

Abstract
A recent report suggested that the thioredoxin-dependent metabolic regulation, which is widespread in all domains of life, existed in methanogenic archaea about 3.5 billion years ago. We now show that the respective electron delivery enzyme (thioredoxin reductase, TrxR), although structurally similar to flavin-containing NADPH-dependent TrxRs (NTR), lacked an NADPH-binding site and was dependent on reduced coenzyme F420 (F420H2), a stronger reductant with a mid-point redox potential (E′0) of −360 mV; E′0 of NAD(P)H is −320 mV. Because F420 is a deazaflavin, this enzyme was named deazaflavin-dependent flavin-containing thioredoxin reductase (DFTR). It transferred electrons from F420H2 to thioredoxin via protein-bound flavin; Km values for thioredoxin and F420H2 were 6.3 and 28.6 μm, respectively. The E′0 of DFTR-bound flavin was approximately −389 mV, making electron transfer from NAD(P)H or F420H2 to flavin endergonic. However, under high partial pressures of hydrogen prevailing on early Earth and present day deep-sea volcanoes, the potential for the F420/F420H2 pair could be as low as −425 mV, making DFTR efficient. The presence of DFTR exclusively in ancient methanogens and mostly in the early Earth environment of deep-sea volcanoes and DFTR's characteristics suggest that the enzyme developed on early Earth and gave rise to NTR. A phylogenetic analysis revealed six more novel-type TrxR groups and suggested that the broader flavin-containing disulfide oxidoreductase family is more diverse than previously considered. The unprecedented structural similarities between an F420-dependent enzyme (DFTR) and an NADPH-dependent enzyme (NTR) brought new thoughts to investigations on F420 systems involved in microbial pathogenesis and antibiotic production.
Keywords: archaea, electron transfer, evolution, redox regulation, thioredoxin reductase, coenzyme F420, Methanocaldococcus jannaschii, deazaflavin, flavin-gated, methanogen
Introduction
The thioredoxin (Trx)2 system is key to the cellular redox homeostasis in almost all organisms examined (1–5). It is composed of Trxs, Trx reductases (TrxR), and a reductant (1–5). Trx reduces Cys-disulfide of target proteins influencing their physical and/or catalytic properties and thereby institutes redox-based regulation of cellular metabolism as well as repairs oxidatively damaged proteins (1–5). It also provides reducing equivalents for select enzymes (2). Oxidized Trx is reduced by two types of TrxRs, one contains flavin and uses NADPH as electron source (NTR) and the other contains an iron-sulfur cluster and is ferredoxin-dependent (FTR).
The Trx system is known for its extensive involvement in the metabolic regulation and defense against oxidative stress in numerous aerobic organisms (1–5). However, it has also been investigated significantly in the anaerobic organisms (6–24) focusing on both the characteristics of the Trxs and TrxRs (6, 8, 9, 11–13, 15–23) and their physiological roles (6, 7, 10, 14, 20). For example, the isolation of the putative Trx targets via Trx-affinity chromatography and the results from activation assays with select purified and deactivated enzymes have established that in Chlorobaculum tepidum, a green sulfur bacterium and an anaerobic phototroph, the thioredoxin system implements post-translational control on the tricarboxylic acid or the TCA cycle and sulfur metabolism and assists in the defense against oxidative stress (7). An analysis of mutant phenotypes and a gene expression study have revealed the critical roles of Trxs and TrxRs in the redox homeostasis and survival during oxidative stress in Bacteroides fragilis and Desulfovibrio vulgaris Hildenborough, respectively (6, 14). Work with Clostridium pasteurianum, an anaerobic fermentative bacterium, has revealed a ferredoxin-dependent flavin containing TrxR (8). Similarly, there have been several reports on the Trx and TrxRs of anaerobic archaea (9, 15, 17–23), including the methanogenic archaea, which are strict anaerobes (9, 17, 18, 21–23). Recently, a proteomics study has shown that there is clear potential for a Trx-based global redox regulation of metabolism, including methanogenesis, in Methanocaldococcus jannaschii, a strictly anaerobic and deeply rooted hyperthermophilic methanogenic archaeon that lives in deep-sea hydrothermal vents (20). Two reports also describe the activation of deactivated methanogen proteins by thioredoxin (10, 20).
In this report, we show that the electron delivery enzyme of the above-mentioned redox regulation system of M. jannaschii is tuned for operation under highly reduced conditions. This archaeon carries two Trxs, Mj-Trx1 which is a typical Trx, and Mj-Trx2 with unknown activity, and a TrxR (Mj-TrxR) that is an NTR homolog yet does not use NAD(P)H (20). We demonstrate that Mj-TrxR uses coenzyme F420, a deazaflavin derivative present in all methanogens, as electron carrier. We reveal novel redox properties of this deazaflavin-dependent flavin-containing thioredoxin reductase (DFTR) and present the respective ecological implications. Also, the structural and phylogenetic analyses of DFTR homologs bring up new possibilities with the long studied and widespread disulfide oxidoreductase family of flavoenzymes and the expanding field of F420-dependent enzymes. Similarly, the properties and distribution of DFTR and the conditions prevailing in M. jannaschii's habitat lead to the proposal that this enzyme developed in the low redox environment of early Earth and gave rise to NTR. This implication fits the hypothesis that the Trx-based redox control developed on Earth long before oxygen appeared (20).
Results
Discovery of a Deazaflavin-dependent Thioredoxin Reductase
The thioredoxin reductase of M. jannaschii (Mj-TrxR) was expressed recombinantly in E. coli with an NH2-terminal His6 tag, and it was soluble. The recombinant protein was purified to homogeneity via Ni2+-affinity chromatography, and the His6 tag was removed via TEV-protease digestion. The UV-visible spectra of the purified protein with absorbance maxima at 380 and 460 nm indicated that it contained flavin (Fig. 1A) (25). However, it had a partial incorporation of this cofactor, and incubation with FAD led to full incorporation. The denatured molecular mass of the protein as determined via SDS-PAGE was found to be 33 kDa (Fig. 1B), and the value of the apparent native molecular mass as estimated via size exclusion chromatography was 63 kDa. Thus, the native Mj-TrxR was likely a homodimer of 33-kDa subunits, which is a typical property of low molecular weight flavin-containing thioredoxin reductases (1–4, 25).
FIGURE 1.

Structural and spectroscopic characteristics of recombinant Mj-DFTR. A, UV-visible spectrum of purified recombinant Mj-DFTR. A solution of the protein (10 μg) in 1 ml of solution containing 100 mm potassium phosphate buffer, pH 7, was analyzed in a cuvette with 1-cm light path. Prior to analysis, the protein was reconstituted with FAD to provide full incorporation of the coenzyme (A280/A460 nm = 4). B, SDS-PAGE profile. A 12% polyacrylamide gel was used. M, unstained protein ladder, broad Range (New England Biolabs, Ipswich, MA), with molecular masses of 250, 150, 100, 80, 60, 50, 40, 30, 25, 20, 15, and 10 kDa; selected bands have been marked to the left of the image. Two micrograms of Mj-DFTR in a solution containing 100 mm potassium phosphate buffer, pH 6.8, was analyzed.
The initial test for the Trx reductase activity in Mj-TrxR was via the commonly used insulin disulfide reduction assay (26), and it was performed under anaerobic conditions (20, 27). It utilized the following electron flow: electron donor → Mj-TrxR → Mj-Trx → insulin. In this assay, Mj-TrxR did not reduce either Mj-Trx1 or Mj-Trx2 with NADH, NADPH, coenzyme M, or reduced ferredoxin as electron donor. However, with dithionite, an effective artificial electron donor for NTR (28), Mj-TrxR reduced Mj-Trx1 but not Mj-Trx2. Thus, Mj-TrxR was a bona fide Trx reductase and recognized Mj-Trx1 as a substrate. UV-visible spectra of Mj-DFTR mixtures incubated with various electron donors are shown in Fig. 2. Then, we found that with reduced coenzyme F420 (F420H2), Mj-TrxR reduced Mj-Trx1. The details for this assay appear below. Coenzyme F420 is a 7,8-didemethyl-8-hydroxy-5-deazaflavin derivative, and accordingly, we called the Mj-TrxR a DFTR (Fig. 3).
FIGURE 2.

UV-visible spectra of Mj-DFTR incubated with various reductants. Oxidized Mj-DFTR (30 μm) was incubated anaerobically in a solution containing 100 mm potassium phosphate buffer, pH 7, with one of the following compounds at the indicated final concentrations: F420H2, 0.1 mm; dithionite, 1 mm; NADPH, 0.1 mm. Oxidized Mj-DFTR-bound flavin showed typical flavin absorption peaks at 380 and 460 nm (32). F420 showed absorbance maxima at 420 nm (32).
FIGURE 3.

Electron flow in the deazaflavin-dependent thioredoxin reductase system of M. jannaschii. F420H2-generating systems: F420-reducing hydrogenase (Frh) (93); Hmd-Mtd cycle, H2-dependent methylenetetrahydromethanopterin dehydrogenase (Hmd) + F420H2-dependent methylenetetrahydromethanopterin dehydrogenase (Mtd) (94). Inset, structures of coenzyme F420 and F420H2. *, the Trx targets and Trx controlled systems were identified in proteomics study (20).
Kinetic Properties of Mj-DFTR
The enzyme activity was assayed by following the oxidation of F420H2 spectrophotometrically at 400 nm; F420, but not F420H2, has a detectable absorbance at the 360–450 nm range with a maximum at 420 nm, and 401 nm is the isosbestic point with respect to pH (29). Concentration of Trx was held constant by coupling the assay to the reduction of oxidized glutathione or GSSG (30). The flow of electrons in the assay was as follows: F420H2 → DFTR → Trx → GSSG. The DFTR activity was not observed in the absence of Trx or TrxR in the assay.
The activity of the enzyme increased with a rise in temperature up to 90 °C; the assays at higher temperatures could not be performed due to technical limitations. The activation energy of the DFTR-catalyzed reaction as determined from the slope of the linear section of the Arrhenius plot (50–90 °C) was 36.14 kJ mol−1 (Fig. 4A). The optimal pH for activity was 8.0 (Fig. 4B).
FIGURE 4.

Effects of temperature and pH on the activity of Mj-DFTR. Concentrations of Mj-Trx1 and F420H2 in the activity assay were 10 and 50 μm, respectively. A, temperature study. Activities at 10 different temperatures ranging from 25 to 90 °C were measured. Inset, re-plot of the temperature data according to the Arrhenius equation, k = A e−Ea/(RT), where k, A, Ea, R, and T are rate constant, frequency factor, energy of activation (kJ mol−1), universal gas constant (8.314 J K−1 mol −1), and temperature (K), respectively. The value of the energy of activation was calculated from the slope (− Ea/R) of the linear segment of the curve (50–90 °C). B, pH Study. Assays were conducted at 11 different pH values ranging from 4 to 9 with 0.5-unit increment in buffers with constant ionic strength (32).
