

Abstract
Dopamine, a key striatal neuromodulator, increases synaptic strength by promoting surface insertion and/or retention of AMPA receptors (AMPARs). This process is mediated by the phosphorylation of the GluA1 subunit of AMPAR by cyclic nucleotide-dependent kinases, making cyclic nucleotide phosphodiesterases (PDEs) potential regulators of synaptic strength. In this study, we examined the role of phosphodiesterase 2 (PDE2), a medium spiny neuron-enriched and cGMP-activated PDE, in AMPAR trafficking. We found that inhibiting PDE2 resulted in enhancement of dopamine-induced surface GluA1 expression in dopamine receptor 1-expressing medium spiny neurons. Using pharmacological and genetic approaches, we found that inhibition of PDE1 resulted in a decrease in surface AMPAR levels because of the allosteric activation of PDE2. The cross-regulation of PDE1 and PDE2 activities results in counterintuitive control of surface AMPAR expression, making it possible to regulate the directionality and magnitude of AMPAR trafficking.
Keywords: AMPA receptor (AMPAR), cAMP, cGMP, dopamine, phosphodiesterases, trafficking, GluA1, PDE1, PDE2
Introduction
Changes to synaptic strength at corticostriatal synapses regulate the excitability of MSNs,3 resulting in coding for motivated behavior (1, 2). Fluctuations in synaptic strength can occur by trafficking of AMPARs in and out of the postsynaptic membrane in response to neuromodulators such as dopamine (DA) (3, 4). This process requires the phosphorylation of GluA1-containing AMPARs by cyclic nucleotide-dependent kinases, making receptor trafficking tightly coupled to local cyclic nucleotide levels (5–10). 3′-5′ cyclic nucleotide phosphodiesterases (PDEs), the enzymes that degrade cAMP and cGMP, are essential in shaping the spatial and temporal dynamics of signaling by maintaining the compartmentalization of cyclic nucleotides to ensure effective signal propagation to downstream effectors (11–15) and thus are poised to be significant regulators of AMPAR trafficking in response to DA stimulation.
One interesting but underappreciated feature of the PDE family is that some members display complex cross-regulation, as there are PDEs whose activities are directly and indirectly regulated by cyclic nucleotides. Several examples of cyclic nucleotide control of PDE activity have been reported, such as allosteric regulation and competitive inhibition by cyclic nucleotide binding and phosphorylation by cyclic nucleotide-dependent kinases (16–18). This leads to instances where cAMP levels can modulate the degradation of cGMP and vice versa, resulting in counterintuitive downstream responses (19, 20). Furthermore, it is possible that, when PDEs are pharmacologically inhibited, these signaling features can result in the cross-regulation of PDEs, meaning that the modulation of one PDE activity could affect the activity of an unintended second PDE regulated by cAMP/cGMP, causing unanticipated downstream signaling. There are a few reported examples of cross-regulation of PDE activities controlling cAMP or cGMP output that lead to counterintuitive signaling in non-neuronal cells, but whether this occurs in MSNs has not been explored (21–23).
An often overlooked target of cGMP in MSNs is PDE2, a highly expressed dual specificity PDE (24–30). PDE2 contains two N-terminal GAF domains, non-catalytic cGMP binding regions that act as allosteric regulatory sites. cGMP binding to the GAF domain of PDE2 induces a conformational change resulting in an enhancement of the hydrolysis of cAMP (16, 30–34). In MSNs, PDE2 is a significant regulator of cellular cyclic nucleotides levels and is readily activated by cGMP, making it a candidate for cross-regulation by other PDEs (35–38). However, whether this type of cross-regulation is part of the cyclic nucleotide signaling repertoire that controls AMPAR trafficking has yet to be determined. We hypothesize that PDE2 is an effector of cGMP signaling to regulate AMPAR dynamics and part of a cross-regulation signaling motif with other PDEs possessing cGMP degradation activity.
MSNs express several PDEs families with a wide range of kinetic properties (24–30). The complexity of cyclic nucleotide signaling makes it difficult to intuit the effect each PDEs has on observed AMPAR trafficking. Here we use a systems pharmacology approach, extending our previous computational model of dopamine signaling and AMPAR trafficking to investigate the role of cGMP on dopamine-induced AMPAR trafficking (39). We present a novel mechanism of how cGMP may be controlling striatal AMPAR trafficking in response to DA via the cross-regulation of PDEs. We show that increases in cGMP (either by enhancing cGMP production or decreasing cGMP degradation) result in PDE2 activation, leading to a reduction in DA-induced cAMP levels and AMPAR insertion. We also show that the interplay of PDE1 and PDE2 activities results in the counterintuitive regulation of cAMP levels and AMPAR response.
Results
PDE2 Inhibition Enhances Surface GluA1 Levels
MSNs express a number of PDEs that could potentially affect surface expression of GluA1-containing AMPAR (24). We have previously shown the involvement of PDE4 (39) in dopamine-mediated AMPAR trafficking in MSNs. Here we examined the role of PDE2, a highly enriched striatal PDE, in DA-induced GluA1 surface insertion using a pHluorin (SEP)-based imaging approach (40–42). First, we explored the contribution of PDE2 activity to DA-induced GluA1 surface insertion. After a stable baseline, we treated neurons with a D1R agonist (A68930, added at t = 0 min) and observed an increase in GluA1 surface insertion (Fig. 1A). After 10 min, we added the PDE2-specific inhibitor BAY60-7550, inducing a further increase in surface GluA1 levels. BAY60-7550 alone had no effect on GluA1 surface insertion, indicating that increasing cAMP levels induced by basal adenylyl cyclase activity may not be sufficient to drive GluA1 surface insertion (Fig. 1B). These data suggest that PDE2, by increasing cAMP signaling (Fig. 1C), is a regulator of dopamine-stimulated GluA1 trafficking in D1 MSNs.
FIGURE 1.
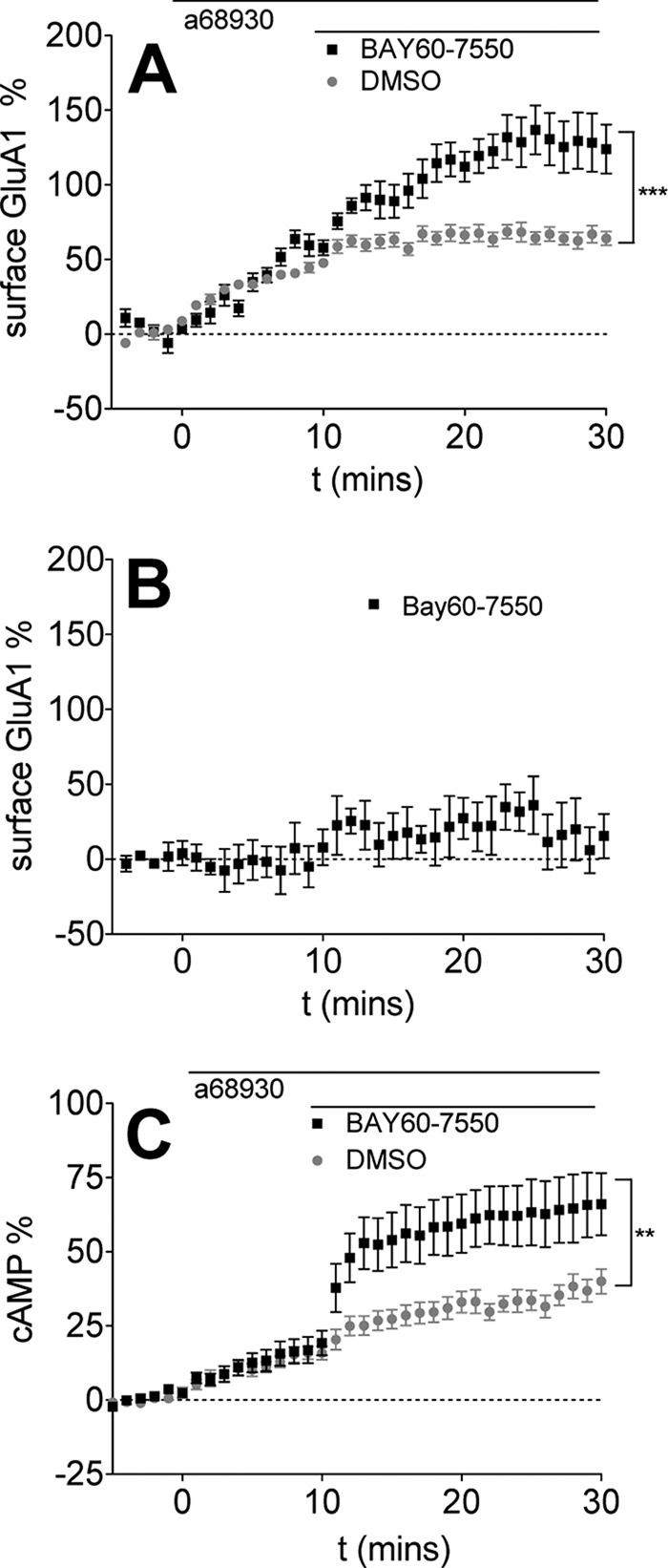
PDE2 inhibition enhances D1R agonist-induced GluA1 surface expression. Data are represented as mean ± S.E. normalized to baseline. A, D1R agonist (0.1 μm A68930) was added for 10 min, followed by DMSO (gray, 27 cells) or PDE2 inhibitor (1.0 μm BAY60-7550, black, 15 cells). ***, p < 0.0001 (repeated measures two-way ANOVA, F1,19 = 24.23). B, BAY60-7550 alone has no effect on GluA1 surface insertion. 1.0 μm BAY60-7550 was added after 5 min of baseline. (four cells). C, PDE2 inhibition increases D1R agonist-induced cAMP production. 0.1 μm A68930 was added after 5 min of baseline, followed by DMSO (gray, 17 cells) or 1.0 μm BAY60-7550 (black, 11 cells). *, p = 0.0041 (repeated measures two-way ANOVA, F1,26 = 9.906).
