR increased numbers of CD3+ TILs, intratumoral CD4+ T cells and CD8+ T cells (Fig. 8A). In contrast, the number of regulatory CD4+ Foxp3+ T cells drastically decreased on WR treatment, this translated into a significant WR-induced increase in the CD8+ T cell/Foxp3+ T cell ratio (Fig. 8B). We also studied the intracellular expressions of tumor necrosis factor-α (TNFα) and interferon-gamma (IFNγ) in intratumoral CD8+ T cells showing an increase of the number of activated T cells into the tumor after the virus treatment (Fig. 8C). Moreover, we observed an upregulation of the expression of the immune checkpoint PD-1 on CD3+ lymphoid cells following WR treatment (Fig. 8D). Overall, IT injection of WR vaccinia virus induced a dramatic infiltration of activated T cells. Nevertheless, there was no significant difference in the measured parameters at the time point of the sampling, between the treatments with WR and WR-mAb1 vaccinia viruses.
Figure 8.

Characterization of the tumor-infiltrating immune cells. MCA 205 tumors were implanted in C57BL/6 (n = 5) and treated as described in Fig. 6. Four days after the last injection of WR, tumors were processed for flow cytometry determination of the number of CD3+, CD4+, and CD8+ T cells (A). The ratio of CD8+ T cells to Treg cells (B) and the number of CD8+ IFNγ+ TNFα+ T cells (C) is also shown. The expression of the immune checkpoint PD-1 was also determined (D). *p < 0.05 by Mann-Whitney test.
Discussion
We report here, the detailed characterization and comparison of scFv, Fab and antibody forms of a monoclonal anti-murine PD-1 (J43) successfully vectorized in an attenuated WR strain of vaccinia virus. The impact of the promoter strength on chains assembly of mAb and Fab was evaluated and showed that, at least for this antibody, the strongest promoter has to drive the heavy chain expression. The selected viruses (WR-mAb1, WR-Fab1 and WR-scFv) had similar replicative and oncolytic properties in vitro compared to the parental “unarmed” virus (i.e., WR). The level of expression of the three forms of anti-mPD-1, mAb1, Fab1 and scFv, were also measured in supernatant of infected CEF, scFv being the most expressed. Recombinant Mab, Fab and scFv were purified to homogeneity with the expected light and heavy chain assembly for the mAb1 (not evaluated on Fab1). In vitro assays, using either recombinant mPD-1 or mPD-1-positive cells, have demonstrated that the three purified forms of blocker were functional, i.e., able to block the binding of mPD-L1 to mPD-1. Moreover, we have demonstrated that mAb1 was expressed and accumulated preferentially in tumor when WR-mAb1 was injected IT in two tumor models. In one model, MCA 205, that responds to both vaccinia virus oncolysis and anti-PD-1 treatment, the IT injection of WR-mAb1 and WR-scFv had a comparable antitumoral efficacy and similar to the one of the combination of unarmed WR combined with systemic administration of monoclonal anti-mPD-1. This latest result indicates that the in vivo expression of the mAb1 and scFv into the tumor met at least the minimum quantity, quality and duration required for an antitumoral activity.
None of the immunological parameters measured, at the time point studied (4 d post-virus injection), and in the MCA 205 model could explain the difference of efficacy between WR and WR-mAb1 virus. More immunological parameters and/or more time point need to be explored to link the better efficacy of WR-mAb1 to an immunological mechanism of action.
We have previously published the expression of functional mAb via the vectorization in either a Copenhagen vaccinia virus 19 or in a modified vaccinia Ankara virus (MVA).20 These two publications reported that mAb could be correctly assembled and secreted in the context of a vaccinia virus-infected cells. Frentzen et al.21 have previously reported the vectorization in vaccinia virus and the in vitro and in vivo expression of a scFv against murine and human vascular endothelial growth factor (VEGF). The anti-VEGF-scFv armed vaccinia virus had a better antitumoral efficacy than the corresponding unarmed vector in two models of human tumor-bearing nude mice. Moreover, the detection in serum of the anti-VEGF-scFv (up to 1.4 µg/mL) was reported up to 37 d post-virus single injection. The main difference between our experimental settings and the ones from Frentzen et al.21 is the use of wild-type immunocompetent versus Nude mice models, respectively. In Nude mice, the multiplication of virus is not restrained by a functional adaptive immune system as in a wild-type immunocompetent mice. Therefore, the expression of the transgene carried by an armed oncolytic vaccinia virus can last longer in Nude than in wild-type mice. Since PD-1 is expressed at the surface of T lymphocytes, Nude mice were not appropriate, in our case, as an animal model to demonstrate the activities of our anti-mPD-1-armed oncolytic vaccinia viruses. Successful vectorization of antibodies or antibody derived forms have been reported for other oncolytic viruses such as measles 22 or adenovirus.23 Engeland et al.22 reported the vectorization of scFv-Fc fusions against either CTLA-4 or PD-L1, with an antitumor activities enhanced compared to the parental virus in a murine syngeneic B16-CD20 model. Dias et al.23 have vectorized an anti-CTLA-4 antibody (light and heavy chains) in an oncolytic adenovirus and tested its antitumoral activity in two xenograft models (i.e., Nude mice-bearing human tumor cells expressing CTLA-4). In these conditions, the anti-CTLA-4 antibody does not have any immunological activity but its binding to tumor cells could enhance the oncolysis or the apoptosis triggered by the virus. Indeed, anti-CTLA-4-armed adenovirus had an improved antitumoral effect in one of the two models tested. Note that vaccinia virus has an advantage over measles or adenovirus as it can tolerate the insertion of large DNA fragments (up to 25 kb) and therefore could express several transgenes (antibodies, enzymes, cytokines…) simultaneously.