At a fixed F420H2 concentration of 60 μm and varying concentrations of Mj-Trx1 (0.5–40 μm), the apparent Km for Mj-Trx1 was found to be 6.3 ± 2 μm (Fig. 5A). Under similar conditions and with Mj-Trx2 concentrations of 25–500 μm, the value of Km for Mj-Trx2 was 371 ± 211 μm (Fig. 5B). Assays with 15 μm Mj-Trx1 and 4–60 μm F420H2, yielded an apparent Km value of 28.6 ± 2.5 μm for F420H2 (Fig. 5C). The specific activity of the enzyme with 60 μm F420H2 and 30 μm Mj-Trx1 was 29.7 ± 1.3 μmol/min/mg. The reactivity of DFTR toward Mj-Trx2 was poor, and the respective Vmax value was 3-fold lower than that observed with Mj-Trx1. Also, unlike most Trxs, Mj-Trx2 does not reduce insulin, and in contrast to Mj-Trx1, it is not recognized by Escherichia coli NTR (20). Thus, it is unclear whether this protein is a true Trx and how it is reduced or oxidized.
FIGURE 5.

Kinetic analysis of Mj-DFTR reaction. Each panel provides specific activities of the enzyme at various concentrations of a substrate as indicated: A, Mj-Trx1; B, Mj-Trx2; C, F420H2; D, DTNB. Assay at each concentration of the varied substrate was performed in triplicate. Each solid curve represents the best fit of the respective data to the Henri-Michaelis-Menten's hyperbola v = Vmax[S]/Km/[S]. ±, standard deviation.
The enzyme reduced 5′,5′-dithiobis(2-nitrobenzoate) (DTNB), an artificial electron acceptor. In this property DFTR followed NTRs from anaerobic and/or thermophilic archaea and bacteria, which exhibit DTNB reductase activity (9, 16, 31). From assays with a fixed concentration of F420H2 of 60 μm and a varied concentration of DTNB (50–1000 μm), an apparent Km value of DFTR for DTNB of 182.2 ± 70 μm was obtained (Fig. 5D). The specific activity of DTNB reduction at F420H2 and DTNB concentrations of 60 and 1000 μm, respectively, was 26.8 ± 0.4 μmol/min/mg. Unlike the high molecular mass NTRs, DFTR did not reduce lipoate and selenite, and low molecular mass NTRs in general behave similarly (2, 3).
Redox Properties of Mj-DFTR, E′0 Values for the Redox Centers
Similar to prokaryotic NTRs that are composed of two identical approximate 35-kDa subunits (32), the DFTR is expected to have two redox centers as follows: a protein-bound FAD and a catalytically active Cys pair residing within a CXXC motif (Fig. 6). We determined the mid-point redox potential (E′0) values for these two centers via redox titration as follows.
FIGURE 6.

Amino acid sequence alignment of Mj-DFTR and other low molecular weight flavin-containing TrxRs. The alignment was performed by the use of PROMALS3D (92) with the three-dimensional structures listed below as guides. % identities and % similarities with Mj-DFTR: Ta-TrxR, 36, 50; Tm-NTR, 37, 53; Ec-NTR, 33, 50; Cp-TrxR, 27, 43. Abbreviations (organism, enzyme, NCBI accession number, PDB code) are as follows: Mj-DFTR (M. jannaschii, DFTR, Q58931); Ta-TrxR (T. acidophilum, non-NAD(P)H-dependent TrxR, WP_010901395, 3CTY); Tm-NTR (T. maritima, NADH-NADPH dual specificity NTR, AAD35951.1); Ec-NTR (E. coli, NTR, WP_001460710, 1CL0); Cp-TrxR (C. pasteurianum, ferredoxin-TrxR, WP_015617437.1). Conserved amino acids are in bold. Consensus predicted secondary structures: red h, α-helix; blue e, β-strand. Gray shading, the redox active CXXC motif. Black bar, conserved motifs as found in low molecular weight NTR and flavoproteins and are labeled as such. DBM, dinucleotide-binding motif.
(i) For the E′0 for the Cys-disulfide/Cys-SH pair of the catalytically active CTMC motif of Mj-DFTR, this value was determined via the measurement of the fluorescence emission intensities of mBBr-labeled forms of the enzyme preparations that were subjected to incubation in a series of redox buffers offering a potential range of −350 to −200 mV (Fig. 7A). The data fit the Nernst equation for a 2-electron reduction reaction and yielded an E′0 value of −279 ± 7 mV at pH 7.0 (Fig. 7A) which compared well to the reported values of 230–277 mV for the E′0 of Trxs from a variety of sources (33).
FIGURE 7.

Redox titration of active site Cys-disulfide-dithiol and bound flavin of Mj-DFTR. Each data point in A, C, and D or each spectrum in B represent an average from three independent measurements. Consequently, each error bar shown is for three independent measurements. In each case the dataset from one biological replicate is shown. The E′0 values derived from two biological replicates (D) were similar (data not shown). A, redox titration of Cys-disulfide-dithiol: Oxidized Mj-DFTR (30 μm) was incubated in a series of solutions containing 100 mm MOPS buffer, pH 7.0, and set at varying potential (E′) values with mixtures of DTT and oxidized DTT at appropriate ratios. The total concentration of DTT and oxidized DTT was 5 mm. % thiol, portion of the Mj-DFTR molecules with the active site Cys residues at the thiol state was quantified via a fluorimetric assay for mBBr-labeled Cys-thiolate; the fluorescence intensity values for the fully oxidized and fully reduced preparations were set to 0 and 100%. B, UV-visible spectra of Mj-DFTR redox titration mixtures with F420H2 as a reductant. The titration of Mj-DFTR-bound FAD involved the addition of various amounts of F420H2 (at final concentrations of 5–115 μm) to a series of solutions containing oxidized Mj-DFTR (25 μm) and 100 mm potassium phosphate buffer, pH 7.0, inside an anaerobic chamber. The labels on the spectra (0.2, 0.5, 0.7, 1, 1.5, 2, and 3) are the amounts of added F420H2 presented in terms of equivalents with respect to the Mj-DFTR-bound FAD; the label “oxidized Mj-DFTR” is for zero F420H2. The isosbestic points that occurred at 445 and 500 are marked with asterisks. C, changes in extinction coefficient values at 480 (filled circles) and 540 nm (open circles) of the Mj-DFTR mixtures as described in B. D, redox titration of Mj-DFTR-bound FAD with dithionite in the presence of methyl viologen, a redox dye, as a reference. The concentration of Mj-DFTR-bound FAD in a mixture was determined from the respective A460 nm value. % FADH2, percentage of the bound flavin in the FADH2 state as calculated from the concentration of Mj-DFTR-bound FAD in a mixture with dithionite and that in a control mixture without dithionite. The E′0 value shown was obtained by fitting the plotted data to the Nernst equation for a 2-electron reduction reaction (n = 2) (see Equation 2).
(ii) For the E′0 of the Mj-DFTR-bound flavin cofactor, our first attempt to obtain this value involved redox titration of Mj-DFTR with F420H2, which revealed a complex picture for the interaction between this substrate and the enzyme that has been elaborated under the “Discussion.” The second attempt involved an artificial reductant and provided the E′0 value. We describe both below.
Titration with F420H2
Because Mj-DFTR carried only one catalytically active Cys pair, it was expected to follow the mechanism reported for E. coli NTR (34) and allow the determination of the E′0 value from the equilibrium constant (Keq) for the Mj-DFTR reaction (Eox + F420H2 → EH2 + F420, where E is Mj-DFTR) and the E′0 of the F420/F420H2 pair, which is −360 mV (36) (Eox, oxidized enzyme; EH2, 2-electron reduced form of the enzyme). We attempted to obtain the Keq value from the changes in the UV-visible spectrum of Mj-DFTR upon redox titration with F420H2 (Fig. 7B). To avoid the wavelengths where all or most of the participating cofactors exhibit significant absorbance values (29, 37), the A480 and A540 values (where A is absorbance) were chosen for the determination of the concentrations of FAD and the FAD-thiolate charge transfer species from which similar information on other species could be calculated. Fig. 7C presents the resulting data in terms of respective molar absorbance values, ϵ480, EH2/Eox and Δϵ540, EH2/Eox. It was observed that both values attained saturation only after the addition of 2 eq of F420H2, and as judged from the 480 nm data, even at this stage only 1 eq of FAD was reduced. As elaborated under the “Discussion,” although these observations brought up interesting aspects of the enzyme, it also highlighted the need for a method that will involve a non-physiological reductant and an artificial reporting system and thereby would allow direct determination of the E′0 value of Mj-DFTR-bound FAD. Accordingly, an alternative method, as presented below, was used.
Titration with Dithionite in the Presence of Methyl Viologen (MV) as Redox Reference
The primary basis for this measurement was that at any point of the titration, the E′ of Mj-DFTR-FAD/Mj-DFTR-FADH2 (E′Mj-DFTR-FAD/Mj-DFTR-FADH2) and the MV+/MV0 (E′MV+/MV0) would be the same. The reduction of FAD and methyl viologen (MV+) to FADH2 and MV0 (1-electron reduced methyl viologen) with various amounts of dithionite was followed spectrophotometrically; the former caused a decrease in A460 and the latter raised A604. The concentrations of reduced MV0 and enzyme-bound FAD were determined from A604 and A460 values, respectively, employing the following extinction coefficient values: MV0, 12 mm−1 cm−1 at 604 nm and pH 7 (a value determined experimentally using the published value of 13.5 mm−1 cm−1 at pH 8 (38)); enzyme-bound FAD, 12.7 mm−1 cm−1 at 460 nm and pH 7, determined with oxidized enzyme carrying one FAD/subunit. With the concentrations of MV+ and MV0, the E′ values of the titrated mixtures were calculated from the Nernst equation for a 1-electron transfer reaction (Equation 1),
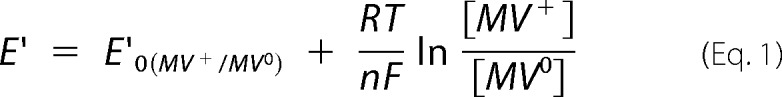 |
where E′0 of the MV+/MV0 pair was −446 mV (38). Then the equilibrium concentrations of FADH2 versus E′ data were fitted to Equation 2,
 |
which considers a 2-electron transfer reaction (n = 2), yielding an E′0 value of −389 ± 24 mV for FAD-bound Mj-DFTR (Fig. 7D).
Primary Structure Features of DFTR
Mj-DFTR was composed of 301 amino acids with theoretical molecular mass and pI values of 33 kDa and 6.86, respectively, which are typical of low molecular mass NTRs (2, 3). At the amino acid sequence level, it was more similar to flavin containing TrxRs of Thermoplasma acidophilum (19), a facultative anaerobic archaeon (39), and Thermotoga maritima (16), an anaerobic bacterium (40), than to E. coli NTR (41) and ferredoxin-dependent and flavin containing TrxR of C. pasteurianum, an anaerobic fermentative bacterium (8) (Fig. 6); the respective percent identity and similarity values appear in the legend of Fig. 6. The nature of the reductant used by TrxR of T. acidophilum is unknown (19), and the enzyme of T. maritima is an NTR of dual specificity for NADH and NADPH (16); both organisms are thermophiles (39, 40). Fig. 6 presents an amino acid sequence alignment of Mj-DFTR with the above-mentioned flavin containing TrxRs (8, 16, 19, 41) that leveraged the x-ray crystallographic structures of the E. coli and T. acidophilum enzymes (PDB codes 1CL0 and 3CTY, respectively (19, 41)).
