

Abstract
Coagulation kinetics are well established for purified blood proteases or human plasma clotting isotropically. However, less is known about thrombin generation kinetics and transport within blood clots formed under hemodynamic flow. Using microfluidic perfusion (wall shear rate, 200 s−1) of corn trypsin inhibitor-treated whole blood over a 250-μm long patch of type I fibrillar collagen/lipidated tissue factor (TF; ∼1 TF molecule/μm2), we measured thrombin released from clots using thrombin-antithrombin immunoassay. The majority (>85%) of generated thrombin was captured by intrathrombus fibrin as thrombin-antithrombin was largely undetectable in the effluent unless Gly-Pro-Arg-Pro (GPRP) was added to block fibrin polymerization. With GPRP present, the flux of thrombin increased to ∼0.5 × 10−12 nmol/μm2-s over the first 500 s of perfusion and then further increased by ∼2–3-fold over the next 300 s. The increased thrombin flux after 500 s was blocked by anti-FXIa antibody (O1A6), consistent with thrombin-feedback activation of FXI. Over the first 500 s, ∼92,000 molecules of thrombin were generated per surface TF molecule for the 250-μm-long coating. A single layer of platelets (obtained with αIIbβ3 antagonism preventing continued platelet deposition) was largely sufficient for thrombin production. Also, the overall thrombin-generating potential of a 1000-μm-long coating became less efficient on a per μm2 basis, likely due to distal boundary layer depletion of platelets. Overall, thrombin is robustly generated within clots by the extrinsic pathway followed by late-stage FXIa contributions, with fibrin localizing thrombin via its antithrombin-I activity as a potentially self-limiting hemostatic mechanism.
Keywords: antithrombin (AT), hemostasis, platelet, thrombin, thrombosis, microfluidic
Introduction
Human thrombin is a multifunctional protease central to coagulation by its enzymatic cleavage of platelet PAR1 and PAR4 receptors (1), cleavage of fibrinogen to fibrin monomer (2), generation of Factor XIIIa (FXIIIa)2 (3, 4), and feedback activation of Factor XIa (FXIa) (5, 6). The extrinsic coagulation pathway is triggered by tissue factor/Factor VIIa (TF/FVIIa). which generates Factor Xa (FXa) and Factor IXa (FIXa) and is essential for hemostasis. In contrast, deficiencies in the contact pathway (Factor XII [FXII] and FXI) are not linked to strong bleeding phenotypes.
The kinetics of the extrinsic tenase (TF/FVIIa), the intrinsic tenase (FIXa/FVIIIa), and prothrombinase (FXa/FVa) have been extensively measured (7–9) and kinetically modeled for plasma (10–13) or purified enzymes with added lipid (14–16) and platelet-rich plasma (17–20). These prior kinetic studies explore rate processes in a closed and isotropic context (i.e. a tube). However, clotting under hemodynamic flow is an open system involving platelet adhesion and activation on a surface as well as rapid build-up of a dense platelet core surrounded by less activated platelets in surrounding shell of the clot. The core of the clot is highly contracted with P-selectin-positive platelets (21, 22), localized thrombin and fibrin (23, 24), and localized disulfide reductase activity (25). The kinetics of thrombin generation are less well understood in this hierarchical structure where intrathrombus transport and binding effects may control reactions (26–30). For example, fibrin is known to inhibit thrombin via its antithrombin-I activity (31–33). Under venous flow, γ'-fibrin has been shown to limit thrombin transport, fibrin production, and clot size (34).
Direct thrombin or Factor Xa (FXa) inhibitors are orally available and clinically approved and do not requiring frequent coagulation monitoring in patients (35). The reduction of thrombotic risk, although still allowing for sufficient hemostasis to prevent undesired bleeding, is central to therapeutic potency and dosing. Calibrated automated thrombinography (CAT) is a calibrated thrombin generation assay that reports thrombin concentration in activated plasma samples as a function of time (36). CAT measurement determines the coagulability of plasma, potentially helpful for clinical diagnosis and drug monitoring. However, the CAT assay provides limited information on the actual dynamics of thrombin generation in platelet-rich clots formed under flow. By recreating hemodynamic flow over procoagulant surfaces, the dynamics and pharmacology of thrombin generation can be studied with human blood ex vivo. In microfluidic clotting assays, fibrin generation is often used as an indirect indicator for thrombin activity (5, 37–40). Our laboratory previously developed a peptide-based platelet targeting biosensor to report platelet associated thrombin activity in both microfluidic and animal thrombosis models (23, 24). To our knowledge there has been no direct quantitative measurement of thrombin flux from a growing thrombus under hemodynamic flow. This lack of measurement may be due to the limited sensitivity of immunoassays to detect released thrombin in the stable complex of thrombin-antithrombin (TAT) as released thrombin would be severely diluted in macroscopic flow systems with ml/min-scale perfusion. In contrast, microfluidics allows detection of clot-released species by limiting their dilution through use of μl/min-scale perfusion.
Theoretical and experimental studies have suggested thrombin generation is sensitive to prevailing shear rate and surface TF levels (41–43). At a venous shear rate condition, thrombin flux is predicted to be within a range of 10−13-10−11 nmol/μm2-s (41, 42, 44). Using plasma perfusion, our group previously demonstrated in a membrane microfluidic system that both wall shear rate and thrombin flux regulate physical structure of deposited fibrin fibers. Under venous shear condition, a wall thrombin flux of 10−11 nmol/μm2-s is required for platelet-free plasma to form a three-dimensional fibrin network (44), which is the fibrin structure that is usually observed in whole blood thrombi formed on the surface with collagen and TF at venous shear rates (5). In blood, antithrombin (AT) is a potent and rapid thrombin inhibitor (45). Thrombin half-life is less than a minute in the presence of AT (46), a reaction accelerated by heparin. TAT complex measurement has been routinely used to estimate thrombin level in plasma samples (17, 47, 48). Here we measure thrombin flux from TF bearing collagen surface and aggregated platelets at a venous shear rate in a human whole blood microfluidic thrombosis assay by collecting effluent at the outlet of the microfluidic system and subsequently measuring TAT complex concentration with enzyme linked immunosorbent assay (ELISA).
