

Abstract
Reduced smooth muscle (SM)‐specific α2 Na+ pump expression elevates basal blood pressure (BP) and increases BP sensitivity to angiotensin II (Ang II) and dietary NaCl, whilst SM‐α2 overexpression lowers basal BP and decreases Ang II/salt sensitivity. Prolonged ouabain infusion induces hypertension in rodents, and ouabain‐resistant mutation of the α2 ouabain binding site (α2R/R mice) confers resistance to several forms of hypertension. Pressure overload‐induced heart hypertrophy and failure are attenuated in cardio‐specific α2 knockout, cardio‐specific α2 overexpression and α2R/R mice. We propose a unifying hypothesis that reconciles these apparently disparate findings: brain mechanisms, activated by Ang II and high NaCl, regulate sympathetic drive and a novel neurohumoral pathway mediated by both brain and circulating endogenous ouabain (EO). Circulating EO modulates ouabain‐sensitive α2 Na+ pump activity and Ca2+ transporter expression and, via Na+/Ca2+ exchange, Ca2+ homeostasis. This regulates sensitivity to sympathetic activity, Ca2+ signalling and arterial and cardiac contraction.
Keywords: artery, cardiac hypertrophy, heart failure, hypertension
Abbreviations
- ACTH
adrenocorticotropic hormone
- Ang II
angiotensin II
- AT1R
angiotensin type‐1 receptor
- BP
blood pressure
- CNS
central nervous system
- CSF
cerebrospinal fluid
- C‐Src
C‐Src kinase
- CTS
cardiotonic steroid
- CV
cardiovascular
- DN
dominant negative
- DOCA
deoxycorticosterone acetate
- EDL
extensor digitorum longus
- EF
ejection fraction
- ENaC
epithelial Na+ channels
- EO
endogenous ouabain
- HF
heart failure
- HH
heart hypertrophy
- i.c.v.
intracerebroventricular
- jS/ER
junctional sarco‐/endoplasmic reticulum
- LV
left ventricular
- MBG
marinobufagenin
- MI
myocardial infarction
- MS
mass spectrometry
- NCLX
mitochondrial Na+/Ca2+ exchanger
- NCX
Na+/Ca2+ exchanger
- NKA
Na+ pump or Na+, K+‐ATPase
- PLM
phospholemman
- PM
plasma membrane
- RAAS
renin–angiotensin–aldosterone system
- ROS
reactive oxygen species
- RyR
ryanodine receptor
- s.c.
subcutaneous
- S/ER
sarco‐/endoplasmic reticulum
- SERCA
sarco‐/endoplasmic reticulum Ca2+ pump
- SkM
skeletal muscle
- SM
smooth muscle
- SNA
sympathetic nerve activity
- SPM
sub‐PM; SR, sarcoplasmic reticulum
- SS
salt‐sensitive
- TAC
trans‐aortic constriction
- TRPC6
transient receptor potential channel‐6
- WT
wild‐type
Introduction
A decade ago, an article in this journal (Zhang et al. 2005), and two contemporary articles (Dostanic et al. 2005; Dostanic‐Larson et al. 2005), supported the hypothesis (Blaustein, 1977) that arterial Na+ pumps, their endogenous ouabain‐like ligand, and Na+/Ca2+ exchangers (NCX), contribute to salt‐sensitive hypertension. The genetically engineered mouse studies implicate the Na+ pump catalytic subunit α2 isoform. Here, we review recent reports that substantiate the seminal role of α2 Na+ pumps in the pathogenesis of hypertension and also in cardiac hypertrophy and failure. Remarkably, these pathologies can be prevented/attenuated by genetically altered α2 expression and/or ouabain resistant mutation of its binding site. This pinpoints α2 Na+ pumps as a key, but largely overlooked, therapeutic target.
Background
Sodium pumps (Na+,K+‐ATPase or NKA) are expressed in nearly all vertebrate cells. They export three Na+ ions and import two K+ ions while hydrolysing one ATP molecule during each transport cycle (Blanco & Mercer, 1998). The Na+ pumps maintain cell and organism Na+ and K+ homeostasis and influence numerous physiological processes. They also serve as cellular signal transducers for cardiotonic steroids (CTSs) (Xie & Askari, 2002).
Four Na+ pump catalytic subunit isoforms (α1–α4) have been cloned (Shull et al. 1985; Shull & Lingrel, 1987; Woo et al. 1999). Pumps with an α1 isoform (‘α1 Na+ pumps’) are expressed in virtually all cells, and are prevalent in most. They maintain the low Na+ and high K+ concentrations in ‘bulk’ cytoplasm, [Na+]CYT and [K+]CYT, respectively, and the resting membrane potential (e.g. McDonough et al. 1992; He et al. 2001; Radzyukevich et al. 2013), and mediate net Na+ transport across epithelia (McDonough et al. 1992; Rajasekaran et al. 2005). α3 is found in neurones, neonatal myocardium, adult human myocardium, and some other tissues; α4 is expressed in sperm (Lingrel, 2010).
α2 Na+ pumps
We focus on Na+ pumps with an α2 catalytic subunit, which are expressed in the cardiovascular (CV) system (Lingrel, 2010), including the endothelium (Zahler et al. 1996), in skeletal muscle (Radzyukevich et al. 2013) and in the brain (McGrail et al. 1991; Arakaki et al. 2013). In rodent cardiac and vascular smooth muscles, the α1:α2 ratio is ∼4:1 (James et al. 1999; Zhang et al. 2005; Berry et al. 2007; Despa & Bers, 2007); in skeletal muscle the α1:α2 ratio is ∼1:6 (He et al. 2001).
The minimal Na+ pump functional unit is an αβ protomer (Blanco & Mercer, 1998). The α subunit contains the Na+, K+ and Mg‐ATP binding sites, the catalytic machinery, and the CTS binding site. Rodents are unusual, however, because their α1 Na+ pumps have very low affinity for CTS (O'Brien et al. 1994). Thus, in rodents, and probably in other orders of mammals too, α2 and α3 Na+ pumps are the receptors for picomolar to nanomolar CTS (Song et al. 2013). CTSs selectively inhibit Na+ pump‐mediated cation transport (Schatzmann, 1953). Therefore, CTSs, and especially ouabain, which is relatively hydrophilic, are widely employed to study the consequences of Na+ pump blockade.
The Na+ pump β subunit (there are 3 isosforms) chaperones α, and is essential for the catalytic activity (Shull et al. 1986; Blanco & Mercer, 1998; Lingrel, 2010). β1 is the most prevalent isoform in cardiac muscle and vascular smooth muscle, where it forms both α1β1 and α2β1 protomers (Hundal et al. 1994; Cougnon et al. 2002; Hauck et al. 2009; Dey et al. 2012).
α2 Na+ pump localization and its significance
Most arterial (Fig. 1) and cardiac (Fig. 2) myocyte α2 Na+ pumps are localized in plasma membrane (PM) microdomains in close proximity to ‘junctional’ elements of the sarco‐/endoplasmic reticulum (jS/ER), i.e. at PM–S/ER junctions (Juhaszova & Blaustein, 1997 a,b; Mohler et al. 2003; Despa & Bers, 2007; Linde et al. 2012). There may, however, be some overlap with α1 in these microdomains (Mohler et al. 2003; Dey et al. 2012). Na+/Ca2+ exchangers (NCX), too, are localized in the PM–jS/ER microdomains (Figs 1 and 2) (Juhaszova & Blaustein, 1997 a; Berry et al. 2007; Lynch et al. 2008; Davis et al. 2009; Jayasinghe et al. 2009; Kuszczak et al. 2010). In cardiomyocytes, α2 pumps and NCX are found at, or adjacent to (Scriven et al. 2000), PM–S/ER junctions in the transverse (t‐) tubules as well as in the surface membrane (Fig. 2) (Mohler et al. 2003; Berry et al. 2007; Despa & Bers, 2007).
Figure 1. Distribution of α2 Na+ pumps and NCX1 in embryonic mouse (A and B) and human (C and D) artery smooth muscle determined by immunocytochemistry .

A, α2 Na+ pumps (green) and the plasma membrane Ca2+ pump (PMCA, red) are not co‐localized (negligible white areas) in this pseudocolour overlay image of a mouse aorta myocyte. B, α2 Na+ pumps (green) and NCX1 (red) exhibit substantial co‐localization (white areas) in a mouse aorta myocyte. C, primary cultured human mesenteric artery smooth muscle cells (hASMCs) were labelled with anti‐α2 polyclonal antibodies (pAb) and anti‐NCX1 monoclonal antibodies (mAb); the SR was then stained with ER‐Tracker, as indicated by the labels. Insets are enlargements of the boxed areas. Pseudocolour images of the enlarged α2 (red) and NCX1 (green) regions, and the overlay, are shown on the right. D, hASMCs were cross‐reacted with anti‐NCX1 mAb and anti‐TRPC6 pAb; the SR was then stained with ER‐Tracker, as indicated. Insets are enlargements of the boxed areas. Pseudocolour images of the enlarged NCX1 (green) and TRPC6 (red) regions, and the overlay, are shown on the right. In C and D, the patterns of staining by both antibodies were very similar to the pattern of ER Tracker (i.e. SR) distribution. Scale bars in C and D = 30 μm. Note that the α2, NCX1 and TRPC6 staining patterns are all very similar to that of ER‐Tracker. This is reflected by the yellow‐orange staining in the C and D overlay panels, and indicates that hASMC α2 Na+ pumps and NCX1 co‐localize (as in the mouse, B) and overlie elements of SR. A and B were kindly provided by Dr Ronald P. Lynch (B is from Lynch et al. 2008 with permission); C and D are from Linde et al. (2012) with permission.
Figure 2. Confocal images of normal adult rat cardiomyocytes immunolabelled with antibodies raised against SERCA2, Na+ pump α1, Na+ pump α2 and NCX1 .

All four antibodies stained the Z‐line/t‐tubule regions. The surface membrane was stained by anti‐α1, anti‐NCX1 and, to a much lesser extent, anti‐α2 antibodies, but not by anti‐SERCA2. Scale bar = 40 μm. Reproduced from Mohler et al. (2003) with permission.
This organization enables privileged communication among the α2 Na+ pumps, NCX and S/ER Ca2+ pumps (SERCA) through the tiny sub‐PM cytosolic compartment, ‘fuzzy space’, at the junctions (Figs 3 and 4) (Juhaszova & Blaustein, 1997 b; Goldhaber et al. 1999; Poburko et al. 2004; Verdonck et al. 2004; Pritchard et al. 2010; Swift et al. 2010; Aronsen et al. 2013). Consequently, the local α2 pump‐generated Na+ electrochemical gradient (Poburko et al. 2007) modulates NCX‐mediated Ca2+ transport, and local Ca2+ sequestration and Ca2+ signalling (Blaustein & Lederer, 1999; Arnon et al. 2000 b; Golovina et al. 2003; Verdonck et al. 2004; Lee et al. 2006; Lingrel, 2010; Despa et al. 2012; Shattock et al. 2015). This α2‐NCX linkage is consistent with the observation that ∼75% knockdown of cardiac NCX1 decreased α2, but not α1, expression by ∼50% (Bai et al. 2013). Also, ∼65% knockdown of α2 decreased NCX1 by ∼65%, but did not affect α1 expression in arteries (Chen et al. 2015 b).
Figure 3. Diagrams illustrating the acute and chronic effects of EO on Ca2+ homeostasis in arteries: roles of α2 Na+ pumps (NKA), NCX1, SERCA2 and inositol trisphosphate receptors (IP3R) .

Other Ca2+ transporters such as L‐type voltage‐gated Ca2+ channels and plasma membrane (PM) Ca2+ pumps (PMCA) are omitted for simplicity. A, basal conditions. In arteries with tone, myocyte NCX1 operates primarily in the Ca2+ entry mode because the membrane potential, V m = −35 to −50 mV, is more positive than the NCX1 ‘reversal potential’, E Na/Ca (Blaustein & Lederer, 1999); i.e. the driving force (V m − E Na/Ca) is positive. B, acute exposure of arteries to low dose ouabain or EO inhibits (a fraction of) arterial myocyte α2 Na+ pumps, raises [Na+] in the sub‐PM restricted cytosolic space between the PM and SR (shaded area; i.e. [Na+]SPM)*, thereby increasing E Na/Ca and the driving force for NCX1‐mediated Ca2+ entry. The consequent rise in [Ca2+]CYT and Ca2+ sequestered in the SR augments Ca2+ signalling and contraction (the vasotonic effect), thereby increasing vascular tone and BP. C, sustained exposure of arterial myocytes to low dose ouabain or EO, in addition to its acute effects, activates an α2 Na+ pump‐mediated protein kinase (PK) signalling cascade that leads to increased expression of Ca2+ transporters including NCX1 and SERCA (green dotted line and ‘+’ sign). This promotes long‐term arterial Ca2+ gain and sequestration in the SR; via increased Ca2+ signalling, this leads to long‐term elevation of BP. D, comparison of approximate acute and chronic EO‐induced relative changes in NCX1 and SERCA2 expression and contraction, and anticipated [Na+]SPM * and [Ca2+]CYT *. The α2 Na+ pump–NCX1 functional coupling acts as an amplifier: small increases in [Na+]SPM translate to large increases in [Ca2+]CYT and contraction because of the 3 Na+:1 Ca2+ stoichiometry of NCX1 (Blaustein & Lederer, 1999). Furthermore, arterial resistance is inversely related to the fourth power of the radius, r 4 (Poiseuille's law), so small decreases in the radii of resistance arteries will greatly increase peripheral vascular resistance and BP. *Note: [Na+]SPM has not been measured in arterial myocytes, nor have the acute and chronic effects of EO/ouabain on [Ca2+]CYT been compared; thus, the relative changes shown in the figure are speculative. The anticipated [Na+]SPM changes are consistent with NCX1‐mediated Ca2+ entry during chronic high EO (Iwamoto et al. 2004) and with the evidence that immuno‐neutralization of EO rapidly decreases BP in mice with chronic Ang II + salt‐induced hypertension (Chen et al. 2015 a).
Figure 4. Diagrams illustrating the acute and chronic effects of EO on Ca2+ homeostasis in the heart: roles of α2 Na+ pumps (NKA), NCX1, SERCA2 and ryanodine receptors (RyR) .