Interestingly, in our case, the accumulation in the tumor of mAb1 produced after WR-mAb1 IT injection was higher and last longer than in the case of IT administration of 10 µg of J43 (at least from D1 to D11 or D5 to D11 according to the considered model). Intratumoral delivery of low doses of some therapeutic monoclonal antibodies have been proven to be as efficient as higher quantities systemically administrated (see ref.24 for review). For example, Marabelle et al.25 have described a similar therapeutic effect of a cocktail of two monoclonal antibodies recognizing OX40 and CTLA-4 in combination with CpG injected IT and at a dose 100-fold lower than the same antibodies injected systemically. Moreover, Palazon et al.26 have demonstrated in a CT26 murine tumor model that three IT injections of 5 µg of an agonist anti-CD137 were sufficient to generate an antitumoral response and an improved survival. This IT treatment led also to the regression of distant tumor nodules indicating the elicitation of systemic rather than local antitumoral immune response. Interestingly, the authors have shown that at these doses, the anti-CD137 mAb did not have the liver toxicity otherwise observed with a systemic administration of 3 ×100 µg of the same antibody. The anti-CTLA-4 monoclonal Ipilimumab, which has some toxicity drawbacks, is currently evaluated in clinic through IT delivery of low doses (10 or 25 mg i.e., ∼10 to 20-fold lower than the intravenously doses usually administered in clinical oncology) and in combination with CpG in patients with low grade B-cell lymphoma and with a second measurable disease site to assess abscopal effects (see clinical trial: NCT02254772 for details). Together these results demonstrate that relatively low doses of IT delivered antibodies can have significant antitumoral activity and could be an alternative to systemic administration in case of toxicity issues.
As described above vectorization into an oncolytic vaccinia virus can be used to deliver into tumor significant therapeutic doses of monoclonal antibodies and therefore could be an alternative to combination therapy (unarmed oncolytic virus + monoclonal antibody). Nevertheless, vectorization strategy has some drawbacks over combinations (systemic or IT combinations): (i) The level and duration of expression is strictly dependent of the virus replication and therefore are not easily controllable. (ii) The target is reached only in tumor vicinity. Targets that need to be neutralized systematically are not suitable for this application. (iii) Some antibody/therapeutic protein could inhibit the viral multiplication when vectorized but not when combined (because in case of combination, administrations of therapeutic molecule and oncolytic virus can be separated). This has been observed by Rojas et al.,13 who have demonstrated that for an optimal combination, an anti-CTLA-4 mAb had to be administrated once the replication of a vaccinia oncolytic virus has begun to wane i.e., 3 d after virus injection.
However, we believe that these drawbacks could be overcome by several key advantages: (i) Vectorization offers a solution for biotherapeutics (monoclonal antibody or protein) with toxicity issue. If IT route is chosen to minimize the toxicity, the vectorization would allow a sustained in situ production of the drug avoiding the need of repeated injections of the biotherapeutic. Moreover, vectorization allows a systemic treatment of biotherapeutic with toxicity issues. Given the natural tropism of oncolytic vaccinia virus for tumor cells, the virus could be injected intravenously and then reach several distant tumor sites where it will selectively replicate. Vectorization allows drug delivery to restricted multiple sites, and therefore limits the toxicity inherent with a systemic administration. (ii) The vectorization can be applied to antibody or proteins at any stage of development and ultimately implies the approval of a single product (i.e., armed oncolytic virus) while combination therapy is restricted to already approved drugs or necessitate the development and approval of two products. Therefore, vectorization strategy could spare some tremendous efforts necessary for the drug approval of a biotherapeutic molecule that is not yet on the market. (iii) Vectorization would decrease the costs of treatment and manufacturing (one drug instead of two).
We believe that vectorization of complex biomolecules such as antibodies will complete, in the near future, the arsenal of payload that can be expressed by an oncolytic vaccinia virus in clinic. New generation of oncolytic vaccinia virus expressing several transgenes simultaneously may also provide enhanced therapeutic/toxicity ratio to the patients.