Results
Device Response Time
Release of a diffusible species from a surface into a flow stream is a classic concentration boundary layer phenomenon that can influence the dynamics of measurement of the species at the system outlet. Additionally, for sampling parabolic flows at the system outlet, the faster streamlines at and near the center of the flow contribute more volume per unit time than the slower streamlines at the wall boundary. The discrete sampling of volume at the system exit flow where the species concentration may be nonuniform is defined as the “mixing-cup” concentration and involves weighting the concentration profile by the velocity profile (49). To calculate the lag time of the microfluidic device and exit line and thus relate surface events to remotely sampled volumes at the exit, a computational model with a rectangular domain was created, and the transport of thrombin was calculated by standard finite element method in COMSOL (Inc., Burlington, MA) (Fig. 1). A flux with constant amplitude of 10−11 nmol/μm2-s and duration of 1 s was applied at the bottom of the channel near the entrance. Within 1 s, the thrombin concentration was high over the entrance (where collagen/TF resides in the experiment) due to the release of soluble thrombin from the bottom boundary; there was no significant thrombin yet reaching the outlet. After 1.6 s, the outlet (mixing-cup average) thrombin concentration began to rise as thrombin was transported advectively by the flow and also by molecular diffusion. By 5 s, there was essentially no thrombin left near the entrance as the imposed thrombin flux only lasted for 1 s, indicating complete and rapid washout at a wall shear rate of 200 s−1. By 7 s the concentration of thrombin collected at the outlet reached a maximum. It took almost 20 s for the concentration of thrombin to decay to zero at the outlet (Fig. 1, B and C). Overall, the simulation demonstrated that the microfluidic assay (Fig. 1A) allows the detection of locally generated thrombin at the collagen site with only a minor lag time of ∼10 s. Also, dilution of thrombin (or TAT) in the prevailing microfluidic flow was predicted to allow detection of nanomolar levels of TAT in the effluent.
FIGURE 1.

Microfluidic setup for measuring thrombin flux during whole blood thrombosis on collagen/TF surface. A, a microfluidic device with 8 channels (each channel: 250-μm in width and 60-μm in height) diverging from a single inlet and converging into a single outlet was used for whole blood perfusion over collagen/TF surface. Blood was collected into CTI (40 μg/ml) and loaded into syringes that were subsequently mounted on a syringe pump. Blood infusion was initiated within 10 min of phlebotomy at a constant flow rate of 16 μl/min (initial wall shear rate = 200 s−1). Device outlet flow was collected into EDTA to quench thrombin generation with EDTA and allow for TAT formation. Blood samples (collected every 120 s) were subsequently centrifuged and analyzed by TAT ELISA. The microfluidic system was characterized with a COMSOL convection-diffusion model (B). To calculate the device response time, thrombin flux signal from a 250-μm-long surface domain with amplitude of 10−11 nmol/μm2-s was imposed as a boundary condition for duration of 1 s. Location of the surface domain is indicated by a red dashed line. Average thrombin concentration at the outlet peaked after a delay of 7 s and leveled off to zero by 20 s (C).
Clotting under Flow: Fibrin Rapidly Captures Thrombin
Platelets immediately adhered and accumulated on the collagen/TF surface (∼1 TF molecule/μm2) after initiation of whole blood flow, whereas fibrin generation was detected after a 250-s lag followed by a nearly linear increase until the end of the experiment at 800 s (Fig. 2A and supplemental Fig. S1). Under these conditions of platelet deposition and fibrin generation on the collagen/TF surface, very little TAT was detected over the 800-s experiment unless Gly-Pro-Arg-Pro (GPRP, 5 mm) was used to prevent fibrin polymerization (Fig. 2B). Collection of effluent into benzamidine (instead of EDTA) to inhibit thrombin prevented the immunodetection of TAT (not shown), indicating that free thrombin was eluted off the clot in the presence of 5 mm GPRP, which then complexed with antithrombin to form TAT in the EDTA-treated sample. Fibrin captured >85% of locally generated thrombin (Fig. 2B). Thus, the fibrin polymerization inhibitor GPRP was added to all subsequent experiments to eliminate fibrin capture of thrombin and allow detection of TAT in the effluent. Prior studies have shown that GPRP has a minor effect on platelet deposition at the low forces of venous flow conditions (40). Also, the contribution of the contact pathway in this microfluidic assay has been shown to be minimal for 40 μg/ml corn trypsin inhibitor (CTI)-treated whole blood when compared with the contribution of the TF-driven extrinsic pathway (50).
FIGURE 2.
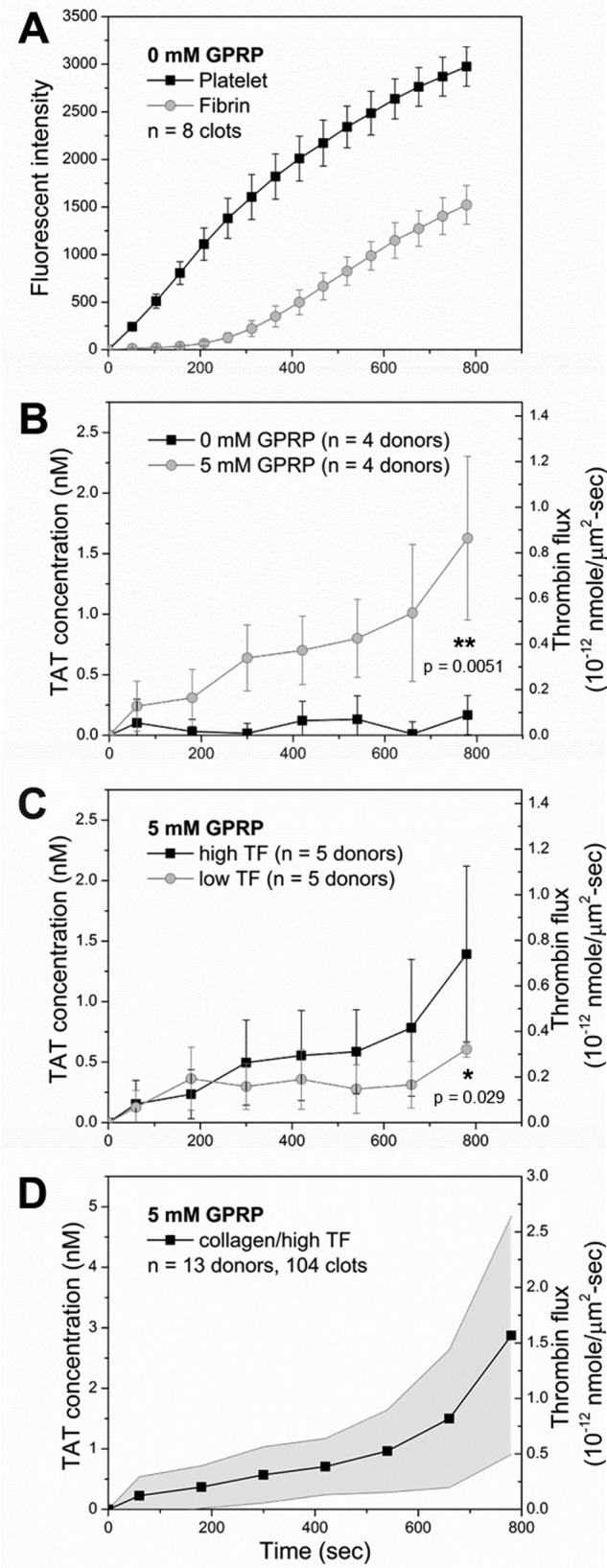
Thrombin flux from clots growing on collagen/TF. A, dynamics of platelet aggregation (■) and fibrin formation (○) during CTI-treated (40 μg/ml) whole blood perfusion over collagen/high TF (1 molecule/μm2) at initial wall shear rate of 200 s−1. B, measured TAT concentration and thrombin flux during blood perfusion over collagen/high TF in the presence (○) and absence (■) of GPRP. C, TAT concentration and thrombin flux for TF surface concentration from 1 molecule/μm2 (■) to 0.1 molecule/μm2 (○). D, averaged TAT concentration and thrombin flux from 13 healthy donors for blood perfusion (40 μg/ml CTI and 5 mm GPRP) over collagen/high TF at 200 s−1. *, p < 0.05; **, p < 0.01.