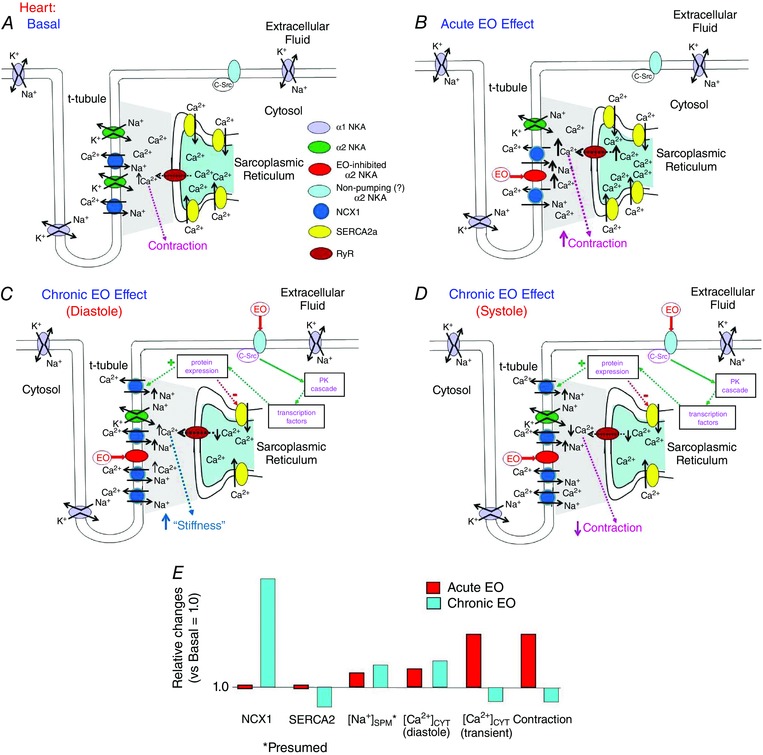
A, basal conditions. In cardiac myocytes, during the major part of the cardiac cycle the NCX1 operates in the Ca2+ exit mode because the diastolic V m, perhaps about −65 to −75 mV, is more negative than E Na/Ca; i.e. the driving force (V m − E Na/Ca) is negative. B, acute exposure of the heart to low dose ouabain or EO inhibits cardiac myocyte α2 Na+ pumps and raises [Na+]SPM. This increases E Na/Ca, but reduces the driving force for Ca2+ extrusion and elevates [Ca2+]SPM. Thus, the net effect, as in arteries, is enhanced Ca2+ signalling and contraction (i.e. the cardiotonic effect). C and D, sustained exposure of cardiac myocytes to low dose ouabain or EO also, as in arteries, activates an α2 Na+ pump‐mediated protein kinase (PK) signalling cascade. In the heart, however, this leads to increased NCX1 expression, but decreased SERCA expression (green and red dotted lines and ‘+’ and ‘−’, respectively). Thus, initially, the cytosolic and SR [Ca2+] are elevated, the cardiotonic effect prevails, increased cardiac contraction is sustained (as in ‘B’), and the heart may hypertrophy from the increased workload. Eventually, however, the sustained Na+ pump inhibition and [Na+]CYT/[Na+]SPM elevation will maintain an elevated diastolic [Ca2+]CYT (C) despite the up‐regulated NCX1. The decreased SERCA2 expression and leakage of Ca2+ from the SR via RyR, however, reduces SR Ca2+ sequestration and [Ca2+]CYT transients (D); thus, cardiac contraction decreases, and the heart fails. E, summary of the acute and chronic EO‐induced approximate relative changes in NCX1 and SERCA2 expression, [Na+]SPM (postulated; see Fig. 3 legend), [Ca2+]CYT and contraction. Note that the acute vasotonic (Fig. 3 B and D) and cardiotonic effects of EO are similar, whereas the chronic effects of EO in the heart (C–E) differ greatly from those in arteries (Fig. 3 C, D).
The special role of α2 also is evident in skeletal muscle: α2 Na+ pumps and NCX are prevalent in t‐tubules, where they may coordinate with junctional sarcoplasmic reticulum (SR) to help regulate the SR Ca2+ concentration, [Ca2+]SR, and contraction (Radzyukevich et al. 2013; Altamirano et al. 2014; DiFranco et al. 2015). Skeletal myocytes (Kristensen et al. 2008) (and choroid epithelial cells; Arakaki et al. 2013), are unusual in that most α2 Na+ pumps are located in intracellular vesicles and are inactive. When translocated to the PM, triggered, e.g., by insulin, muscle contraction or cyclic stretching, they become active (Therien & Blostein, 2000; Yuan et al. 2007; Benziane & Chibalin, 2008; Kristensen et al. 2008; Zhang et al. 2012).
Regulation of α2 Na+ pumps; role of phospholemman
Na+ pumps, including α2 pumps, are regulated by multiple factors, including substrates, hormones (e.g. aldosterone, insulin and catecholamines) and protein phosphorylation (Therien & Blostein, 2000; Phakdeekitcharoen et al. 2011). Importantly, the Na+ and K+ affinities are modulated by the regulatory protein phospholemman (PLM), also called FXYD1 (Crambert et al. 2002; Bibert et al. 2008; Bossuyt et al. 2009; Han et al. 2010; Mishra et al. 2015). Surprisingly, this small molecule with a single transmembrane helix (Geering, 2006) also regulates NCX1 (Wang et al. 2011; Hafver et al. 2016).
Unphosphorylated PLM binds to α2β and reduces α2 affinity for intracellular Na+ and extracellular K+ (Han et al. 2009; Pavlovic et al. 2013 a). Phosphorylation of cardiac or arterial PLM by protein kinase A or C relieves the pump inhibition by altering PLM‐α2β1 interaction and restoring the Na+ high affinity (Bossuyt et al. 2006, 2009; Pavlovic et al. 2007, 2013 a; Han et al. 2010; Dey et al. 2012; Shattock et al. 2015).
Activation of the renin–angiotensin (Ang)–aldosterone system (RAAS), as in hypertension and heart failure (see below), stimulates reactive oxygen species (ROS) generation. This leads to glutathionylation of β1 and pump inhibition (Figtree et al. 2009; Liu et al. 2013). PLM promotes de‐glutathonylation and protects against oxidative inhibition of the pumps in arteries and heart (Liu et al. 2013; Chia et al. 2015).
Endogenous cardiotonic steroids
High affinity CTS binding is observed in all vertebrates (Pressley, 1992; Lingrel, 2010). Ouabain, digoxin and bufalin (a bufadienolide CTS) all block the pump's cation transport pathway (Laursen et al. 2013, 2015). The idea of an endogenous ligand for the Na+ pump CTS binding site (Szent‐Gyorgi, 1953) fostered the proposal that an endogenous ouabain‐like compound contributes to the pathogenesis of hypertension (Haddy & Overbeck, 1976; Blaustein, 1977). Studies in mice with an α2 null mutation, α2+/− and, especially, with ouabain‐resistant α2 Na+ pumps, α2R/R, provide definitive evidence that α2 and its endogenous ligand(s) have a physiological role in mammals (Dostanic et al. 2005; Dostanic‐Larson et al. 2005; Zhang et al. 2005; Lingrel, 2010; Van Huysse et al. 2011; Wansapura et al. 2011).
A CTS was isolated from human plasma and was identified by mass spectrometry (MS) as endogenous ouabain (EO) or a ouabain isomer (Hamlyn et al. 1991; Mathews et al. 1991). Nuclear magnetic resonance (NMR) and MS studies on human, bovine and rodent plasma and tissues verified that the endogenous substance is ouabain (Schneider et al. 1998; Kawamura et al. 1999; Komiyama et al. 2000; Tashko et al. 2010; Jacobs et al. 2012; Hamlyn et al. 2014); reviewed in Hamlyn & Blaustein, 2016). Moreover, recent studies identified two novel EO isomers that are not seen in commercial (plant) ouabain (Jacobs et al. 2012; Hamlyn et al. 2014). The isomers, which may also be present in human plasma (Hamlyn & Blaustein, 2016), apparently are physiologically regulated, but their relative affinities for α2 and their significance are unknown.
Another CTS, marinobufagenin (MBG), was identified in human plasma and urine by immunoassay (Bagrov et al. 1996, 2009). Both EO and MBG reportedly play a role in the pathogenesis of hypertension and heart failure (HF) (Schoner & Scheiner‐Bobis, 2007; Bagrov et al. 2009; Blaustein et al. 2012; Pavlovic, 2014). Prolonged administration of ouabain (Doursout et al. 1992; Yuan et al. 1993; Huang et al. 1994) or MBG (Kennedy et al. 2006) induces hypertension in normal rats, but digoxin and digitoxin do not (Manunta et al. 2000). This implies that Na+ pump inhibition per se does not cause the ouabain‐ or MBG‐induced hypertension. Experiments on α2R/R mice (see Table 1), and studies with fab fragments that immunoneutralize EO and MBG, demonstrate that elevated endogenous CTS levels contribute to CV pathophysiology (Schoner & Scheiner‐Bobis, 2007; Bagrov et al. 2009; Blaustein & Hamlyn, 2010). This review focuses on EO and not MBG because: (i) DigiBind and DigiFab, commercial fab fragments used to immunoneutralize endogenous CTS in vivo, bind ouabain with much higher affinity than MBG (Pullen et al. 2004, 2008); (ii) MBG preferentially binds to α1 rather than α2 subunits (Wansapura et al. 2009); and (iii) several clinical and animal studies on the functions of EO in CV physiology and pathophysiology are backed by analytical (MS) measurements, e.g. Stella et al. (2008), Jacobs et al. (2012) and Hamlyn et al. (2014).
Table 1.
Cardiovascular and skeletal muscle manifestations of genetically modified mouse α2 Na+ pumps
| Genetic modification | Vascular effects | Cardiac effects | Skeletal muscle effects | References |
|---|---|---|---|---|
| Globally reduced α2 (α2+/−) |
|
|
↑Contractile force | |
| Smooth muscle α2 dominant negative (α2SM‐DN) |
|
— | — |
|
| Smooth muscle α2 transgenic over‐expressor (SM‐α2Tg/Tg) |
|
— | — | |
| CV α2 null (CV‐α2−/−) |
|
|
— |
|
| Cardiac α2 null (Cardiac‐α2−/−) |
|
|
— |
|
| Cardiac α2 (Tg) transgenic over‐expressor (Cardiac‐α2Tg) | — |
|
— |
|
| Cardiac α1 (Tg) transgenic over‐expressor (Cardiac‐α1Tg) | — |
|
— |
|
| Skeletal muscle α2 null (α2SkM−/−) | — | — |
|
|
| Global ouabain‐resistant α2 (α2R/R = α1R/R‐α2R/R) |
|
|
↓Sensitivity to fatigue | |
| Global SWAP (α1S/S‐α2R/R vs. WT = α1R/R‐α2S/S) | — |
|
— |
Ouabain‐triggered, Na+ transport‐independent cell signalling mediated by Na+ pumps
Prolonged treatment with ouabain activates multiple intracellular signalling pathways independent of effects on Na+ transport in rat heart and other tissues (Xie & Askari, 2002; Tian & Xie, 2008; Li & Xie, 2009; Zulian et al. 2013). The ouabain‐activated signalling may be mediated by a separate, ‘non‐pumping pool’ of pumps (Liang et al. 2007), perhaps located in caveolae (Liu et al. 2003; Wang et al. 2004; Kristensen et al. 2008). Most investigations employed 1–100 μm ouabain, and emphasized the participation of rodent α1 Na+ pumps, which are relatively ouabain resistant (Liang et al. 2006; Tian et al. 2006). The effectiveness of submicromolar ouabain in rat tissues (Liu et al. 2000; Pulina et al. 2010; Zulian et al. 2013), however, implies mediation by ouabain‐sensitive α2 (or α3), and not resistant α1 Na+ pumps. Ouabain‐induced cell signalling was not observed in immortalized α1‐deficient cells transfected with rat α2 (Xie et al. 2015), but there is no evidence that α2 was linked to the appropriate signalling molecules in those cells, which do not normally express α2. This requires direct comparison of low dose ouabain in native cells from wild‐type (WT) and α2R/R mice.
Ouabain‐triggered intracellular signalling involves protein kinase cascades such as C‐Src kinase (C‐Src), ERK1/2, MAPK, phosphatidylinositide 3‐kinase 1A, protein kinase B (Akt) and NF‐κB, and may be cell‐type specific (Xie & Askari, 2002; Li & Xie, 2009; Wu et al. 2013). C‐Src can be activated by ouabain‐induced ROS generation and carbonylation of the pump (Yan et al. 2013).
Functions of α2 Na+ pumps in arterial physiology and pathophysiology
Arterial myocyte α2 Na+ pumps, modulation of vasoconstriction and blood pressure
Most, if not all, α2 pumps in vascular smooth muscle co‐localize with NCX1 at the PM–jS/ER where, together, they help regulate Ca2+ homeostasis and influence Ca2+ signalling and vasoconstriction (Juhaszova & Blaustein, 1997 a; Lynch et al. 2008; Linde et al. 2012). Normally, most arteries maintain myogenic or vasoconstrictor‐induced (mainly sympathetic nerve‐mediated) ‘basal’ tone (Hill et al. 2001; Zhang et al. 2010 a). Myocyte membrane potential is in the order of −35 to −50 mV (Knot & Nelson, 1998), and the electrochemical driving force on NCX (Blaustein & Lederer, 1999) favours net Ca2+ entry (Iwamoto et al. 2004; Zhang et al. 2010 b; Wang et al. 2015) (Fig. 3 A). Consequently, reduced α2 Na+ pump activity (e.g. ouabain inhibition or reduced expression) should raise the local, sub‐PM Na+ concentration ([Na+]SPM) and promote net Ca2+ gain via NCX, thereby enhancing Ca2+ stores and signalling, and increasing vascular tone and BP (Fig. 3 B) (Zhang et al. 2005, 2009; Chen et al. 2015 b).
In fact, ouabain induces hypertension in most strains of rats; the few negative reports (Ghadhanfar et al. 2014) are consistent with the evidence that ouabain sensitivity is genetically controlled (Aileru et al. 2001). Ouabain also induces hypertension in WT, but not α2R/R mice (Dostanic et al. 2005). Further, α2R/R mice are resistant to adrenocorticotropic hormone (ACTH)‐induced hypertension, and ACTH hypertension is prevented by DigiBind and by the NCX antagonist KB‐R7942 (Dostanic‐Larson et al. 2005; Lorenz et al. 2008). Mice in which CV α2 pumps are selectively knocked out (CV‐α2−/− mice) are also resistant to ACTH‐induced hypertension (Rindler et al. 2011). Because ACTH stimulates EO secretion (Laredo et al. 1994), the implication is that EO‐induced inhibition of α2 raises [Na+]SPM and promotes NCX1‐mediated net gain of Ca2+ and increased arterial constriction.
Mice with genetically reduced α2 pump expression, whether global α2 heterozygous null mutants, α2+/− (Zhang et al. 2005), or smooth muscle (SM)‐specific dominant negative (DN) knockdown, α2SM‐DN (Chen et al. 2015 b), also have elevated BP. (Global α2−/− is embryonic lethal; James et al. 1999.) The α2SM‐DN mice have increased BP sensitivity to subcutaneous (s.c.) Ang II and high dietary salt (vs. WT; not tested in α2+/− mice), presumably because there are fewer available α2 EO receptors and a larger fraction are inhibited by the elevated EO (Blaustein et al. 2015; Chen et al. 2015 b). Conversely, mice with SM‐specific α2 overexpression (α2SM‐Tg) and excess α2 EO binding sites, have low basal BP (Pritchard et al. 2007; Chen et al. 2015 b) and reduced BP sensitivity to s.c. Ang II and high dietary salt (Chen et al. 2015 b).