PDE2 Activation Results in a Decrease in Surface GluA1
Because the pool of cAMP controlled by PDE2 could modulate surface GluA1 levels, we explored whether the cGMP-dependent allosteric activation of PDE2 activity could result in removal of surface GluA1. However, this cGMP regulation of PDE2 depends on a number of factors: the cellular levels of cGMP and cAMP, the affinity of cGMP for the allosteric site on PDE2, the cellular concentration of PDE2, and the catalytic activity of PDE2. Furthermore, reports that recombinant GAF domains can bind cGMP with high affinity, the activation constant for PDE2 is much higher (∼0.2–0.5 μm) (43, 44). Based on all of these variables, it is challenging to intuitively predict the effect of cGMP on PDE2 activity and GluA1 trafficking. In other words, given the affinity of cGMP for the allosteric site on PDE2, could biologically relevant changes to cGMP levels affect PDE2 activity sufficiently to have an impact on GluA1 trafficking? To answer this, we developed a dynamic model of PDE2 regulation of AMPAR trafficking in D1 MSNs by building upon our previous model (39). The novel connectivity (consisting of a detailed representation of PDE2 and its regulation by cGMP and PDE1 activity) is depicted in Fig. 2A (16, 30, 31, 34). First, we constrained any unknown parameters to recapitulate the PDE2 inhibition increase in GluA1 surface insertion (supplemental Fig. S2A) and the cGMP-dependent activation of PDE2 (supplemental Fig. S2B) (22). Then we used the model to explore the effect of cGMP activation of PDE2 on surface GluA1 levels. We first probed the contribution of cGMP on surface GluA1 levels by simultaneously varying the concentration of PDE2 and cGMP. We calculated and plotted the area under the curve (AUC) from the resulting time course of GluA1 surface insertion (Fig. 2B) as a function of PDE2 and cGMP concentration (Fig. 2C). The AUC value was color-coded, with white representing no change from CT (dopamine stimulation of D1R), red representing an increase in surface AMPAR levels above CT levels (surface insertion), and blue representing a decrease in surface AMPAR levels below CT (surface removal). The resulting simulations show that, because of the activation of PDE2 by cGMP, an increase in cGMP results in a robust decrease in surface GluA1 (Fig. 2C). Moreover, a decrease in PDE2 levels (comparable with PDE2 inhibition) can reverse this effect on surface GluA1. This suggests that the balance between the cellular cGMP levels and PDE2 activity can determine the directionality of AMPAR trafficking.
FIGURE 2.

Allosteric PDE2 activation by cGMP modulates GluA1 surface expression. A, diagram of model topology of PDE1/2 regulation of GluA1 surface insertion in D1R MSNs. Arrows and plungers indicate activation/production and inhibition/degradation, respectively. Species in red represent pharmacological agents used. B, cGMP level controls GluA1 surface insertion by enhancing PDE2 activity. Shown is a simulation comparison of GluA1 surface insertion time courses. C, varying cGMP and PDE2 concentrations affects GluA1 surface insertion. cGMP (x axis) and PDE2 (y axis) concentrations were varied from 0.0–1.0 μm. The resulting GluA1 surface insertion time courses were plotted, and the AUC value was calculated. AUC values were color-coded so that an increase over control (DA alone) is red, a decrease is blue, and a value equal to control is white. D, varying PDE1 and PDE2 concentrations affects GluA1 surface insertion. PDE1 (x axis) and PDE2 (y axis) concentrations were varied from 0.0–1.0 μm. AUC values are color-coded.
Next we wanted to gain insight into whether inhibiting a PDE with cGMP degradation activity could activate PDE2 activity and induce a decrease in surface AMPAR levels. This is an important question, as enhancing the basal cellular levels of cGMP may not be sufficient to support PDE2 activation. A possible candidate is PDE1, an MSN-enriched PDE with a preference for cGMP (24, 45). We explored the role of PDE1 in AMPAR trafficking by varying the concentrations of PDE1 and PDE2 and plotting the AUC of the resulting time courses of GluA1 surface insertion as a function of PDE2 and PDE1 concentration (Fig. 2D). In these simulations, decreasing PDE1 levels resulted in a loss of surface GluA1. This suggested that inhibition of PDE1 could be a possible mechanism to enhance basal levels of cGMP and activate PDE2 to decrease surface GluA1 levels.
We implemented two parallel approaches to modulate cellular levels of cGMP to experimentally test whether the predicted cGMP allosteric activation of PDE2 does indeed regulate GluA1 trafficking: increasing cGMP production by directly activating soluble guanylyl cyclase using NO donors and inhibiting cGMP degradation using the PDE1 inhibitor, MMPX. Based on the simulation results, we expected that cGMP-activated PDE2, because of its substrate specificity, would promote the preferential degradation of cAMP, resulting in a decrease in PKA activity and DA-induced GluA1 insertion. First, we increased cGMP levels using S-nitroso-N-acetylpenicillamine (SNAP), an NO donor. As expected, SNAP treatment of D1 MSNs led to a rapid increase in cGMP (Fig. 3A). SNAP treatment also decreased cAMP levels, and this effect could be reversed with co-treatment with a PDE2 inhibitor (Fig. 3B), suggesting that cGMP enhanced the degradation activity of PDE2 as described previously (36, 37). Furthermore, SNAP induced the removal of D1-induced surface GluA1 (Fig. 3C), and co-treatment with a PDE2 inhibitor rescued the SNAP-induced deficit, suggesting that PDE2 activation by cGMP contributes to the action of SNAP on GluA1 trafficking. The change in cAMP displayed similar temporal dynamics as those observed with GluA1 trafficking, further supporting the notion that the mode of action of SNAP on GluA1 trafficking is by augmenting cAMP degradation because of PDE2 activation. SNAP alone failed to affect surface GluA1 levels, indicating that G-protein signaling is required for cGMP regulation of GluA1 trafficking.
FIGURE 3.

Increasing cGMP production reduces D1R agonist-induced cAMP levels and GluA1 surface expression by activating PDE2. Data are represented as mean ± S.E. normalized to baseline. A, SNAP significantly increases cGMP levels. 0.1 μm A68930 was added after 5 min of baseline, followed by DMSO (gray, eight cells) or 50 μm SNAP (black, eight cells). ***, p = 0.0001 (repeated measures two-way ANOVA, F1,19 = 27.00). B, increasing cGMP production decreases D1R agonist-induced cAMP levels via modulation of PDE2. After 5 min of baseline, 0.1 μm A68930 was added for 10 min, followed by DMSO (replotted from Fig. 1 for comparison) or 50 μm SNAP (black, 21 cells) or 1.0 μm BAY60-7550 + 50 μm SNAP (black open square, seven cells). ***, p = 0.0009 (repeated measures two-way ANOVA, DMSO versus SNAP, F1,36 = 25.84); p < 0.0001 (repeated measures two-way ANOVA, SNAP versus SNAP + BAY60-7550, F1,26 = 74.03). C, increasing cGMP reduces GluA1 surface expression by activating PDE2. After 5 min of baseline, the D1R agonist A68930 (0.1 μm) was added for 10 min, followed by DMSO (replotted from Fig. 1 for comparison) or 50 μm SNAP (black squares, 11 cells) or 1.0 μm BAY60-7550 + 50 μm SNAP (black open squares, 10 cells). ***, p < 0.0001 (repeated measures two-way ANOVA, DMSO versus SNAP, F1,36 = 26.31). **, p = 0.0061 (two-way ANOVA, SNAP versus BAY60-7550 + SNAP, F1,19 = 9.50). D, SNAP alone has no effect on GluA1 surface insertion. 50 μm SNAP was added after 5 min of baseline (six cells).
The Cross-regulation of PDE1 and PDE2 Induces a Decrease in Surface GluA1 Levels
Based on our simulation results, we predicted that PDE1 inhibition could enhance basal cGMP levels and activate PDE2, bringing about a reduction of cAMP levels and GluA1 surface insertion. Indeed, as expected, treatment with the PDE1 inhibitor MMPX increased cGMP levels (Fig. 4A) with a concomitant decrease in D1R-induced cAMP levels (Fig. 4B). Co-application of a PDE2 inhibitor reversed the PDE1 inhibition effect on cAMP (Fig. 4B), suggesting that the PDE1-induced increase in cGMP resulted in an activation of PDE2, and thus the interplay of PDE1 and PDE2 activities could regulate cAMP levels. Moreover, this cross-regulation of PDE1 and PDE2 also affected GluA1 surface expression, as PDE1 inhibition resulted in a decrease in GluA1 surface levels that was rescued by co-application of a PDE2 inhibitor (Fig. 4C).