Increased Surface TF Concentration Promotes Thrombin Flux
Because fibrin formation requires a threshold concentration of surface TF (43), we tested if the amount of immunodetected TAT complex in the effluent was dependent on TF concentration on the collagen surface. At ∼1 TF mol/μm2 (collagen/high TF), the TAT concentration displayed a slow increase during the first 500 s and then an accelerated increase during the following 300 s. By 800 s, the thrombin flux reached ∼0.8 × 10−12 nmol/μm2-s (Fig. 2C). At ∼0.1 TF mol/μm2 (collagen/low TF), there was a brief initial increase of TAT concentration within the first 200 s. Between 200 and 600 s, TAT concentration remained largely constant. After 600 s, accelerated increase of the TAT concentration was observed. By 800 s, thrombin flux from collagen/low TF surface reached ∼0.4 × 10−12 nmol/μm2-s, which was only half that of the thrombin flux from collagen/high TF surface (Fig. 2C).
The collagen/high TF surface was used as the test surface for all subsequent experiments. A master thrombin generation curve (Fig. 2D) was developed from 13 individual experiments with blood from 13 donors, representing the average dynamics of human thrombin generation from clots formed on collagen/high TF surface (∼1 molecule TF/μm2, 5 mm GPRP, 40 μg/ml CTI): a slow increase in thrombin flux during the first 500 s to 0.5 × 10−12 nmol/μm2-s followed by an accelerated increase (∼4× higher than the rate of increase during the first 500 s), reaching a thrombin flux of ∼1.5 × 10−12 nmol/μm2-s by 800 s at the end point of the experiment. The standard error was attributed to interdonor variation in thrombin generation of about ±50%, as has been previously observed with measurements of thrombin generation by the CAT assay (47). Given this observed variation, subsequent experiments were conducted with at least four donors under matched conditions.
Thrombin Flux Amplification via Thrombin Feedback Activation of FXI
The large and late stage increase in thrombin flux detected between 500 and 800 s was consistent with the late stage fibrin and thrombin activity previously detected as a result of platelet polyphosphate enhancement of the thrombin-mediated FXIa feedback pathway (5, 51–53). This late stage participation of thrombin-mediated activation of FXI has also been theoretically predicted (54). The FXI antibody O1A6 inhibits FXIIa activation of FXI and disrupts the FXI-dependent thrombin amplification loop by inhibiting FXIa generation of FIXa and subsequent FXa activation (Fig. 3A). Adding O1A6 (20 μg/ml) into the blood sample abolished the accelerated increase of thrombin flux from the collagen/TF surface after 500 s (Fig. 3B). Thrombin flux increased to 0.2 × 10−12 nmol/μm2-s during the first 400 s and remained relatively constant after that.
FIGURE 3.
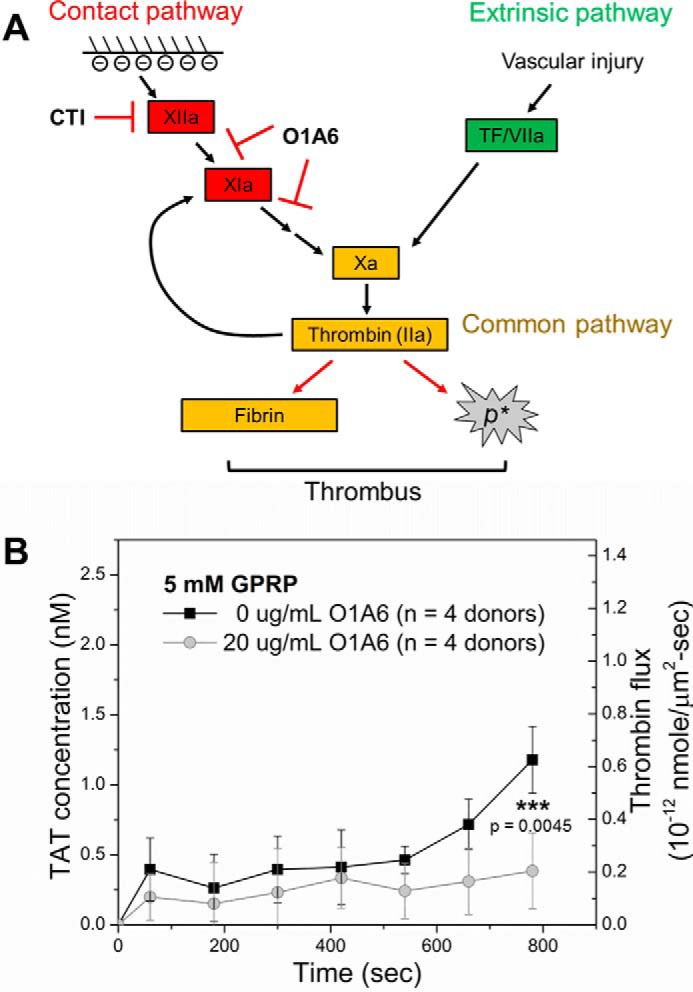
Thrombin flux is amplified after 500 s of clotting via thrombin feedback generation of FXIa. A, FXI antibody O1A6 disrupts thrombin feedback amplification loop by inhibiting FXIIa activation of FXI and blocking thrombin feedback activation of FXI. B, O1A6 (20 μg/ml, ○) abolished late stage increase in thrombin flux from growing thrombi during blood perfusion (40 μg/ml CTI, 5 mm GPRP) over collagen/high TF and caused significant reduction in final thrombin flux when compared with clotting without O1A6 (■). ***, p < 0.005.