The fact that CV‐α2−/− mice have normal basal BP despite the nearly complete absence of arterial SM α2 Na+ pumps (Rindler et al. 2011) seems inconsistent with these other reports. In CV‐α2−/− mice, however, the α2‐NCX1 coupling at PM–S/ER junctions is disrupted and NCX1‐mediated Ca2+ transport is stabilized by overexpression of α1 Na+ pumps (Rindler et al. 2011) which maintain a constant, low global [Na+]CYT and are resistant to ouabain/EO.
Digibind lowers BP in deoxycorticosterone acetate (DOCA)–salt hypertensive rats (Krep et al. 1995), and their arteries overexpress the Ca2+ transporter transient receptor potential channel‐6 (TRPC6) (NCX1 and SERCA2 were not tested) (Bae et al. 2007), suggesting that EO is involved. However, α2R/R mice develop DOCA–salt hypertension (Lorenz et al. 2012), implying that EO and MBG are not involved. Whether this is a species or technical difference is unknown.
Collectively, the above reports demonstrate that arterial SM α2 Na+ pumps and EO, along with arterial NCX1, modulate arterial tone and BP, and play an important role in some forms of hypertension (Table 1 and Fig. 3).
Brain α2 Na+ pumps and hypertension
The role of the central nervous system (CNS) in the pathogenesis of essential hypertension and salt‐sensitive (SS‐) hypertension is well documented, albeit incompletely understood. There is broad agreement that central sympathetic drive is a major contributor to BP elevation (Fisher & Fadel, 2010; Allen, 2011; Gabor & Leenen, 2012; Stocker et al. 2015). In addition, EO in the hypothalamus (‘brain ouabain’) and brain α2 Na+ pumps play a role in the pathogenesis of rodent SS‐hypertension: In Wistar rats (Leenen, 2010; Gabor & Leenen, 2012) and WT mice, but not in α2R/R mice (Van Huysse et al. 2011), prolonged intracerebroventricular (i.c.v.) infusion of Na+‐rich cerebrospinal fluid (CSF) or very low dose ouabain elevates BP. These effects are augmented in α2+/− mice (Hou et al. 2009), presumably because there are fewer available α2 EO receptors and a larger fraction are inhibited. Further, i.c.v. infusion of anti‐ouabain, but not control, fab fragments prevents the Na+‐rich CSF‐induced BP elevation (Huang et al. 2006; Van Huysse et al. 2011). Clearly, α2 Na+ pumps, their CTS binding site, and the endogenous ligand are all critical for SS‐hypertension.
Salt‐sensitive hypertension is also attenuated by i.c.v. infusion of the epithelial Na+ channel (ENaC) inhibitor benzamil (Gomez‐Sanchez et al. 1996; Leenen, 2010; Gabor & Leenen, 2012; Van Huysse et al. 2012; Osborn et al. 2014). Brain ENaCs are expressed in neurones and glia, and in the choroid plexus and ependyma (Amin et al. 2005; Leenen, 2010; Miller & Loewy, 2013; Miller et al. 2013; Oshima et al. 2013). Knockout of the ubiquitin ligase Nedd4‐2, a regulator of ENaC expression, enhances ENaC activity in the kidney and brain, and Nedd4‐2−/− mice develop moderate SS‐hypertension (Shi et al. 2008; Van Huysse et al. 2012). When crossed with α2R/R mice, the double mutants (Nedd4‐2−/−‐α2R/R mice) had a markedly attenuated BP elevation, compared to Nedd4‐2−/− mice, in response to either Na+‐rich CSF (i.c.v.) or high dietary salt (Leenen et al. 2015). Thus, both arterial and brain α2 Na+ pumps, and their endogenous ligand, contribute to SS‐hypertension. The locus of the relevant brain α2 pumps is unknown, but α2 is expressed in meningeal capillary endothelia, in the choroid epithelial cell cytoplasm (Arakaki et al. 2013), and in neurones and glia (McGrail et al. 1991; Moseley et al. 2003). The cytoplasmic pumps may be cycled to the PM (Benziane & Chibalin, 2008) under conditions yet to be determined.
Ouabain‐triggered, α2‐mediated cell signalling and hypertension
Prolonged exposure to nanomolar ouabain increases expression of several arterial Ca2+ transporters, both in rats in vivo (i.e. during hypertension induction), and in primary cultured human and rat artery myocytes. Ca2+ signalling is then augmented even after ouabain washout (Pulina et al. 2010; Linde et al. 2012; Zulian et al. 2013). The same proteins, most notably NCX1 and SERCA2, are also up‐regulated in several rodent hypertension models, including Dahl‐salt‐sensitive, Milan, and spontaneously hypertensive rats, and the Ang II, Ang II + salt and DOCA + salt models (Blaustein et al. 2012, 2015; Pulina et al. 2013). This is consistent with the idea that circulating EO is elevated in these models and that it initiates these changes in protein expression. Arterial NCX1 is also up‐regulated in human primary pulmonary hypertension (Zhang et al. 2007). Prolonged ouabain/EO–α2 interaction triggers arterial myocyte C‐Src phosphorylation, reduces ERK1/2 phosphorylation, and leads to the Ca2+ transporter reprogramming (Fig. 3 C) (Zulian et al. 2013). Importantly, both the acute and chronic actions of EO and ouabain augment arterial Ca2+ entry and signalling. Both should therefore foster vasoconstriction and BP elevation in essential hypertension, primary aldosteronism, and other forms of hypertension in which plasma EO is elevated (Fig. 3 B–D) (Blaustein & Hamlyn, 2010; Blaustein et al. 2012).
CTS–α2 Na+ pump interactions are more complex than anticipated. All CTSs inhibit α2 (and α3) Na+ pumps, augment Ca2+ signalling (Song et al. 2013) and have similar acute vasotonic effects (Song et al. 2014). In contrast, ouabain‐like CTSs (e.g. Strophanthus steroids), but not digoxin‐like CTSs (e.g. Digitalis steroids), also activate downstream signalling cascades that modify protein expression (Zulian et al. 2013). These chronic ouabain‐induced effects are blocked by digoxin (Zulian et al. 2013), which is a ouabain antagonist (Huang et al. 1999; Manunta et al. 2000; Song et al. 2014). This suggests that novel digoxin analogues might block the actions of ouabain without inhibiting Na+ transport; such agents might be therapeutically useful. Indeed, one such agent, rostafuroxin, was synthesized from digoxigenin (Quadri et al. 1997). It blocks the actions of ouabain at concentrations that do not inhibit the Na+,K+‐ATPase (Ferrari et al. 1998; Song et al. 2014), and lowers BP in ouabain hypertensive rats and Milan hypertensive rats (Ferrari et al. 1998, 1999). Unfortunately, rostafuroxin's affinity for the vascular myocyte Na+ pump may be too low to be clinically useful (Song et al. 2014).
Functional linkage of arterial α2 Na+ pumps and NCX1
The above findings emphasize the functional (and structural) linkage between α2 and NCX1 in arterial myocytes. This cross‐talk probably occurs via alterations in [Na+]SPM, which may also be influenced by other adjacent channels and transporters such as TRPC6 (Fig 1 D) (Arnon et al. 2000 a; Poburko et al. 2007, 2008).
Genetic engineering studies illustrate a crucial difference between primary alteration of α2 expression and of NCX1 (and SERCA2) expression. Reduction of α2 by heterozygous null mutation elevates cell Ca2+ and induces hypertension and secondary reduction of NCX1 and SERCA2 expression; the latter is, presumably, a compensatory effect (Zhang et al. 2005; Chen et al. 2015 b). Conversely, transgenic overexpression of SM‐α2 lowers BP and causes a secondary increase in NCX1 and SERCA2 expression, probably to compensate, partially, for the BP reduction which may be due to a fall in [Ca2+]CYT (Pritchard et al. 2007; Chen et al. 2015 b). In contrast, primary SM‐NCX1 overexpression increases [Ca2+]CYT and elevates BP (Iwamoto et al. 2004), whilst SM‐specific knockout of NCX1 lowers [Ca2+]CYT and BP (Zhang et al. 2010 b; Wang et al. 2015). We infer that the EO‐induced, α2‐mediated increase in arterial NCX1 and SERCA2 expression, observed in many types of hypertension (Blaustein & Hamlyn, 2010; Blaustein et al. 2012; Pulina et al. 2013), contributes directly to the elevation of BP (Chen et al. 2015 b).
How the brain talks to the arteries
In many forms of hypertension, including salt‐sensitive hypertension, a central angiotensinergic pathway (‘brain RAAS’) is activated (Allen, 2011; Gabor & Leenen, 2012; Takahashi, 2012). Circulating Ang II, which is elevated in some forms of hypertension, also stimulates the brain RAAS via circumventricular organs such as the subfornical organ (SFO) (Huang et al. 2010; Biancardi et al. 2014; Ufnal & Skrzypecki, 2014). This increases CNS driven arterial sympathetic nerve activity (SNA) and α‐adrenergic arterial constriction (Fink & Bruner, 1985; Osborn et al. 2007, 2011; Gabor & Leenen, 2012; Leenen, 2014), and contributes to BP elevation (Wang et al. 2013). Persistent activation of this central angiotensinergic mechanism appears to depend upon a novel neurohumoral pathway that is triggered by high dietary salt/Na+‐rich CSF (Huang et al. 2006), as well as Ang II (Huang et al. 2010). The hypothalamic component of the neurohumoral pathway involves local aldosterone production, mineralocorticoid receptors, ENaCs, local EO release and α2 Na+ pumps (Huang & Leenen, 1999; Van Huysse & Hou, 2004; Leenen, 2010; Gabor & Leenen, 2012; Van Huysse et al. 2012; Takahashi, 2012). This ‘brain EO’ enhances hypothalamic Ang type 1 receptor (AT1R) signalling (Huang et al. 2011).
Sustained neurohumoral pathway activation raises circulating EO, which increases arterial expression of NCX1 and SERCA2 (Hamlyn et al. 2014); this should enhance arterial responses to sympathetic drive. Elevation of plasma EO and up‐regulation of arterial Ca2+ transporters, as well as the elevation of BP, are prevented by directly blocking the central neurohumoral pathway (Hamlyn et al. 2014). This implies that the increased SNA and the neurohumoral pathway that enhances arterial Ca2+ signalling operate jointly to raise BP chronically when the brain angiotensinergic mechanisms are activated.
α2 Na+ pumps and cardiac function
α2 Na+ pumps mediate the cardiotonic response to CTS
The positive inotropic effect of CTS on the heart (Fig. 4 A, B and E), analogous to the previously mentioned vasotonic effect, requires both ouabain‐sensitive Na+ pumps (see Table 1) and NCX1 (Reuter et al. 2002; Dostanic et al. 2003, 2004; Altamirano et al. 2006). Inhibition of cardiac Na+ pumps by CTS, the presumed consequent rise in [Na+]SPM (see Swift et al. 2010), and the Na+‐dependent, NCX‐mediated net gain of intracellular Ca2+ and enhanced Ca2+ signalling (Swift et al. 2007, 2010) are widely accepted as the basis of the cardiotonic response. Both α1 and α2 Na+ pumps are located in rodent cardiac muscle t‐tubules at or near PM–SR junctions (Mohler et al. 2003; Dostanic et al. 2004; Berry et al. 2007), but which isoform mediates this cardiotonic response? To obtain a definitive answer, engineered ‘SWAP’ mice (with ouabain‐sensitive α1 and resistant α2 pumps, α1S/S‐α2R/R) and WT mice (with ouabain‐resistant α1 and sensitive α2 pumps, α1R/R‐α2S/S) were compared. SWAP mice exhibited a positive inotropic response to ouabain that was mediated by the mutated, ouabain‐sensitive α1 pumps (Dostanic et al. 2004). Nevertheless, comparable inhibition (∼25%) of total Na+ pump activity by low dose ouabain in WT and SWAP mice demonstrates that the α2 isoform preferentially modulates SR Ca2+ release and Ca2+ transients in cardiomyocytes (Fig. 4 A and B) (Despa et al. 2012). Swift and colleagues came to the same conclusion by showing that cardiac α2 pumps and NCX1 are functionally coupled via [Na+]SPM (Swift et al. 2007, 2010). Importantly, despite this compelling evidence, low dose ouabain‐induced elevation of [Na+]SPM (Fig. 4 B) has not yet been measured (Swift et al. 2010). Furthermore, these observations imply that Na+ diffusion between the sub‐PM microdomains and bulk cytosol is restricted (Wendt‐Gallitelli et al. 1993; Arnon et al. 2000 b; Silverman et al. 2003; Poburko et al. 2007; Swift et al. 2010; Aronsen et al. 2013), but definitive data are still lacking.
Cardiac hypertrophy and failure induced by pressure overload: role of α2 Na+ pumps
Mouse models with genetically engineered α2 Na+ pumps (Table 1) or altered pump regulation elucidate the link between Na+ pump expression/activity and cardiac function, and provide new clues to the pathogenesis of heart hypertrophy (HH) and HF. Pressure overload induced by trans‐aortic constriction (TAC) is a common model for inducing HH and HF. TAC induces progressive HH and left ventricular (LV) dysfunction in WT mice that depends on the extent and duration of the TAC (Liao et al. 2002). Cardio‐specific knockout of α2 delays the development of TAC‐induced cardiac dysfunction, i.e. increased end‐diastolic and systolic volumes, and decreased ejection fraction (EF) (Rindler et al. 2013). However, cardio‐specific α2, but not α1, overexpression also attenuates TAC‐induced HH (Correll et al. 2014). How can we reconcile these contradictory results?
First, consider the effects of TAC in mice with altered Na+ pump ouabain sensitivity. SWAP (α1S/S‐α2R/R) mice are more susceptible to HH following TAC than are WT (α1R/R‐α2S/S) or α1R/R‐α2R/R mice, even though the latter two lines have higher LV systolic pressures (Wansapura et al. 2011). Heart weight was greatly increased in SWAP mice, but only modestly in WT and α1R/R‐α2R/R mice, after 4 weeks of TAC. SWAP mice also had substantial LV enlargement, and a reduced EF, indicating cardiac decompensation (HF), i.e. the pathophysiological processes were accelerated. Remarkably, banded α1R/R‐α2R/R mice had no LV enlargement and no echocardiographic evidence of cardiac dysfunction (vs. sham) after 4 weeks of TAC (Wansapura et al. 2011). Clearly, TAC‐induced HH and HF depend, in part, upon ouabain sensitivity. Further, the cardiac changes are attenuated by anti‐ouabain fab fragments (Wansapura et al. 2011). Thus, Na+ pumps and their endogenous ligand contribute to the pathogenesis of HH and HF. More rapid TAC‐induced cardiac dysfunction is therefore anticipated in SWAP mice because low CTS concentrations inhibit only cardiac α2 pumps in WT mice, and ouabain‐sensitive α1 Na+ pumps in SWAP mice, and the α1:α2 ratio is ∼4:1 in both strains (James et al. 1999; Berry et al. 2007; Despa & Bers, 2007). Thus, at submaximal EO, more pumps will be inhibited in SWAP than in WT mice. In other words, the TAC‐induced cardiac dysfunction correlates with the proportion of Na+ pumps that is EO sensitive. These considerations also explain why both cardiac‐α2 knockout (Rindler et al. 2013) and overexpression (Correll et al. 2014) delay/attenuate TAC‐induced cardiac dysfunction. Neither α2 nor its ouabain receptor is expressed in knockouts. In over‐expressors, more ‘reserve’ α2 pumps/EO receptors are available to keep [Na+]SPM low when a fraction is blocked by EO.