FIGURE 4.

PDE1 inhibition decreases D1R agonist-induced cAMP levels and GluA1 surface insertion by activating PDE2. Data are represented as mean ± S.E. normalized to baseline. A, PDE1 inhibition increases cGMP levels. 0.1 μm A68930 was added after 5 min of baseline, followed by DMSO (replotted from Fig. 3 for comparison) or 10 μm MMPX (black, 11 cells). *, p = 0.0373 (repeated measures two-way ANOVA, F1,17 = 5.11). B, PDE1 inhibition by MMPX decreases D1R agonist-induced cAMP levels. 0.1 μm A68930 was added for 10 min, followed by DMSO (replotted from Fig. 1 for comparison) or 10 μm MMPX (black, 14 cells) or 1.0 μm BAY60-7550 + 10 μm MMPX (black open squares, 11 cells). **, p = 0.0015 (repeated measures two-way ANOVA, DMSO versus MMPX, F1,29 = 12.39). ***, p < 0.0001 (repeated measures two-way ANOVA, MMPX versus MMPX + BAY60-7550, F1,22 = 25.61). C, PDE1 inhibition reduces GluA1 surface levels. The D1R agonist A68930 (0.1 μm) was added for 10 min, followed by DMSO (gray, 12 cells) or the PDE1 inhibitor MMPX (10 μm, black filled squares, 10 cells), or 1.0 μm BAY60-7550 + 10 μm MMPX (black open squares, 11 cells). **, p = 0.0016 (repeated measures two-way ANOVA, DMSO versus MMPX, F1,20 = 13.40). ***, p = 0.0005 (repeated measures two-way ANOVA, F1,19 = 17.29). D, expression of PDE2 mutants abolished PDE1 regulation of GluA1 surface insertion. Expression of PDE2 lacking catalytic activity (PDE2DN, gray squares, 15 cells) or allosteric regulation by cGMP (PDE2D485A, open circles; 14 cells) increases GluA1 surface insertion in the presence of 10 μm MMPX. ***, p < 0.0001 (repeated measures two-way ANOVA, MMPX versus PDE2DN, F1,23 = 27.70). ***, p = 0.0002 (repeated measures two-way ANOVA, MMPX versus PDE2D485A, F1,22 = 20.15).
Most of our data that implicate PDE1 and PDE2 activity in the regulation of AMPAR trafficking were obtained using pharmacological agents that may have off-target actions. To address this concern, we expressed mutated forms of PDE2 lacking cGMP regulation (D485A) or catalytic activity (PDE2DN) in lieu of PDE2 pharmacological inhibition. Mutation D485A abolishes the ability of the GAF domain to bind cGMP and displays no allosteric regulation (21). The PDE2DN mutant consists of two point mutations (D685A and D796A) in the catalytic region, rendering PDE2 catalytically inactive (21). Both mutants retain the N-terminal region of PDE2 so that subcellular targeting signals are intact, and overexpression of these mutants displaces endogenous local PDE2 from its subdomain (46). Representative images of expression PDE2 mutants are shown in supplemental Fig. S6. We applied a PDE1 inhibitor while overexpressing mutant forms of PDE2 in D1 MSNs to determine whether the PDE1-dependent regulation of GluA1 trafficking requires PDE2 activity. We found that expression of either PDE2 mutant prevented the deficit in GluA1 trafficking observed with PDE1 inhibitor treatment and allowed GluA1 insertion to reach levels comparable with those observed with D1R stimulation (Fig. 4D). To control for the overexpression of a PDE activity, we overexpressed PDE4A5, a PDE not enriched in MSNs (47). Overexpression of PDE4A5 did not affect surface GluA1 levels despite having significant effects on global cAMP levels (supplemental Fig. S7, A and B). These results support the notion that the rise in cGMP levels because of PDE1 inhibition is sufficient to activate PDE2 and decrease local cAMP levels, resulting in a decrease in AMPAR surface levels.
Based on all of these data, we conclude that increasing cGMP levels results in a dose-dependent decrease in D1-induced cAMP and GluA1 surface expression. This is summarized in Fig. 5A, where we relate changes in cGMP levels because of SNAP or MMPX treatment to decreases in D1-induced surface GluA1 expression. To further confirm that activation of PDE2 is sufficient to decrease surface GluA1 levels, we utilized an optogenetics approach. Recently, Möglich and co-workers (48) developed a light-activated PDE2 by replacing the GAF domains of PDE2 with the photosensor module of Deinococcus radiodurans bacterial phytochrome (originally named LAPD; we termed it LA-PDE2 for clarity). Upon red light exposure, the photosensors dimerize and induce a conformational change that results in up-regulation of LA-PDE2 enzymatic activity by 6-fold (similar to the allosteric activation by cGMP). Thus, light stimulation can result in PDE2 activation independent of cGMP levels. We tested whether light activation of LA-PDE2 could modulate the levels of surface GluA1 in MSNs to determine the exclusive contribution of PDE2 activation to AMPAR function. We overexpressed LA-PDE2 in MSNs to determine whether exposure to 650-nm light could decrease GluA1 surface expression. MSNs exposed to red light for 2 min resulted in significant degradation of cAMP (Fig. 5A) and was accompanied by removal of surface GluA1 (Fig. 5B). These data support that PDE2 activation, independent of cGMP signaling, is sufficient to decrease cAMP levels and AMPAR surface expression.
FIGURE 5.
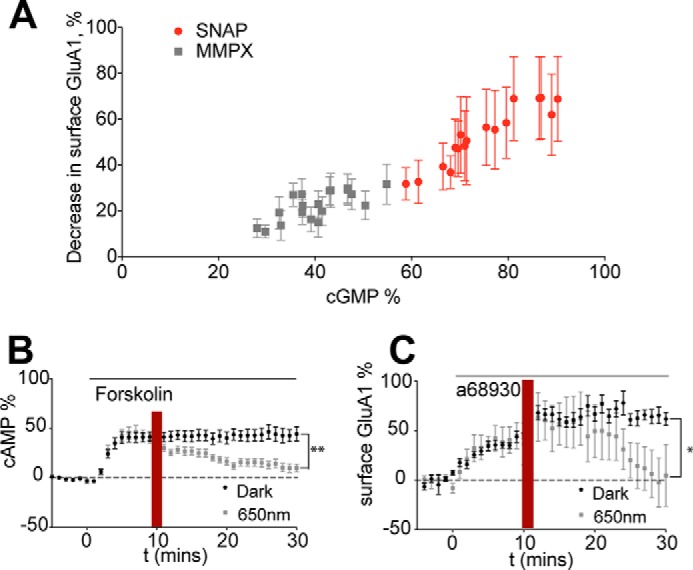
Activation of PDE2 decreases surface GluA1 levels. Data are represented as mean ± S.E. normalized to baseline. A, increasing cGMP levels is inversely proportional to surface GluA1 expression in D1 MSNs. The graph relates cGMP levels induced by SNAP or MMPX (Figs. 3A and 4A) and surface GluA1 (Figs. 3C and 4C). B, LA-PDE2 activation by 650-nm light induces a decrease in cAMP levels. 12.5 μm forskolin was added after a 5-min baseline, followed by exposure to no light (black circles, 10 cells) or to red light (min 10–12, gray squares, 12 cells). **, p = 0.0011 (repeated measures two-way ANOVA, F1,20 = 14.65). C, LA-PDE2 activation by 650-nm light induces a decrease in surface GluA1 levels. 0.1 μm A68930 was added after a 5-min baseline, followed by exposure to no light (black circles, 12 cells) or to red light (min 10–12, gray squares, six cells). *, p = 0.0294 (repeated measures two-way ANOVA, F1,20 = 7.13).
Next we explored the specificity of the PDE1/PDE2 cross-talk in regulating surface GluA1 levels. First we tested whether other MSN-enriched PDEs with cGMP and cAMP activity, such as PDE10A, could have a comparable role to PDE1 (24, 49–54). Application of the PDE10 inhibitor TC-E5005 did not affect surface GluA1 levels (Fig. 6A), suggesting that this cross-regulation was specific to PDE1 and that modulating any PDEs with cGMP activity does not result in the activation of PDE2. Next we tested whether PDE4, a PDE that has been implicated in AMPAR trafficking, could modulate the deficit in surface GluA1 expression induced by PDE1 inhibition (38). Treatment with the PDE4 inhibitor rolipram produced the reported enhancement in D1-induced GluA1 surface expression (Fig. 6B). We reasoned that, if PDE4 controlled the same local pool of cAMP as PDE2, then co-application of a PDE4 inhibitor could rescue the PDE1-induced deficit in an analogous fashion as PDE2 inhibition did, as shown in Fig. 4C. However, when rolipram was applied along with a PDE1 inhibitor, it failed to recover the deficit in surface expression of GluA1 induced by PDE1 inhibition alone (Fig. 6C), suggesting that the pool of cAMP controlled by rolipram cannot overcome the deficit in trafficking of GluA1 induced by PDE2 activation.
FIGURE 6.