The defining features of NTR are the FAD- and NADPH-binding elements and an active site CXXC motif with two redox-active cysteine residues (X, non-conserved amino acid residue), and Mj-DFTR mimicked NTR in the FAD-binding elements but not in the rest (Fig. 6). The NTR interacts with FAD via a dinucleotide-binding motif (DBMFAD) encompassing three conserved elements (GXGXXG, GG, and ATG) and two additional COOH-terminal binding sequences (GD and a G-helix) (42). Mj-DFTR contained all these features (Fig. 6). The NADPH-binding elements of NTR (DBMNADPH) are GXGXX(G/A), GGGXXA, and HRR motifs, and the last element is for binding the 2′-phosphate group of the coenzyme. In Mj-DFTR, the sequence elements equivalent to GXGXX(G/A) and GGGXXA of NTR were found to be GRGISY and GRDTPA, respectively, representing a replacement of the glycine/alanine residue by a larger aromatic and hydrophobic amino acid for the first and a substitution of two glycine residues with two charged residues for the second (Fig. 6). In addition, the 2′-phosphate-binding HRR residues of NTRs were absent in Mj-DFTR. Another special feature was found at the active site of Mj-DFTR, which carried a CTMC motif, whereas E. coli NTR carries CATC at this location (41); the respective element in T. acidophilum TrxR is CSTC (19).
As true for the majority of F420-dependent enzymes (43), the coenzyme F420-binding amino acid residues of Mj-DFTR could not be identified from primary and secondary sequence alignments. It is known that the features of the F420-binding sites of different groups of F420-dependent enzymes are different (43–45).
Distribution and Phylogenetic Clades of DFTR Homologs
Because of high sequence similarities between flavin-containing thioredoxin reductases and other pyridine dinucleotide oxidoreductase family proteins (46), especially the alkyl hydroperoxide reductases, we employed a two-step method involving the following for the identification of DFTR homologs: identification of candidates via Psi-BLAST searches (47) using Mj-DFTR (locus number, MJ1536) as a query and with stringent parameters (low e-value, 1e−25; 10 iterations); phylogenetic analysis-based screening of the candidates. Such an analysis of the extracted methanogen proteins suggested that within this group the DFTR homologs were limited to the phylogenetically deeply rooted methanogens belonging to the class of Methanococci under the euryarchaeal phylum, and these organisms lacked NTR and ferredoxin-thioredoxin reductase (FTR) homologs (Fig. 8 and supplemental Table S1). Other methanogens carried NTR, and some of these harbored one or more FTR homologs as well (Fig. 8 and supplemental Table S1). A close homolog of DFTR was not found in the genomes of bacteria and non-methanogenic archaea. However, as discussed below, the study suggested that some of the yet to be studied TrxRs have the potential of using electron donors other than NADPH.
FIGURE 8.

Thioredoxins and thioredoxin reductases in representative methanogens. The information is presented on a maximum likelihood inference-based 16S-rRNA phylogenetic tree. Dash, not detectable in homology based searches; black bullet at the branches, a confidence value >70%; scale bar, number of base substitution per site; Trx, thioredoxin; NTR, FTR, and DFTR, NADPH-, ferredoxin-, and deazaflavin-dependent thioredoxin reductase, respectively. The NCBI accession numbers for Trxs and Trx reductases are listed in supplemental Table S1.
Fig. 9 presents the results from a phylogenetic analysis of the NTR homologs (including DFTR) of all methanogens, selected non-methanogenic archaea, known non-canonical (non-NAD(P)H utilizing) flavin containing TrxRs, and selected TrxRs of bacteria and archaea that have been studied via direct assays; some information about these proteins appear in supplemental Table S2. In this analysis the homologs of DFTR and ferredox in-dependent clostridial TrxRs formed two tightly clustered groups (marked with F420H2 and Ferredoxin, respectively, in Fig. 9) that seemed to have diverged recently from a common ancestor. The homologs of the non-canonical TrxR of T. acidophilum with unknown electron donor specificity (19) also formed a cohesive clade (Non-NAD(P)H I, Fig. 9). However, it was closely related to an NTR group, which included NADPH-dependent TrxR of Lactobacillus casei (protein accession number, WP_012491289.1), which is a facultative anaerobic bacterium (Fig. 9). The study revealed four more clades of potentially non-canonical enzymes. The non-NAD(P)H II group contained TrxR of D. vulgaris (D.vulgaris_3 in Fig. 9; protein accession number AAS95935.1) that does not utilize NAD(P)H and uncharacterized TrxRs of halophilic archaea, whereas the non-NAD(P)H III clade included experimentally validated Sulfolobus solfataricus TrxR-1 (S. solfa_1, protein accession number WP_010923960.1) and certain unstudied enzymes of methanogens from the Methanomicrobia class (Fig. 9). Two additional groups of methanogen TrxRs of non-canonical types (Unknown I and Unknown II, Fig. 9) were also identified, and none of these has been examined experimentally. However, their relationships with the phylogenetically nearest neighbors suggested that the TrxRs of the Unknown I group are likely to be NTRs. The T. maritima TrxR that can use both NADH and NADPH (16) belonged to an NTR group containing bona fide enzymes from D. vulgaris (D.vulgaris_2, accession number AAS94860) (6) and Methanosarcina acetivorans (accession number, AAM04784) (9).
FIGURE 9.

Maximum likelihood phylogenetic analysis of DFTR and selected flavin-containing TrxRs. The phylogenetic tree was constructed as described previously (91). Name in boldface, bona fide TrxR validated by direct activity assay; details are in supplemental Table S1. Black and white bullets near branches: bootstrap values >70 (calculated from 100 replicates). Each label appearing on the outer side of the tree; electron donor is utilized by the respective clade. Unknown, TrxR with unknown reductant. Non-NAD(P)H, inability to use NAD(P)H as electron donor as determined via direct activity assay of representative members (supplemental Table S1). Symbols for the departures in the amino acid sequences at select conserved elements of TrxRs as described in Fig. 6: filled black and gray circles, unrecognizable HRR and GGG motifs, respectively; open circle, absence of the GG motif. Ec-DLD, dihydrolipoamide dehydrogenase of E. coli used as an outgroup. Details for the abbreviations for the host organism names and the accession numbers of respective TrxRs are listed in supplemental Table S2.
All known and putative non-canonical TrxRs lacked the phosphate-binding HRR motif, and most of them contained non-E. coli NTR-type CXXC motifs. However, the candidate non-canonical TrxRs could not be analyzed for their potentials of utilizing F420 as electron carrier, because the F420-binding residues of DFTR are not known, and the F420-binding residues do not show broad conservation across F420-dependent enzymes families (43).
The case of Methanopyrus kandleri, a strictly hydrogenotrophic methanogen that lives in deep-sea hydrothermal vents and grows at temperatures as high as 110 °C (48), raised an intriguing question. It does not carry a Trx or DFTR but an NTR-type TrxR (Fig. 8), offering an opportunity of discovering a new TrxR reductase substrate. For evolutionary deductions, this enzyme presents a problematic case, because in terms of 16S rRNA sequence M. kandleri appears closely related to the Methanococci (Fig. 8), and the whole genome-based analysis contradicts this position (49).
Discussion
The results described above revealed a novel type of thioredoxin reductase and provided the mechanistic basis for the dependence of the enzyme on coenzyme F420 and the respective ecological relevance. A comparative study with this enzyme provided new information about the catalytic properties and evolutionary biology of a long-studied and broadly important family of enzymes and the evolutionary development of the widely distributed NTR. It also presents leads for the expanding field of F420-dependent enzymes in a broad range of organisms. We elaborate these outcomes below.
Our study began with a bioinformatic analysis that showed that every methanogen carried at least one NTR homolog, and FTR was present only in late evolving and nutritionally diverse species that live in close association with anaerobic bacteria (Fig. 8); only three species contained more than one NTR homolog (Fig. 8). FTR is thought to have originated in bacteria and acquired by the late evolving methanogens through horizontal gene transfer (50). Thus, NTR homologs could be considered a fundamental and ancient component of methanogens. An analysis of the structural features identified the NTR homologs of late evolving methanogens as potentially true NTRs, whereas those of Methanococci, such as M. jannaschii, were non-canonical types (Figs. 6 and 9); as mentioned under the “Results,” Methanococci lacked FTR (Fig. 8). M. jannaschii and all other Methanocaldococcus species carried one non-canonical TrxR and 2–4 Trxs, presenting a minimal system among the methanogens (Fig. 8; supplemental Table S1), and these organisms live in the deep-sea hydrothermal vents presenting early Earth-like environments that are characterized by low redox potential values and high temperatures (51, 52). Therefore, we hypothesized that Methanocaldococcus species carry the history of the development of NTR, and to explore this possibility further, we have investigated the catalytic properties of M. jannaschii TrxR (Mj-TrxR).
Matching the predictions from the structural features (Fig. 6), Mj-TrxR was found to contain bound flavin and did not utilize NAD(P)H. It utilized coenzyme F420H2, a deazaflavin derivative, an electron donor, and this is the first report of a DFTR. F420 is present in high abundance in all methanogenic archaea tested (29, 53), and the gene for F0-synthase, the key F420-biosynthesis enzyme (54, 55), is present in all methanogen genomes (54, 55). The coenzyme is present in trace amounts in Halobacteria and Thermoplasma (56), which are non-methanogenic archaea, and among the bacteria, it is found mostly in the Actinobacteria (57). In methanogens, F420 is the electron carrier for 12 previously described enzymes (36), and six of these utilize F420H2 (43, 44, 58, 59). The observed Km value for F420H2 (28.6 μm) was similar to that recorded for several F420H2-oxidizing enzymes (5.4–150 μm) (58, 59), and a similar agreement was seen between the Km values of DFTR for Mj-Trx1 DFTR (6.3 μm) and that of other thioredoxin reductases for their cognate Trxs (0.6–86 μm) (9, 15). These values were also physiologically meaningful; the F420 contents of methanogen cells fall in the range 200–4000 pmol/mg dry cell weight (36), which roughly translates to an intercellular concentrations of 60–1200 μm, assuming that 70% of the mass of a cell is due to water.
The chemistry of the other analogous F420-dependent enzymatic reactions suggested that DFTR would use F420 for hydride transfer, a role also served by NAD(P)+ (60). Because the Mj-DFTR-bound FAD could be reduced with F420H2 (Fig. 3) and reduced enzyme could be oxidized with Mj-Trx1, the enzyme was expected to exhibit a Ping-Pong mechanism involving the above-mentioned two independent half-reactions. A preliminary bi-substrate kinetic analysis (Fig. 10) supports this expectation as the reciprocal plots for the initial velocity data collected with varied concentrations of F420H2 (5–60 μm) and Mj-Trx1 at two fixed concentrations (2 and 15 μm) produced two parallel lines. Because there were indications of complications arising from inhibition of the enzyme by F420H2 and the generation of single-electron reduced deazaflavin species (37) in the presence of EDTA and visible light used in spectrophotometry, albeit in very minor amounts, the detailed mechanistic studies will be pursued and reported in the future. It should be noted that to avoid such a problem, all redox titrations were performed in the absence of EDTA.