Material and methods
Animal models
This study was conducted in compliance with CEE directive 2010/63/UE of 22th September 2010 and the French décret n° 2013–118 of 1st February 2013.
Six weeks old female C57BL/6 were obtained from Charles River. Animals were acclimatized in the study room of a specific pathogen free (SPF) animal facility, at minima one week before the start of the experiment in order to ensure their suitability for the study.
WR constructions
Sequences coding for the hamster J43 monoclonal antibody anti-PD-1 were partially disclosed in patent US 7,858,746 B2 which showed the sequence of the variable domain (VH) of the heavy chain (HC) and the sequence of the light chain (LC). The sequence of VH showed high homology with the HC of anti-CD79b hamster IgG (accession number AGH06135.1). Thus, the sequence retained for cloning of the HC was the variable domain of J43 and the constant domains of anti CD79b. The light chain of J43 was cloned with signal sequence from the light chain (LC) of anti CD79b antibody (accession number AGH06134.1). Both chains were generated by synthetic way (Geneart: Regensburg, Germany) and were put under the control of the vaccinia viral promoters pH5R 27 or p7.5K 28 which have slightly different strengths (as defined by the level of protein expression). Further to “whole” antibody constructs (mAb), antigen binding fragments (Fab) with a 6 His tag at the C-terminus of the HC fragment as well as a C-terminus His-tagged single-chain fragment variable (scFv) were constructed. In the case of scFv, the VH was fused to the variable domain (VL) of the LC with a GS spacer between them. For mAb and Fab, two versions were generated that differ by the promoter used to express each chain (either pH5R or p7.5K; Fig. 1).
All recombinant WRs are derivatives of Western Reserve strain deleted in ribonucleotide reductase (RR) I4L gene. The WR shuttle plasmid, pTG18496, contains the pH5R promoter, surrounded by portions of the vaccinia virus J1R and J3R genes (sequences from nt 80219 to 80723 and from nt 81258 to 81756 relative to WR sequence, Genbank: NC_006998); which allows homologous recombination into these loci and deletion of J2R (TK: thymidine kinase) gene. The expression cassettes were cloned in head to-tail manner into the pTG18496 transfer vector by standard cloning techniques. Recombinant WR was then generated as previously described.6 Briefly, CEF (Chicken Embryo Fibroblasts) cells were infected with WRTG18674 (RR deleted) at a multiplicity of infection (MOI) of 0.05 and transfected with the recombinant shuttle plasmid. Selection of TK- plaques was done after infection of the TK- deficient 143B cells in selection medium containing 5-bromo-2′-deoxyuridine at final concentration of 150 µg/mL (Sigma). Positive TK- plaques were isolated and selected for a second cycle in 143B cells in presence of 5-bromo-2´-dexoyuridine. Final recombinant WR viruses were amplified in BHK-21 cells and virus stocks were titrated on CEF by plaque assay. The features of the five recombinant WR vaccinia virus generated in this publication are summarized below (see also Fig. 1): WR-mAb1 corresponding to pH5R-HC/p7.5K-LC; WR-mAb2 corresponding to pH5R-LC/p7.5K-HC; WR-Fab1 corresponding to pH5R-VH-CH1-6His/p7.5K-LC; WR-Fab2 corresponding to pH5R-LC/p7.5K-VH-CH1-6His; WR-scFv corresponding to pH5R-VH-gs-VL-6His. WR corresponding to parental TK- and RR- vector (i.e., “empty” virus) was also used in this publication as benchmark. The recombinant proteins encoded by the transgenes are named respectively mAb1 for WR-mAb1, Fab2 for WR-Fab2 etc.
In vitro and in vivo virus titration by q-PCR
For in vitro experiments: 105 cells per well of MCA 205, B16F10 and BHK-21 cells were plated in six-well culture dishes in 2 mL of culture medium and infected with WR, WR-mAb1, WR-Fab1 or WR-scFv at an MOI of 10−2. At indicated time points, cells and medium were harvested and frozen at −80°C until use. Cell suspensions were thawed and sonicated, and 100 µL was treated with 5 units of benzonase (Novagen) to eliminate non-encapsidated DNA. Benzonase was then inactivated by addition of EDTA (27 mM final) and the virus capsids were disrupted by addition of an equal volume of Tris 20 mM, EDTA 10 mM, SDS 1 % pH7.4 followed by 160 µg of Proteinase K (Qiagen) and an incubation of 30 min at 65°C. Proteinase K was then inactivated by an incubation of 15 min at 95°C. The samples were stored at −20°C until q-PCR analysis.
For in vivo experiments: Tumors were collected, frozen in liquid N2 and stored at −80°C until use. Tumors were thawed and transferred in GentleMACS M–type tubes (Miltenyi) containing 600 µL/30 mg of tumor of lysis Buffer (i.e., RLT buffer) from QIAGEN kit (Allprep DNA/RNA/Protein Mini kit). Tumors were mechanically dissociated and the lysates were kept at −80°C until analysis. Lysates were thawed and centrifuged at 10,000 g during 3 min, and the DNA was extracted from the supernatants following the protocol provided by QIAGEN's kit. The samples were stored at −20°C until q-PCR analysis.