The First Layer of Collagen-activated Platelets Generates the Majority of Thrombin
We tested the thrombin generating capacity of a monolayer of collagen-adherent platelets to that of a thick and dense platelet deposit. As previously observed (55), the glycoprotein IIb/IIIa (αIIbβ3) antagonist, GR144053 (500 nm), abolished secondary platelet deposition on the collagen/high TF surface. For the first 500 s of blood perfusion, the thrombin production was essentially the same for a platelet monolayer and a thick platelet deposit, indicating that the thick platelet mass did not diminish thrombin production by limiting FX transport or by additional coverage/hindrance of surface TF. Interestingly, the accelerated increase of thrombin flux from 600 to 800 s was largely prevented by GR144053 (Fig. 4A), indicating a role for the thickened platelet deposit in thrombin-feedback activation of FXIa (Fig. 3B) that was operative in this late-stage time regime (Fig. 4A) and also consistent with platelet-dependent release of polyphosphate (56). Additionally, in the absence of GPRP, a platelet monolayer was sufficient to support localized fibrin formation (Fig. 4B).
FIGURE 4.
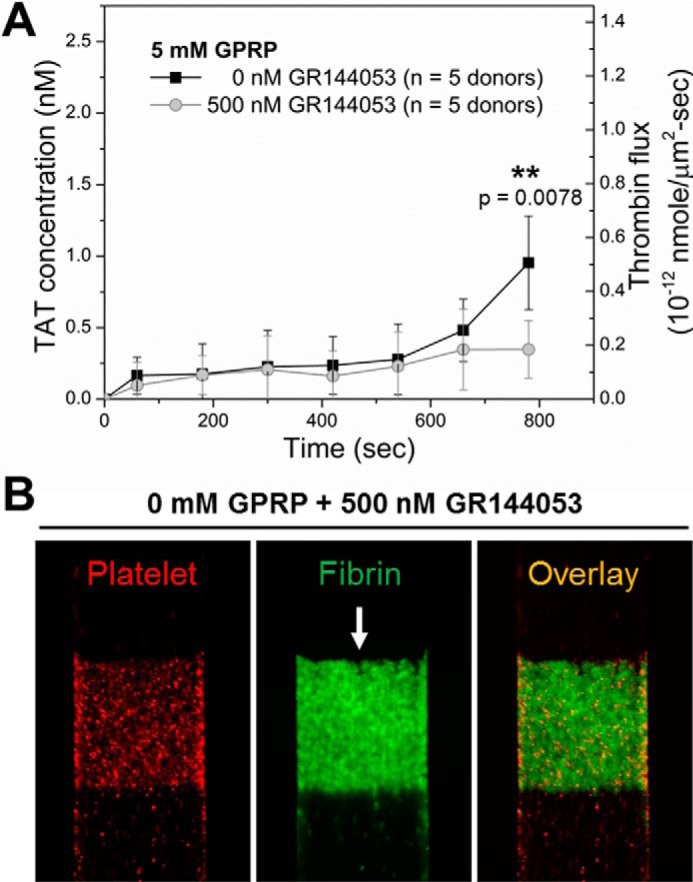
The first layer of collagen-activated platelets support initial thrombin production. A, dynamics of measured TAT concentration and thrombin flux from growing thrombi (■) or from the first layer of collagen-adherent platelets (○) during blood (40 μg/ml CTI, 5 mm GPRP) perfusion over collagen/high TF. The αIIbβ3 inhibitor, GR144053 (500 nm) was added to abolish platelet secondary aggregation and achieve a monolayer of platelet on collagen/high TF. Initial thrombin production during the first 500 s of blood perfusion was intact, whereas the final thrombin production on collagen/high TF was significantly reduced by GR144053 (A). A platelet monolayer was sufficient to support localized fibrin formation when fibrin was allowed to polymerize in the absence of GPRP (B). **, p < 0.01.
Longer Collagen/TF Zones Are Less Efficient in Thrombin Production
The TAT concentration in effluent collected downstream of 1000-μm long collagen/high TF was ∼2× higher than the detected concentration in effluent from 250-μm-long collagen/high TF (Fig. 5A). However, the 1000-μm zone was less efficient in supporting overall thrombin generation on a per unit area basis. The overall thrombin flux from the entire 1000-μm-long zone of collagen/high TF was only half that of the overall flux from the 250-μm-long zone of collagen/high TF (Fig. 5B). On a 1000-μm zone, most of the platelet accumulation was observed on the first 250 μm, with significantly less platelet deposition between 250 and 1000 μm (Fig. 5, C and D). This is consistent with boundary layer depletion of depositing platelets in the red blood cell-free layer of plasma nearest the collagen coating. Roughly half of detected thrombin flux from the 1000-μm collagen/high TF zone originated from the first 250-μm subregion of the 1000-μm-long zone, assuming the first 250 μm had the exact efficiency in supporting thrombin generation as for the 250-μm-long zone (Fig. 5, E and F).
FIGURE 5.

Longer collagen/TF zones were less efficient in thrombin production. Measured TAT concentration (A) and thrombin flux (B) from blood perfusion (40 μg/ml CTI, 5 mm GPRP) over 250-μm-long or 1000-μm-long collagen/high TF (∼1 molecule/μm2). Longer 1000-μm zones supported more thrombin production but less thrombin flux on a per unit area basis (A and B). Platelet accumulation was observed over the first 250 μm on 1000-μm collagen/high TF with far fewer platelets between 250 μm and 1000 μm along the flow direction (C and D). About half of the generated thrombin originated from the first 250 μm of the 1000-μm zone (E and F). ***, p < 0.005.
Discussion
In this study ELISA allowed direct measurement of TAT in plasma isolated from whole blood samples collected from the outlet of a microfluidic thrombosis assay. Numerical simulation demonstrated the delay in thrombin detection in this setup was small (∼10 s) compared with the time interval (120 s) between sample collections. Thus, TAT measurements can be directly converted to thrombin fluxes within each time interval. Previous theoretical and experimental studies suggested alterations in shear rates and surface TF affect coagulation reaction dynamics and cause substantial changes in thrombin flux (41, 42).