FIGURE 10.
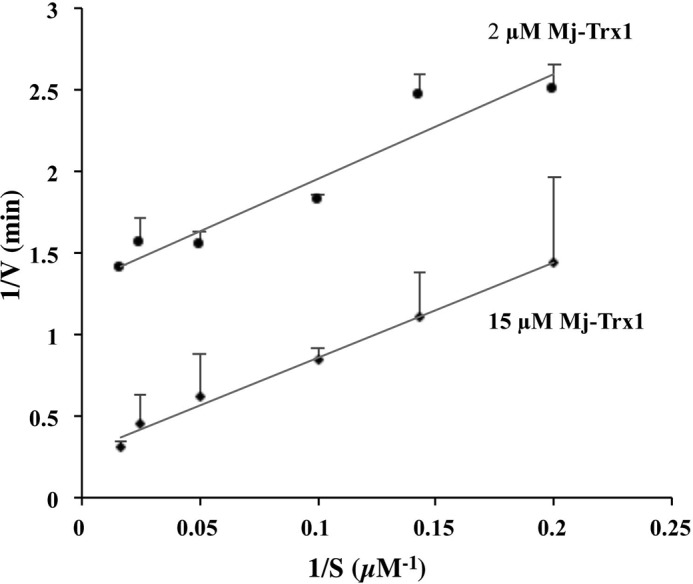
Catalytic mechanism of Mj-DFTR. The assays were performed anaerobically in a mixture containing 100 mm potassium phosphate, pH 7.0, 2 mm EDTA, 1 mm oxidized glutathione, and varying concentrations of F420H2 (5, 7, 10, 20, 40, and 60 μm) at two fixed concentrations of Mj-Trx1 (2 or 15 μm) at 70 °C. Each initial rate value is an average over three replicates. Error bar, standard deviation of samples.
The observed conservation of several key residues of NTR in DFTR (Fig. 6) brought up the following question. Why can't DFTR utilize NAD(P)H and need a low potential electron carrier such as F420 for activity? To answer this question, we determined the E′0 values for the Cys-disulfide/Cys-SH pair at the catalytic 130CXXC133 unit and the protein-bound FAD/FADH2 system. The former value (E′0, −279 mV) did not yield a surprise, as it was similar to the E′0 of the Cys-disulfide/Cys pair of E. coli NTR (−270 mV) (41).
In contrast, the results from the efforts to measure the E′0 value of Mj-DFTR-bound FAD provided the illuminating information about the redox properties of DFTR and the ecophysiological implications for the organisms carrying this enzyme. The first attempt to measure the E′0 value of Mj-DFTR-bound FAD involved redox titration of the oxidized form of the enzyme with F420H2. It presented several challenges. One was due to overlapping UV-visible spectra of the flavin and deazaflavin species that made measurement at the most responsive wavelengths (400–470 mm) rather difficult (Fig. 7B). When we opted for analysis at less sensitive wavelengths of 480 and 540 nm, which represented oxidized form of FAD and the charge transfer between FAD and Cys-thiolate at the active site, the results presented a complex picture. The observations at both 480 and 540 nm showed that the full reduction of the enzyme-bound flavin required 2 eq of F420H2 (Fig. 7C). This was surprising, as even the eukaryotic NTR from Drosophila melanogaster that carries two catalytically active Cys pairs attains the EH2 state with only 1 eq of NADH, and the consumption of 2 eq amounts of NADH converts this enzyme to an EH4 form (35). The D. melanogaster NTR cycles between EH2 and EH4 states during catalysis, and the UV-visible spectra gathered after redox titration of the protein with NADH exhibit four isosbestic points (35). In contrast, the titration of Mj-DFTR with F420H2 produced two isosbestic points (at 445 and 500 nm; Fig. 7B), which is indicative of an EH2 species (35). The consumption of 2 eq of F420H2 in part was certainly due to the reduction of the enzyme-bound FAD and the active site Cys-disulfide and was most likely a part of the added F420H2 bound to the enzyme. This is because the enzyme will be loaded with reducing equivalents at all sites, the F420H2-binding site, FAD, and the active site Cys pair, and as the equilibrium principle dictates, none of these sites will be saturated with the addition of 2 eq of F420H2. Even if it were possible to measure the concentration of these species, the analysis of the resultant data would be further challenged by the possibility that in the enzyme-bound form the deazaflavin would present an E′0 value that is different from that measured with the free forms of F420 and F420H2 (−360 mV) (36). We also found that the changes in the flavin spectrum at 540 nm during the titration with F420H2 (Fig. 7B) are not characteristics of a charge transfer between FAD and Cys thiolate (35). For these reasons, in our second attempt, we used dithionite as the reductant in the titration on Mj-DFTR, and the MV (the MV+/MV0 redox pair) was the redox reference. Both dithionite and methyl viologen are not expected to bind the enzyme. This analysis provided an E′0 value of −389 mV for the Mj-DFTR-bound FAD. This property makes the transfer of electrons from NAD(P)H to Mj-DFTR-bound FAD highly endergonic, as E′0 of NAD(P)+/NAD(P)H pair is −320 mV (60). Even with F420H2 as the electron source, a similar but less severe limitation is expected as the E′0 of F420/F420H2 is −360 mV (36). As discussed below, it would restrict the operation of the enzyme in an environment represented by the habitat of M. jannaschii. However, the FADH2 so generated would readily reduce the Cys-disulfide formed at the 130CMTC133 element that has an E′0 value of −279 mV. This design is consistent with the life style of M. jannaschii as described above and discussed below (51, 52, 61).
Although much remains to be learned about the redox properties of Mj-DFTR, including the structural elements that determine the redox properties of protein-bound flavins, the above-described redox characteristics could be rationalized in light of the conditions prevailing in the habitat of M. jannaschii. Within the deep-sea hydrothermal vents, the partial pressure of hydrogen varies from 4 Pa to 233 kPa (52), which translated to E′ values of −284 to −425 mV for the H+/H2 pair (calculated from the E′0 value of −414 mV of H+/H2 (62)). Because the reduction of F420 in a methanogen cell is well coordinated with the environmental partial pressure of hydrogen (63, 64), E′ of F420/F420H2 will also range between −284 and −425 mV and allow facile reduction of Mj-DFTR-bound FAD and efficient catalysis at high partial pressures of hydrogen. These observations call for a detailed mechanistic study on this novel thioredoxin reductase.
To examine the distribution of DFTR, we performed phylogenetic analysis of DFTR homologs and identified six groups of potentially non-canonical TrxRs (Fig. 9). These proteins carried non-NTR type CXXC motifs, did not contain a fully conserved nicotinamide cofactor-binding site, and fully lacked the 2′-phosphate-specific HRR motif (Fig. 6). In almost all these TrxRs, the CXXC center was preceded not by a GXGXX(G/A) motif as in an NTR but by GXGXXY (Fig. 9). The presence of Tyr, a large aromatic and hydrophobic residue, instead of a smaller Gly/Ala residue, at a position intervening an NADPH binding-type site and the CXXC motif could influence either the redox potential of the catalytic Cys residues or the electron donor specificity or both. In few cases, a Thr residue appeared in place of the Tyr (data not shown), suggesting a hydroxyl group and hydrophobicity as the common denominator for a residue at this location in non-canonical TrxRs. However, these departures from the NTR structure did not suggest the nature of the electron donors used by the non-canonical enzymes. In fact, the structure-guided comparative analysis presented in Fig. 6 indicated that this aspect would likely be determined by a limited number of changes within rather well conserved active site architecture. The difficulty of identifying these signatures is underscored by the DFTR as well as the following additional examples. The non-canonical TrxR of C. pasteurianum (9), an anaerobic bacterium, that uses ferredoxin with E′0 of approximately −400 mV (48), is a close relative of DFTR (Fig. 11), and it is likely that similar to DFTR the E′0 of the FAD bound to this clostridial enzyme is low enough to exclude the participation of NAD(P)H as an electron source and to justify the use of ferredoxin as electron carrier. However, none of these features could have been predicted from the primary structure of this enzyme. This case is further complicated by the fact that the sequence of the CXXC motif of this enzyme is same as that of E. coli NTR (Fig. 6). An opposite situation is presented by the T. maritima TrxR, which, despite having significant structural differences with the E. coli NTR (Fig. 6), is still a nicotinamide-dependent enzyme, using both NADH and NADPH (16). Another surprise is offered by the flavin-containing TrxR of T. acidophilum, an anaerobic archaeon, where the E′0 of the protein-bound FAD is −305 mV, and yet the enzyme cannot use NAD(P)H as a reductant (65). A recent report describes a TrxR from an aerobic cyanobacterium that is a close structural homolog of NTR and cannot use nicotinamides (66). The overall situation is reminiscent of the enolase superfamily, where few changes within a generally conserved active site have allowed transformation of a diversity of substrates (67). Therefore, it would be rather difficult if not impossible to identify new non-NTR flavin-dependent TrxRs and predict the natures of their electron donors solely via computational analysis. This caution aside, the report opens up the long studied flavin-dependent thioredoxin reductase family for further exploration. These enzymes belong to a broader disulfide oxidoreductase family of flavoenzymes (lipoamide dehydrogenase, glutathione reductase, trypanothione reductase, mercuric reductase, and NADH peroxidase), which are known for the diversity of their substrates (46). However, the members that have been studied thus far are nicotinamide-dependent, and the possibility that some of the uncharacterized members use other types of electron carriers has not been explored.
FIGURE 11.
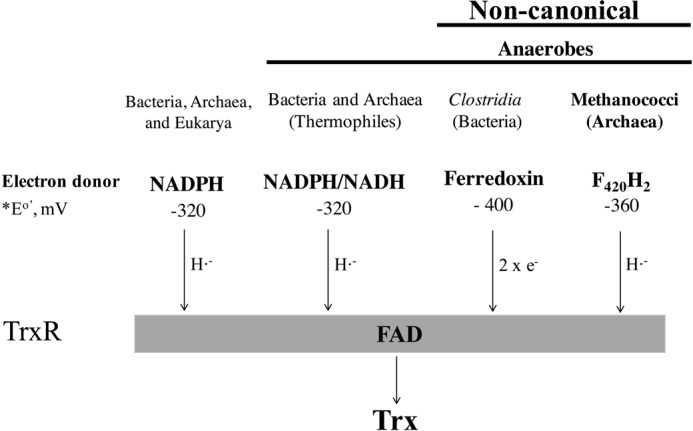
Diversity of electron donors of low molecular weight flavin-containing thioredoxin reductase (TrxR). e−, electron; H˙̄, hydride. General electron flow scheme: electron donor → bound FAD → disulfide of TrxR → disulfide of Trx. The E′0 standard mid-point redox potential for a reduced electron carrier/oxidized electron carrier pair.