The q-PCR assay used, was an absolute quantification using purified and quantified MVA DNA as standard. Standard curve was performed by a series of dilutions from 106 to 102 copies of MVA genome. A set of oligonucleotides primers and probe that hybridize into the gene encoding for the secreted chemokine-binding protein (SCBP) of MVA were designed as following: forward primer 5´-CGATGATGGAGTAATAAGTGGTAGGA-3´; reverse primer 5´-CACCGACCGATGATAAGATTTG-3´; probe 5´FAM—ACTGATTCCACCTCGGG—3´MGB/NFQ. The Quantitect Multiplex PCR kit from Qiagen was used to perform the q-PCR reactions on either 7500 real time system from Applied Biosystem (SDS software v2.0.6.) or on BIORAD q PCR CFX96 model.
Viability assay
To determine the oncolytic activity of the different WR viruses over MCA 205, B16F10 and BHK-21 cells, a total of 5 × 104 cells per well were plated in six-well culture dishes in 2 mL of culture medium and infected with either WR, WR-mAb1, WR-mAb2 or WR-scFv at an MOI of either 10−2 or 10−3. Each condition was performed in triplicate. Five days after infection, cell viability was determined by trypan blue exclusion with a cell counter (Vi-Cell, Beckman coulter). The condition of untreated cells was used to set the 100% of viability. At least the results of two experiments were pooled.
Western Blot analysis
Monolayers of CEF cells were infected at a MOI of 0.2 with WR-mAb1, WR-mAb2, WR-Fab1, WR-Fab2, WR-scFv or WR. Twenty four hours after infection, cell culture supernatants were centrifuged 5 min at 16,000 g. Twenty five microliters of supernatant were prepared in Laemmli buffer in either reducing (5% β-mercaptoethanol) or non-reducing condition. Monoclonal commercial J43 (BioXcell) was used as reference molecule. Samples were loaded onto a Criterion TGX Stain free PrecastGel 4–15% polyacrylamide gel. Proteins were then transferred onto a PVDF membrane using the Trans-blot Turbo system (Transblot Turbo Transfer pack Biorad) with the preprogrammed protocol: High MW: 10 min; 2.5 A constant; up to 25 V. Membranes were saturated overnight at 4°C in blocking solution (PBS: 8 mM NaPO4, 2 mM KPO4, 154 mM NaCl pH 7.2, supplemented with 0.05% Tween20, 5% Nonfat dry milk Biorad). Horseradish peroxidase (HRP) conjugated goat anti Armenian Hamster IgG (Jackson Immunoresearch) at 80 ng/mL was used to immunodetect J43 and mAb1. PentaHis (recognizing 5 histidine tag, Qiagen) at 100 ng/mL followed by a HRP conjugated Polyclonal Rabbit anti mouse Ig (Dako) were used to immunodetect Fab or scFv. Amersham ECL Prime Western Blotting detection reagents and Molecular Imager ChemiDOC™ XRS were used to capture chemiluminescence.
mAb, Fab and scFv purifications
Eighteen F175 flasks containing about 8 × 107 CEF/flask were infected at a MOI of 0.2 with either WR-mAb1, WR-Fab1 or WR-scFv for 48 h. The pool of culture media (≈350 mL) was centrifuged at 12,000 g, 30 min at 4°C to remove most of debris and viruses. The supernatant was then filtered through 0.2 µm filters and stored at −20°C until use. After thawing, the supernatant was filtered again as described above. The purification was performed on either 1 mL HisTrap™ FF (GE healthcare) for Fab and scFv or on 1 mL HiTrap rProtein A FF (GE healthcare) for mAb. Before loading, both HisTrap and HiTrap rProtein A columns were equilibrated in PBS. Before column-loading, 500 mM NaCl and 20 mM Imidazole were added to Fab and scFv supernatants. The loaded columns were washed with 10 column volumes (CV) of PBS, 20 mM Imidazole (in case of Fab and scFv) or PBS (in case of mAb) until OD280 nm reached base line. Captured material was eluted in 0.25 mL fractions with 250–500 mM Imidazole (scFv); 100–500 mM Imidazole (Fab); or 0.1 M citric acid/ 0.2 M Na2HPO4 pH 3 (mAb). The proteins were further purified by SEC using either Superdex 75 10/300 (GE Healthcare) for Fab and scFv or Superdex 200 10/300 for mAb, equilibrated in PBS. The protein concentration of the pools (mAb, Fab, scFv) were determined by measuring absorbance at 280 nm (Nanodrop 2000 spectrophotometer) and by using an extinction coefficient calculated from primary structure (Protparam program, at expazy.org: 1.51, 1.63 and 1.73 mL.mg−1.cm−1 for, respectively, mAb, Fab and scFv).