The pressure overload data suggest that EO, via its cardiotonic effect, contributes to HH with preserved, or even enhanced, cardiac performance, e.g. increased EF (Wansapura et al. 2011). Human and rodent HF data infer, however, that the impaired contractility and reduced EF also are linked to high plasma EO (Gottlieb et al. 1992; Pitzalis et al. 2006; Stella et al. 2008; Blaustein et al. 2015). How can this be reconciled?
The fact that prolonged ouabain treatment activates protein kinase cascades that modulate cardiac protein expression (Tian & Xie, 2008; Li & Xie, 2009) suggests an explanation. In cultured cardiomyocytes, 30–100 μm ouabain (24–48 h) increases NCX1 expression (Vemuri et al. 1989; Müller‐Ehmsen et al. 2003); indeed, 50 nm ouabain (72 h) is sufficient, but 100 nm digoxin is ineffective (Blaustein et al. 2015). Increased cardiac NCX1 and decreased SERCA2 expression, which are common findings in human HF and animal models (Studer et al. 1994; O'Rourke et al. 1999), contribute to the reduced SR Ca2+ stores and attenuated Ca2+ signals (Bers & Despa, 2006; Lehnart et al. 2009). Therefore, while its acute effect is cardiotonic, chronically elevated ouabain/EO and the enhanced NCX1 and reduced SERCA2 expression should accelerate Ca2+ extrusion, and promote [Ca2+]SR decline and progression to hypocontractility and HF (Fig. 4 B and C) (Rodriguez et al. 2014). Indeed, partial NCX inhibition restores Ca2+ signalling in myocytes from failing hearts (Hobai et al. 2004). Importantly, these conclusions need to be tested in other HH and HF models, e.g. coronary artery ligation/myocardial infarction (MI), in α2R/R and SWAP mice.
Regulation of α2 pumps in HH and HF
In some forms of HF, expression of α1, α2 and PLM, and Na+ pump activity, are all reduced, and [Na+]CYT is elevated, in left ventricular myocytes (Bossuyt et al. 2005; Pavlovic et al. 2013 a), although PLM transcription is up‐regulated (Gronich et al. 2010). Also, in HF, oxidative stress (Burgoyne et al. 2012) inhibits cardiomyocyte Na+ pumps by inducing β1 subunit glutathionylation; this can be reversed by phosphorylated PLM (Bibert et al. 2011).
The link between α2 activity and cardiac pathophysiology is affirmed by two models of PLM dysregulation. In PLM knockout mice, total Na+,K+‐ATPase activity and α2 Na+ pump expression are reduced by 50–60% (Jia et al. 2005). This should sustain a high [Na+]SPM and a large cardiotonic effect (Golovina et al. 2003) to account for the hypertrophic hearts and increased LV EF (Jia et al. 2005).
In the second model, mice with non‐phosphorylatable PLM (PLM35A) have normal cardiac function under basal conditions. Following TAC, however, PLM35A mice exhibit accelerated cardiac hypertrophy and dysfunction with increased NCX and decreased SERCA2a expression (Boguslavskyi et al. 2014). The inability to phosphorylate PLM and augment pump‐mediated Na+ extrusion and, thus, NCX‐mediated Ca2+ extrusion, when the heart is stressed (e.g. by TAC) enhances Ca2+ dysregulation and accelerates the cardiomyopathy. These models reinforce the view that cardiac α2 Na+ pumps and NCX1 conjointly contribute to the pathogenesis of HH and HF. An important caveat, however, is that in the rat heart in HF, expression of α2 declines and α3, the fetal isoform, increases (Semb et al. 1998; Verdonck et al. 2003), but the significance of this isoform switch and the localization of α3 are unknown.
Myocardial [Na+]CYT and NCX1 in HF
Elevated myocardial [Na+]CYT (Pogwizd et al. 2003; Murphy & Eisner, 2009; Bay et al. 2013; Pavlovic et al. 2013 a) fosters the NCX‐mediated Ca2+ dysregulation in HF (Bers & Despa, 2006; Despa & Bers, 2013; Shattock et al. 2015). Multiple mechanisms may contribute to the high [Na+]CYT, including: (i) reduced α2 pump expression; (ii) Na+ pump dysregulation due to reduced PLM expression (Bossuyt et al. 2005); (iii) increased late Na+ current due to altered CaMKII regulation of cardiac Na+ channels (Grandi & Herren, 2014); (iv) direct inhibition of α2 by the elevated plasma EO (Gottlieb et al. 1992; Pitzalis et al. 2006; Stella et al. 2008; Hamlyn & Manunta, 2015); (v) increased Na+ entry via Na+/H+ exchange (Baartscheer et al. 2003; Karmazyn et al. 2008); (vi) dysregulation of the Ca2+‐dependent, nitric oxide (NO)‐mediated mechanism that stimulates Na+ pumps by phosphorylating PLM (Pavlovic et al. 2013 b); and (vii) increased oxidative stress and ROS generation (Munzel et al. 2015; Zuo et al. 2015) that not only reduces NO availability and PLM phosphorylation, but also increases β1 subunit glutathionylation (Figtree et al. 2009); both mechanisms depress pump‐mediated cation transport.
Elevated [Na+]CYT promotes Ca2+ export by the mitochondrial Na+/Ca2+ exchanger, NCLX, which lowers intra‐mitochondrial [Ca2+] and increases oxidation of mitochondrial NADH (Murphy & Eisner, 2009; Liu et al. 2010; De Marchi et al. 2014; Nita et al. 2015). Thus, elevated [Na+]CYT and/or [Ca2+]CYT (which should limit NCLX‐mediated Ca2+ export) can not only enhance Ca2+ signalling, but also increase oxidative stress and ROS production (Li et al. 2014) and further depress Na+ pump function (Figtree et al. 2009).
The preceding two paragraphs focus on ‘global’ [Na+]CYT. However, [Na+]SPM, which apparently modulates cardiac [Ca2+]CYT transients and excitation–contraction coupling, may be independently affected (Su et al. 2001; Verdonck et al. 2004; Swift et al. 2010; Aronsen et al. 2013). Further, [Na+]SPM may also be modified by Na+ channels associated with these microdomains (Verdonck et al. 2004; Aronsen et al. 2013).
Genetically induced cardiac NCX1 overexpression in mice, itself, accelerates HH and HF induced by stresses (TAC, intense exercise, or pregnancy) known to activate the RAAS (Roos et al. 2007). The NCX1 over‐abundance and enhanced Ca2+ removal are manifested by reduced SR Ca2+, Ca2+ transients, and excitation–contraction coupling gain (Reuter et al. 2004; Ottolia et al. 2013), also observed in myocytes from failing human and rat hearts (Gomez et al. 1997; Piacentino et al. 2003). In contrast, genetically reduced (50%) cardiac NCX1 expression confers tolerance to pressure overload and attenuates HF development, perhaps by reducing Ca2+ overload (Takimoto et al. 2002; Jordan et al. 2010). Nevertheless, ∼20% of WT NCX1 is essential for cardiac function: nearly complete knockout causes HH and accelerates stress‐induced progression to HF (Jordan et al. 2010), presumably because of Ca2+ overload due to impaired Ca2+ clearance. NCX1 apparently also plays a role in some other HH and HF models. For example, cardiac‐specific NCX1 knockout (by 80–90%) mitigates chronic intermittent hypoxia‐induced LV hypertrophy and contractile dysfunction in mice (Chen et al. 2010).
Cardiomyocyte Ca2+ dysregulation in HF
In HF, cardiac Ca2+ dysregulation is usually manifested by elevated diastolic [Ca2+]CYT but reduced SR Ca2+ content (Fig. 4 C–E) (Lehnart et al. 2009; Reuter & Schwinger, 2012). The latter, which may be the result of reduced SERCA2 expression and activity (Lehnart et al. 2009) and increased Ca2+ leakage through ryanodine receptors (RyRs) (Marx & Marks, 2013), probably explains the attenuated peak systolic [Ca2+]CYT transients and cardiac hypocontractility (Fig. 4 D and E). The elevated diastolic (quasi‐steady state) [Ca2+]CYT can be attributed largely to the previously mentioned high [Na+]CYT and reduced driving force for Ca2+ extrusion via NCX, although reduced SR Ca2+ uptake and increased RyR leak may also contribute. The high [Na+]CYT and thus [Ca2+]CYT may help explain the impaired relaxation and increased stiffness of cardiac muscle in HF (Kass et al. 2004; Louch et al. 2010; Li et al. 2012).
Central and peripheral mechanisms in the progression from hypertrophy to failure
In HH and HF, as in hypertension, central angiotensinergic mechanisms are usually stimulated, and sympathetic drive is increased (Yu et al. 2008; Westcott et al. 2009; Lymperopoulos et al. 2013; Zucker et al. 2014). Blockers of these mechanisms are therefore used to treat both hypertension and HF (Leenen et al. 2012; Krum & Driscoll, 2013; James et al. 2014). The angiotensinergic mechanisms activate the neurohumoral pathway that elevates circulating EO (Hamlyn et al. 2014): e.g. both MI and s.c. Ang II + high dietary salt raise plasma EO and increase both cardiac and arterial NCX1 expression (Blaustein et al. 2015). Because arterial NCX1 operates primarily in the Ca2+ entry mode, both acute and chronic high EO should enhance vasoconstriction and foster hypertension, but why do high EO and increased NCX1 also lead to cardiac hypocontractility and failure? The main cardiac Ca2+ extrusion mechanism, NCX1, exports Ca2+ during most of the cardiac cycle because, during diastole, the membrane potential is about −65 to −75 mV (Eisner et al. 2013; Eisner, 2014). Acutely elevated plasma EO therefore induces ‘classic’ positive inotropy, but markedly increased cardiac NCX1 expression, due to chronically elevated EO, promotes Ca2+ extrusion, reduces [Ca2+]CYT and causes negative inotropy (Fig. 4 B–D).
Comparison of WT, α2R/R and SWAP mouse data (Wansapura et al. 2011) lead us to postulate that TAC activates the brain RAAS–neurohumoral pathway, raises plasma EO, and in WT, and even more so in SWAP mice (α2R/R mice are EO‐resistant), induces a positive inotropic response. This is initially amplified as NCX1 expression increases. The consequent, sustained hypercontractility, as well as other, possibly EO‐triggered, changes in protein programming contribute to hypertrophy and, at least initially, to enhanced cardiac performance. With progressive increase in NCX1 and decrease in SERCA2 expression, however, the NCX1‐mediated Ca2+ extrusion mode starts to prevail, and [Ca2+]CYT falls, thereby reducing cardiac performance and leading to HF; i.e. the cardiac changes in HH and HF are a continuum. Indeed, HF with preserved EF (Kamimura et al. 2012; Gladden et al. 2014; Sharma & Kass, 2014; Zuo et al. 2015) might be an intermediate stage in this continuum. Further, following MI, even the RAAS‐stimulated initial tendency to induce a positive inotropic response may be circumvented rapidly if there is much damaged and unresponsive myocardium. The altered NCX1 and SERCA2 expression may then dominate early on, leading rapidly to a negative ionotropic response and HF (Fig. 4 D).
Exercise intolerance in HF: role of skeletal muscle α2 Na+ pumps in fatigue resistance
Skeletal muscle (SkM) α2 plays a negligible role in quiescent muscle, but is activated by the rise in t‐tubule [K+] and [Na+]CYT during exercise, and is needed to attenuate fatigue (DiFranco et al. 2015; Manoharan et al. 2015). As in the heart, phosphorylation of SkM PLM enhances Na+ pump activity, but PLM knockout mice show that PLM is not needed for acute exercise‐induced SkM α2 activation (Manoharan et al. 2015). Nevertheless, intense exercise increases PLM phosphorylation, and α2 pump expression and activity in human type II (fast twitch, fatigable) SkM fibres, which express more α2 than do type I (slow twitch, fatigue‐resistant) fibres (Kristensen et al. 2008; Thomassen et al. 2010, 2013; Benziane et al. 2011). Reduced α2 Na+ pump expression in ageing humans may decrease muscle strength and increase fatigability (Chibalin et al. 2012).
Mice with targeted knockout of SkM α2 pumps, α2SkM−/−, fatigue faster than WT mice on a treadmill (Radzyukevich et al. 2013). Also, extensor digitorum longus (EDL) muscles isolated from SkM‐α2−/− mice have reduced twitch and tetanic force compared to WT EDL. Selective block of α2 by ouabain in WT EDL mimics the results in α2SkM−/− EDL (Radzyukevich et al. 2013). Resistance to fatigue apparently is due in part to the rapid increase in α2‐mediated cation transport triggered by the rise in t‐tubule [K+] during stimulation (DiFranco et al. 2015).
α2 ouabain binding sites and EO play a role in SkM: α2R/R mice exhibit fewer exercise failures on a treadmill than do WT mice (Radzyukevich et al. 2009). Also, 86Rb (K+ surrogate) uptake is reduced following high frequency contractile activation (vs. rest) in EDL from WT mice. In contrast, following muscle stimulation, 86Rb uptake is increased in EDL from α2R/R mice and in EDL from WT mice pre‐infused with DigiBind prior to euthanasia (Radzyukevich et al. 2009).
Clearly, susceptibility to fatigue is inversely related to skeletal muscle α2 Na+ pump expression/activity and is modulated by EO, but why? A clue is that knockout of the predominant NCX isoform in SkM, NCX3, also reduces endurance and increases fatigue, although it increases both twitch and tetanus tension (Sokolow et al. 2004). We postulate that reduced NCX3‐mediated Ca2+ extrusion, due to decreased NCX3 expression or diminished Na+ extrusion by α2 pumps when exercise elevates t‐tubule [K+], enhances fatigability.
The above findings imply that the high EO levels observed in hypertension and HF contribute to the reduced SkM α2 Na+ pump activity (contrast Barr et al. 2005), increased fatigability (Carlsen et al. 1996; Helwig et al. 2003; Okita et al. 2013; Tzanis et al. 2014) and reduced hand grip strength (Mainous et al. 2015). Exercise may enhance SkM α2 Na+ pump activity by increasing α2 expression or translocation to the sarcolemma, or PLM phosphorylation, and thereby reduce fatigability and improve muscle strength (Thomassen et al. 2010; Rasmussen et al. 2011).