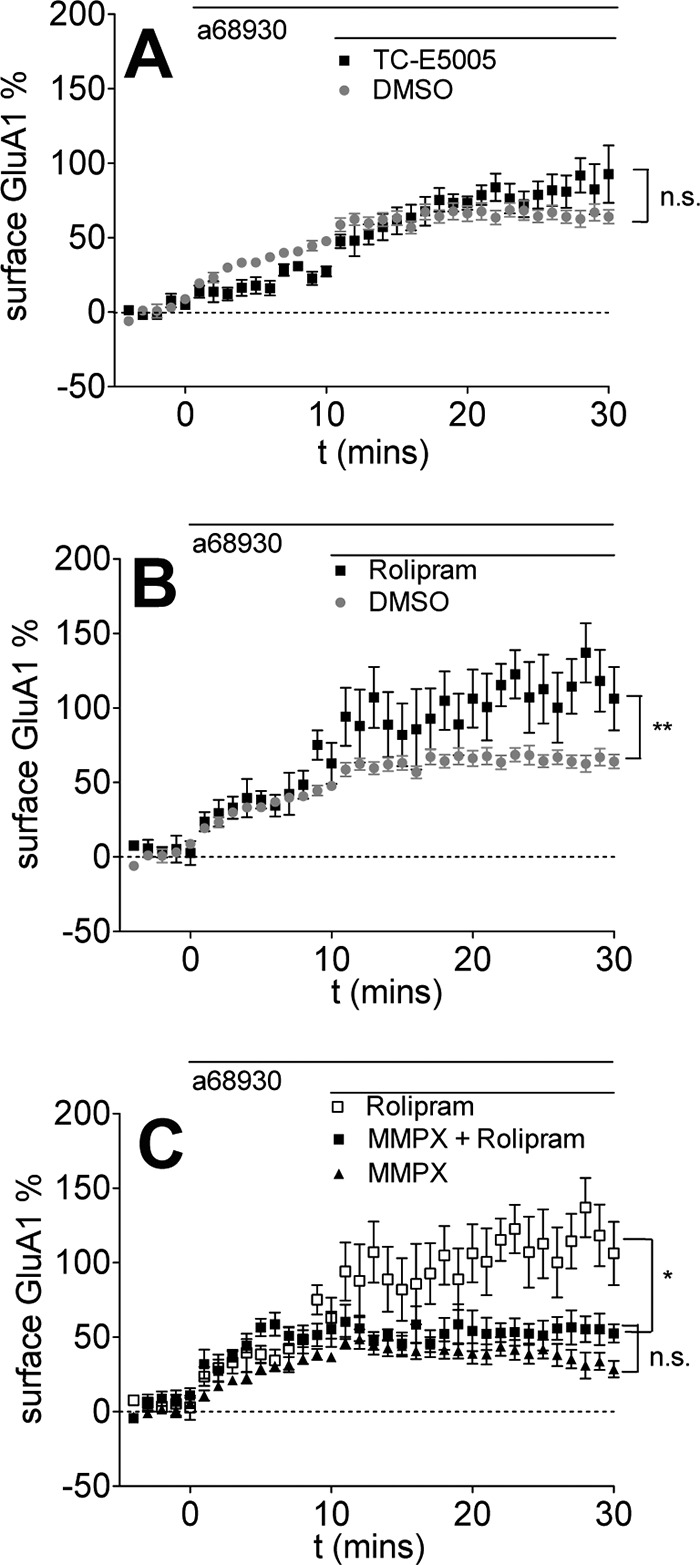
PDE1/PDE2 cross-talk regulation of surface GluA1 levels is not dependent on PDE10 or PDE4 and is specific for GluA1. Data are represented as mean ± S.E. normalized to baseline. A, PDE10 inhibition has no effect on D1R agonist-induced GluA1 surface insertion. 0.1 μm A68930 was added for 10 min, followed by DMSO (replotted from Fig. 1 for comparison) or the PDE10 inhibitor TC-E5005 (10 μm, black triangles, seven cells). n.s., p = 0.3658 (repeated measures two-way ANOVA, F1,32 = 0.84). B, PDE4 inhibition enhances surface GluA1 levels induced by D1R agonist treatment. 0.1 μm A68930 was added for 10 min, followed by DMSO (replotted from Fig. 1 for comparison) or the PDE4 inhibitor rolipram (10 μm, black squares, six cells). **, p = 0.0019 (repeated measures two-way ANOVA, DMSO versus rolipram, F1,31 =11.47). C, PDE4 inhibition fails to rescue the PDE1 inhibition-induced decrease in surface GluA1 levels. 0.1 μm A68930 was added for 10 min, followed by MMPX (replotted from Fig. 4 for comparison), rolipram (replotted from Fig. 6B for comparison), or MMPX + rolipram (black triangles, 10 cells). n.s., p = 0.1708 (repeated measures two-way ANOVA, F1,20 = 2.02).
The Cross-regulation of PDE1 and PDE2 Results in a Decrease in Surface GluA1 Levels in Intact Adult Striatal Tissue
So far we have presented compelling evidence of the cross-regulation of PDE1 and PDE2 controlling GluA1 surface levels in primary MSNs. Thus, we explored whether the cross-regulation of PDE1 and PDE2 occurs in adult tissue using biotinylation assays to monitor surface GluA1-containing AMPAR. We treated slices with the D1 agonist A68930 in the presence and absence of the PDE2 inhibitor BAY60-7550 and determined the proportion of surface GluA1 versus total GluA1 (surface and intracellular pool of GluA1). In agreement with our primary MSNs data, we found that PDE2 inhibition significantly enhanced D1R-mediated GluA1 surface expression (Fig. 7, A and B).
FIGURE 7.

PDE1/PDE2 cross-talk regulates surface GluA1 surface expression in adult striatal tissue. A, treatment with a PDE2 inhibitor potentiates the D1-induced increase in surface GluA1 expression. Acute mouse striatal slices were treated for 30 min under the following conditions: DMSO, 1.0 μm A68930, or 1.0 μm A68930 + 10 μm BAY60-7550. Surface GluA1 and total GluA1 expression are shown. GAPDH was used as a loading CT. B, densitometry of the immunoblots shown in A. Significance was analyzed using one-way ANOVA followed by Tukey's post hoc test (DMSO versus A68930: *, p = 0.0154; DMSO versus A68930 + BAY60-7550: **, p = 0.0071; A68930 versus A68930 + BAY60-7550: *, p = 0.0402; n = 7). C, treatment with a PDE1 inhibitor decreases the D1-induced increase in surface GluA1 expression. Acute mouse striatal slices were treated for 30 min under the following conditions: DMSO, 0.1 μm A68930, 1.0 μm A68930 + 100 μm MMPX, or 1.0 μm A68930 + 10 μm BAY60-7550 + 100 μm MMPX for 30 min. Surface GluA1 and total GluA1 expression are shown. GAPDH was used as a loading CT. D, densitometry of the immunoblots shown in C. Significance was analyzed using one-way ANOVA followed by Tukey's post hoc test (DMSO versus A68930: *, p = 0.0496; DMSO versus A68930 + MMPX: n.s., p = 0.9683; DMSO versus A68930 + MMPX + BAY60-7550: *, p = 0.0438; A68930 versus A68930 + MMPX: *, p = 0.0336; A68930 versus A68930 + MMPX + BAY60-7550: n.s., p = 0.8937; A68930 + MMPX versus A68930 + MMPX + BAY60-7550: *, p = 0.0347).
Next we tested whether the interplay of PDE1 and PDE2 can affect surface GluA1 levels in tissue. Treatment with a PDE1 inhibitor, MMPX, resulted in a significant decrease in D1R-induced GluA1 surface expression (Fig. 7, C and D), and this was reversed by co-application of a PDE2 inhibitor. These results further support our MSN culture data and indicate that the cross-regulation of PDE1 and PDE2 occurs in intact striatal adult tissue and is part of the signaling repertoire of adult D1 MSNs.
Discussion
We propose PDE cross-regulation as a novel mechanism shaping cyclic nucleotide control of AMPAR trafficking. Our computational model simulations indicate that the cGMP activation of PDE2 is capable of inducing changes in cAMP levels that are sufficient to decrease AMPAR surface expression. Despite having activity toward cAMP, PDE1 inhibition resulted in a counterintuitive decrease in cAMP levels. Moreover, these simulations predict that the interplay of PDE1 and PDE2 activities can alter synaptic strength, as PDE1 inhibition allows basal cGMP levels to accumulate to levels sufficient to activate PDE2, resulting in the modulation of AMPAR surface expression. Experiments using SEP-GluA1, cAMP, and cGMP imaging and biotinylation assays of surface GluA1 in adult intact striatal tissue confirmed these predictions. Co-application of PDE1 and PDE2 inhibitors resulted in a complete reversal of the deficit in AMPAR trafficking observed when PDE1 activity was impaired. There was a quantitative difference between the observed results of this reversal in the primary neurons and the intact tissue slices. In primary neurons, this reversal resulted in surface AMPAR levels that surpassed those observed with a D1 agonist alone. In tissue slices, the reversal resulted in levels that matched those obtained with a D1 agonist alone. This difference may be because of the inclusion of signaling originating from D2 MSNs in the immunoblots of intact tissue. PDE10 inhibition did not have an effect on GluA1 trafficking, and this is congruent with a previous report of the role of PDE10 in cAMP signaling in D1 MSNs (55). We showed that, when in combination with a PDE1 inhibitor, PDE4 inhibition fails to rescue the deficit in surface expression of GluA1 induced by MMPX. This points to a high degree of compartmentalization, as these PDEs may be controlling the trafficking of distinct pools of cAMP, although the nature of this compartmentalization is still unknown.