Quantification of mAb, Fab and scFv in supernatant of infected CEF
The supernatants of infected CEF (i.e., corresponding to starting material of purification) were loaded on Criterion TGX Stain free PrecastGel 4–15%. The trihalo compounds contained in gels reacts with tryptophan (W) residues in a UV-induced reaction to produce fluorescence signal proportional to the quantity of the loaded protein (if the protein contains at least one W). After electrophoresis the fluorescence profiles were visualized with the Molecular imager ChemiDOCTM XRS using Image Lab Software (Biorad). For each culture supernatant (i.e., mAb, Fab, scFv), an appropriate standard range prepared with the purified molecule was loaded on the same gel at different known concentrations. The volume and intensity of fluorescence signals of sample bands were measured and protein quantity was deduced using the standard curve.
ELISA
Quantitation ELISA
For measurement of J43 a quantitative sandwich ELISA was developed. Ninety six wells plates (Nunc immune plate Maxisorp) were coated with 80 ng of goat anti-hamster IgG (Southern Biotech) in 100 µL of coating solution (0.05 M Na carbonate pH 9.6, Sigma), overnight at 4°C. Plates were then incubated 1h at RT with 200 µL/well of blocking solution (PBS/0.05% Tween20, 5% Non-Fat Dry Milk). A standard curve of J43 (BioXcell, gel filtrated in PBS) was prepared from 500 to 0.244 ng/mL in 50% mouse serum and 100 µL/well were added in duplicate to the microplate. Samples were diluted at least 2-fold in the same diluent as the standards and 100 µL were added to the plates and were incubated 2 h at 37°C. Microplates were developed by 100 µL of HRP conjugated goat anti-armenian Hamster IgG at 80 ng/mL followed by 100 µL of 3,3´,5,5´-tétraméthylbenzidine (TMB, Sigma). The reaction was stopped with 100 µL/well of 2 M H2SO4 and the absorbance was measured at 450 nm with a plate reader (TECAN Infinite M200 PRO). The absorbance values were transferred into the software GraphPadPrism and samples concentrations were back-calculated using the standard curve fitted with five parameters. The limit of quantification (LOQ = 2-fold the mean of blank) of the ELISA was 2 ng/mL.
Competition ELISA
Solutions and conditions, not otherwise specified, were the same as for quantitation ELISA described above. Plates were coated with 100 µL/well of 1 µg/mL of mPD-1-Fc as described above. Two-fold serial dilutions of the competitors (i.e., commercial J43, isotype control or samples) were performed into the plate and in blocking buffer. PD-L1-Biot (R&D Systems protein biotinylated in house) diluted to 0.2 µg/mL in blocking buffer was added to all wells (100 µL/well) and incubated 1.5 h at 37°C. One hundred µL of streptavidin-conjugated horseradish peroxidase (Southern Biotech) diluted 1/5,000 were added to the wells and incubated 1 h at 37°C. The plates were developed and read as above.
Flow cytometry
To assess the binding of the different WR-encoded molecules to cell-surface PD-1, 1 or 5 × 105 murine T lymphoma EL4 cells were incubated for 45 min on ice with 100 µL of either mAb1 (5 µg/mL), commercially available J43 as positive control (5 µg/mL, BioXCell), hamster IgG as negative control (5 µg/mL, BioXCell), Fab1 (5 µg/mL), monomeric scFv (2.5 µg/mL), mCD80-hFc-6xHis as His-tagged positive control(10 µg/mL, R&D Systems) or hErbB2-hFc-6xHis as His-tagged negative control (20 µg/mL, R&D Systems) and washed. Binding of mAbs and 6 x His-tagged proteins was detected by incubating cells with either 100 µL of FITC-conjugated mouse anti-hamster IgG antibody (10 µg/mL, BD PharMingen) or PE-conjugated mouse anti-His tag antibody (1/10 dilution, Miltenyi Biotec) for 45 min on ice. Fluorescence intensity was measured on a Navios™ flow cytometer (Beckman Coulter). Data were analyzed using Kaluza 1.2 software (Beckman Coulter).
To assess the blocking activity of WR-encoded molecules on PD-L1 binding to cell-surface PD-1, 105 EL4 cells were co-incubated with mPD-L1-hFc (2 µg/mL, R&D Systems) and increasing concentrations of the different competitive proteins (mAb1, Fab1, monomeric or dimeric scFv, positive control J43 or negative control hamster IgG) in 100 µL for 45 min on ice. After washing, PD-L1 binding was detected using a PE-labeled mouse anti-hIgG Fc (5 µg/mL, BioLegend) and the mean fluorescence intensity (MFI) was measured for duplicate samples as described above.