Summary and conclusions
Mice with genetically engineered α2 Na+ pumps, PLM and NCX1 provide novel insight into the central role of α2 and its endogenous ligand, EO, in regulating Ca2+ homeostasis and the function of cardiac, skeletal and vascular muscles. The juxtaposition of these findings enables us to recognize the striking similarities and key differences between the mechanisms involved in the pathogenesis of hypertension, HH and HF. In all three situations, brain angiotensinergic mechanisms are activated; this triggers the CNS rapid sympathetic and slower neurohumoral (EO‐mediated) pathways. Acutely increased nerve frequency is often attenuated by self‐tuning (Greengard, 2001; Turrigiano, 2008), but EO may potentiate peripheral synaptic transmission and sympathetic nerve responses (Aileru et al. 2001). Also, the chronic, protein kinase cascade‐mediated effects of elevated plasma EO on arteries and heart may amplify the cardiac and vascular responses to sympathetic drive. Initially, the cardio‐ and vasotonic actions of EO enhance Ca2+ signalling and contractility and thus elevate BP and heighten cardiac function, and lead to hypertrophy. Slow, EO‐mediated up‐regulation of NCX1 (and SERCA2) in arteries favours Ca2+ entry and further fosters vasoconstriction. In the heart, however, EO‐mediated NCX1 up‐regulation (and SERCA2 decline) eventually tips the balance toward Ca2+ exit, hypocontractility and HF. It is worth emphasizing that, when the brain RAAS is activated in hypertension, the elevated plasma EO is expected to influence cardiac function simultaneously. Likewise, when an MI activates the brain RAAS, simultaneously altered arterial function is expected (Blaustein et al. 2015). Thus, elevated plasma EO is likely to contribute to the increased peripheral vascular resistance often observed in HF post‐MI (Zelis et al. 1968; Ledoux et al. 2003).
Despite this compelling evidence for the key roles of α2 and EO, numerous challenges remain. First of all, details of the CNS pathways are poorly understood. For example, brain α2 pumps are important in SS‐hypertension (Leenen et al. 2015), but the cellular location of the relevant pumps is unknown. Also, the proposed role of brain α2 in HH and HF must be verified. Further, while EO is synthesized in the brain, and is a critical link in both hypertension and HF (Leenen et al. 1995; Huang et al. 2010), precisely where in the CNS pathways it participates is unresolved.
Circulating EO comes from the adrenals (Hamlyn et al. 1991; Boulanger et al. 1993; Manunta et al. 2010), but what is the biosynthetic pathway? Also, how does brain RAAS regulate plasma EO? Is it via increased sympathetic traffic to the adrenals (Shah et al. 1998), or some other mechanism? And, what role, if any, does ACTH play (Laredo et al. 1994)?
We suggest that α2 pumps mediate both the acute and chronic effects of nanomolar ouabain/EO in rodents (Dostanic et al. 2005; Despa et al. 2012; Zulian et al. 2013), but others suggest that α1 pumps are responsible for the chronic effects (Liu & Xie, 2010; Xie et al. 2015). Comparison of the acute and chronic effects of nanomolar ouabain on WT and α2R/R cardiac and arterial myocytes could resolve this controversy. Further, since human α1 is ouabain sensitive, do human α2 pumps play the same key role as in rodents? A clue is that human, like rodent, arterial α2 is localized in PM microdomains at PM–S/ER junctions (Linde et al. 2012).
We postulate that the [Na+]SPM at PM–S/ER junctions is a crucial factor in the EO‐dependent modulation of Ca2+ signalling and contractility in the arteries and heart (Figs 3 and 4). More precise information about the structural organization of the junctions and their resident transporters, e.g. using super‐resolution imaging, should improve our understanding of ion regulation in these regions. Critically, direct measurement of the effects of nanomolar ouabain on [Na+]SPM with Na+‐sensitive fluorochromes and, e.g., ‘total internal reflection fluorescence’ (TIRF) imaging, is needed to validate our inferences.
The effect of sustained ouabain/EO exposure on signalling cascades is established, but the precise time course of these responses, and all of the contributors (e.g. the complete range of affected Ca2+ transporter and signalling molecules), are unknown. For example, does ouabain/EO, per se, trigger down‐regulation of cardiac SERCA2 expression? Measurement of ouabain/EO‐ and disease‐dependent gene activation (quantitative PCR analysis of mRNA) or changes in protein expression (MS) would provide important new clues to underlying pathogenic mechanisms.
Finally, a fundamental implication of the work reviewed above is that novel agents that interfere with the biosynthesis, release and/or peripheral actions of EO should be therapeutically beneficial in hypertension, HH and HF. Such agents might also be useful in attenuating the renal damage, often linked to these CV diseases, that has been attributed to elevated circulating EO (Bignami et al. 2013; Ferrandi et al. 2014; Hamlyn & Manunta, 2015). Indeed, application of these agents would provide a critical test of many of the ideas summarized here.
Additional information
Competing interests
None declared.
Funding
This work was supported by NIH grants HL‐45215 and HL‐107555 to M.P.B. and J.M.H., AM‐063710 to J.B.L., HL‐091969 and OD‐018315 to W.G.W., and HL‐107654 to J.Z., by Canadian Institutes of Health Research grant FRN:MOP‐74432 to F.H.H.L., and by American Heart Association Grant‐in‐Aid 24940022 to J.Z.
Acknowledgements
We thank Drs Ronald M. Lynch and Peter J. Mohler for providing figures for reproduction, and Dr Judith A. Heiny for discussion about skeletal α2 Na+ pumps.
Biographies
Baltimore group (left to right; key mentors in parentheses): Mordecai Blaustein, a discoverer of Na+/Ca2+ exchange (NCX), studies arterial Ca2+ regulation, arterial tone and hypertension (Daniel Tosteson and Alan Hodgkin).
![]()
Ling Chen investigates cardiovascular mechanisms of hypoxia and ischaema (Morris Karmazyn and Steven Scharf).
John Hamlyn identified (with Blaustein) endogenous ouabain and its role in hypertension and heart failure (Thomas Duffy and Alan Senior).
Gil Wier, pioneer of cardiovascular Ca2+ signalling in vitro, in situ, and in awake mice in vivo (John Blinks).
Jin Zhang performed seminal studies on arteries from genetically altered arterial α2 and NCX mice (Xiaoliang Wang and Blaustein). Individual photos. Frans Leenen (Ottawa, left) discovered brain ouabain's role in the novel slow neuromodulatory pathway in hypertension and heart failure (Wybren deJong and Alvin Shapiro). Jerry Lingrel (Cincinatti), who cloned the Na+ pump isoforms, studies their physiological roles in genetically engineered mice (Harry Bosook and John Gurdon).
This is an Editor's Choice article from the 1 November 2016 issue.
References
- Aileru AA, De Albuquerque A, Hamlyn JM, Manunta P, Shah JR, Hamilton MJ & Weinreich D (2001). Synaptic plasticity in sympathetic ganglia from acquired and inherited forms of ouabain‐dependent hypertension. Am J Physiol Regul Integr Comp Physiol 281, R635–R644. [DOI] [PubMed] [Google Scholar]
- Allen AM (2011). Role of angiotensin in the rostral ventrolateral medulla in the development and maintenance of hypertension. Curr Opin Pharmacol 11, 117–123. [DOI] [PubMed] [Google Scholar]
- Altamirano F, Eltit JM, Robin G, Linares N, Ding X, Pessah IN, Allen PD & Lopez JR (2014). Ca2+ influx via the Na+/Ca2+ exchanger is enhanced in malignant hyperthermia skeletal muscle. J Biol Chem 289, 19180–19190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altamirano J, Li Y, DeSantiago J, Piacentino V 3rd, Houser SR & Bers DM (2006). The inotropic effect of cardioactive glycosides in ventricular myocytes requires Na+–Ca2+ exchanger function. J Physiol 575, 845–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin MS, Wang HW, Reza E, Whitman SC, Tuana BS & Leenen FH (2005). Distribution of epithelial sodium channels and mineralocorticoid receptors in cardiovascular regulatory centers in rat brain. Am J Physiol Regul Integr Comp Physiol 289, R1787–R1797. [DOI] [PubMed] [Google Scholar]
- Arakaki X, McCleary P, Techy M, Chiang J, Kuo L, Fonteh AN, Armstrong B, Levy D & Harrington MG (2013). Na,K‐ATPase alpha isoforms at the blood‐cerebrospinal fluid‐trigeminal nerve and blood‐retina interfaces in the rat. Fluids Barriers CNS 10, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnon A, Hamlyn JM & Blaustein MP (2000. a). Na+ entry via store‐operated channels modulates Ca2+ signaling in arterial myocytes. Am J Physiol Cell Physiol 278, C163–C173. [DOI] [PubMed] [Google Scholar]
- Arnon A, Hamlyn JM & Blaustein MP (2000. b). Ouabain augments Ca2+ transients in arterial smooth muscle without raising cytosolic Na+ . Am J Physiol Heart Circ Physiol 279, H679–H691. [DOI] [PubMed] [Google Scholar]
- Aronsen JM, Swift F & Sejersted OM (2013). Cardiac sodium transport and excitation‐contraction coupling. J Mol Cell Cardiol 61, 11–19. [DOI] [PubMed] [Google Scholar]
- Baartscheer A, Schumacher CA, van Borren MM, Belterman CN, Coronel R & Fiolet JW (2003). Increased Na+/H+‐exchange activity is the cause of increased [Na+]i and underlies disturbed calcium handling in the rabbit pressure and volume overload heart failure model. Cardiovasc Res 57, 1015–1024. [DOI] [PubMed] [Google Scholar]
- Bae YM, Kim A, Lee YJ, Lim W, Noh YH, Kim EJ, Kim J, Kim TK, Park SW, Kim B, Cho SI, Kim DK & Ho WK (2007). Enhancement of receptor‐operated cation current and TRPC6 expression in arterial smooth muscle cells of deoxycorticosterone acetate‐salt hypertensive rats. J Hypertens 25, 809–817. [DOI] [PubMed] [Google Scholar]
- Bagrov AY, Dmitrieva RI, Fedorova OV, Kazakov GP, Roukoyatkina NI & Shpen VM (1996). Endogenous marinobufagenin‐like immunoreactive substance. A possible endogenous Na,K‐ATPase inhibitor with vasoconstrictor activity. Am J Hypertens 9, 982–990. [DOI] [PubMed] [Google Scholar]
- Bagrov AY, Shapiro JI & Fedorova OV (2009). Endogenous cardiotonic steroids: physiology, pharmacology, and novel therapeutic targets. Pharmacol Rev 61, 9–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Y, Morgan EE, Giovannucci DR, Pierre SV, Philipson KD, Askari A & Liu L (2013). Different roles of the cardiac Na+/Ca2+‐exchanger in ouabain‐induced inotropy, cell signaling, and hypertrophy. Am J Physiol Heart Circ Physiol 304, H427–H435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr DJ, Green HJ, Lounsbury DS, Rush JW & Ouyang J (2005). Na+‐K+‐ATPase properties in rat heart and skeletal muscle 3 mo after coronary artery ligation. J Appl Physiol (1985) 99, 656–664. [DOI] [PubMed] [Google Scholar]
- Bay J, Kohlhaas M & Maack C (2013). Intracellular Na+ and cardiac metabolism. J Mol Cell Cardiol 61, 20–27. [DOI] [PubMed] [Google Scholar]
- Benziane B & Chibalin AV (2008). Frontiers: skeletal muscle sodium pump regulation: a translocation paradigm. Am J Physiol Endocrinol Metab 295, E553–E558. [DOI] [PubMed] [Google Scholar]
- Benziane B, Widegren U, Pirkmajer S, Henriksson J, Stepto NK & Chibalin AV (2011). Effect of exercise and training on phospholemman phosphorylation in human skeletal muscle. Am J Physiol Endocrinol Metab 301, E456–E466. [DOI] [PubMed] [Google Scholar]
- Berry RG, Despa S, Fuller W, Bers DM & Shattock MJ (2007). Differential distribution and regulation of mouse cardiac Na+/K+‐ATPase α1 and α2 subunits in T‐tubule and surface sarcolemmal membranes. Cardiovasc Res 73, 92–100. [DOI] [PubMed] [Google Scholar]
- Bers DM & Despa S (2006). Cardiac myocytes Ca2+ and Na+ regulation in normal and failing hearts. J Pharmacol Sci 100, 315–322. [DOI] [PubMed] [Google Scholar]
- Biancardi VC, Son SJ, Ahmadi S, Filosa JA & Stern JE (2014). Circulating angiotensin II gains access to the hypothalamus and brain stem during hypertension via breakdown of the blood‐brain barrier. Hypertension 63, 572–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibert S, Liu CC, Figtree GA, Garcia A, Hamilton EJ, Marassi FM, Sweadner KJ, Cornelius F, Geering K & Rasmussen HH (2011). FXYD proteins reverse inhibition of the Na+‐K+ pump mediated by glutathionylation of its β1 subunit. J Biol Chem 286, 18562–18572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibert S, Roy S, Schaer D, Horisberger JD & Geering K (2008). Phosphorylation of phospholemman (FXYD1) by protein kinases A and C modulates distinct Na,K‐ATPase isozymes. J Biol Chem 283, 476–486. [DOI] [PubMed] [Google Scholar]
- Bignami E, Casamassima N, Frati E, Lanzani C, Corno L, Alfieri O, Gottlieb S, Simonini M, Shah KB, Mizzi A, Messaggio E, Zangrillo A, Ferrandi M, Ferrari P, Bianchi G, Hamlyn JM & Manunta P (2013). Preoperative endogenous ouabain predicts acute kidney injury in cardiac surgery patients. Crit Care Med 41, 744–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco G & Mercer RW (1998). Isozymes of the Na‐K‐ATPase: heterogeneity in structure, diversity in function. Am J Physiol 275, F633–F650. [DOI] [PubMed] [Google Scholar]
- Blaustein MP (1977). Sodium ions, calcium ions, blood pressure regulation, and hypertension: a reassessment and a hypothesis. Am J Physiol Cell Physiol 232, C165–C173. [DOI] [PubMed] [Google Scholar]
- Blaustein MP, Chen L, Song H, Leenen FHH & Hamlyn JM (2015). How does the brain talk to the arteries and heart? FASEB J 29, 984.3. (Abstract.) [Google Scholar]