In non-neuronal cell types, the activities of PDE2, PDE3 (a cGMP-inhibited cAMP PDE), and PDE5 (a cGMP-activated cGMP PDE) have the potential to be cross-regulated, leading to counterintuitive cAMP/cGMP downstream responses (56). Because each cell type displays a distinct PDE expression profile, there have been several reports of different PDE combinations partaking in cross-regulation. For instance, in platelets, the interplay of PDE3 and PDE5 activities modulates platelet aggregation, whereas the cross-regulation of PDE2 and PDE3 activities affects cellular permeability in endothelial cells (21–23). To our knowledge, our study is the first report of PDE cross-regulation in neuronal signaling. Additionally, the PDE1 and PDE2 combination as a PDE cross-regulatory motif has not, until now, been described as controlling downstream signaling. Our results suggest that the interplay of PDE1 and PDE2 activities is a significant novel feature of MSNs cyclic nucleotide signaling and AMPAR trafficking dynamics. These types of PDE cross-regulation should be fully characterized, as PDEs show great promise as therapeutic targets for a number of pathologies but have fared poorly in clinical trials. Elucidating the indirect signaling effects of modulating a PDE activity may help understand some of the factors that have precluded PDE inhibitors from reaching the clinic.
There are several considerations that we must point out regarding this study. Here we are solely exploring the cyclic nucleotide signaling that occurs in D1 MSNs. Whether the interplay of PDE1 and PDE2 activities also occurs in D2 MSNs is an active area of research in our laboratory. One aspect that we did not explore is whether Ca2+ signaling could modulate the net effect of PDE1/PDE2 interplay on AMPAR trafficking. This is especially pertinent, as PDE1 is a Ca2+/calmodulin-activated PDE (57–59), and this regulatory feature could potentially disrupt PDE2 cross-activation and affect AMPAR trafficking dynamics. We speculate that, under high Ca2+ conditions, PDE1 is fully activated, reducing the cGMP pool that controls PDE2 activity and resulting in a less active form of PDE2 and perhaps modulating surface GluA1 expression. Additionally, it has been reported that high Ca2+ conditions can also suppress cAMP production by inhibiting AC5, the main cAMP production activity of MSNs (60, 61). More recently, the identification of Cdk5 and calmodulin-activated kinase II as additional regulators of PDE4 activity has highlighted the central role of PDEs in the integration of cAMP/Ca2+ signaling (62, 63). Thus, future extensions of the dopamine-induced AMPAR trafficking model may require inclusion of Ca2+ signaling and its effect on cAMP.
Furthermore, high Ca2+ conditions (such as those induced by synaptic glutamate stimulation) activate soluble guanylyl cyclase to produce cGMP accumulation to sufficient levels to activate cGMP-dependent protein kinase II, leading to the phosphorylation and surface insertion of GluA1 (64–66). DA stimulation alone without an additional Ca2+ signal fails to activate cGMP-dependent kinase, resulting in GluA1 trafficking that is solely PKA-dependent (64). All of this points to a dichotomy in cyclic nucleotide control of AMPAR trafficking where DA stimulation alone induces cAMP/PKA-driven GluA1 insertion, and small increases in cGMP levels can counteract this, whereas glutamate produces significant Ca2+ signaling that results in significant cGMP levels to activate cGMP-dependent protein kinase II and promotes GluA1 insertion (64). Whether PDE1 and PDE2 play any role in cGMP-dependent protein kinase II-dependent GluA1 insertion has still to be determined. Additionally, this type of cGMP signaling dichotomy may explain some of the discrepancies found when examining the role of cGMP in corticostriatal synaptic plasticity in in vitro versus in vivo models (67). For instance, in vivo recordings display increases in synaptic potentiation in response to NO/cGMP signaling (67–69), whereas slice experiments have demonstrated a contradictory role for the cGMP pathway in plasticity of corticostriatal synapses. In acute tissue slices, agents that elevate cGMP levels induce long-term synaptic depression (LTD), implying a rapid removal of surface AMPAR (70). Similar to this study, this type of LTD induction is achieved with intracellular application of non-degradable cGMP analogues, treatment with SNAP, or inhibition of cGMP degradation using PDE1 inhibitors. It is possible that, in these in vitro conditions, there is sufficient dopaminergic tone but a lack of adequate glutamatergic/Ca2+ signals found in vivo, resulting in a failure to activate PKG but permitting the activation of PDE2, giving rise to the rapid removal of surface AMPAR to promote an LTD state.
Collectively, the simulations and experimental validation presented here shed light on how the interplay of PDE1 and PDE2 activities gives rise to cross-talk between cAMP and cGMP signaling, resulting in control of DA-induced AMPAR trafficking. These results suggest that PDE2 regulates forward trafficking and/or maintenance of AMPAR on the plasma membrane in D1 MSNs. Our work shows how PDE1 inhibition results in unintended and counterintuitive downstream signaling. Future extensions of this quantitative framework will allow analysis of more complex signaling, such as the inclusion of Ca2+ signals that can regulate PDE1 and PDE4 activity and cGMP and cAMP production. Furthermore, our mechanistic representation of cyclic nucleotide signaling in MSNs provides a quantitative framework to explore how targeting the interplay of PDE1/PDE2 activities could modulate the magnitude and directionality of synaptic responsiveness. This approach could have therapeutic value in a number of striatal pathologies characterized by hyperactivation of D1 signaling, such as levodopa-induced dyskinesia.
Experimental Procedures
This study conforms with the Guide for the Care and Use of Laboratory Animals by the National Institutes of Health (publication no. 85-23, revised 1996). All experimental protocols were approved by the Institutional Animal Care and Use Committee of the Icahn School of Medicine at Mount Sinai.
Cell Culture and Transfection
HEK-293T cells (ATCC) were maintained in Dulbecco's modified Eagle's medium (Life Technologies) supplemented with 10% fetal bovine serum (Life Technologies) and 1% penicillin-streptomycin (Life Technologies).
Live-cell Imaging
Primary MSNs were obtained as described previously (39). To monitor GluA1 trafficking, MSNs (days in vitro 7–16) were transfected with PCI-SEP-GluR1. pCI-SEP-GluR1 was a gift from Robert Malinow (Addgene plasmid 24000) (42). pCI-SEP-GluR1 and pmCherry (Clontech) plasmids were transfected using Lipofectamine 2000 (Life Technologies). In some experiments, MSNs were also transfected with pcDNA3-PDE2-D485A-mcherry or PDE2DN-mcherry plasmids (21) (kindly provided by Manuela Zaccolo) in addition to the SEP-GluA1 plasmid. For cAMP imaging, neurons were transfected with the pICUE3 plasmid (71) (kindly provided by Jin Zhang). To monitor intracellular cGMP levels, we used FlincG3. The plasmid pTriEx4-H6-FGAm (FlincG3) was a gift from John Garthwaite (Addgene plasmid 49202). 24 h after transfections, MSNs were incubated with Neurobasal medium containing nifedipine (10 μm, Sigma-Aldrich) and tetrodotoxin (1 μm, Tocris) for 1 h to control for depolarization and calcium-mediated signaling. MSNs were then mounted into an imaging chamber maintained at 32 °C and continually perfused with 1× Hanks Buffered Saline Solution (Life Technologies) medium containing 25 mm HEPES, 10 mm glucose, and 0.5 mm Trolox (Sigma-Aldrich). MSNs were imaged on an inverted Axio-Observer Z1 microscope (Zeiss) with an automated stage using AxioVision 4.8 software using a Plan-Apochromat ×40/1.3 oil objective and a quantEM electron-multiplying charge-coupled device camera (Photometrics) illuminated by a Colibri controlled LED system (Zeiss). For SEP-GluA1, FlingG3, or mCherry channels, images were acquired every 1 min with the following filters: excitation (ex) LED 470 nm and emission (em) 527/54 nm for SEP-GluA1 and ex LED 590 nm and em 615 nm for mCherry. For cAMP measurements, images from the Cerulean and FRET channels were acquired every 1 min with the following settings: ex LED 455 nm and em 475/40 nm for Cerulean and ex LED 455 nm and em 535/25 nm for FRET. There have been reports of experimental conditions changing the cellular pH of neurons, affecting the interpretation of SEP-trafficking data and putting into question the utility of SEP-based approaches (72). Because of this concern, we ruled out any nonspecific effects pharmacological agents may have on intracellular pH that may affect SEP fluorescence using non-tagged cytoplasmic SEP (supplemental Fig. S1). Only compounds that did not affect non-tagged SEP fluorescence were further employed. To differentiate between D1 versus D2 MSNs, we treated the neuron with the D1 agonist A68930, followed by an agonists targeting adenosine A2A receptors, a Gs-GPCR subtype exclusively expressed in D2 MSNs. This type of sequential drug treatment allowed us to identify D1 versus D2 MSNs, and in this study we concentrate on signaling of D1 MSNs. After 10 min of receptor stimulation and depending on the experimental conditions, DMSO or the following treatments were added: the PDE1 inhibitor 8-methoxymethyl-3-isobutyl-1-methylxanthine (MMPX, 10 μm, Tocris), the PDE2 inhibitor 2-[(3,4-dimethoxyphenyl)methyl]-7-[(1R)-1-hydroxyethyl]-4-phenylbutyl]-5-methyl-imidazo[5, 1-f][1,2,4]triazin-4(1H)-one (BAY60-7550, 1 μm, Cayman Chemicals), or the NO donor SNAP (50 μm, Tocris). These pharmacological treatments were applied at concentrations utilized in previous studies. The adenylyl cyclase activator forskolin (FK, 50 μm, Tocris) and the nonspecific PDE inhibitor isobutylmethylxanthine (IBMX, 100 μm, Tocris) were added after 20 min to saturate the response and assess the health of the neuron. In the case of the cGMP measurements, SNAP (50 μm) and IBMX (100 μm) were used to saturate the response. We analyzed the resulting time series of images as follows. All images were background-corrected. Only measurements from dendritic segments were used. Dendritic fluorescent intensities for SEP-GluA1 were normalized to corresponding mCherry fluorescence intensities. The ratios of donor to acceptor dendritic intensities were calculated for FRET experiments. All time points were scaled to FK/IBMX or SNAP/IBMX and normalized to the baseline and are presented as time courses of averaged percent change of basal levels. Any cell that lacked a stable baseline, displayed photobleaching, or did not display a robust D1 or FK/IBMX response was not included in our dataset. All time courses are presented as averages of the mean of multiple cells (obtained from at least three independent preparations of primary neurons) and corresponding standard error.