In vivo expression of vectorized mAb
B16F10 (murine melanoma) cells (3 × 105) or MCA 205 (fibrosarcoma) cells (8 × 105) were injected subcutaneously (SC) into the right flank. When the tumor surface reached 40–60 (MCA 205) or 50–70 mm2 (B16F10), WR-mAb1 (107 pfu), WR-scFv (107 pfu), WR (107 pfu, for MCA 205 model only), anti-PD1 antibody (J43, BioXcell, 10 µg), or Vehicle (for B16F10 model only) were injected into the tumor. For MCA 205 model only, a second injection of the virus and/or of the antibody was performed 3 d after the first injection. For mice without tumor, virus was injected SC at the same time points.
Blood, and tumors of three mice were sampled at different time points (day 1, 3, 5, 7 and 11 in the MCA 205 model and day 1, 5 and 11 in the B16F10 model). Blood was stored at 4°C during 8 h and the serum was collected after two centrifugations at 10,000 g during 2 min and stored at −20°C until analysis. Tumors were isolated, cut into small pieces and transferred in GentleMACS C–type tubes (Miltenyi) containing 2 mL (MCA 205 model) or 3 mL (B16F10 model) of PBS. Tumors were mechanically dissociated and after centrifugation at 300 g for 7 min, supernatant was recovered and kept at −20°C. Before analysis, samples were centrifuged at 10,000 g for 20 min and supernatants were recovered.
In vivo therapeutic activity of vectorized mAb
For the MCA 205 tumor model, 8 × 105 MCA 205 cells (in 100 µL PBS) were injected SC into the right flanks of mice. When tumor surface reached between 40 and 60 mm2, mice were injected with vehicle (IT, 50 µL), WR-mAb1 (IT, 107 pfu/50 µL), WR-scFv (IT, 107 pfu/50 µL), WR (IT, 107 pfu/50 µL), or J43 antibody (IP, 250 µg/100 µL) or with the combination of WR-empty (IT, 107 pfu/50 µL) and J43 (IP, 250 µg/100 µL). Injections of virus were repeated once 3 d after the first injection (at D3) and injections of antibody were repeated twice at D3 and at D6. Tumor surfaces were monitored two or three times per week by caliper measurements. Mice were sacrificed when tumor size reached 300 mm2.
Statistics on tumor volumes and survival (n =12 mice/group) were performed. A log-rank test was used to compare the effects of different viruses, pairwise comparison were adjusted with Tukey's correction and a mixed model was used to evaluate the impact of the viruses on the evolution over time of the tumor size. Hochberg's multiple tests correction was used.
Intratumoral infiltration of immune cells
Seven days after the start of the treatment, tumors were harvested, cut into small pieces and digested in RPMI-1640 medium containing Liberase at 25 μg/mL (Roche, Boulogne-Billancourt, France) and DNase1 at 150 UI/mL (Roche) for 30 min at 37°C. The mixture was subsequently passaged through a 100 μm cell strainer. 2 × 106 tumor cells were preincubated with purified anti-mouse CD16/CD32 (93; eBioscience, San Diego, CA, USA) for 15 min at 4°C, before surface staining. For surface staining, cell suspensions were stained with anti-CD45 (30-F11; eBioscience), anti-CD3 (145-2C11; BD Biosciences), anti-CD4 (GK1.5; eBioscience), anti-CD8 (53–6.7; eBioscience), anti-CD25 (PC61.5.3; eBioscience), anti-PD1 (29F.1A12; Biolegend), and respective isotype antibodies. For intracellular transcription factor staining, surface-stained cells were fixed and permeabilized using the Foxp3/Transcription Factor Staining Buffer Set (eBioscience), according to the manufacturer's protocol, and stained using anti-Foxp3 (FJK-16s, eBioscience). For intracellular staining of IFNγ and TNFα, cells were stimulated in vitro with 50 ng/mL phorbol 12-myristate 13-acetate (PMA; Sigma-Aldrich) and 1 μg/mL ionomycin (Sigma-Aldrich) in the presence of Golgi Plug (BD Biosciences) for 4 h and then surface stained as aforementioned. Surface-stained cells were then fixed and permeabilized using BD Cytofix/Cytoperm (BD Biosciences) according to the manufacturer's protocol and stained with anti-IFNγ (XMG1.2; eBioscience) and anti-TNFα (MP6-XT22; Biolegend) and respective isotype antibodies. A FACS® Canto flow cytometer (BD Biosciences) was used for eight-color flow cytometry acquisition. Analyses were performed using FACS® Diva software.
Supplementary Material
Disclosure of potential conflicts of interest
All authors but Laurence Zitvogel are employees of Transgene. Laurence Zitvogel is a member of the management Board of Transgene.
Acknowledgments
The authors thank Jean-Marc Strub (Laboratoire de Spectrométrie de Masse BioOrganique, Strasbourg, France) for the analysis of glycosylation-profiles of the different mAbs, Berangere Marie-Bastien for the statistical analysis of the data, Johann Foloppe for its kind help with the cell viability assay and Caroline Shenkels for proofreading.