The microfluidic experiments were operated at a constant flow rate mode resulting in an initial wall shear rate of 200 s−1. The thrombin flux released from growing thrombi on collagen/high TF (∼1 molecule/μm2) increased over time and reached a level of ∼10−12 nmol/μm2-s by 800 s at the end of the experiment. This final thrombin flux falls within the suggested range from previous computational models for similar shear conditions (41, 42, 44). In our flow system, fibrin localized >85% of thrombin within the thrombus and traveled downstream, indicating that free thrombin lasted less than a second or two before its capture by fibrin. The TAT ELISA-based measurement might underestimate the actual thrombin flux due to the following factors: (i) other protease inhibitors such as α2-macroglobulin, even though far less effective than AT, also complex thrombin (45), (ii) thrombin bound to the clot by fibrin-independent mechanisms would not be detected in the plasma TAT-ELISA assay, and (iii) robust thrombin generation at later time points boosted fibrin polymerization, which was not fully quenched by 5 mm GPRP (supplemental Fig. S2).
A thrombin generation curve from 13 individual measurements provides the average dynamic change of thrombin flux from growing thrombi on 250-μm-long collagen/high TF. We calculate that each TF molecule supported generation of ∼92,000 thrombin molecule via the extrinsic pathway during the first 500-s time period. This curve displayed an accelerated increase in thrombin flux starting at ∼500 s. This accelerated increase was prevented by FXI antibody O1A6. Our previous work indicated that participation of the TF-triggered extrinsic pathway was required to generate enough thrombin to initiate the FXIa-dependent thrombin amplification mechanism, which can be interrupted by O1A6 (5). This FXI function-blocking antibody also reduced platelet aggregation downstream of thrombi formed on collagen/TF, suggesting a role of the FXI-thrombin axis in distal thrombi formation (57). Therefore, thrombin flux after 500 s is most likely augmented by the FXIa-dependent thrombin amplification loop. FXI inhibition did not significantly interfere with thrombin flux within the first 500 s, indicating the extrinsic pathway is the major contributor of initial thrombin generation in CTI-inhibited whole blood clotting. By 500 s, ∼70% of collagen/TF had been covered with accumulated platelets (supplemental Fig. S1), potentially reducing access to surface-immobilized TF and enhancing platelet-dependent pathways (such as polyphosphate pathways).
It is known that platelets provide lipid surface for prothrombinase assembly and, therefore, are essential for promoting coagulation (58, 59). Interestingly, a monolayer of platelets provided enough lipid surfaces for robust thrombin generation in the initial phase. The prevention of amplified thrombin generation during later times suggests the importance of aggregated platelets in aiding the FXI-dependent thrombin feedback loop. It is possible that platelet aggregates promote thrombin generation by limiting flow dilution and active transport of activated coagulation factors. Additionally, the accumulated platelets release polyphosphate to act as a cofactor and promote thrombin activation of FXI (5, 51, 53) at later times.
We found increased collagen/TF patch length reduced the overall efficiency of thrombin generation on a per unit area basis, most likely due to the decay in platelet deposition along the flow direction on 1000-μm long collagen/high TF patch. Furthermore, we have shown in our previous study that a single collagen/TF fiber (patch length <1 μm) prompts a clotting response of multiple layers of platelet deposition and thrombin generation at a venous shear condition (60). We suspect that the total thrombin generation might be greater on larger thrombotic patches, but thrombin generation on a per unit area basis decays once the patch size gets too large, which is probably due to boundary layer platelet depletion of the near-wall platelet layer and consequently reduced platelet deposition in downstream regions of larger patches.
In summary, we made direct measurements of TAT complex concentration in effluent from microfluidic thrombosis assay. We estimated thrombin flux from growing thrombi on the collagen/TF surface can reach up to 10−12 nmol/μm2-s at a venous shear condition. Our results suggest polymerized fibrin fibers consume most of the free thrombin and likely serve as a mechanism of localizing clotting response near the injury site and potentially promoting a hierarchical structure of self-limiting clots. Furthermore, aggregated platelets were found to play pivotal roles in amplifying thrombin generation via the FXI-dependent feedback loop.
Experimental Procedures
Materials
Anti-human CD61 (BD Biosciences), Alexa Fluor®647 conjugated human fibrinogen (Life Technologies), H-Gly-Pro-Arg-Pro-OH (GPRP, EMD Chemicals, San Diego, CA), CTI (Hematologic Technologies, Essex Junction, VT), GR144053 trihydrochloride (Tocris Bioscience, Minneapolis, MN), EDTA (Sigma), Sigmacote® siliconizing reagent (Sigma), human thrombin-antithrombin complex ELISA kit (Abcam, Cambridge, MA), Dade® Innovin® PT reagent (Siemens, Malvern, PA), collagen Type I Chrono-ParTM aggregation reagent (Chrono-log, Havertown, PA), and Sylgard® 184 Silicone Elastomer kit (Dow Corning, Auburn, MI). O1A6 FXI antibody was a gift from Dr. Andras Gruber (Department of Biomedical Engineering, Oregon Health and Science University).
Polydimethylsiloxane (PDMS) Patterning and Flow Devices
PDMS devices were fabricated as previously described (61, 62). Protein patches with defined dimensions were patterned on glass slides with single channel devices (250 or 1000 μm in width, 60 μm in height). Multichannel flow chambers with 8 evenly spaced flow channels (250 μm in width, 10000 μm in length, and 60 μm in height) diverging from a single inlet and converging into a single outlet were used for microfluidic thrombosis assay. All PDMS devices have features on their bottom allowing them to be reversibly vacuum-bonded onto glass slides.
Analysis of System Response Time
A model microfluidic system was characterized in COMSOL Multiphysics® Modeling Software (COMSOL). A rectangular domain (10,000 μm by 60 μm) was created to represent one lane of the actual microfluidic device. The local wall shear rate was maintained at 200 s−1, consistent with the experiments. The thrombin flux was imposed between 100 μm and 350 μm, and the transport was mainly governed by the convection and diffusion assuming no hindrance from the developing clot. To calibrate the system response time, a rectangular flux signal with amplitude of 10−11 nmol/μm2-s was applied for duration of 1 s. The system was propagated with a constant time step of 0.05 s to obtain the average thrombin concentration at the outlet. Similarly, an empirical thrombin flux fitted with the experimental measures was applied to obtain the resultant mixing-cup average concentration at the outlet.