Considering the possibility that the electron donor choices for the non-NAD(P)H-dependent enzymes could be diverse, we have proposed a naming strategy for this group where the identity of the electron donor (for example, deazaflavin or D) and the presence of flavin (F) are indicated. The four letter abbreviations, such as DFTR for deazaflavin-dependent flavin-containing thioredoxin reductase, will distinguish these enzymes from FTR (ferredoxin thioredoxin reductase), which contains iron-sulfur cluster and utilize ferredoxin (F) as electron carrier (5).
The DFTR brought a major surprise in the area of the structure-function relationships in deazaflavin-dependent enzymes. Although both F420 and NAD+(P) catalyze hydride transfer and facilitate chemically similar reactions, none of the previously described F420-dependent enzymes show meaningful similarities to functionally analogous or other type NAD+(P)-dependent enzymes (27, 43–45). In contrast, at the primary structural level DFTR exhibits 33% identities and 50% similarities to E. coli NTR, and they share several conserved sequence features (Fig. 6). This is highly significant, as F420 is key to biological production of methane by methanogens (36), antibiotic production by Actinobacteria (36, 57), and diseases such as tuberculosis (68–70). Thus, for the research on F420 metabolism in these areas and others, one now needs to consider the possibility of encountering deazaflavin-dependent enzymes that are significantly homologous to cognate nicotinamide-dependent enzymes. The discovery of DFTR provided a new perspective on the evolution of NTR. The distribution of DFTR homologs led to the hypothesis that the development of NTR occurred in concert with the environmental changes on Earth. It is striking that although coenzyme F420 is present in all characterized methanogens, DFTR homologs were limited to the most phylogenetically deeply rooted group euryarchaeal class of Methanococci (Figs. 8 and 9), which utilize primarily hydrogen as energy source (51, 71), carry limited numbers (2–4) of Trx homologs, and lack NTR and FTR. The more ancient members of this group, the Methanocaldococcus species live in the early Earth environment of deep-sea hydrothermal vents (51, 52). In contrast, late evolving methanogens, such as Methanobacteria and Methanomicrobia, were devoid of DFTR homologs, carried 1–9 Trxs, 1–3 NTRs, and 0–3 FTRs (9, 20). The Methanococci are more sensitive to oxygen than the late evolving methanogens (72). Thus, DFTR seems to be part of the simpler Trx systems that exist in more fastidious and phylogenetically and deeply rooted methanogens inhabiting niches presenting early Earth type reduced environment with an abundant supply of hydrogen, and a transition to the NTR and FTR systems brought about more tolerance to oxygen exposure or it is a consequence of an adaptation to niches that experience occasional oxygen exposure and/or less reduced conditions. Hence, DFTR likely represents one of the early forms of flavin-containing thioredoxin reductases from which NTR developed.
The Trx-based metabolic regulation has been studied extensively in aerobic organisms and was considered less relevant to the strictly anaerobic microorganisms (20). This is likely because the focus has been on the catalytic Cys residues (E′0, −279 mV), which are unlikely to be oxidized and consequently of less use in metabolic regulation in the anaerobic microorganisms living not only in the absence of oxygen but also in a highly reduced environment. The Trx reductase with a flavin-gating system described in this report changes this situation. By virtue of carrying a low potential flavin, the enzyme would function only under highly reduced conditions, leaving target proteins with cysteine disulfides even under moderate anaerobic conditions. This control on catalysis will allow the thioredoxin system to be responsive to redox changes within reduced environments. Because flavins provide a wider range of possibilities in terms of redox potentials (E′0, +150 to −500 mV) (73), the gating system would also offer a diversity of redox potential ranges for the thioredoxin system to operate. Thus, our findings open up a new area of research on methanogens and other anaerobes, which are highly relevant in bioenergy production, bioremediation, climate change, and gut metabolism in relation to beef and dairy production and the development of type 2 diabetes and obesity in human (20).
Experimental Procedures
Materials
E. coli NiCo21(DE3)® competent cells (74) were obtained from New England Biolabs (Ipswich, MA). F420 was purified from Methanothermobacter thermoautotrophicus and reduced to generate F420H2 employing previously described methods (59, 75). C. pasteurianum ferredoxin and bidirectional hydrogenase were generous gifts from Dr. J. H. Chen of Virginia Tech (76). All other chemicals were purchased from standard suppliers.
Generation of Recombinant and Homogeneous Preparation of M. jannaschii Thioredoxins and DFTR
Heterologous expression and purification of Mj-Trx1 and Mj-Trx2 were performed as described previously (20). An overexpression vector for Mj-DFTR, pUL206, was constructed by amplifying the respective open reading frame (locus tag number MJ1536) from M. jannaschii chromosomal DNA and cloning it into the NdeI and BamHI sites of pTev5, a T7-based expression vector (77). It was designed to generate the recombinant protein with an NH2-terminal His6 tag (MSYYHHHHHHDYDIPTSENLYFQGASH). The plasmid was then transformed into E. coli NiCo21(DE3). The resulting strain was grown at 37 °C in 20 liters of Luria Bertani media containing 100 and 34 μg/ml ampicillin and chloramphenicol, respectively. After 5 h of growth, cells were harvested at 10,000 × g using a Beckman Coulter Avanti J-E centrifuge (Beckman Coulter, Brea, CA) for 20 min at 4 °C and stored at −20 °C until used.
DFTR purification was performed under air at 4 °C as described previously (20). Briefly, 20 g of frozen cell pellets were thawed on ice and resuspended in 50 ml of lysis solution containing 50 mm sodium phosphate buffer, pH 7.0, 300 mm NaCl, 10 mm imidazole, and a cOmpleteTM EDTA-free protease inhibitor mixture tablet (Roche Applied Science). The cells in the suspension were lysed by three passages through a French pressure cell operating at a pressure of 1.28 × 108 Pa and the cell lysate was centrifuged at 20,000 × g at 4 °C for 30 min. From the resulting supernatant, recombinant Mj-DFTR was purified via Ni2+-nitrilotriacetic acid chromatography (20) where the protein was eluted at an imidazole concentration of 150 mm. The eluted fractions that were found to contain homogeneous DFTR via SDS-PAGE were pooled and concentrated using an Amicon ultracentrifugal filter, 3-kDa molecular mass cutoff (EMD Millipore, Bedford, MA). The NH2-terminal His6 tag of the purified protein was cleaved by incubation at 4 °C with recombinant His6-TEV (recombinant TEV-protease carrying a His6 tag) (78), at an Mj-DFTR to protease ratio of 20:1 under overnight dialysis against a solution containing Tris-Cl buffer, pH 8.0, NaCl, and DTT at final concentrations of 50, 200, and 5 mm, respectively. DTT was then removed by three more rounds of dialysis against the same solution but without DTT and containing a higher final concentration of NaCl (400 mm). From this preparation the recombinant His6-TEV, Mj-DFTR carrying uncleaved His6 tag, and the cleaved His6 tag were removed via a passage through an Ni2+-nitrilotriacetic acid column. The protein concentrations in Mj-TrxR and Trx solutions were determined via the Bradford assay (79) using a protein assay kit (Bio-Rad) following the manufacturer's protocol.
The yield of Mj-DFTR was 0.8 mg of purified protein/liter of culture. This low protein yield was due to two factors. First, the protein was expressed in E. coli deliberately at a basal level in the absence of isopropyl 1-thio-β-d-galactopyranoside-driven induction of the T7lac-promoter. Second, the culture was harvested at a relatively low optical density (0.8 at 600 nm, as measured with a DU-800 UV-visible spectrophotometer, Beckman Coulter, Inc., Brea, CA). These cultivation and expression conditions were chosen to increase the purity of Mj-TrxR protein preparation. The results from early trials showed that Mj-TrxR purified from E. coli cells overexpressing the protein under isopropyl 1-thio-β-d-galactopyranoside induction contained several tightly bound proteins.
Size Exclusion Chromatography
The size exclusion chromatography was performed employing a 7.8-mm × 30-cm TSK-GEL G3000SWXL column (TosoHaas, Montgomeryville, PA), a 6-mm × 4-cm SWxl guard column (TosoHaas), and a Shimadzu Prominence HPLC system consisting of LC-20AD dual pumps, SIL-20A autosampler, SPD-M20A diode array detector, and CBM 20A controller system (Shimadzu Scientific Instruments, Columbia, MD) as described previously (80). An aqueous mobile phase composed of 100 mm potassium phosphate buffer, pH 7, and 150 mm NaCl was used. The flow rate was 1 ml/min. Before applying a sample, the column was equilibrated with the mobile phase for 1 h. The elution was monitored at 280 nm. A mixture of protein standards (Bio-Rad) containing the following components (protein or molecule, estimated molecular mass) was used for the calibration of the column: vitamin B12, 1,350 Da; myoglobin, 17,000 Da; ovalbumin, 44,000 Da; γ-globulin, 158,000; thyroglobulin, 670,000 Da. Then, 160 μg of Mj-DFTR dissolved in a 75-μl mobile phase was analyzed.
Reconstitution of Purified DFTR with Flavin Adenine Dinucleotide
The purified Mj-DFTR was yellowish in color, and the UV-visible spectra of the protein exhibited absorbance maxima at 380 and 460 nm, which are typical characteristics of flavin-containing proteins (25). The ratio of the absorbance of freshly purified Mj-TrxR at 280 and 460 nm (A280/A460) was typically 11, suggesting a partial incorporation of flavin into the protein; full incorporation of FAD in E. coli NTR leads to an A280/A460 value of 4 (32). To achieve full incorporation of FAD, the purified preparation of DFTR was incubated in a solution containing 100 mm potassium phosphate buffer, pH 7, 100 mm KCl, and 1 mm FAD at 4 °C for an hour. The excess FAD was removed by extensively washing the protein with the same solution but without FAD on the membrane of an Amicon ultracentrifugal filter, 3-kDa molecular mass cutoff (EMD Millipore, Bedford, MA). This final preparation exhibited an A280/A460 value of 4.1.
Insulin Disulfide Reduction Assay
The insulin disulfide reduction assay was performed as described previously (20, 26), except it occurred under anaerobic conditions (20). The assay temperature was 25 °C. The following electron flow scheme was used: electron donor → Mj-TrxR → Mj-Trx → insulin. NADH, NADPH, dithionite, coenzyme M, and ferredoxin at final concentrations of 0.1, 0.1, 1, and 6 mm and 5 μm, respectively, were examined as potential electron donors. The assays were performed in round glass cuvettes sealed with cutoff butyl rubber stoppers with N2 (1.3 × 105 Pa) in the headspace (27), except with ferredoxin as electron carrier H2 (1.3 × 105 Pa) was used. Ferredoxin was from C. pasteurianum, and it was reduced in situ with bidirectional hydrogenase from the same organism, and the respective final concentrations in the assay were 5 μm and 50 nm, respectively (76).