References
- 1.Pol J, Bloy N, Obrist F, Eggermont A, Galon J, Cremer I, Erbs P, Limacher JM, Preville X, Zitvogel L et al.. Trial Watch:: Oncolytic viruses for cancer therapy. Oncoimmunology 2014; 3:e28694; PMID:25097804; http://dx.doi.org/ 10.4161/onci.28694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andtbacka RH, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, Delman KA, Spitler LE, Puzanov I, Agarwala SS et al.. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J clin Oncol 2015; 33:2780-8; PMID:26014293; http://dx.doi.org/ 10.1200/JCO.2014.58.3377 [DOI] [PubMed] [Google Scholar]
- 3.Lusky M, Erbs P, Foloppe J, Acres RB. Oncolytic vaccinia virus: a silver bullet? Exp Rev Vaccines 2010; 9:1353-6; PMID:21105770; http://dx.doi.org/25839047 10.1586/erv.10.137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chan WM, McFadden G. Oncolytic poxviruses. Annu Rev Virol 2014; 1:119-41; PMID:25839047; http://dx.doi.org/ 10.1146/annurev-virology-031413-085442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Erbs P, Regulier E, Kintz J, Leroy P, Poitevin Y, Exinger F, Jund R, Mehtali M. In vivo cancer gene therapy by adenovirus-mediated transfer of a bifunctional yeast cytosine deaminase/uracil phosphoribosyltransferase fusion gene. Cancer Res 2000; 60:3813-22; PMID:10919655 [PubMed] [Google Scholar]
- 6.Foloppe J, Kintz J, Futin N, Findeli A, Cordier P, Schlesinger Y, Hoffmann C, Tosch C, Balloul JM, Erbs P. Targeted delivery of a suicide gene to human colorectal tumors by a conditionally replicating vaccinia virus. Gene Ther 2008; 15:1361-71; PMID:18480846; http://dx.doi.org/ 10.1038/gt.2008.82 [DOI] [PubMed] [Google Scholar]
- 7.Parato KA, Breitbach CJ, Le Boeuf F, Wang J, Storbeck C, Ilkow C, Diallo JS, Falls T, Burns J, Garcia V et al.. The oncolytic poxvirus JX-594 selectively replicates in and destroys cancer cells driven by genetic pathways commonly activated in cancers. Mol Ther 2012; 20:749-58; PMID:22186794; http://dx.doi.org/ 10.1038/mt.2011.276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Breitbach CJ, Arulanandam R, De Silva N, Thorne SH, Patt R, Daneshmand M, Moon A, Ilkow C, Burke J, Hwang TH et al.. Oncolytic vaccinia virus disrupts tumor-associated vasculature in humans. Cancer Res 2013; 73:1265-75; PMID:23393196; http://dx.doi.org/ 10.1158/0008-5472.CAN-12-2687 [DOI] [PubMed] [Google Scholar]
- 9.Kirn DH, Thorne SH. Targeted and armed oncolytic poxviruses: a novel multi-mechanistic therapeutic class for cancer. Nat Rev Cancer 2009; 9:64-71; PMID:19104515; http://dx.doi.org/ 10.1038/nrc2545 [DOI] [PubMed] [Google Scholar]
- 10.Sistigu A, Yamazaki T, Vacchelli E, Chaba K, Enot DP, Adam J, Vitale I, Goubar A, Baracco EE, Remedios C et al.. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med 2014; 20:1301-9; PMID:25344738; http://dx.doi.org/ 10.1038/nm.3708 [DOI] [PubMed] [Google Scholar]
- 11.Guo ZS, Liu Z, Bartlett DL. Oncolytic Immunotherapy: Dying the Right Way is a Key to Eliciting Potent Antitumor Immunity. Front Oncol 2014; 4:74; PMID:24782985; http://dx.doi.org/ 10.3389/fonc.2014.00074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.John LB, Howland LJ, Flynn JK, West AC, Devaud C, Duong CP, Stewart TJ, Westwood JA, Guo ZS, Bartlett DL et al.. Oncolytic virus and anti-4-1BB combination therapy elicits strong antitumor immunity against established cancer. Cancer Res 2012; 72:1651-60; PMID:22315352; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-2788 [DOI] [PubMed] [Google Scholar]