Preparation of Thrombotic Patches
Glass slides were treated with Sigmacote® to retard surface-triggered clotting. To generate collagen patches on glass slides, collagen type I (1 mg/ml, 5 μl) followed by bovine serum albumin (0.5% BSA in Hepes-buffered saline, 20 μl) was perfused through the main channel on a single channel patterning device (62). The length of a collagen patch (250 μm or 1000 μm) is defined by the dimension of the main channel. Tissue factor can be added by subsequent perfusion of 10 μl of Dade Innovin® PT reagent through the main channel and incubation over patterned collagen for 30 min before a BSA wash. The PT reagent stock (23 nm recombinant human TF and synthetic phospholipids) was diluted 10-fold and 200-fold to achieve high and low surface TF surface densities of ∼1 and ∼0.1 TF molecule/μm2, respectively. Sorbed TF lipid vesicles were stained with FITC-annexin V. TF surface densities were estimated by fluorescent imaging of stained vesicles, as previously described (5, 39).
Blood Collection and Sample Preparation
Blood was collected via venipuncture from healthy donors who provided consent under approval of University of Pennsylvania Institutional Review Board and were free of any medication or alcohol for at least 72 h before donation. Blood was collected into syringes with a high dosage of CTI (40 μg/ml) and was transferred into Eppendorf tubes where it was labeled by CD61 antibody (2%v/v) and fluorescent fibrinogen (1.3% v/v) for platelet and fibrin epifluorescence detection, respectively. In some of the microfluidic experiments, additional treatments were added to the blood sample before perfusion to block platelet aggregation or fibrin polymerization. These additional treatments are indicated under “Results.”
Microfluidic Thrombosis Assay
After protein patterning, the single channel patterning device was replaced with the multichannel flow device. The flow channels were placed perpendicularly to patterned TF bearing collagen patches. Labeled whole blood was transferred to syringes which were then mounted on a PHD 2000 syringe pump (Harvard Apparatus, Holliston, MA) operating on an infusing mode. Blood was infused into the flow device at a constant flow rate of 16 μl/min, which corresponds to an initial wall shear rate of 200 s−1. Platelet aggregation and fibrin formation were simultaneously monitored with a fluorescence microscope (IX81, Olympus America Inc., Center Valley, PA). Images were captured with a CCD camera (Hamamasu, Bridgewater, NJ) and analyzed with ImageJ (NIH, Bethesda, MD). The outlet of the flow device was blocked with 10 μl of EDTA. Calcium-dependent thrombin generation in blood was immediately quenched by EDTA once blood exits the device. Blood samples were collected from the outlet every other minute. Another 10 μl of EDTA was added to block the outlet immediately after sample collection. Collected blood samples were allowed to sit for at least 10 min for TAT complex formation before subsequent steps.
TAT-ELISA Assay
Collected blood samples were centrifuged at 1300 × g for 15 min. TAT complex concentration in isolated platelet poor plasma was detected in a sandwich TAT-ELISA assay. Background TAT level was determined by measuring TAT concentration in plasma sample isolated from blood that was quenched with EDTA right after phlebotomy. Because AT reacts with thrombin in a 1:1 stoichiometric ratio, measured average TAT concentration (C̄) within the 2-min time interval between sample collections was converted to an average thrombin flux (J̄) using the equation,
 |
where Q is flow rate (16 μl/min), and A is total thrombotic area in each device.
Author Contributions
S. Z. and S. L. D. designed the experiments. S. Z. conducted the experiments. Y. L. performed numerical simulation. S. Z., Y. L., T. S., and S. L. D. analyzed the data and wrote the manuscript.
Supplementary Material
This work was supported by National Institutes of Health Grants R01 HL103419 and U01-HL131053 (to S. L. D.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Figs. S1 and S2.
- FXI-
- Factor XI
- TF
- tissue factor
- CAT
- calibrated automated thrombinography
- AT
- antithrombin
- TAT
- thrombin-antithrombin
- CTI
- corn trypsin inhibitor
- GPRP
- Gly-Pro-Arg-Pro
- PDMS
- polydimethylsiloxane.
References
- 1. Coughlin S. R. (2000) Thrombin signalling and protease-activated receptors. Nature 407, 258–264 [DOI] [PubMed] [Google Scholar]
- 2. Wolberg A. S. (2007) Thrombin generation and fibrin clot structure. Blood Rev. 21, 131–142 [DOI] [PubMed] [Google Scholar]
- 3. Hornyak T. J., Bishop P. D., and Shafer J. A. (1989) α-Thrombin-catalyzed activation of human platelet factor XIII: relationship between proteolysis and factor XIIIa activity. Biochemistry 28, 7326–7332 [DOI] [PubMed] [Google Scholar]
- 4. Schwartz M. L., Pizzo S. V., Hill R. L., and McKee P. A. (1973) Human Factor XIII from plasma and platelets: Molecular weights, subunit structures, proteolytic activation, and cross-linking of fibrinogen and fibrin. J. Biol. Chem. 248, 1395–1407 [PubMed] [Google Scholar]
- 5. Zhu S., Travers R. J., Morrissey J. H., and Diamond S. L. (2015) FXIa and platelet polyphosphate as therapeutic targets during human blood clotting on collagen/tissue factor surfaces under flow. Blood 126, 1494–1502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kravtsov D. V., Matafonov A., Tucker E. I., Sun M. F., Walsh P. N., Gruber A., and Gailani D. (2009) Factor XI contributes to thrombin generation in the absence of factor XII. Blood 114, 452–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Krishnaswamy S., Church W. R., Nesheim M. E., and Mann K. G. (1987) Activation of human prothrombin by human prothrombinase: influence of factor Va on the reaction mechanism. J. Biol. Chem. 262, 3291–3299 [PubMed] [Google Scholar]
- 8. Krishnaswamy S., Jones K. C., and Mann K. G. (1988) Prothrombinase complex assembly: kinetic mechanism of enzyme assembly on phospholipid vesicles. J. Biol. Chem. 263, 3823–3834 [PubMed] [Google Scholar]
- 9. Walker R. K., and Krishnaswamy S. (1994) The activation of prothrombin by the prothrombinase complex: the contribution of the substrate-membrane interaction to catalysis. J. Biol. Chem. 269, 27441–27450 [PubMed] [Google Scholar]
- 10. Butenas S., Orfeo T., Gissel M. T., Brummel K. E., and Mann K. G. (2004) The significance of circulating factor IXa in blood. J. Biol. Chem. 279, 22875–22882 [DOI] [PubMed] [Google Scholar]