Oxidation of Mj-DFTR with 2-Aldrithiol
This task was accomplished with the addition of 2-aldrithiol to a final concentration of 1 mm to a solution (total volume of 4 ml) containing 100 mm potassium phosphate, pH 7, and 50 μm enzyme and incubation of the mixture at 4 °C for 90 min. The excess aldrithiol was removed by three rounds of filtration in an Amicon Ultra-0.5 centrifugal filter device 3-kDa molecular mass cutoff (Millipore, Billerica, MA). Each round involved centrifugation at 10,000 × g for 10 min followed by resuspension of the retentate in 0.5 ml of 100 mm MOPS buffer, pH 7.
Reduction of DFTR-bound Flavin with Various Reductants
A solution containing 30 μm oxidized Mj-DFTR, 100 mm potassium phosphate buffer, pH 6.8, and one of the following reductants (final concentration) was incubated at 70 °C for 30 min: NADPH (0.1 mm), dithionite (1 mm), and F420H2 (0.1 mm). Then the UV-visible spectrum of the mixture was recorded using a Beckman Coulter DU800 spectrophotometer (Brea, CA). In each case, a solution containing all components but without Mj-DFTR was used as controls.
Determination of the Midpoint Redox Potential of the Catalytic Cys-Disulfide/Cys-SH Pair of the Mj-DFTR
A series of solutions containing 30 μm oxidized DFTR, varying concentrations of DTT (threo-1,4-dimercapto-2,3-butanediol), and oxidized DTT (trans-1,2-dithiane-4,5-diol) giving a total concentration of 5 mm for these two compounds and 100 mm MOPS buffer, pH 7.0, were incubated inside an anaerobic chamber at 25 °C for 2 h. Then to each of these mixtures mBBr, a thiol-specific labeling reagent, was added to a final concentration of 4 mm, and the incubation was continued for another 45 min but in the dark. The labeling reaction was stopped by the addition of trichloroacetic acid to a final concentration of 10% (v/v), and the mixtures were incubated on ice for 1 h. The resulting protein pellet was recovered by centrifugation at 10,000 × g for 10 min, washed with ice-cold acetone, and air-dried and dissolved in a 200-μl solution containing 100 mm Tris-HCl buffer, pH 7, and 1% SDS. The solution was then analyzed via fluorometry by the use of an Infinite® M200 plate reader (Tecan Group Ltd., Männedorf, Switzerland) with the excitation and emission wavelengths set to 380 and 450 nm, respectively. Three mixtures of the following compositions served as controls: lacking both the DTT and oxidized DTT (providing background fluorescence for subtraction from all other values); only 5 mm DTT (providing a fully reduced Mj-DFTR preparation); only 5 mm oxidized DTT or ox-DTT (providing a fully oxidized Mj-DFTR preparation). From the collected fluorescence emission intensity data, the relative amounts (%) of the DFTR molecules with fully reduced thiol groups were calculated by setting the values for the fully oxidized and fully reduced preparations to 0 and 100%, respectively, and these data were plotted against the redox potential values for the incubation mixtures that were obtained from the following equation, where R and F are universal gas and Faraday constants, respectively; T and n are temperature and number of electrons involved in the reaction, and E′0 for the DTT/DTTox is −332 mV (81).Then the data in the plot were fitted to the Nernst equation for a 2-electron transfer reaction (n = 2), see Equation 3.
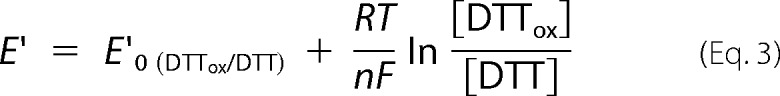 |
Determination of the E′0 of the Mj-DFTR-bound Flavin Cofactor
This task involved redox titration of Mj-DFTR-bound FAD with F420H2 or dithionite as reductant. A series of aqueous solutions containing oxidized Mj-DFTR (25 μm), F420H2 (5–115 μm), or dithionite (0–500 μm) and 100 mm potassium phosphate buffer, pH 7.0 were prepared from anaerobic stocks inside an anaerobic chamber (Coy Lab Products, Grass Lake, MI) and incubated there for 2 h at 25 °C. The internal gas atmosphere of the chamber was composed of N2 and H2 (ratio 98:2, v/v); hydrogen did not have any influence on the spectral characteristic of the mixtures (data not shown). A solution lacking the reductant (F420H2 or dithionite)was used as control. After incubation, each mixture was transferred into a UV-transparent cuvette with a 1-cm light path (BrandTech® Scientific, Inc., Essex, CT), and the cuvette was sealed with a butyl rubber stopper (size 00 with of the bottom cut off) and brought outside from the anaerobic chamber. Then the respective absorption spectrum (200–800 nm) was collected on a Beckman Coulter DU800 spectrophotometer (Brea, CA) and analyzed as described under “Results.” With F420H2 as the reductant, the F420/F420H2 system served as the reference for calculating the redox potential (E′) values for the mixtures. For the titration with dithionite, MV, a redox dye, was added as the reference, and the resultant MV+/MV0 system allowed calculations for system E′ values.
Kinetic Assay of F420H2-dependent Trx Reductase (DFTR)
The assay was performed anaerobically with nitrogen (1.3 × 105 Pa) in the headspace (20, 82). The assay mixture (total volume, 1000 μl) contained 100 mm potassium phosphate buffer, pH 7.0, 2 mm EDTA, 1 mm oxidized glutathione, F420H2, and Mj-Trx or DTNB at desired levels. The reaction was initiated by the addition of the enzyme and monitored using a Beckman Coulter DU800 spectrophotometer by following an increase in absorbance at 400 nm. For assay with DTNB, the increase in absorbance at 400 nm was due to the formation of both F420 and TNB. Accordingly, the initial rate of TNB production was calculated using composite extinction coefficient values of 24.4 mm−1 cm−1 (ϵ400 nm, TNB + 0.5 × ϵ400 nm, F420, ϵ400 nm, TNB = 11.9 mm−1 cm−1). A reaction without Mj-DFTR served as a control. The initial velocity was calculated using an extinction coefficient value of 25 mm−1 cm−1 for F420 at 400 nm (83). For pH studies, the potassium phosphate buffer was replaced with constant ionic strength buffers composed of 60 mm MES, 120 mm Tris, and 60 mm glacial acetic acid and adjusted to the desired pH values (4–9.5) with HCl or NaOH (84).
For kinetic analysis, the assay mixture was supplemented with oxidized glutathione (1 mm) to maintain a constant level of Mj-Trx (30). Each assay was performed in triplicate. The apparent kinetic constants were calculated by fitting the initial velocity data to the Henri-Michaelis-Menten's equation using Solver function in the Microsoft excel (85).
For examining activities with the following alternative substrates, Trx was replaced with one of them (entity (final concentration)): DTNB (0.2 mm) and Na2SeO3 (50 μm) which are reduced by high-molecular weight TrxR (86); oxidized l-glutathione (1 mm) for glutathione reductase activity (87); and lipoate (1 mm) for lipoamide dehydrogenase activity (88).
Bioinformatic Analysis of DFTR Homologs
The candidate proteins were identified via PSI-BLAST searches (47) into the non-redundant protein database of the NCBI using the protein sequence of Mj-DFTR (locus number, MJ1536) as a query with 10 iterations and an e value threshold of 1e−25. The identified homologs were screened via exploratory phylogenetic analysis using the following proteins (protein and accession number): E. coli NTR, EGJ07907.1; T. maritima NTR, AAD35951.1; T. acidophilum TrxR, WP_010901395; Aeropyrum pernix NTR, BAA80046.2; Pyrococcus horikoshii NTR, WP_048053388.1; S. solfataricus NTR, WP_009989411.1; C. pasteurianum ferredoxin-dependent flavin-containing TrxR, WP_015617437.1; and E. coli dihydrolipoamide dehydrogenase, AIZ85699.1. The amino acid sequences of the retrieved DFTR homologs and selected TrxR were aligned and trimmed by the use of Muscle (89) and Gblocks (90), respectively. A phylogenetic tree was constructed using “proml,” a maximum likelihood-based phylogenetic reconstruction program, in the Phylip 3.67 package (91) with 100 bootstraps replicates. The tree was viewed with FigTree version 1.4.2.
A multiple sequence alignment was performed by the use of PROMALS3D (92). Three-dimensional structures of E. coli NTR and T. acidophilum TrxR (PDB codes 1CL0 and 3CTY, respectively) served as the references.
Author Contributions
D. S. and B. M. designed the research; D. S. and U. L. performed the research; D. S. and B. M. analyzed the data; and D. S. and B. M. wrote the paper.
Supplementary Material
Acknowledgments
We thank Prof. Bob. B. Buchanan for bringing a paper bearing the NH2-terminal sequence of C. pasteurianum TrxR to our attention. We thank two anonymous reviewers for their helpful comments on our manuscript.
This work was supported by National Aeronautics and Space Administration Astrobiology: Exobiology and Evolutionary Biology Grant NNX13AI05G (to B. M.) and Virginia Tech Agricultural Experiment Station Hatch Program, CRIS Project VA-160021. The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Tables S1 and S2.
- Trx
- thioredoxin
- TrxR
- thioredoxin reductase
- NTR
- NADPH-dependent TrxR
- DFTR
- deazaflavin-dependent flavin-containing thioredoxin reductase
- FTR
- ferredoxin thioredoxin reductase
- TEV
- tobacco etch virus
- PDB
- Protein Data Bank
- MV
- methyl viologen
- Pa
- pascal
- mBBr
- monobromobimane.