- 13.Rojas J, Sampath P, Hou W, Thorne SH. Defining effective combinations of immune checkpoint blockade and oncolytic virotherapy. Clin Cancer Res 2015. 24:5543-51; PMID:26187615; http://dx.doi.org/26060497 10.1158/1078-0432.CCR-14-2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khan H, Gucalp R, Shapira I. Evolving concepts: immunity in oncology from targets to treatments. J Oncol 2015; 2015:847383; PMID:26060497; http://dx.doi.org/ 10.1155/2015/847383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell 2015; 27:450-61; PMID:25858804; http://dx.doi.org/ 10.1016/j.ccell.2015.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaplan B, Livneh A, Sela BA. Immunoglobulin free light chain dimers in human diseases. Scient World J 2011; 11:726-35; PMID:21442150; http://dx.doi.org/23751726 10.1100/tsw.2011.65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li C, Rossomando A, Wu SL, Karger BL. Comparability analysis of anti-CD20 commercial (rituximab) and RNAi-mediated fucosylated antibodies by two LC-MS approaches. mAbs 2013; 5:565-75; PMID:23751726; http://dx.doi.org/ 10.4161/mabs.24814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ansari MJ, Salama AD, Chitnis T, Smith RN, Yagita H, Akiba H, Yamazaki T, Azuma M, Iwai H, Khoury SJ et al.. The programmed death-1 (PD-1) pathway regulates autoimmune diabetes in nonobese diabetic (NOD) mice. J Exp Med 2003; 198:63-9; PMID:12847137; http://dx.doi.org/ 10.1084/jem.20022125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Paul S, Bizouarne N, Dott K, Ruet L, Dufour P, Acres RB, Kieny MP. Redirected cellular cytotoxicity by infection of effector cells with a recombinant vaccinia virus encoding a tumor-specific monoclonal antibody. Cancer Gene Ther 2000; 7:615-23; PMID:10811480; http://dx.doi.org/ 10.1038/sj.cgt.7700161 [DOI] [PubMed] [Google Scholar]
- 20.Paul S, Regulier E, Rooke R, Stoeckel F, Geist M, Homann H, Balloul JM, Villeval D, Poitevin Y, Kieny MP et al.. Tumor gene therapy by MVA-mediated expression of T-cell-stimulating antibodies. Cancer Gene Ther 2002; 9:470-7; PMID:11961670; http://dx.doi.org/ 10.1038/sj.cgt.7700461 [DOI] [PubMed] [Google Scholar]
- 21.Frentzen A, Yu YA, Chen N, Zhang Q, Weibel S, Raab V, Szalay AA. Anti-VEGF single-chain antibody GLAF-1 encoded by oncolytic vaccinia virus significantly enhances antitumor therapy. Proc Natl Acad Sci U S A 2009; 106:12915-20; PMID:19617539; http://dx.doi.org/ 10.1073/pnas.0900660106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Engeland CE, Grossardt C, Veinalde R, Bossow S, Lutz D, Kaufmann JK, Shevchenko I, Umansky V, Nettelbeck DM, Weichert W et al.. CTLA-4 and PD-L1 checkpoint blockade enhances oncolytic measles virus therapy. Mol Thera 2014; 22:1949-59; PMID:25156126; http://dx.doi.org/22071969 10.1038/mt.2014.160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dias JD, Hemminki O, Diaconu I, Hirvinen M, Bonetti A, Guse K, Escutenaire S, Kanerva A, Pesonen S, Loskog A et al.. Targeted cancer immunotherapy with oncolytic adenovirus coding for a fully human monoclonal antibody specific for CTLA-4. Gene Ther 2012; 19:988-98; PMID:22071969; http://dx.doi.org/ 10.1038/gt.2011.176 [DOI] [PubMed] [Google Scholar]
- 24.Marabelle A, Kohrt H, Caux C, Levy R. Intratumoral immunization: a new paradigm for cancer therapy. Clin Cancer Res 2014; 20:1747-56; PMID:24691639; http://dx.doi.org/ 10.1158/1078-0432.CCR-13-2116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marabelle A, Kohrt H, Levy R. Intratumoral anti-CTLA-4 therapy: enhancing efficacy while avoiding toxicity. Clin Cancer Res 2013; 19:5261-3; PMID:23965900; http://dx.doi.org/ 10.1158/1078-0432.CCR-13-1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Palazon A, Martinez-Forero I, Teijeira A, Morales-Kastresana A, Alfaro C, Sanmamed MF, Perez-Gracia JL, Penuelas I, Hervas-Stubbs S, Rouzaut A et al.. The HIF-1alpha hypoxia response in tumor-infiltrating T lymphocytes induces functional CD137 (4-1BB) for immunotherapy. Cancer Dis 2012; 2:608-23; PMID:22719018; http://dx.doi.org/3021979 10.1158/2159-8290.CD-11-0314 [DOI] [PubMed] [Google Scholar]
- 27.Rosel JL, Earl PL, Weir JP, Moss B. Conserved TAAATG sequence at the transcriptional and translational initiation sites of vaccinia virus late genes deduced by structural and functional analysis of the HindIII H genome fragment. J Virol 1986; 60:436-49; PMID:3021979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cochran MA, Puckett C, Moss B. In vitro mutagenesis of the promoter region for a vaccinia virus gene: evidence for tandem early and late regulatory signals. J Virol 1985; 54:30-7; PMID:3973982 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.