- Blaustein MP & Hamlyn JM (2010). Signaling mechanisms that link salt retention to hypertension: Endogenous ouabain, the Na+ pump, the Na+/Ca2+ exchanger and TRPC proteins. Biochim Biophys Acta 1082, 1219–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaustein MP & Lederer WJ (1999). Sodium/calcium exchange: its physiological implications. Physiol Rev 79, 763–854. [DOI] [PubMed] [Google Scholar]
- Blaustein MP, Leenen FH, Chen L, Golovina VA, Hamlyn JM, Pallone TL, Van Huysse JW, Zhang J & Wier WG (2012). How NaCl raises blood pressure: a new paradigm for the pathogenesis of salt‐dependent hypertension. Am J Physiol Heart Circ Physiol 302, H1031–H1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boguslavskyi A, Pavlovic D, Aughton K, Clark JE, Howie J, Fuller W & Shattock MJ (2014). Cardiac hypertrophy in mice expressing unphosphorylatable phospholemman. Cardiovasc Res 104, 72–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossuyt J, Ai X, Moorman JR, Pogwizd SM & Bers DM (2005). Expression and phosphorylation of the Na‐pump regulatory subunit phospholemman in heart failure. Circ Res 97, 558–565. [DOI] [PubMed] [Google Scholar]
- Bossuyt J, Despa S, Han F, Hou Z, Robia SL, Lingrel JB & Bers DM (2009). Isoform specificity of the Na/K‐ATPase association and regulation by phospholemman. J Biol Chem 284, 26749–26757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossuyt J, Despa S, Martin JL & Bers DM (2006). Phospholemman phosphorylation alters its fluorescence resonance energy transfer with the Na/K‐ATPase pump. J Biol Chem 281, 32765–32773. [DOI] [PubMed] [Google Scholar]
- Boulanger BR, Lilly MP, Hamlyn JM, Laredo J, Shurtleff D & Gann DS (1993). Ouabain is secreted by the adrenal gland in awake dogs. Am J Physiol 264, E413–E419. [DOI] [PubMed] [Google Scholar]
- Burgoyne JR, Mongue‐Din H, Eaton P & Shah AM (2012). Redox signaling in cardiac physiology and pathology. Circ Res 111, 1091–1106. [DOI] [PubMed] [Google Scholar]
- Carlsen RC, Gray SD & Pickar JG (1996). Na+,K+‐pump activity and skeletal muscle contractile deficits in the spontaneously hypertensive rat. Acta Physiol Scand 156, 237–245. [DOI] [PubMed] [Google Scholar]
- Chen L, Blaustein MP & Hamlyn JM (2015. a). Immuno‐neutralization of endogenous ouabain lowers blood pressure in angiotensin II‐dependent models. Hypertension 66, AP145. [Google Scholar]
- Chen L, Song H, Wang Y, Lee JC, Kotlikoff MI, Pritchard TJ, Paul RJ, Zhang J & Blaustein MP (2015. b). Arterial α2‐Na+ pump expression influences blood pressure: lessons from novel, genetically engineered smooth muscle‐specific α2 mice. Am J Physiol Heart Circ Physiol 309, H958–H968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Zhang J, Hu X, Philipson KD & Scharf SM (2010). The Na+/Ca2+ exchanger‐1 mediates left ventricular dysfunction in mice with chronic intermittent hypoxia. J Appl Physiol (1985) 109, 1675–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chia KK, Liu CC, Hamilton EJ, Garcia A, Fry NA, Hannam W, Figtree GA & Rasmussen HH (2015). Stimulation of the cardiac myocyte Na+‐K+ pump due to reversal of its constitutive oxidative inhibition. Am J Physiol Cell Physiol 309, C239–C250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chibalin AV, Heiny JA, Benziane B, Prokofiev AV, Vasiliev AV, Kravtsova VV & Krivoi II (2012). Chronic nicotine modifies skeletal muscle Na,K‐ATPase activity through its interaction with the nicotinic acetylcholine receptor and phospholemman. PLoS One 7, e33719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correll RN, Eder P, Burr AR, Despa S, Davis J, Bers DM & Molkentin JD (2014). Overexpression of the Na+/K+ ATPase α2 but not α1 isoform attenuates pathological cardiac hypertrophy and remodeling. Circ Res 114, 249–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cougnon MH, Moseley AE, Radzyukevich TL, Lingrel JB & Heiny JA (2002). Na,K‐ATPase α‐ and β‐isoform expression in developing skeletal muscles: α2 correlates with t‐tubule formation. Pflugers Arch 445, 123–131. [DOI] [PubMed] [Google Scholar]
- Crambert G, Fuzesi M, Garty H, Karlish S & Geering K (2002). Phospholemman (FXYD1) associates with Na,K‐ATPase and regulates its transport properties. Proc Natl Acad Sci USA 99, 11476–11481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis KA, Samson SE, Hammel KE, Kiss L, Fulop F & Grover AK (2009). Functional linkage of Na+‐Ca2+‐exchanger to sarco/endoplasmic reticulum Ca2+ pump in coronary artery: comparison of smooth muscle and endothelial cells. J Cell Mol Med 13, 1775–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Marchi U, Santo‐Domingo J, Castelbou C, Sekler I, Wiederkehr A & Demaurex N (2014). NCLX protein, but not LETM1, mediates mitochondrial Ca2+ extrusion, thereby limiting Ca2+‐induced NAD(P)H production and modulating matrix redox state. J Biol Chem 289, 20377–20385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Despa S & Bers DM (2007). Functional analysis of Na+/K+‐ATPase isoform distribution in rat ventricular myocytes. Am J Physiol Cell Physiol 293, C321–C327. [DOI] [PubMed] [Google Scholar]
- Despa S & Bers DM (2013). Na+ transport in the normal and failing heart – remember the balance. J Mol Cell Cardiol 61, 2–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Despa S, Lingrel JB & Bers DM (2012). Na+/K+‐ATPase α2‐isoform preferentially modulates Ca2+ transients and sarcoplasmic reticulum Ca2+ release in cardiac myocytes. Cardiovasc Res 95, 480–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey K, Roy S, Ghosh B & Chakraborti S (2012). Role of protein kinase C in phospholemman mediated regulation of α2β1 isozyme of Na+/K+‐ATPase in caveolae of pulmonary artery smooth muscle cells. Biochimie 94, 991–1000. [DOI] [PubMed] [Google Scholar]
- DiFranco M, Hakimjavadi H, Lingrel JB & Heiny JA (2015). Na,K‐ATPase α2 activity in mammalian skeletal muscle T‐tubules is acutely stimulated by extracellular K+ . J Gen Physiol 146, 281–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dostanic‐Larson I, Van Huysse JW, Lorenz JN & Lingrel JB (2005). The highly conserved cardiac glycoside binding site of Na,K‐ATPase plays a role in blood pressure regulation. Proc Natl Acad Sci USA 102, 15845–15850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dostanic I, Lorenz JN, Schultz JJ, Grupp IL, Neumann JC, Wani MA & Lingrel JB (2003). The α2 isoform of Na,K‐ATPase mediates ouabain‐induced cardiac inotropy in mice. J Biol Chem 278, 53026–53034. [DOI] [PubMed] [Google Scholar]
- Dostanic I, Paul RJ, Lorenz JN, Theriault S, Van Huysse JW & Lingrel JB (2005). The α2‐isoform of Na‐K‐ATPase mediates ouabain‐induced hypertension in mice and increased vascular contractility in vitro. Am J Physiol Heart Circ Physiol 288, H477–H485. [DOI] [PubMed] [Google Scholar]
- Dostanic I, Schultz JJ, Lorenz JN & Lingrel JB (2004). The α1 isoform of Na,K‐ATPase regulates cardiac contractility and functionally interacts and co‐localizes with the Na/Ca exchanger in heart. J Biol Chem 279, 54053–54061. [DOI] [PubMed] [Google Scholar]
- Doursout MF, Chelly JE, Liang YY & Buckley JP (1992). The ouabain‐dependent Na+‐K+ pump and the brain renin‐angiotensin system. Clin Exp Hypertens A 14, 393–411. [DOI] [PubMed] [Google Scholar]
- Eisner D (2014). Calcium in the heart: from physiology to disease. Exp Physiol 99, 1273–1282. [DOI] [PubMed] [Google Scholar]
- Eisner D, Bode E, Venetucci L & Trafford A (2013). Calcium flux balance in the heart. J Mol Cell Cardiol 58, 110–117. [DOI] [PubMed] [Google Scholar]
- Ferrandi M, Molinari I, Rastaldi MP, Ferrari P, Bianchi G & Manunta P (2014). Rostafuroxin protects from podocyte injury and proteinuria induced by adducin genetic variants and ouabain. J Pharmacol Exp Ther 351, 278–287. [DOI] [PubMed] [Google Scholar]
- Ferrari P, Ferrandi M, Tripodi G, Torielli L, Padoani G, Minotti E, Melloni P & Bianchi G (1999). PST 2238: A new antihypertensive compound that modulates Na,K‐ATPase in genetic hypertension. J Pharmacol Exp Ther 288, 1074–1083. [PubMed] [Google Scholar]
- Ferrari P, Torielli L, Ferrandi M, Padoani G, Duzzi L, Florio M, Conti F, Melloni P, Vesci L, Corsico N & Bianchi G (1998). PST2238: A new antihypertensive compound that antagonizes the long‐term pressor effect of ouabain. J Pharmacol Exp Ther 285, 83–94. [PubMed] [Google Scholar]
- Figtree GA, Liu CC, Bibert S, Hamilton EJ, Garcia A, White CN, Chia KK, Cornelius F, Geering K & Rasmussen HH (2009). Reversible oxidative modification: a key mechanism of Na+‐K+ pump regulation. Circ Res 105, 185–193. [DOI] [PubMed] [Google Scholar]
- Fink GD & Bruner CA (1985). Hypertension during chronic peripheral and central infusion of angiotensin III. Am J Physiol 249, E201–E208. [DOI] [PubMed] [Google Scholar]
- Fisher JP & Fadel PJ (2010). Therapeutic strategies for targeting excessive central sympathetic activation in human hypertension. Exp Physiol 95, 572–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabor A & Leenen FH (2012). Central neuromodulatory pathways regulating sympathetic activity in hypertension. J Appl Physiol (1985) 113, 1294–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geering K (2006). FXYD proteins: new regulators of Na‐K‐ATPase. Am J Physiol Renal Physiol 290, F241–F250. [DOI] [PubMed] [Google Scholar]
- Ghadhanfar E, Al‐Bader M & Turcani M (2014). Wistar rats resistant to the hypertensive effects of ouabain exhibit enhanced cardiac vagal activity and elevated plasma levels of calcitonin gene‐related peptide. PLoS One 9, e108909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladden JD, Linke WA & Redfield MM (2014). Heart failure with preserved ejection fraction. Pflugers Arch 466, 1037–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldhaber JI, Lamp ST, Walter DO, Garfinkel A, Fukumoto GH & Weiss JN (1999). Local regulation of the threshold for calcium sparks in rat ventricular myocytes: role of sodium‐calcium exchange. J Physiol 520, 431–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golovina VA, Song H, James PF, Lingrel JB & Blaustein MP (2003). Na+ pump α2‐subunit expression modulates Ca2+ signaling. Am J Physiol Cell Physiol 284, C475–C486. [DOI] [PubMed] [Google Scholar]
- Gomez AM, Valdivia HH, Cheng H, Lederer MR, Santana LF, Cannell MB, McCune SA, Altschuld RA & Lederer WJ (1997). Defective excitation‐contraction coupling in experimental cardiac hypertrophy and heart failure. Science 276, 800–806. [DOI] [PubMed] [Google Scholar]
- Gomez‐Sanchez EP, Zhou M & Gomez‐Sanchez CE (1996). Mineralocorticoids, salt and high blood pressure. Steroids 61, 184–188. [DOI] [PubMed] [Google Scholar]
- Gottlieb SS, Rogowski AC, Weinberg M, Krichten CM, Hamilton BP & Hamlyn JM (1992). Elevated concentrations of endogenous ouabain in patients with congestive heart failure. Circulation 86, 420–425. [DOI] [PubMed] [Google Scholar]
- Grandi E & Herren AW (2014). CaMKII‐dependent regulation of cardiac Na+ homeostasis. Front Pharmacol 5, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greengard P (2001). The neurobiology of slow synaptic transmission. Science 294, 1024–1030. [DOI] [PubMed] [Google Scholar]
- Gronich N, Kumar A, Zhang Y, Efimov IR & Soldatov NM (2010). Molecular remodeling of ion channels, exchangers and pumps in atrial and ventricular myocytes in ischemic cardiomyopathy. Channels 4, 101–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddy FJ & Overbeck HW (1976). The role of humoral agents in volume expanded hypertension. Life Sci 19, 935–947. [DOI] [PubMed] [Google Scholar]
- Hafver TL, Hodne K, Wanichawan P, Aronsen JM, Dalhus B, Lunde PK, Lunde M, Martinsen M, Enger UH, Fuller W, Sjaastad I, Louch WE, Sejersted OM & Carlson CR (2016). Protein phosphatase 1c associated with the cardiac sodium calcium exchanger 1 regulates its activity by dephosphorylating serine 68‐phosphorylated phospholemman. J Biol Chem 291, 4561–4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamlyn JM & Blaustein MP (2016). Endogenous ouabain: recent advances and controversies. Hypertension DOI:10.1161/HYPERTENSIONAHA.116.06599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamlyn JM, Blaustein MP, Bova S, DuCharme DW, Harris DW, Mandel F, Mathews WR & Ludens JH (1991). Identification and characterization of a ouabain‐like compound from human plasma. Proc Natl Acad Sci USA 88, 6259–6263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamlyn JM, Linde CI, Gao J, Huang BS, Golovina VA, Blaustein MP & Leenen FH (2014). Neuroendocrine humoral and vascular components in the pressor pathway for brain angiotensin II: A new axis in long term blood pressure control. PLoS One 9, e108916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamlyn JM & Manunta P (2015). Endogenous cardiotonic steroids in kidney failure: a review and an hypothesis. Adv Chron Kidney Dis 22, 232–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han F, Bossuyt J, Martin JL, Despa S & Bers DM (2010). Role of phospholemman phosphorylation sites in mediating kinase‐dependent regulation of the Na+‐K+‐ATPase. Am J Physiol Cell Physiol 299, C1363–C1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han F, Tucker AL, Lingrel JB, Despa S & Bers DM (2009). Extracellular potassium dependence of the Na+‐K+‐ATPase in cardiac myocytes: isoform specificity and effect of phospholemman. Am J Physiol Cell Physiol 297, C699–C705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauck C, Potter T, Bartz M, Wittwer T, Wahlers T, Mehlhorn U, Scheiner‐Bobis G, McDonough AA, Bloch W, Schwinger RH & Muller‐Ehmsen J (2009). Isoform specificity of cardiac glycosides binding to human Na+,K+‐ATPase α1β1, α2β1 and α3β1 . Eur J Pharmacol 622, 7–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, Shelly DA, Moseley AE, James PF, James JH, Paul RJ & Lingrel JB (2001). The α1‐ and α2‐isoforms of Na‐K‐ATPase play different roles in skeletal muscle contractility. Am J Physiol Regul Integr Comp Physiol 281, R917–R925. [DOI] [PubMed] [Google Scholar]