Surface GluA1 Biotinylation in Tissue Slices
250-μm-thick coronal slices were obtained using a Leica vibratome from 3- to 5-month old C57BL/6 mice (The Jackson Laboratory). Signature anatomical landmarks (e.g. the anterior commissure, corpus callosum) were used to identify the striatum. Slices were allowed to recover for 1 h at room temperature in oxygenated ACSF (119 mm NaCl, 2.5 mm KCl, 1 mm NaH2PO4-H2O, 26.2 mm NaHCO3, 1.3 mm MgCl2, 2.5 mm CaCl2, and 11 mm glucose). After recovery, slices were treated for 30 min in oxygenated ACSF at room temperature with 1 μm A68930 in the presence and absence of 10 μm BAY60–7550 or 100 μm MMPX. Following the drug treatment, the slices were rapidly chilled by washing three times with ice-cold ACSF and incubated with 1.0 mg/ml sulfo-NHS-SS-biotin (Thermo Scientific) for 45 min on ice. Tissue slices were washed with ice-cold ACSF, followed by washes with cold 100 mm glycine and flash-frozen on dry ice. When frozen, the striata were carefully dissected from each slice. Striatal tissue was mechanically dissociated in radioimmune precipitation assay lysis buffer (10 mm Tris (pH 7.4), 150 mm NaCl, 1.0 mm EDTA, 1% Triton-X-100, 0.1% SDS, and 1% sodium deoxycholate) supplemented with protease and phosphate inhibitors (Thermo Scientific). The protein concentration of each lysate was calculated using Bradford reagent (Bio-Rad). Protein lysates were incubated overnight with streptavidin-coated Dynabeads (Life Technologies) at 4 °C to isolate biotinylated surface proteins. Biotinylated proteins were eluted from streptavidin beads with radioimmune precipitation assay buffer containing 1× SDS-PAGE reducing sample buffer with DTT at 95 °C for 5 min. Eluted proteins were resolved by SDS-PAGE as described previously (39, 73). Nitrocellulose membranes were incubated with anti-GluA1 N-terminal antibody (MAB2263, EMD Millipore). GAPDH (2275, Trevigen) staining was used as loading control, normalized to Ponceau S staining as recommended in Ref. 74. Full images of the representative immunoblots are shown in supplemental Fig. S8.
Dynamical Modeling
We expanded our previous computational model of dopamine-induced AMPAR trafficking (39) by adding the following reactions: basal production of cGMP by soluble guanylyl cyclase from GTP, degradation of cAMP and cGMP by PDE1, allosteric activation of PDE2 by cGMP, and degradation of cAMP and cGMP by PDE2. The model was initially developed in VCell and was exported to MATLAB (Mathworks) for ease of computing and optimization. All simulations were conducted using the ordinary differential equation solver of MATLAB, ODE15s, with relative and absolute tolerance set to 1e–6. The initial concentrations and kinetic reaction parameters for the reactions listed above are provided in the (supplemental Tables S1 and S2). The full MATLAB scripts are available upon request. As we have done in our previous work (39, 75, 76), most of the model parameters were obtained from the literature. Unknown parameters were constrained using published data, such as the cGMP-dependent activation of PDE2 (22), or data generated by us, such as the cAMP or surface GluA1 increases because of PDE2 inhibition (supplemental Fig. S2 or Fig. 1A). Sensitivity analysis was performed as described in (supplemental Figs. S3–S5).
Author Contributions
R. S. S. and R. T. performed the experiments and analyzed the data. R. S. S. developed the model and performed the simulations. S. R. N. Z. designed and supervised the experiments and the simulations. E. A. S. supervised the sensitivity analysis. R. S. S. and S. R. N. Z. wrote the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Dr. Masago Ishikawa and Dr. Eric Sweet for technical assistance and Dr. Manuela Zaccolo, Dr. Robbert Havekes, and Dr. Jin Zhang for reagents. We also thank Dr. Joe Beavo, Dr. Ravi Iyengar, Dr. Paul Kenny, and Dr. Robert Blitzer for helpful discussions. VCell is funded by NIGMS, National Institutes of Health Grant P41 GM103313.
This work was supported by National Institutes of Health Grant 5R01DK087650 and by Systems Biology Center of New York Grant PSDGM071558. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Experimental Procedures, References, Figs. S1–S8, and Tables S1–S3.
- MSN
- medium spiny neuron
- DA
- dopamine
- AMPAR
- AMPA receptor
- PDE
- phosphodiesterase
- D1R
- dopamine receptor 1
- AUC
- area under the curve
- SNAP
- S-nitroso-N-acetylpenicillamine
- LTD
- long-term depression
- ex
- excitation
- em
- emission
- MMPX
- 8-methoxymethyl-3-isobutyl-1-methylxanthine
- BAY60-7550
- 2-[(3,4-dimethoxyphenyl)methyl]-7-[(1R)-1-hydroxyethyl]-4-phenylbutyl]-5-methyl-imidazo[5, 1-f][1,2,4]triazin-4(1H)-one
- FK
- forskolin
- ACFS
- artificial cerebralspinal fluid
- ANOVA
- analysis of variance
- CT
- control
- LED
- light emitting diodes
- IBMX
- isobutylmethylxanthine
- SEP
- pHluorin.