References
- 1. Arnér E. S., and Holmgren A. (2000) Physiological functions of thioredoxin and thioredoxin reductase. Eur. J. Biochem. 267, 6102–6109 [DOI] [PubMed] [Google Scholar]
- 2. Holmgren A. (1989) Thioredoxin and glutaredoxin systems. J. Biol. Chem. 264, 13963–13966 [PubMed] [Google Scholar]
- 3. Lu J., and Holmgren A. (2014) The thioredoxin antioxidant system. Free Radic. Biol. Med. 66, 75–87 [DOI] [PubMed] [Google Scholar]
- 4. Hirt R. P., Müller S., Embley T. M., and Coombs G. H. (2002) The diversity and evolution of thioredoxin reductase: new perspectives. Trends Parasitol. 18, 302–308 [DOI] [PubMed] [Google Scholar]
- 5. Buchanan B. B., Schürmann P., Wolosiuk R. A., and Jacquot J. P. (2002) The ferredoxin/thioredoxin system: from discovery to molecular structures and beyond. Photosynth. Res. 73, 215–222 [DOI] [PubMed] [Google Scholar]
- 6. Pieulle L., Stocker P., Vinay M., Nouailler M., Vita N., Brasseur G., Garcin E., Sebban-Kreuzer C., and Dolla A. (2011) Study of the thiol/disulfide redox systems of the anaerobe Desulfovibrio vulgaris points out pyruvate:ferredoxin oxidoreductase as a new target for thioredoxin 1. J. Biol. Chem. 286, 7812–7821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hosoya-Matsuda N., Inoue K., and Hisabori T. (2009) Roles of thioredoxins in the obligate anaerobic green sulfur photosynthetic bacterium Chlorobaculum tepidum. Mol. Plant 2, 336–343 [DOI] [PubMed] [Google Scholar]
- 8. Hammel K. E., Cornwell K. L., and Buchanan B. B. (1983) Ferredoxin/flavoprotein-linked pathway for the reduction of thioredoxin. Proc. Natl. Acad. Sci. U.S.A. 80, 3681–3685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. McCarver A. C., and Lessner D. J. (2014) Molecular characterization of the thioredoxin system from Methanosarcina acetivorans. FEBS J. 281, 4598–4611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sheehan R., McCarver A. C., Isom C. E., Karr E. A., and Lessner D. J. (2015) The Methanosarcina acetivorans thioredoxin system activates DNA binding of the redox-sensitive transcriptional regulator MsvR. J. Ind. Microbiol. Biotechnol. 42, 965–969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sarin R., and Sharma Y. D. (2006) Thioredoxin system in obligate anaerobe Desulfovibrio desulfuricans: identification and characterization of a novel thioredoxin 2. Gene 376, 107–115 [DOI] [PubMed] [Google Scholar]
- 12. Johnson T. C., Crawford N. A., and Buchanan B. B. (1984) Thioredoxin system of the photosynthetic anaerobe Chromatium vinosum. J. Bacteriol. 158, 1061–1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Harms C., Meyer M. A., and Andreesen J. R. (1998) Fast purification of thioredoxin reductases and of thioredoxins with an unusual redox-active centre from anaerobic, amino acid-utilizing bacteria. Microbiology 144, 793–800 [DOI] [PubMed] [Google Scholar]
- 14. Reott M. A., Parker A. C., Rocha E. R., and Smith C. J. (2009) Thioredoxins in redox maintenance and survival during oxidative stress of Bacteroides fragilis. J. Bacteriol. 191, 3384–3391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kashima Y., and Ishikawa K. (2003) A hyperthermostable novel protein-disulfide oxidoreductase is reduced by thioredoxin reductase from hyperthermophilic archaeon Pyrococcus horikoshii. Arch. Biochem. Biophys. 418, 179–185 [DOI] [PubMed] [Google Scholar]
- 16. Yang X., and Ma K. (2010) Characterization of a thioredoxin-thioredoxin reductase system from the hyperthermophilic bacterium Thermotoga maritima. J. Bacteriol. 192, 1370–1376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lee D. Y., Ahn B. Y., and Kim K. S. (2000) A thioredoxin from the hyperthermophilic archaeon Methanococcus jannaschii has a glutaredoxin-like fold but thioredoxin-like activities. Biochemistry 39, 6652–6659 [DOI] [PubMed] [Google Scholar]
- 18. Amegbey G. Y., Monzavi H., Habibi-Nazhad B., Bhattacharyya S., and Wishart D. S. (2003) Structural and functional characterization of a thioredoxin-like protein (Mt0807) from Methanobacterium thermoautotrophicum. Biochemistry 42, 8001–8010 [DOI] [PubMed] [Google Scholar]
- 19. Hernandez H. H., Jaquez O. A., Hamill M. J., Elliott S. J., and Drennan C. L. (2008) Thioredoxin reductase from Thermoplasma acidophilum: a new twist on redox regulation. Biochemistry 47, 9728–9737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Susanti D., Wong J. H., Vensel W. H., Loganathan U., DeSantis R., Schmitz R. A., Balsera M., Buchanan B. B., and Mukhopadhyay B. (2014) Thioredoxin targets fundamental processes in a methane-producing archaeon, Methanocaldococcus jannaschii. Proc. Natl. Acad. Sci. U.S.A. 111, 2608–2613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. McFarlan S. C., Terrell C. A., and Hogenkamp H. P. (1992) The purification, characterization, and primary structure of a small redox protein from Methanobacterium thermoautotrophicum, an archaebacterium. J. Biol. Chem. 267, 10561–10569 [PubMed] [Google Scholar]
- 22. Kumar A. K., Kumar R. S., Yennawar N. H., Yennawar H. P., and Ferry J. G. (2015) Structural and biochemical characterization of a ferredoxin:thioredoxin reductase-like enzyme from Methanosarcina acetivorans. Biochemistry 54, 3122–3128 [DOI] [PubMed] [Google Scholar]
- 23. Kumar A. K., Yennawar N. H., Yennawar H. P., and Ferry J. G. (2011) Expression, purification, crystallization and preliminary x-ray crystallographic analysis of a novel plant-type ferredoxin/thioredoxin reductase-like protein from Methanosarcina acetivorans. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 67, 775–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hocking W. P., Roalkvam I., Magnussen C., Stokke R., and Steen I. H. (2015) Assessment of the carbon monoxide metabolism of the hyperthermophilic sulfate-reducing archaeon Archaeoglobus fulgidus VC-16 by comparative transcriptome analyses. Archaea 2015, 235384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Thelander L. (1967) Thioredoxin reductase: characterization of a homogeneous preparation from Escherichia coli B. J. Biol. Chem. 242, 852–859 [PubMed] [Google Scholar]
- 26. Holmgren A. (1977) Bovine thioredoxin system. Purification of thioredoxin reductase from calf liver and thymus and studies of its function in disulfide reduction. J. Biol. Chem. 252, 4600–4606 [PubMed] [Google Scholar]
- 27. Johnson E. F., and Mukhopadhyay B. (2005) A new type of sulfite reductase, a novel coenzyme F420-dependent enzyme, from the methanarchaeon Methanocaldococcus jannaschii. J. Biol. Chem. 280, 38776–38786 [DOI] [PubMed] [Google Scholar]
- 28. Thelander L. (1968) Studies on thioredoxin reductase from Escherichia coli B-relation of structure and function. Eur. J. Biochem. 4, 407–419 [DOI] [PubMed] [Google Scholar]
- 29. Eirich L. D., Vogels G. D., and Wolfe R. S. (1979) Distribution of coenzyme F420 and properties of its hydrolytic fragments. J. Bacteriol. 140, 20–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kanzok S. M., Rahlfs S., Becker K., and Schirmer R. H. (2002) Thioredoxin, thioredoxin reductase, and thioredoxin peroxidase of malaria parasite Plasmodium falciparum. Methods Enzymol. 347, 370–381 [DOI] [PubMed] [Google Scholar]
- 31. Jeon S. J., and Ishikawa K. (2002) Identification and characterization of thioredoxin and thioredoxin reductase from Aeropyrum pernix K1. Eur. J. Biochem. 269, 5423–5430 [DOI] [PubMed] [Google Scholar]
- 32. Moore E. C., Reichard P., and Thelander L. (1964) Enzymatic synthesis of deoxyribonucleotides. V. purification and properties of thioredoxin reductase from Escherichia Coli B. J. Biol. Chem. 239, 3445–3452 [PubMed] [Google Scholar]
- 33. Holmgren A. (1985) Thioredoxin. Annu. Rev. Biochem. 54, 237–271 [DOI] [PubMed] [Google Scholar]
- 34. Prongay A. J., and Williams C. H. Jr. (1992) Oxidation-reduction properties of Escherichia coli thioredoxin reductase altered at each active site cysteine residue. J. Biol. Chem. 267, 25181–25188 [PubMed] [Google Scholar]
- 35. Cheng Z., Arscott L. D., Ballou D. P., and Williams C. H. Jr. (2007) The relationship of the redox potentials of thioredoxin and thioredoxin reductase from Drosophila melanogaster to the enzymatic mechanism: reduced thioredoxin is the reductant of glutathione in Drosophila. Biochemistry 46, 7875–7885 [DOI] [PubMed] [Google Scholar]
- 36. DiMarco A. A., Bobik T. A., and Wolfe R. S. (1990) Unusual coenzymes of methanogenesis. Annu. Rev. Biochem. 59, 355–394 [DOI] [PubMed] [Google Scholar]
- 37. Massey V., and Hemmerich P. (1978) Photoreduction of flavoproteins and other biological compounds catalyzed by deazaflavins. Biochemistry 17, 9–16 [DOI] [PubMed] [Google Scholar]
- 38. Mayhew S. G. (1978) The redox potential of dithionite and SO−2 from equilibrium reactions with flavodoxins, methyl viologen and hydrogen plus hydrogenase. Eur. J. Biochem. 85, 535–547 [DOI] [PubMed] [Google Scholar]
- 39. Segerer A., Langworthy T. A., and Stetter K. O. (1988) Thermoplasma acidophilum and Thermoplasma volcanium sp. nov. from Solfatara fields. Syst. Appl. Microbiol. 10, 161–171 [Google Scholar]
- 40. Huber R., Langworthy T. A., Konig H., Thomm M., Woese C. R., Sleytr U. B., and Stetter K. O. (1986) Thermotoga maritima sp. nov. represents a new genus of unique extremely thermophilic eubacteria growing up to 90 °C. Arch. Microbiol. 144, 324–333 [Google Scholar]
- 41. Lennon B. W., Williams C. H. Jr., and Ludwig M. L. (1999) Crystal structure of reduced thioredoxin reductase from Escherichia coli: structural flexibility in the isoalloxazine ring of the flavin adenine dinucleotide cofactor. Protein Sci. 8, 2366–2379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dym O., and Eisenberg D. (2001) Sequence-structure analysis of FAD-containing proteins. Protein Sci. 10, 1712–1728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ceh K., Demmer U., Warkentin E., Moll J., Thauer R. K., Shima S., and Ermler U. (2009) Structural basis of the hydride transfer mechanism in F420-dependent methylenetetrahydromethanopterin dehydrogenase. Biochemistry 48, 10098–10105 [DOI] [PubMed] [Google Scholar]
- 44. Aufhammer S. W., Warkentin E., Ermler U., Hagemeier C. H., Thauer R. K., and Shima S. (2005) Crystal structure of methylenetetrahydromethanopterin reductase (Mer) in complex with coenzyme F420: architecture of the F420/FMN binding site of enzymes within the nonprolyl cis-peptide containing bacterial luciferase family. Protein Sci. 14, 1840–1849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Seedorf H., Hagemeier C. H., Shima S., Thauer R. K., Warkentin E., and Ermler U. (2007) Structure of coenzyme F420H2 oxidase (FprA), a di-iron flavoprotein from methanogenic archaea catalyzing the reduction of O2 to H2O. FEBS J. 274, 1588–1599 [DOI] [PubMed] [Google Scholar]