- Helwig B, Schreurs KM, Hansen J, Hageman KS, Zbreski MG, McAllister RM, Mitchell KE & Musch TI (2003). Training‐induced changes in skeletal muscle Na+‐K+ pump number and isoform expression in rats with chronic heart failure. J Appl Physiol (1985) 94, 2225–2236. [DOI] [PubMed] [Google Scholar]
- Hill MA, Zou J, Potocnik SJ, Meininger GA & Davis MJ (2001). Arteriolar smooth muscle mechanotransduction: Ca2+ signaling pathways underlying myogenic reactivity. J Appl Physiol (1985) 91, 973–983. [DOI] [PubMed] [Google Scholar]
- Hobai IA, Maack C & O'Rourke B (2004). Partial inhibition of sodium/calcium exchange restores cellular calcium handling in canine heart failure. Circ Res 95, 292–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou X, Theriault SF, Dostanic‐Larson I, Moseley AE, Lingrel JB, Wu H, Dean S & Van Huysse JW (2009). Enhanced pressor response to increased CSF sodium concentration and to central ANG I in heterozygous α2 Na+‐K+‐ATPase knockout mice. Am J Physiol Regul Integr Comp Physiol 296, R1427–R1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang BS, Ahmadi S, Ahmad M, White RA & Leenen FH (2010). Central neuronal activation and pressor responses induced by circulating ANG II: role of the brain aldosterone‐‘ouabain’ pathway. Am J Physiol Heart Circ Physiol 299, H422–H430. [DOI] [PubMed] [Google Scholar]
- Huang BS, Cheung WJ, Wang H, Tan J, White RA & Leenen FH (2006). Activation of brain renin‐angiotensin‐aldosterone system by central sodium in Wistar rats. Am J Physiol Heart Circ Physiol 291, H1109–H1117. [DOI] [PubMed] [Google Scholar]
- Huang BS, Huang X, Harmsen E & Leenen FH (1994). Chronic central versus peripheral ouabain, blood pressure, and sympathetic activity in rats. Hypertension 23, 1087–1090. [DOI] [PubMed] [Google Scholar]
- Huang BS, Kudlac M, Kumarathasan R & Leenen FH (1999). Digoxin prevents ouabain and high salt intake‐induced hypertension in rats with sinoaortic denervation. Hypertension 34, 733–738. [DOI] [PubMed] [Google Scholar]
- Huang BS & Leenen FH (1999). Brain renin‐angiotensin system and ouabain‐induced sympathetic hyperactivity and hypertension in Wistar rats. Hypertension 34, 107–112. [DOI] [PubMed] [Google Scholar]
- Huang BS, Zheng H, Tan J, Patel KP & Leenen FH (2011). Regulation of hypothalamic renin‐angiotensin system and oxidative stress by aldosterone. Exp Physiol 96, 1028–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hundal HS, Maxwell DL, Ahmed A, Darakhshan F, Mitsumoto Y & Klip A (1994). Subcellular distribution and immunocytochemical localization of Na,K‐ATPase subunit isoforms in human skeletal muscle. Mol Membr Biol 11, 255–262. [DOI] [PubMed] [Google Scholar]
- Iwamoto T, Kita S, Zhang J, Blaustein MP, Arai Y, Yoshida S, Wakimoto K, Komuro I & Katsuragi T (2004). Salt‐sensitive hypertension is triggered by Ca2+ entry via Na+/Ca2+ exchanger type‐1 in vascular smooth muscle. Nat Med 10, 1193–1199. [DOI] [PubMed] [Google Scholar]
- Jacobs BE, Liu Y, Pulina MV, Golovina VA & Hamlyn JM (2012). Normal pregnancy: mechanisms underlying the paradox of a ouabain‐resistant state with elevated endogenous ouabain, suppressed arterial sodium calcium exchange, and low blood pressure. Am J Physiol Heart Circ Physiol 302, H1317–H1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James PA, Oparil S, Carter BL, Cushman WC, Dennison‐Himmelfarb C, Handler J, Lackland DT, LeFevre ML, MacKenzie TD, Ogedegbe O, Smith SC Jr, Svetkey LP, Taler SJ, Townsend RR, Wright JT Jr, Narva AS & Ortiz E (2014). 2014 evidence‐based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA 311, 507–520. [DOI] [PubMed] [Google Scholar]
- James PF, Grupp IL, Grupp G, Woo AL, Askew GR, Croyle ML, Walsh RA & Lingrel JB (1999). Identification of a specific role for the Na,K‐ATPase α2 isoform as a regulator of calcium in the heart. Mol Cell 3, 555–563. [DOI] [PubMed] [Google Scholar]
- Jayasinghe ID, Cannell MB & Soeller C (2009). Organization of ryanodine receptors, transverse tubules, and sodium‐calcium exchanger in rat myocytes. Biophys J 97, 2664–2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia LG, Donnet C, Bogaev RC, Blatt RJ, McKinney CE, Day KH, Berr SS, Jones LR, Moorman JR, Sweadner KJ & Tucker AL (2005). Hypertrophy, increased ejection fraction, and reduced Na‐K‐ATPase activity in phospholemman‐deficient mice. Am J Physiol Heart Circ Physiol 288, H1982–H1988. [DOI] [PubMed] [Google Scholar]
- Jordan MC, Henderson SA, Han T, Fishbein MC, Philipson KD & Roos KP (2010). Myocardial function with reduced expression of the sodium‐calcium exchanger. J Card Fail 16, 786–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juhaszova M & Blaustein MP (1997. a). Distinct distribution of different Na+ pump α subunit isoforms in plasmalemma. Physiological implications. Ann NY Acad Sci 834, 524–536. [DOI] [PubMed] [Google Scholar]
- Juhaszova M & Blaustein MP (1997. b). Na+ pump low and high ouabain affinity α subunit isoforms are differently distributed in cells. Proc Natl Acad Sci USA 94, 1800–1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamimura D, Ohtani T, Sakata Y, Mano T, Takeda Y, Tamaki S, Omori Y, Tsukamoto Y, Furutani K, Komiyama Y, Yoshika M, Takahashi H, Matsuda T, Baba A, Umemura S, Miwa T, Komuro I & Yamamoto K (2012). Ca2+ entry mode of Na+/Ca2+ exchanger as a new therapeutic target for heart failure with preserved ejection fraction. Eur Heart J 33, 1408–1416. [DOI] [PubMed] [Google Scholar]
- Karmazyn M, Kilic A & Javadov S (2008). The role of NHE‐1 in myocardial hypertrophy and remodelling. J Mol Cell Cardiol 44, 647–653. [DOI] [PubMed] [Google Scholar]
- Kass DA, Bronzwaer JG & Paulus WJ (2004). What mechanisms underlie diastolic dysfunction in heart failure? Circ Res 94, 1533–1542. [DOI] [PubMed] [Google Scholar]
- Kawamura A, Guo J, Itagaki Y, Bell C, Wang Y, Haupert GT Jr, Magil S, Gallagher RT, Berova N & Nakanishi K (1999). On the structure of endogenous ouabain. Proc Natl Acad Sci USA 96, 6654–6659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy DJ, Vetteth S, Periyasamy SM, Kanj M, Fedorova L, Khouri S, Kahaleh MB, Xie Z, Malhotra D, Kolodkin NI, Lakatta EG, Fedorova OV, Bagrov AY & Shapiro JI (2006). Central role for the cardiotonic steroid marinobufagenin in the pathogenesis of experimental uremic cardiomyopathy. Hypertension 47, 488–495. [DOI] [PubMed] [Google Scholar]
- Knot HJ & Nelson MT (1998). Regulation of arterial diameter and wall Ca2+ in cerebral arteries of rat by membrane potential and intravascular pressure. J Physiol 508 199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komiyama Y, Nishimura N, Dong XH, Hirose S, Kosaka C, Masaki H, Masuda M & Takahashi H (2000). Liquid chromatography mass spectrometric analysis of ouabainlike factor in biological fluid. Hypertens Res 23, Suppl., S21–27. [DOI] [PubMed] [Google Scholar]
- Krep H, Price DA, Soszynski P, Tao QF, Graves SW & Hollenberg NK (1995). Volume sensitive hypertension and the digoxin‐like factor. Reversal by a Fab directed against digoxin in DOCA‐salt hypertensive rats. Am J Hypertens 8, 921–927. [DOI] [PubMed] [Google Scholar]
- Kristensen M, Rasmussen MK & Juel C (2008). Na+‐K+ pump location and translocation during muscle contraction in rat skeletal muscle. Pflugers Arch 456, 979–989. [DOI] [PubMed] [Google Scholar]
- Krum H & Driscoll A (2013). Management of heart failure. Med J Aust 199, 334–339. [DOI] [PubMed] [Google Scholar]
- Kuszczak I, Kuner R, Samson SE & Grover AK (2010). Proximity of Na+‐Ca2+‐exchanger and sarco/endoplasmic reticulum Ca2+ pump in pig coronary artery smooth muscle: fluorescence microscopy. Mol Cell Biochem 339, 293–300. [DOI] [PubMed] [Google Scholar]
- Laredo J, Hamilton BP & Hamlyn JM (1994). Ouabain is secreted by bovine adrenocortical cells. Endocrinology 135, 794–797. [DOI] [PubMed] [Google Scholar]
- Laursen M, Gregersen JL, Yatime L, Nissen P & Fedosova NU (2015). Structures and characterization of digoxin‐ and bufalin‐bound Na+,K+‐ATPase compared with the ouabain‐bound complex. Proc Natl Acad Sci USA 112, 1755–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laursen M, Yatime L, Nissen P & Fedosova NU (2013). Crystal structure of the high‐affinity Na+K+‐ATPase‐ouabain complex with Mg2+ bound in the cation binding site. Proc Natl Acad Sci USA 110, 10958–10963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledoux J, Gee DM & Leblanc N (2003). Increased peripheral resistance in heart failure: new evidence suggests an alteration in vascular smooth muscle function. Br J Pharmacol 139, 1245–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MY, Song H, Nakai J, Ohkura M, Kotlikoff MI, Kinsey SP, Golovina VA & Blaustein MP (2006). Local subplasma membrane Ca2+ signals detected by a tethered Ca2+ sensor. Proc Natl Acad Sci USA 103, 13232–13237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leenen FH (2010). The central role of the brain aldosterone‐‘ouabain’ pathway in salt‐sensitive hypertension. Biochim Biophys Acta 1802, 1132–1139. [DOI] [PubMed] [Google Scholar]
- Leenen FH (2014). Actions of circulating angiotensin II and aldosterone in the brain contributing to hypertension. Am J Hypertens 27, 1024–1032. [DOI] [PubMed] [Google Scholar]
- Leenen FH, Hou X, Wang HW & Ahmad M (2015). Enhanced expression of epithelial sodium channels causes salt‐induced hypertension in mice through inhibition of the α2‐isoform of Na+, K+‐ATPase. Physiol Rep 3, e12383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leenen FH, Huang BS, Yu H & Yuan B (1995). Brain ‘ouabain’ mediates sympathetic hyperactivity in congestive heart failure. Circ Res 77, 993–1000. [DOI] [PubMed] [Google Scholar]
- Leenen FH, Ruzicka M & Floras JS (2012). Central sympathetic inhibition by mineralocorticoid receptor but not angiotensin II type 1 receptor blockade: are prescribed doses too low? Hypertension 60, 278–280. [DOI] [PubMed] [Google Scholar]
- Lehnart SE, Maier LS & Hasenfuss G (2009). Abnormalities of calcium metabolism and myocardial contractility depression in the failing heart. Heart Fail Rev 14, 213–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Louch WE, Niederer SA, Aronsen JM, Christensen G, Sejersted OM & Smith NP (2012). Sodium accumulation in SERCA knockout‐induced heart failure. Biophys J 102, 2039–2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Pogwizd SM, Prabhu SD & Zhou L (2014). Inhibiting Na+/K+ ATPase can impair mitochondrial energetics and induce abnormal Ca2+ cycling and automaticity in guinea pig cardiomyocytes. PLoS One 9, e93928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z & Xie Z (2009). The Na/K‐ATPase/Src complex and cardiotonic steroid‐activated protein kinase cascades. Pflugers Arch 457, 635–644. [DOI] [PubMed] [Google Scholar]
- Liang M, Cai T, Tian J, Qu W & Xie ZJ (2006). Functional characterization of Src‐interacting Na/K‐ATPase using RNA interference assay. J Biol Chem 281, 19709–19719. [DOI] [PubMed] [Google Scholar]
- Liang M, Tian J, Liu L, Pierre S, Liu J, Shapiro J & Xie ZJ (2007). Identification of a pool of non‐pumping Na/K‐ATPase. J Biol Chem 282, 10585–10593. [DOI] [PubMed] [Google Scholar]
- Liao Y, Ishikura F, Beppu S, Asakura M, Takashima S, Asanuma H, Sanada S, Kim J, Ogita H, Kuzuya T, Node K, Kitakaze M & Hori M (2002). Echocardiographic assessment of LV hypertrophy and function in aortic‐banded mice: necropsy validation. Am J Physiol Heart Circ Physiol 282, H1703–H1708. [DOI] [PubMed] [Google Scholar]
- Linde CI, Antos LK, Golovina VA & Blaustein MP (2012). Nanomolar ouabain increases NCX1 expression and enhances Ca2+ signaling in human arterial myocytes: a mechanism that links salt to increased vascular resistance? Am J Physiol Heart Circ Physiol 303, H784–H794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingrel JB (2010). The physiological significance of the cardiotonic steroid/ouabain‐binding site of the Na,K‐ATPase. Ann Rev Physiol 72, 395–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CC, Karimi Galougahi K, Weisbrod RM, Hansen T, Ravaie R, Nunez A, Liu YB, Fry N, Garcia A, Hamilton EJ, Sweadner KJ, Cohen RA & Figtree GA (2013). Oxidative inhibition of the vascular Na+‐K+ pump via NADPH oxidase‐dependent β1‐subunit glutathionylation: Implications for angiotensin II‐induced vascular dysfunction. Free Radic Biol Med 65, 563–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Tian J, Haas M, Shapiro JI, Askari A & Xie Z (2000). Ouabain interaction with cardiac Na+/K+‐ATPase initiates signal cascades independent of changes in intracellular Na+ and Ca2+ concentrations. J Biol Chem 275, 27838–27844. [DOI] [PubMed] [Google Scholar]
- Liu J & Xie ZJ (2010). The sodium pump and cardiotonic steroids‐induced signal transduction protein kinases and calcium‐signaling microdomain in regulation of transporter trafficking. Biochim Biophys Acta 1802, 1237–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Mohammadi K, Aynafshar B, Wang H, Li D, Liu J, Ivanov AV, Xie Z & Askari A (2003). Role of caveolae in signal‐transducing function of cardiac Na+/K+‐ATPase. Am J Physiol Cell Physiol 284, C1550–C1560. [DOI] [PubMed] [Google Scholar]