- 11. Brummel-Ziedins K., Vossen C. Y., Rosendaal F. R., Umezaki K., and Mann K. G. (2005) The plasma hemostatic proteome: thrombin generation in healthy individuals. J. Thromb. Haemost. 3, 1472–1481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brummel-Ziedins K. E., Orfeo T., Gissel M., Mann K. G., and Rosendaal F. R. (2012) Factor Xa generation by computational modeling: an additional discriminator to thrombin generation evaluation. PloS ONE 7, e29178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Willems G. M., Lindhout T., Hermens W. T., and Hemker H. C. (1991) Simulation model for thrombin generation in plasma. Haemostasis 21, 197–207 [DOI] [PubMed] [Google Scholar]
- 14. Hockin M. F., Jones K. C., Everse S. J., and Mann K. G. (2002) A model for the stoichiometric regulation of blood coagulation. J. Biol. Chem. 277, 18322–18333 [DOI] [PubMed] [Google Scholar]
- 15. Nesheim M. E., Tracy R. P., and Mann K. G. (1984) “Clotspeed,” a mathematical simulation of the functional properties of prothrombinase. J. Biol. Chem. 259, 1447–1453 [PubMed] [Google Scholar]
- 16. Bungay S. D., Gentry P. A., and Gentry R. D. (2003) A mathematical model of lipid-mediated thrombin generation. Math Med. Biol. 20, 105–129 [DOI] [PubMed] [Google Scholar]
- 17. Chatterjee M. S., Denney W. S., Jing H., and Diamond S. L. (2010) Systems biology of coagulation initiation: kinetics of thrombin generation in resting and activated human blood. PLoS Comput. Biol. 6, e1000950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sorensen E. N., Burgreen G. W., Wagner W. R., and Antaki J. F. (1999) Computational simulation of platelet deposition and activation: I. Model development and properties. Ann. Biomed. Eng. 27, 436–448 [DOI] [PubMed] [Google Scholar]
- 19. Sorensen E. N., Burgreen G. W., Wagner W. R., and Antaki J. F. (1999) Computational simulation of platelet deposition and activation: II. Results for Poiseuille flow over collagen. Ann. Biomed. Eng. 27, 449–458 [DOI] [PubMed] [Google Scholar]
- 20. Hoffman M., and Monroe D. M. 3rd. (2003) The action of high-dose factor VIIa (FVIIa) in a cell-based model of hemostasis. Dis. Mon. 49, 14–21 [DOI] [PubMed] [Google Scholar]
- 21. Stalker T. J., Traxler E. A., Wu J., Wannemacher K. M., Cermignano S. L., Voronov R., Diamond S. L., and Brass L. F. (2013) Hierarchical organization in the hemostatic response and its relationship to the platelet-signaling network. Blood 121, 1875–1885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Stalker T. J., Welsh J. D., Tomaiuolo M., Wu J., Colace T. V., Diamond S. L., and Brass L. F. (2014) A systems approach to hemostasis: 3. Thrombus consolidation regulates intrathrombus solute transport and local thrombin activity. Blood 124, 1824–1831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Welsh J. D., Colace T. V., Muthard R. W., Stalker T. J., Brass L. F., and Diamond S. L. (2012) Platelet-targeting sensor reveals thrombin gradients within blood clots forming in microfluidic assays and in mouse. J. Thromb. Haemost. 10, 2344–2353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Welsh J. D., Stalker T. J., Voronov R., Muthard R. W., Tomaiuolo M., Diamond S. L., and Brass L. F. (2014) A system approach to hemostasis: 1. The interdependence of thrombus architecture and agonist movements in the gaps between platelets. Blood 124, 1808–1815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhu S., Welsh J. D., Brass L. F., and Diamond S. L. (2016) Platelet-targeting thiol reduction sensor detects thiol isomerase activity on activated platelets in mouse and human blood under flow. J. Thromb. Haemost. 14, 1070–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Voronov R. S., Stalker T. J., Brass L. F., and Diamond S. L. (2013) Simulation of intrathrombus fluid and solute transport using in vivo clot structures with single platelet resolution. Ann. Biomed. Eng. 41, 1297–1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Leiderman K., and Fogelson A. L. (2013) The influence of hindered transport on the development of platelet thrombi under flow. Bull. Math. Biol. 75, 1255–1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kim O. V., Xu Z., Rosen E. D., and Alber M. S. (2013) Fibrin networks regulate protein transport during thrombus development. PLoS Comput. Biol. 9, e1003095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xu Z., Lioi J., Mu J., Kamocka M. M., Liu X., Chen D. Z., Rosen E. D., and Alber M. (2010) A multiscale model of venous thrombus formation with surface-mediated control of blood coagulation cascade. Biophys. J. 98, 1723–1732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tomaiuolo M., Stalker T. J., Welsh J. D., Diamond S. L., Sinno T., and Brass L. F. (2014) A systems approach to hemostasis: 2. Computational analysis of molecular transport in the thrombus microenvironment. Blood 124, 1816–1823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Meh D. A., Siebenlist K. R., and Mosesson M. W. (1996) Identification and characterization of the thrombin binding sites on fibrin. J. Biol. Chem. 271, 23121–23125 [DOI] [PubMed] [Google Scholar]
- 32. Lovely R. S., Moaddel M., and Farrell D. H. (2003) Fibrinogen γ' chain binds thrombin exosite II. J. Thromb. Haemost. 1, 124–131 [DOI] [PubMed] [Google Scholar]
- 33. Fredenburgh J. C., Stafford A. R., Pospisil C. H., and Weitz J. I. (2004) Modes and consequences of thrombin's interaction with fibrin. Biophys. Chem. 112, 277–284 [DOI] [PubMed] [Google Scholar]
- 34. Muthard R. W., Welsh J. D., Brass L. F., and Diamond S. L. (2015) Fibrin, gamma'-fibrinogen, and transclot pressure gradient control hemostatic clot growth during human blood flow over a collagen/tissue factor wound. Arterioscler. Thromb. Vasc. Biol. 35, 645–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yeh C. H., Fredenburgh J. C., and Weitz J. I. (2012) Oral direct factor Xa inhibitors. Circ. Res. 111, 1069–1078 [DOI] [PubMed] [Google Scholar]
- 36. Hemker H. C., Giesen P., AlDieri R., Regnault V., de Smed E., Wagenvoord R., Lecompte T., and Béguin S. (2002) The calibrated automated thrombogram (CAT): a universal routine test for hyper- and hypocoagulability. Pathophysiol. Haemost. Thromb. 32, 249–253 [DOI] [PubMed] [Google Scholar]