References
- 1. Carlezon W. A. Jr., and Thomas M. J. (2009) Biological substrates of reward and aversion: a nucleus accumbens activity hypothesis. Neuropharmacology 56, 122–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Graybiel A. M. (2005) The basal ganglia: learning new tricks and loving it. Curr. Opin. Neurobiol. 15, 638–644 [DOI] [PubMed] [Google Scholar]
- 3. Surmeier D. J., Ding J., Day M., Wang Z., and Shen W. (2007) D1 and D2 dopamine-receptor modulation of striatal glutamatergic signaling in striatal medium spiny neurons. Trends Neurosci. 30, 228–235 [DOI] [PubMed] [Google Scholar]
- 4. Tritsch N. X., and Sabatini B. L. (2012) Dopaminergic modulation of synaptic transmission in cortex and striatum. Neuron 76, 33–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Snyder G. L., Allen P. B., Fienberg A. A., Valle C. G., Huganir R. L., Nairn A. C., and Greengard P. (2000) Regulation of phosphorylation of the GluR1 AMPA receptor in the neostriatum by dopamine and psychostimulants in vivo. J. Neurosci. 20, 4480–4488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gao C., Sun X., and Wolf M. E. (2006) Activation of D1 dopamine receptors increases surface expression of AMPA receptors and facilitates their synaptic incorporation in cultured hippocampal neurons. J. Neurochem. 98, 1664–1677 [DOI] [PubMed] [Google Scholar]
- 7. Mangiavacchi S., and Wolf M. E. (2004) D1 dopamine receptor stimulation increases the rate of AMPA receptor insertion onto the surface of cultured nucleus accumbens neurons through a pathway dependent on protein kinase A. J. Neurochem. 88, 1261–1271 [DOI] [PubMed] [Google Scholar]
- 8. Sun X., Zhao Y., and Wolf M. E. (2005) Dopamine receptor stimulation modulates AMPA receptor synaptic insertion in prefrontal cortex neurons. J. Neurosci. 25, 7342–7351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Esteban J. A., Shi S. H., Wilson C., Nuriya M., Huganir R. L., and Malinow R. (2003) PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nat. Neurosci. 6, 136–143 [DOI] [PubMed] [Google Scholar]
- 10. Serulle Y., Zhang S., Ninan I., Puzzo D., McCarthy M., Khatri L., Arancio O., and Ziff E. B. (2007) A GluR1-cGKII interaction regulates AMPA receptor trafficking. Neuron 56, 670–688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Taylor S. S., Zhang P., Steichen J. M., Keshwani M. M., and Kornev A. P. (2013) PKA: lessons learned after twenty years. Biochim. Biophys. Acta 1834, 1271–1278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sample V., DiPilato L. M., Yang J. H., Ni Q., Saucerman J. J., and Zhang J. (2012) Regulation of nuclear PKA revealed by spatiotemporal manipulation of cyclic AMP. Nat. Chem. Biol. 8, 375–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Perino A., Ghigo A., Scott J. D., and Hirsch E. (2012) Anchoring proteins as regulators of signaling pathways. Circ. Res. 111, 482–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tsvetanova N. G., and von Zastrow M. (2014) Spatial encoding of cyclic AMP signaling specificity by GPCR endocytosis. Nat. Chem. Biol. 10, 1061–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Conti M., Mika D., and Richter W. (2014) Cyclic AMP compartments and signaling specificity: role of cyclic nucleotide phosphodiesterases. J. Gen. Physiol. 143, 29–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Noyama K., and Maekawa S. (2003) Localization of cyclic nucleotide phosphodiesterase 2 in the brain-derived Triton-insoluble low-density fraction (raft). Neurosci. Res. 45, 141–148 [DOI] [PubMed] [Google Scholar]
- 17. Leroy M. J., Degerman E., Taira M., Murata T., Wang L. H., Movsesian M. A., Meacci E., and Manganiello V. C. (1996) Characterization of two recombinant PDE3 (cGMP-inhibited cyclic nucleotide phosphodiesterase) isoforms, RcGIP1 and HcGIP2, expressed in NIH 3006 murine fibroblasts and Sf9 insect cells. Biochemistry 35, 10194–10202 [DOI] [PubMed] [Google Scholar]
- 18. Omori K., and Kotera J. (2007) Overview of PDEs and their regulation. Circ. Res. 100, 309–327 [DOI] [PubMed] [Google Scholar]
- 19. Zhao C. Y., Greenstein J. L., and Winslow R. L. (2015) Interaction between phosphodiesterases in the regulation of the cardiac β-adrenergic pathway. J. Mol. Cell Cardiol. 88, 29–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhao C. Y., Greenstein J. L., and Winslow R. L. (2016) Roles of phosphodiesterases in the regulation of the cardiac cyclic nucleotide cross-talk signaling network. J. Mol. Cell Cardiol. 91, 215–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stangherlin A., Gesellchen F., Zoccarato A., Terrin A., Fields L. A., Berrera M., Surdo N. C., Craig M. A., Smith G., Hamilton G., and Zaccolo M. (2011) cGMP signals modulate cAMP levels in a compartment-specific manner to regulate catecholamine-dependent signaling in cardiac myocytes. Circ. Res. 108, 929–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Surapisitchat J., Jeon K. I., Yan C., and Beavo J. A. (2007) Differential regulation of endothelial cell permeability by cGMP via phosphodiesterases 2 and 3. Circ. Res. 101, 811–818 [DOI] [PubMed] [Google Scholar]
- 23. Méry P. F., Pavoine C., Belhassen L., Pecker F., and Fischmeister R. (1993) Nitric oxide regulates cardiac Ca2+ current: involvement of cGMP-inhibited and cGMP-stimulated phosphodiesterases through guanylyl cyclase activation. J. Biol. Chem. 268, 26286–26295 [PubMed] [Google Scholar]
- 24. Heiman M., Schaefer A., Gong S., Peterson J. D., Day M., Ramsey K. E., Suárez-Fariñas M., Schwarz C., Stephan D. A., Surmeier D. J., Greengard P., and Heintz N. (2008) A translational profiling approach for the molecular characterization of CNS cell types. Cell 135, 738–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kelly M. P., Adamowicz W., Bove S., Hartman A. J., Mariga A., Pathak G., Reinhart V., Romegialli A., and Kleiman R. J. (2014) Select 3′,5′-cyclic nucleotide phosphodiesterases exhibit altered expression in the aged rodent brain. Cell. Signal. 26, 383–397 [DOI] [PubMed] [Google Scholar]
- 26. Stephenson D. T., Coskran T. M., Kelly M. P., Kleiman R. J., Morton D., O'Neill S. M., Schmidt C. J., Weinberg R. J., and Menniti F. S. (2012) The distribution of phosphodiesterase 2A in the rat brain. Neuroscience 226, 145–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Repaske D. R., Corbin J. G., Conti M., and Goy M. F. (1993) A cyclic GMP-stimulated cyclic nucleotide phosphodiesterase gene is highly expressed in the limbic system of the rat brain. Neuroscience 56, 673–686 [DOI] [PubMed] [Google Scholar]
- 28. Van Staveren W. C., Steinbusch H. W., Markerink-Van Ittersum M., Repaske D. R., Goy M. F., Kotera J., Omori K., Beavo J. A., and De Vente J. (2003) mRNA expression patterns of the cGMP-hydrolyzing phosphodiesterases types 2, 5, and 9 during development of the rat brain. J. Comp. Neurol. 467, 566–580 [DOI] [PubMed] [Google Scholar]
- 29. Erneux C., Couchie D., Dumont J. E., Baraniak J., Stec W. J., Abbad E. G., Petridis G., and Jastorff B. (1981) Specificity of cyclic GMP activation of a multi-substrate cyclic nucleotide phosphodiesterase from rat liver. Eur. J. Biochem. 115, 503–510 [DOI] [PubMed] [Google Scholar]
- 30. Martins T. J., Mumby M. C., and Beavo J. A. (1982) Purification and characterization of a cyclic GMP-stimulated cyclic nucleotide phosphodiesterase from bovine tissues. J. Biol. Chem. 257, 1973–1979 [PubMed] [Google Scholar]
- 31. Rosman G. J., Martins T. J., Sonnenburg W. K., Beavo J. A., Ferguson K., and Loughney K. (1997) Isolation and characterization of human cDNAs encoding a cGMP-stimulated 3′,5′-cyclic nucleotide phosphodiesterase. Gene 191, 89–95 [DOI] [PubMed] [Google Scholar]
- 32. Martinez S. E., Wu A. Y., Glavas N. A., Tang X. B., Turley S., Hol W. G., and Beavo J. A. (2002) The two GAF domains in phosphodiesterase 2A have distinct roles in dimerization and in cGMP binding. Proc. Natl. Acad. Sci. U.S.A. 99, 13260–13265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pandit J., Forman M. D., Fennell K. F., Dillman K. S., and Menniti F. S. (2009) Mechanism for the allosteric regulation of phosphodiesterase 2A deduced from the X-ray structure of a near full-length construct. Proc. Natl. Acad. Sci. U.S.A. 106, 18225–18230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Francis S. H., Blount M. A., and Corbin J. D. (2011) Mammalian cyclic nucleotide phosphodiesterases: molecular mechanisms and physiological functions. Physiol. Rev. 91, 651–690 [DOI] [PubMed] [Google Scholar]
- 35. Wykes V., Bellamy T. C., and Garthwaite J. (2002) Kinetics of nitric oxide-cyclic GMP signalling in CNS cells and its possible regulation by cyclic GMP. J. Neurochem. 83, 37–47 [DOI] [PubMed] [Google Scholar]
- 36. Lin D. T., Fretier P., Jiang C., and Vincent S. R. (2010) Nitric oxide signaling via cGMP-stimulated phosphodiesterase in striatal neurons. Synapse 64, 460–466 [DOI] [PubMed] [Google Scholar]
- 37. Polito M., Klarenbeek J., Jalink K., Paupardin-Tritsch D., Vincent P., and Castro L. R. (2013) The NO/cGMP pathway inhibits transient cAMP signals through the activation of PDE2 in striatal neurons. Front. Cell. Neurosci. 7, 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Russwurm C., Koesling D., and Russwurm M. (2015) Phosphodiesterase 10A is tethered to a synaptic signaling complex in striatum. J. Biol. Chem. 290, 11936–11947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Song R. S., Massenburg B., Wenderski W., Jayaraman V., Thompson L., and Neves S. R. (2013) ERK regulation of phosphodiesterase 4 enhances dopamine-stimulated AMPA receptor membrane insertion. Proc. Natl. Acad. Sci. U.S.A. 110, 15437–15442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Miesenböck G., De Angelis D. A., and Rothman J. E. (1998) Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature 394, 192–195 [DOI] [PubMed] [Google Scholar]