- 46. Ojha S., Meng E. C., and Babbitt P. C. (2007) Evolution of function in the “two dinucleotide binding domains'” flavoproteins. PLoS Comput. Biol. 3, e121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Altschul S. F., Madden T. L., Schäffer A. A., Zhang J., Zhang Z., Miller W., and Lipman D. J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kurr M., Huber R., König H., Jannasch H., Fricke H., Trincone A., Kristjansson J., and Stetter K. (1991) Methanopyrus kandleri, gen., and sp. nov. represents a novel group of hyperthermophilic methanogens, growing at 110 °C. Arch. Microbiol. 156, 239–247 [Google Scholar]
- 49. Brochier C., Forterre P., and Gribaldo S. (2004) Archaeal phylogeny based on proteins of the transcription and translation machineries: tackling the Methanopyrus kandleri paradox. Genome Biol. 5, R17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Balsera M., Uberegui E., Susanti D., Schmitz R. A., Mukhopadhyay B., Schürmann P., and Buchanan B. B. (2013) Ferredoxin:thioredoxin reductase (FTR) links the regulation of oxygenic photosynthesis to deeply rooted bacteria. Planta 237, 619–635 [DOI] [PubMed] [Google Scholar]
- 51. Jones W. J., Leigh J. A., Mayer F., Woese C. R., and Wolfe R. S. (1983) Methanococcus jannaschii sp. nov., an extreme thermophilic methanogen from a submarine hydrothermal vent. Arch. Microbiol. 136, 254–261 [Google Scholar]
- 52. Jannasch H. W., and Mottl M. J. (1985) Geomicrobiology of deep-sea hydrothermal vents. Science 229, 717–725 [DOI] [PubMed] [Google Scholar]
- 53. Graham D. E., and White R. H. (2002) Elucidation of methanogenic coenzyme biosyntheses: from spectroscopy to genomics. Nat. Prod. Rep. 19, 133–147 [DOI] [PubMed] [Google Scholar]
- 54. Choi K. P., Kendrick N., and Daniels L. (2002) Demonstration that fbiC is required by Mycobacterium bovis BCG for coenzyme F420 and F0 biosynthesis. J. Bacteriol. 184, 2420–2428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Graham D. E., Xu H., and White R. H. (2003) Identification of the 7,8-didemethyl-8-hydroxy-5-deazariboflavin synthase required for coenzyme F420 biosynthesis. Arch. Microbiol. 180, 455–464 [DOI] [PubMed] [Google Scholar]
- 56. Lin X. L., and White R. H. (1986) Occurrence of coenzyme-F420 and its γ-monoglutamyl derivative in nonmethanogenic archaebacteria. J. Bacteriol. 168, 444–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Purwantini E., Gillis T. P., and Daniels L. (1997) Presence of F420-dependent glucose-6-phosphate dehydrogenase in Mycobacterium and Nocardia species, but absence from Streptomyces and Corynebacterium species and methanogenic archaea. FEMS Microbiol. Lett. 146, 129–134 [DOI] [PubMed] [Google Scholar]
- 58. Berk H., and Thauer R. K. (1998) F420H2:NADP oxidoreductase from Methanobacterium thermoautotrophicum: identification of the encoding gene via functional overexpression in Escherichia coli. FEBS Lett. 438, 124–126 [DOI] [PubMed] [Google Scholar]
- 59. Haase P., Deppenmeier U., Blaut M., and Gottschalk G. (1992) Purification and characterization of F420H2-dehydrogenase from Methanolobus tindarius. Eur. J. Biochem. 203, 527–531 [DOI] [PubMed] [Google Scholar]
- 60. Walsh C. (1986) Naturally occurring 5-deazaflavin coenzymes: biological redox roles. ACC Chem. Res. 19, 216–221 [Google Scholar]
- 61. Bult C. J., White O., Olsen G. J., Zhou L., Fleischmann R. D., Sutton G. G., Blake J. A., FitzGerald L. M., Clayton R. A., Gocayne J. D., Kerlavage A. R., Dougherty B. A., Tomb J. F., Adams M. D., Reich C. I., et al. (1996) Complete genome sequence of the methanogenic archaeon, Methanococcus jannaschii. Science 273, 1058–1073 [DOI] [PubMed] [Google Scholar]
- 62. Thauer R. K., Jungermann K., and Decker K. (1977) Energy conservation in chemotrophic anaerobic bacteria. Bacteriol. Rev. 41, 100–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. de Poorter L. M., Geerts W. J., and Keltjens J. T. (2005) Hydrogen concentrations in methane-forming cells probed by the ratios of reduced and oxidized coenzyme F420. Microbiology 151, 1697–1705 [DOI] [PubMed] [Google Scholar]
- 64. Hendrickson E. L., Haydock A. K., Moore B. C., Whitman W. B., and Leigh J. A. (2007) Functionally distinct genes regulated by hydrogen limitation and growth rate in methanogenic archaea. Proc. Natl. Acad. Sci. U.S.A. 104, 8930–8934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hamill M. J., Chobot S. E., Hernandez H. H., Drennan C. L., and Elliott S. J. (2008) Direct electrochemical analyses of a thermophilic thioredoxin reductase: interplay between conformational change and redox chemistry. Biochemistry 47, 9738–9746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Buey R. M., Galindo-Trigo S., Lopez-Maury L., Velazquez-Campoy A., Revuelta J. L., Florencio F. J., de Pereda J. M., Schurmann P., Buchanan B. B., and Balsera M. (2016) A new member of the thioredoxin reductase family from early oxygenic photosynthetic organisms. Mol. Plant 10.1016/j.molp.2016.06.019 [DOI] [PubMed] [Google Scholar]
- 67. Hicks M. A. II, Barber A. E. II, and Babbitt P. C. (2014) in Protein Families: Relating Protein Sequence, Structure, and Function (Orengo C., and Bateman A., eds) pp. 127–158, John Wiley & Sons, New York [Google Scholar]
- 68. Purwantini E., and Mukhopadhyay B. (2013) Rv0132c of Mycobacterium tuberculosis encodes a coenzyme F420-dependent hydroxymycolic acid dehydrogenase. PLoS ONE 8, e81985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Purwantini E., Daniels L., and Mukhopadhyay B. (2016) F420H2 is required for phthiocerol dimycocerosate synthesis in mycobacteria. J. Bacteriol. 198, 2020–2028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Purwantini E., and Daniels L. (1996) Purification of a novel coenzyme F420-dependent glucose-6-phosphate dehydrogenase from Mycobacterium smegmatis. J. Bacteriol. 178, 2861–2866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Boone D. R., Whitman W. B., and Rouviere P. (1993) in Methanogenesis: Ecology, Physiology, Biochemistry and Genetics (Ferry J. G., ed) pp. 35–80, Chapman and Hall, New York [Google Scholar]
- 72. Kiener A., and Leisinger T. (1983) Oxygen sensitivity of methanogenic bacteria. Syst. Appl. Microbiol. 4, 305–312 [DOI] [PubMed] [Google Scholar]
- 73. Palfey B. A., and Massey V. (1998) in Comprehensive Biological Catalysis (Sinnot M., ed) pp. 88–100, Elsevier, Ltd., Oxford, UK [Google Scholar]
- 74. Robichon C., Luo J., Causey T. B., Benner J. S., and Samuelson J. C. (2011) Engineering Escherichia coli BL21(DE3) derivative strains to minimize E. coli protein contamination after purification by immobilized metal affinity chromatography. Appl. Environ. Microbiol. 77, 4634–4646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Purwantini E., Mukhopadhyay B., Spencer R. W., and Daniels L. (1992) Effect of temperature on the spectral properties of coenzyme F420 and related compounds. Anal. Biochem. 205, 342–350 [DOI] [PubMed] [Google Scholar]
- 76. Chen J. S., and Mortenson L. E. (1974) Purification and properties of hydrogenase from Clostridium pasteurianum W5. Biochim. Biophys. Acta 371, 283–298 [DOI] [PubMed] [Google Scholar]
- 77. Rocco C. J., Dennison K. L., Klenchin V. A., Rayment I., and Escalante-Semerena J. C. (2008) Construction and use of new cloning vectors for the rapid isolation of recombinant proteins from Escherichia coli. Plasmid 59, 231–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Blommel P. G., and Fox B. G. (2007) A combined approach to improving large-scale production of tobacco etch virus protease. Protein Expr. Purif. 55, 53–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 80. Mukhopadhyay B., and Purwantini E. (2000) Pyruvate carboxylase from Mycobacterium smegmatis: stabilization, rapid purification, molecular and biochemical characterization and regulation of the cellular level. Biochim. Biophys. Acta 1475, 191–206 [DOI] [PubMed] [Google Scholar]
- 81. Cleland W. W. (1964) Dithiothreitol, a new protective reagent for SH groups. Biochemistry 3, 480–482 [DOI] [PubMed] [Google Scholar]
- 82. Johnson E. F., and Mukhopadhyay B. (2007) in Proceedings of the International Symposium on Microbial Sulfur Metabolism (Dahl C., and Friedrich C. G., eds), pp. 202–214, Springer, New York [Google Scholar]
- 83. Jacobson F. S., Daniels L., Fox J. A., Walsh C. T., and Orme-Johnson W. H. (1982) Purification and properties of an 8-hydroxy-5-deazaflavin-reducing hydrogenase from Methanobacterium thermoautotrophicum. J. Biol. Chem. 257, 3385–3388 [PubMed] [Google Scholar]
- 84. Ellis K. J., and Morrison J. F. (1982) Buffers of constant ionic-strength for studying pH-dependent processes. Methods Enzymol. 87, 405–426 [DOI] [PubMed] [Google Scholar]
- 85. Kemmer G., and Keller S. (2010) Nonlinear least-squares data fitting in Excel spreadsheets. Nat. Protoc. 5, 267–281 [DOI] [PubMed] [Google Scholar]
- 86. Kumar S., Björnstedt M., and Holmgren A. (1992) Selenite is a substrate for calf thymus thioredoxin reductase and thioredoxin and elicits a large non-stoichiometric oxidation of NADPH in the presence of oxygen. Eur. J. Biochem. 207, 435–439 [DOI] [PubMed] [Google Scholar]
- 87. Patel M. P., Marcinkeviciene J., and Blanchard J. S. (1998) Enterococcus faecalis glutathione reductase: purification, characterization and expression under normal and hyperbaric O2 conditions. FEMS Microbiol. Lett. 166, 155–163 [DOI] [PubMed] [Google Scholar]
- 88. Arnér E. S., Nordberg J., and Holmgren A. (1996) Efficient reduction of lipoamide and lipoic acid by mammalian thioredoxin reductase. Biochem. Biophys. Res. Commun. 225, 268–274 [DOI] [PubMed] [Google Scholar]
- 89. Edgar R. C. (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Castresana J. (2000) Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552 [DOI] [PubMed] [Google Scholar]
- 91. Susanti D., and Mukhopadhyay B. (2012) An intertwined evolutionary history of methanogenic archaea and sulfate reduction. PLoS ONE 7, e45313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Pei J., and Grishin N. V. (2007) PROMALS: towardaccurate multiple sequence alignments of distantly related proteins. Bioinformatics 23, 802–808 [DOI] [PubMed] [Google Scholar]
- 93. Muth E., Mörschel E., and Klein A. (1987) Purification and characterization of an 8-hydroxy-5-deazaflavin-reducing hydrogenase from the archaebacterium Methanococcus voltae. Eur. J. Biochem. 169, 571–577 [DOI] [PubMed] [Google Scholar]
- 94. Hendrickson E. L., and Leigh J. A. (2008) Roles of coenzyme F420-reducing hydrogenases and hydrogen- and F420-dependent methylenetetrahydromethanopterin dehydrogenases in reduction of F420 and production of hydrogen during methanogenesis. J. Bacteriol. 190, 4818–4821 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.