- 37. Zhu S., and Diamond S. L. (2014) Contact activation of blood coagulation on a defined kaolin/collagen surface in a microfluidic assay. Thromb. Res. 134, 1335–1343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Colace T. V., Fogarty P. F., Panckeri K. A., Li R., and Diamond S. L. (2014) Microfluidic assay of hemophilic blood clotting: distinct deficits in platelet and fibrin deposition at low factor levels. J. Thromb. Haemost. 12, 147–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Colace T. V., Jobson J., and Diamond S. L. (2011) Relipidated tissue factor linked collagen surfaces potentiates platelet adhesion and fibrin formation in a microfluidic model of vessel injury. Bioconjug. Chem. 22, 2104–2109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Colace T. V., Muthard R. W., and Diamond S. L. (2012) Thrombus growth and embolism on tissue factor-bearing collagen surfaces under flow: role of thrombin with and without fibrin. Arterioscl. Thromb. Vasc. Biol. 32, 1466–1476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Folie B. J., and McIntire L. V. (1989) Mathematical analysis of mural thrombogenesis: concentration profiles of platelet-activating agents and effects of viscous shear flow. Biophys. J. 56, 1121–1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kuharsky A. L., and Fogelson A. L. (2001) Surface-mediated control of blood coagulation: the role of binding site densities and platelet deposition. Biophys. J. 80, 1050–1074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Okorie U. M., Denney W. S., Chatterjee M. S., Neeves K. B., and Diamond S. L. (2008) Determination of surface tissue factor thresholds that trigger coagulation at venous and arterial shear rates: amplification of 100 fM circulating tissue factor requires flow. Blood 111, 3507–3513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Neeves K. B., Illing D. A., and Diamond S. L. (2010) Thrombin flux and wall shear rate regulate fibrin fiber deposition state during polymerization under flow. Biophys. J. 98, 1344–1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Downing M. R., Bloom J. W., and Mann K. G. (1978) Comparison of the inhibition of thrombin by three plasma protease inhibitors. Biochemistry 17, 2649–2653 [DOI] [PubMed] [Google Scholar]
- 46. Rühl H., Müller J., Harbrecht U., Fimmers R., Oldenburg J., Mayer G., and Pötzsch B. (2012) Thrombin inhibition profiles in healthy individuals and thrombophilic patients. Thromb. Haemost. 107, 848–853 [DOI] [PubMed] [Google Scholar]
- 47. Chandler W. L., and Velan T. (2003) Estimating the rate of thrombin and fibrin generation in vivo during cardiopulmonary bypass. Blood 101, 4355–4362 [DOI] [PubMed] [Google Scholar]
- 48. Rivard G. E., Brummel-Ziedins K. E., Mann K. G., Fan L., Hofer A., and Cohen E. (2005) Evaluation of the profile of thrombin generation during the process of whole blood clotting as assessed by thrombelastography. J. Thromb. Haemost. 3, 2039–2043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bird R. B., Stewart W. E., and Lightfoot E. N. (2006) Transport Phenomena, 2nd Ed., pp. 543–568, John Wiley & Sons, Inc., New York [Google Scholar]
- 50. Li R., Panckeri K. A., Fogarty P. F., and Diamond S. L. (2015) Recombinant factor VIIa enhances platelet deposition from flowing haemophilic blood but requires the contact pathway to promote fibrin deposition. Haemophilia 21, 266–274 [DOI] [PubMed] [Google Scholar]
- 51. Choi S. H., Smith S. A., and Morrissey J. H. (2011) Polyphosphate is a cofactor for the activation of factor XI by thrombin. Blood 118, 6963–6970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Choi S. H., Smith S. A., and Morrissey J. H. (2015) Polyphosphate accelerates factor V activation by factor XIa. Thromb. Haemost. 113, 599–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Smith S. A., Choi S. H., Davis-Harrison R., Huyck J., Boettcher J., Rienstra C. M., Reinstra C. M., and Morrissey J. H. (2010) Polyphosphate exerts differential effects on blood clotting, depending on polymer size. Blood 116, 4353–4359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Fogelson A. L., Hussain Y. H., and Leiderman K. (2012) Blood clot formation under flow: the importance of factor XI depends strongly on platelet count. Biophys. J. 102, 10–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Colace T. V., and Diamond S. L. (2013) Direct observation of von Willebrand factor elongation and fiber formation on collagen during acute whole blood exposure to pathological flow. Arterioscler. Thromb. Vasc. Biol. 33, 105–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ruiz F. A., Lea C. R., Oldfield E., and Docampo R. (2004) Human platelet dense granules contain polyphosphate and are similar to acidocalcisomes of bacteria and unicellular eukaryotes. J. Biol. Chem. 279, 44250–44257 [DOI] [PubMed] [Google Scholar]
- 57. Zilberman-Rudenko J., Itakura A., Wiesenekker C. P., Vetter R., Maas C., Gailani D., Tucker E. I., Gruber A., Gerdes C., and McCarty O. J. (2016) Coagulation factor XI promotes distal platelet activation and single platelet consumption in the bloodstream under shear flow. Arterioscler. Thromb. Vasc. Biol. 36, 510–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Swords N. A., and Mann K. G. (1993) The assembly of the prothrombinase complex on adherent platelets. Arterioscler. Thromb. 13, 1602–1612 [DOI] [PubMed] [Google Scholar]
- 59. Tracy P. B., and Mann K. G. (1983) Prothrombinase complex assembly on the platelet surface is mediated through the 74,000-dalton component of factor Va. Proc. Natl. Acad. Sci. U.S.A. 80, 2380–2384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zhu S., Tomaiuolo M., and Diamond S. L. (2016) Minimum wound size for clotting: flowing blood coagulates on a single collagen fiber presenting tissue factor and von Willebrand factor. Integr. Biol. (Camb.) 8, 813–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Neeves K. B., Maloney S. F., Fong K. P., Schmaier A. A., Kahn M. L., Brass L. F., and Diamond S. L. (2008) Microfluidic focal thrombosis model for measuring murine platelet deposition and stability: PAR4 signaling enhances shear-resistance of platelet aggregates. J. Thromb. Haemost. 6, 2193–2201 [DOI] [PubMed] [Google Scholar]
- 62. Maloney S. F., Brass L. F., and Diamond S. L. (2009) P2Y12 or P2Y1 inhibitors reduce platelet deposition in a microfluidic model of thrombosis while apyrase lacks efficacy under flow conditions. Integr. Biol. (Camb.) 2, 183–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Duckers C., Simioni P., Spiezia L., Radu C., Dabrilli P., Gavasso S., Rosing J., and Castoldi E. (2010) Residual platelet factor V ensures thrombin generation in patients with severe congenital factor V deficiency and mild bleeding symptoms. Blood 115, 879–886 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.