- 41. Sankaranarayanan S., De Angelis D., Rothman J. E., and Ryan T. A. (2000) The use of pHluorins for optical measurements of presynaptic activity. Biophys. J. 79, 2199–2208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kopec C. D., Li B., Wei W., Boehm J., and Malinow R. (2006) Glutamate receptor exocytosis and spine enlargement during chemically induced long-term potentiation. J. Neurosci. 26, 2000–2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Charbonneau H., Prusti R. K., LeTrong H., Sonnenburg W. K., Mullaney P. J., Walsh K. A., and Beavo J. A. (1990) Identification of a noncatalytic cGMP-binding domain conserved in both the cGMP-stimulated and photoreceptor cyclic nucleotide phosphodiesterases. Proc. Natl. Acad. Sci. U.S.A. 87, 288–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Martinez S. E. (2007) in Cyclic Nucleotide Phosophodiesterases in Health and Disease (Beavo J. A., Francis S. H., and Houslay M. D., eds) pp. 55–78, CRC Press, Boca Raton, FL [Google Scholar]
- 45. Sharma R. K., Das S. B., Lakshmikuttyamma A., Selvakumar P., and Shrivastav A. (2006) Regulation of calmodulin-stimulated cyclic nucleotide phosphodiesterase (PDE1): review. Int. J. Mol. Med. 18, 95–105 [PubMed] [Google Scholar]
- 46. McCahill A., McSorley T., Huston E., Hill E. V., Lynch M. J., Gall I., Keryer G., Lygren B., Tasken K., van Heeke G., and Houslay M. D. (2005) In resting COS1 cells a dominant negative approach shows that specific, anchored PDE4 cAMP phosphodiesterase isoforms gate the activation, by basal cyclic AMP production, of AKAP-tethered protein kinase A type II located in the centrosomal region. Cell Signal. 17, 1158–1173 [DOI] [PubMed] [Google Scholar]
- 47. Cherry J. A., and Davis R. L. (1999) Cyclic AMP phosphodiesterases are localized in regions of the mouse brain associated with reinforcement, movement, and affect. J. Comp. Neurol. 407, 287–301 [PubMed] [Google Scholar]
- 48. Gasser C., Taiber S., Yeh C. M., Wittig C. H., Hegemann P., Ryu S., Wunder F., and Möglich A. (2014) Engineering of a red-light-activated human cAMP/cGMP-specific phosphodiesterase. Proc. Natl. Acad. Sci. U.S.A. 111, 8803–8808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Fujishige K., Kotera J., Michibata H., Yuasa K., Takebayashi S., Okumura K., and Omori K. (1999) Cloning and characterization of a novel human phosphodiesterase that hydrolyzes both cAMP and cGMP (PDE10A). J. Biol. Chem. 274, 18438–18445 [DOI] [PubMed] [Google Scholar]
- 50. Fujishige K., Kotera J., and Omori K. (1999) Striatum- and testis-specific phosphodiesterase PDE10A isolation and characterization of a rat PDE10A. Eur. J. Biochem. 266, 1118–1127 [DOI] [PubMed] [Google Scholar]
- 51. Loughney K., Snyder P. B., Uher L., Rosman G. J., Ferguson K., and Florio V. A. (1999) Isolation and characterization of PDE10A, a novel human 3′, 5′-cyclic nucleotide phosphodiesterase. Gene 234, 109–117 [DOI] [PubMed] [Google Scholar]
- 52. Soderling S. H., Bayuga S. J., and Beavo J. A. (1999) Isolation and characterization of a dual-substrate phosphodiesterase gene family: PDE10A. Proc. Natl. Acad. Sci. U.S.A. 96, 7071–7076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nishi A., Kuroiwa M., Miller D. B., O'Callaghan J. P., Bateup H. S., Shuto T., Sotogaku N., Fukuda T., Heintz N., Greengard P., and Snyder G. L. (2008) Distinct roles of PDE4 and PDE10A in the regulation of cAMP/PKA signaling in the striatum. J. Neurosci. 28, 10460–10471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Jäger R., Russwurm C., Schwede F., Genieser H. G., Koesling D., and Russwurm M. (2012) Activation of PDE10 and PDE11 phosphodiesterases. J. Biol. Chem. 287, 1210–1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Polito M., Guiot E., Gangarossa G., Longueville S., Doulazmi M., Valjent E., Herve D., Girault J. A., Paupardin-Tritsch D., Castro L. R., and Vincent P. (2015) Selective effects of PDE10A inhibitors on striatopallidal neurons require phosphatase inhibition by DARPP-32(1,2,3). eNeuro 10.1523/ENEURO.0060-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Castro L. R., Verde I., Cooper D. M., and Fischmeister R. (2006) Cyclic guanosine monophosphate compartmentation in rat cardiac myocytes. Circulation 113, 2221–2228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kakiuchi S., and Yamazaki R. (1970) Calcium-dependent phosphodiesterase activity and its activating factor (PAF) from brain studies on cyclic 3′,5′-nucleotide phosphodiesterase (3). Biochem. Biophys. Res. Commun. 41, 1104–1110 [DOI] [PubMed] [Google Scholar]
- 58. Goraya T. A., Masada N., Ciruela A., and Cooper D. M. (2004) Sustained entry of Ca2+ is required to activate Ca2+-calmodulin-dependent phosphodiesterase 1A. J. Biol. Chem. 279, 40494–40504 [DOI] [PubMed] [Google Scholar]
- 59. Goraya T. A., and Cooper D. M. (2005) Ca2+-calmodulin-dependent phosphodiesterase (PDE1): current perspectives. Cell Signal. 17, 789–797 [DOI] [PubMed] [Google Scholar]
- 60. Kawabe J., Iwami G., Ebina T., Ohno S., Katada T., Ueda Y., Homcy C. J., and Ishikawa Y. (1994) Differential activation of adenylyl cyclase by protein kinase C isoenzymes. J. Biol. Chem. 269, 16554–16558 [PubMed] [Google Scholar]
- 61. Mons N., and Cooper D. M. (1994) Selective expression of one Ca2+-inhibitable adenylyl cyclase in dopaminergically innervated rat brain regions. Brain Res. Mol. Brain Res. 22, 236–244 [DOI] [PubMed] [Google Scholar]
- 62. Plattner F., Hayashi K., Hernández A., Benavides D. R., Tassin T. C., Tan C., Day J., Fina M. W., Yuen E. Y., Yan Z., Goldberg M. S., Nairn A. C., Greengard P., Nestler E. J., Taussig R., et al. (2015) The role of ventral striatal cAMP signaling in stress-induced behaviors. Nat. Neurosci. 18, 1094–1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mika D., Richter W., and Conti M. (2015) A CaMKII/PDE4D negative feedback regulates cAMP signaling. Proc. Natl. Acad. Sci. U.S.A. 112, 2023–2028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Tukey D. S., and Ziff E. B. (2013) Ca2+-permeable AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) receptors and dopamine D1 receptors regulate GluA1 trafficking in striatal neurons. J. Biol. Chem. 288, 35297–35306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Rameau G. A., Chiu L. Y., and Ziff E. B. (2004) Bidirectional regulation of neuronal nitric-oxide synthase phosphorylation at serine 847 by the N-methyl-d-aspartate receptor. J. Biol. Chem. 279, 14307–14314 [DOI] [PubMed] [Google Scholar]
- 66. Rameau G. A., Tukey D. S., Garcin-Hosfield E. D., Titcombe R. F., Misra C., Khatri L., Getzoff E. D., and Ziff E. B. (2007) Biphasic coupling of neuronal nitric oxide synthase phosphorylation to the NMDA receptor regulates AMPA receptor trafficking and neuronal cell death. J. Neurosci. 27, 3445–3455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. West A. R., and Tseng K. Y. (2011) Nitric oxide-soluble guanylyl cyclase-cyclic GMP signaling in the striatum: new targets for the treatment of Parkinson's disease? Front. Syst. Neurosci. 5, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sammut S., Park D. J., and West A. R. (2007) Frontal cortical afferents facilitate striatal nitric oxide transmission in vivo via a NMDA receptor and neuronal NOS-dependent mechanism. J. Neurochem. 103, 1145–1156 [DOI] [PubMed] [Google Scholar]
- 69. West A. R., and Grace A. A. (2004) The nitric oxide-guanylyl cyclase signaling pathway modulates membrane activity states and electrophysiological properties of striatal medium spiny neurons recorded in vivo. J. Neurosci. 24, 1924–1935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Calabresi P., Gubellini P., Centonze D., Sancesario G., Morello M., Giorgi M., Pisani A., and Bernardi G. (1999) A critical role of the nitric oxide/cGMP pathway in corticostriatal long-term depression. J. Neurosci. 19, 2489–2499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Nikolaev V. O., Bünemann M., Hein L., Hannawacker A., and Lohse M. J. (2004) Novel single chain cAMP sensors for receptor-induced signal propagation. J. Biol. Chem. 279, 37215–37218 [DOI] [PubMed] [Google Scholar]
- 72. Rathje M., Fang H., Bachman J. L., Anggono V., Gether U., Huganir R. L., and Madsen K. L. (2013) AMPA receptor pHluorin-GluA2 reports NMDA receptor-induced intracellular acidification in hippocampal neurons. Proc. Natl. Acad. Sci. U.S.A. 110, 14426–14431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Hwangpo T. A., Jordan J. D., Premsrirut P. K., Jayamaran G., Licht J. D., Iyengar R., and Neves S. R. (2012) G Protein-regulated inducer of neurite outgrowth (GRIN) modulates Sprouty protein repression of mitogen-activated protein kinase (MAPK) activation by growth factor stimulation. J. Biol. Chem. 287, 13674–13685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Rivero-Gutiérrez B., Anzola A., Martínez-Augustin O., and de Medina F. S. (2014) Stain-free detection as loading control alternative to Ponceau and housekeeping protein immunodetection in Western blotting. Anal. Biochem. 467, 1–3 [DOI] [PubMed] [Google Scholar]
- 75. Neves S. R., Tsokas P., Sarkar A., Grace E. A., Rangamani P., Taubenfeld S. M., Alberini C. M., Schaff J. C., Blitzer R. D., Moraru I. I., and Iyengar R. (2008) Cell shape and negative links in regulatory motifs together control spatial information flow in signaling networks. Cell 133, 666–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Neves-Zaph S. R., and Song R. S. (2015) Development of computational models of cAMP signaling. Methods Mol. Biol. 1294, 203–217 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.