- Liu T, Brown DA & O'Rourke B (2010). Role of mitochondrial dysfunction in cardiac glycoside toxicity. J Mol Cell Cardiol 49, 728–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz JN, Loreaux EL, Dostanic‐Larson I, Lasko V, Schnetzer JR, Paul RJ & Lingrel JB (2008). ACTH‐induced hypertension is dependent on the ouabain‐binding site of the α2‐Na+‐K+‐ATPase subunit. Am J Physiol Heart Circ Physiol 295, H273–H280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz JN, Oshiro N, Loreaux EL & Lingrel JB (2012). DOCA‐salt hypertension does not require the ouabain‐sensitive binding site of the α2 Na,K‐ATPase. Am J Hypertens 25, 421–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louch WE, Hougen K, Mørk HK, Swift F, Aronsen JM, Sjaastad I, Reims HM, Roald B, Andersson KB, Christensen G & Sejersted OM (2010). Sodium accumulation promotes diastolic dysfunction in end‐stage heart failure following Serca2 knockout. J Physiol 588, 465–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lymperopoulos A, Rengo G & Koch WJ (2013). Adrenergic nervous system in heart failure: pathophysiology and therapy. Circ Res 113, 739–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch RM, Weber CS, Nullmeyer KD, Moore ED & Paul RJ (2008). Clearance of store‐released Ca2+ by the Na+‐Ca2+ exchanger is diminished in aortic smooth muscle from Na+‐K+‐ATPase α2‐isoform gene‐ablated mice. Am J Physiol Heart Circ Physiol 294, H1407–H1416. [DOI] [PubMed] [Google Scholar]
- McDonough AA, Azuma KK, Lescale‐Matys L, Tang MJ, Nakhoul F, Hensley CB & Komatsu Y (1992). Physiologic rationale for multiple sodium pump isoforms. Differential regulation of α1 vs α2 by ionic stimuli. Ann NY Acad Sci 671, 156–168. [DOI] [PubMed] [Google Scholar]
- McGrail KM, Phillips JM & Sweadner KJ (1991). Immunofluorescent localization of three Na,K‐ATPase isozymes in the rat central nervous system: Both neurons and glia can express more than one Na,K‐ATPase. J Neurosci 11, 381–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mainous AG 3rd, Tanner RJ, Anton SD & Jo A (2015). Grip strength as a marker of hypertension and diabetes in healthy weight adults. Am J Prev Med 49, 850–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manoharan P, Radzyukevich TL, Hakim Javadi H, Stiner CA, Landero Figueroa JA, Lingrel JB & Heiny JA (2015). Phospholemman is not required for the acute stimulation of Na+‐K+‐ATPase α2‐activity during skeletal muscle fatigue. Am J Physiol Cell Physiol 309, C813–C822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manunta P, Hamilton J, Rogowski AC, Hamilton BP & Hamlyn JM (2000). Chronic hypertension induced by ouabain but not digoxin in the rat: antihypertensive effect of digoxin and digitoxin. Hypertens Res 23, Suppl., S77–85. [DOI] [PubMed] [Google Scholar]
- Manunta P, Messaggio E, Casamassima N, Gatti G, Carpini SD, Zagato L & Hamlyn JM (2010). Endogenous ouabain in renal Na+ handling and related diseases. Biochim Biophys Acta 1802, 1214–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx SO & Marks AR (2013). Dysfunctional ryanodine receptors in the heart: new insights into complex cardiovascular diseases. J Mol Cell Cardiol 58, 225–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews MR, DuCharme DW, Hamlyn JM, Harris DW, Mandel F, Clark MA & Ludens JH (1991). Mass spectral characterization of an endogenous digitalis‐like factor from human plasma. Hypertension 17, 930–935. [DOI] [PubMed] [Google Scholar]
- Miller RL & Loewy AD (2013). ENaC γ‐expressing astrocytes in the circumventricular organs, white matter, and ventral medullary surface: sites for Na+ regulation by glial cells. J Chem Neuroanat 53, 72–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RL, Wang MH, Gray PA, Salkoff LB & Loewy AD (2013). ENaC‐expressing neurons in the sensory circumventricular organs become c‐Fos activated following systemic sodium changes. Am J Physiol Regul Integr Comp Physiol 305, R1141–R1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra NK, Habeck M, Kirchner C, Haviv H, Peleg Y, Eisenstein M, Apell HJ & Karlish SJ (2015). Molecular mechanisms and kinetic effects of FXYD1 and phosphomimetic mutants on purified human Na,K‐ATPase. J Biol Chem 290, 28746–28759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohler PJ, Schott JJ, Gramolini AO, Dilly KW, Guatimosim S, duBell WH, Song LS, Haurogne K, Kyndt F, Ali ME, Rogers TB, Lederer WJ, Escande D, Le Marec H & Bennett V (2003). Ankyrin‐B mutation causes type 4 long‐QT cardiac arrhythmia and sudden cardiac death. Nature 421, 634–639. [DOI] [PubMed] [Google Scholar]
- Moseley AE, Lieske SP, Wetzel RK, James PF, He S, Shelly DA, Paul RJ, Boivin GP, Witte DP, Ramirez JM, Sweadner KJ & Lingrel JB (2003). The Na,K‐ATPase α2 isoform is expressed in neurons, and its absence disrupts neuronal activity in newborn mice. J Biol Chem 278, 5317–5324. [DOI] [PubMed] [Google Scholar]
- Müller‐Ehmsen J, Nickel J, Zobel C, Hirsch I, Bölck B, Brixius K & Schwinger RH (2003). Longer term effects of ouabain on the contractility of rat isolated cardiomyocytes and on the expression of Ca and Na regulating proteins. Basic Res Cardiol 98, 90–96. [DOI] [PubMed] [Google Scholar]
- Munzel T, Gori T, Keaney JF Jr, Maack C & Daiber A (2015). Pathophysiological role of oxidative stress in systolic and diastolic heart failure and its therapeutic implications. Eur Heart J 36, 2555–2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy E & Eisner DA (2009). Regulation of intracellular and mitochondrial sodium in health and disease. Circ Res 104, 292–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nita II, Hershfinkel M, Lewis EC & Sekler I (2015). A crosstalk between Na+ channels, Na+/K+ pump and mitochondrial Na+ transporters controls glucose‐dependent cytosolic and mitochondrial Na+ signals. Cell Calcium 57, 69–75. [DOI] [PubMed] [Google Scholar]
- O'Brien WJ, Lingrel JB & Wallick ET (1994). Ouabain binding kinetics of the rat alpha two and alpha three isoforms of the sodium‐potassium adenosine triphosphate. Arch Biochem Biophys 310, 32–39. [DOI] [PubMed] [Google Scholar]
- O'Rourke B, Kass DA, Tomaselli GF, Kaab S, Tunin R & Marban E (1999). Mechanisms of altered excitation‐contraction coupling in canine tachycardia‐induced heart failure, I: experimental studies. Circ Res 84, 562–570. [DOI] [PubMed] [Google Scholar]
- Okita K, Kinugawa S & Tsutsui H (2013). Exercise intolerance in chronic heart failure – skeletal muscle dysfunction and potential therapies. Circ J 77, 293–300. [DOI] [PubMed] [Google Scholar]
- Osborn JW, Fink GD & Kuroki MT (2011). Neural mechanisms of angiotensin II‐salt hypertension: implications for therapies targeting neural control of the splanchnic circulation. Curr Hypertens Rep 13, 221–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborn JW, Fink GD, Sved AF, Toney GM & Raizada MK (2007). Circulating angiotensin II and dietary salt: converging signals for neurogenic hypertension. Curr Hypertens Rep 9, 228–235. [DOI] [PubMed] [Google Scholar]
- Osborn JW, Olson DM, Guzman P, Toney GM & Fink GD (2014). The neurogenic phase of angiotensin II‐salt hypertension is prevented by chronic intracerebroventricular administration of benzamil. Physiol Rep 2, e00245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshima N, Onimaru H, Takechi H, Yamamoto K, Watanabe A, Uchida T, Nishida Y, Oda T & Kumagai H (2013). Aldosterone is synthesized in and activates bulbospinal neurons through mineralocorticoid receptors and ENaCs in the RVLM. Hypertens Res 36, 504–512. [DOI] [PubMed] [Google Scholar]
- Oshiro N, Dostanic‐Larson I, Neumann JC & Lingrel JB (2010). The ouabain‐binding site of the α2 isoform of Na,K‐ATPase plays a role in blood pressure regulation during pregnancy. Am J Hypertens 23, 1279–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottolia M, Torres N, Bridge JH, Philipson KD & Goldhaber JI (2013). Na/Ca exchange and contraction of the heart. J Mol Cell Cardiol 61, 28–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlovic D (2014). The role of cardiotonic steroids in the pathogenesis of cardiomyopathy in chronic kidney disease. Nephron Clin Pract 128, 11–21. [DOI] [PubMed] [Google Scholar]
- Pavlovic D, Fuller W & Shattock MJ (2007). The intracellular region of FXYD1 is sufficient to regulate cardiac Na/K ATPase. FASEB J 21, 1539–1546. [DOI] [PubMed] [Google Scholar]
- Pavlovic D, Fuller W & Shattock MJ (2013. a). Novel regulation of cardiac Na pump via phospholemman. J Mol Cell Cardiol 61, 83–93. [DOI] [PubMed] [Google Scholar]
- Pavlovic D, Hall AR, Kennington EJ, Aughton K, Boguslavskyi A, Fuller W, Despa S, Bers DM & Shattock MJ (2013. b). Nitric oxide regulates cardiac intracellular Na+ and Ca2+ by modulating Na/K ATPase via PKCε and phospholemman‐dependent mechanism. J Mol Cell Cardiol 61, 164–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phakdeekitcharoen B, Kittikanokrat W, Kijkunasathian C & Chatsudthipong V (2011). Aldosterone increases Na+‐K+‐ATPase activity in skeletal muscle of patients with Conn's syndrome. Clin Endocrinol 74, 152–159. [DOI] [PubMed] [Google Scholar]
- Piacentino V 3rd, Weber CR, Chen X, Weisser‐Thomas J, Margulies KB, Bers DM & Houser SR (2003). Cellular basis of abnormal calcium transients of failing human ventricular myocytes. Circ Res 92, 651–658. [DOI] [PubMed] [Google Scholar]
- Pitzalis MV, Hamlyn JM, Messaggio E, Iacoviello M, Forleo C, Romito R, de Tommasi E, Rizzon P, Bianchi G & Manunta P (2006). Independent and incremental prognostic value of endogenous ouabain in idiopathic dilated cardiomyopathy. Eur J Heart Fail 8, 179–186. [DOI] [PubMed] [Google Scholar]
- Poburko D, Fameli N, Kuo KH & van Breemen C (2008). Ca2+ signaling in smooth muscle: TRPC6, NCX and LNats in nanodomains. Channels 2, 10–12. [DOI] [PubMed] [Google Scholar]
- Poburko D, Kuo KH, Dai J, Lee CH & van Breemen C (2004). Organellar junctions promote targeted Ca2+ signaling in smooth muscle: why two membranes are better than one. Trends Pharmacol Sci 25, 8–15. [DOI] [PubMed] [Google Scholar]
- Poburko D, Liao CH, Lemos VS, Lin E, Maruyama Y, Cole WC & van Breemen C (2007). Transient receptor potential channel 6‐mediated, localized cytosolic Na+ transients drive Na+/Ca2+ exchanger‐mediated Ca2+ entry in purinergically stimulated aorta smooth muscle cells. Circ Res 101, 1030–1038. [DOI] [PubMed] [Google Scholar]
- Pogwizd SM, Sipido KR, Verdonck F & Bers DM (2003). Intracellular Na in animal models of hypertrophy and heart failure: contractile function and arrhythmogenesis. Cardiovasc Res 57, 887–896. [DOI] [PubMed] [Google Scholar]
- Pressley TA (1992). Phylogenetic conservation of isoform‐specific regions within α‐subunit of Na+‐K+‐ATPase. Am J Physiol 262, C743–C751. [DOI] [PubMed] [Google Scholar]
- Pritchard TJ, Bowman PS, Jefferson A, Tosun M, Lynch RM & Paul RJ (2010). Na+‐K+‐ATPase and Ca2+ clearance proteins in smooth muscle: a functional unit. Am J Physiol Heart Circ Physiol 299, H548–H556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard TJ, Bullard DP, Lynch RM, Lorenz JN & Paul RJ (2007). Transgenic mice expressing Na+‐K+ ATPase in smooth muscle decreases blood pressure. Am J Physiol Heart Circ Physiol 293, H1172–H1182. [DOI] [PubMed] [Google Scholar]
- Pulina MV, Zulian A, Baryshnikov SG, Linde CI, Karashima E, Hamlyn JM, Ferrari P, Blaustein MP & Golovina VA (2013). Cross talk between plasma membrane Na+/Ca2+ exchanger‐1 and TRPC/Orai‐containing channels: key players in arterial hypertension. Adv Exp Med Biol 961, 365–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulina MV, Zulian A, Berra‐Romani R, Beskina O, Mazzocco‐Spezzia A, Baryshnikov SG, Papparella I, Hamlyn JM, Blaustein MP & Golovina VA (2010). Upregulation of Na+ and Ca2+ transporters in arterial smooth muscle from ouabain‐induced hypertensive rats. Am J Physiol Heart Circ Physiol 298, H263–H274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pullen MA, Brooks DP & Edwards RM (2004). Characterization of the neutralizing activity of digoxin‐specific Fab toward ouabain‐like steroids. J Pharmacol Exp Ther 310, 319–325. [DOI] [PubMed] [Google Scholar]
- Pullen MA, Harpel MR, Danoff TM & Brooks DP (2008). Comparison of non‐digitalis binding properties of digoxin‐specific Fabs using direct binding methods. J Immunol Methods 336, 235–241. [DOI] [PubMed] [Google Scholar]
- Quadri L, Bianchi G, Cerri A, Fedrizzi G, Ferrari P, Gobbini M, Melloni P, Sputore S & Torri M (1997). 17β‐(3‐furyl)‐5β‐Androstane‐3β,14β,17α‐triol (PST 2238). A very potent antihypertensive agent with a novel mechanism of action. J Med Chem 40, 1561–1564. [DOI] [PubMed] [Google Scholar]
- Radzyukevich TL, Lingrel JB & Heiny JA (2009). The cardiac glycoside binding site on the Na,K‐ATPase α2 isoform plays a role in the dynamic regulation of active transport in skeletal muscle. Proc Natl Acad Sci USA 106, 2565–2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radzyukevich TL, Neumann JC, Rindler TN, Oshiro N, Goldhamer DJ, Lingrel JB & Heiny JA (2013). Tissue‐specific role of the Na,K‐ATPase α2 isozyme in skeletal muscle. J Biol Chem 288, 1226–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajasekaran SA, Barwe SP & Rajasekaran AK (2005). Multiple functions of Na,K‐ATPase in epithelial cells. Semin Nephrol 25, 328–334. [DOI] [PubMed] [Google Scholar]
- Rasmussen MK, Juel C & Nordsborg NB (2011). Exercise‐induced regulation of muscular Na+‐K+ pump, FXYD1, and NHE1 mRNA and protein expression: importance of training status, intensity, and muscle type. Am J Physiol Regul Integr Comp Physiol 300, R1209–R1220. [DOI] [PubMed] [Google Scholar]
- Reuter H, Han T, Motter C, Philipson KD & Goldhaber JI (2004). Mice overexpressing the cardiac sodium–calcium exchanger: defects in excitation–contraction coupling. J Physiol 554, 779–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter H, Henderson SA, Han T, Ross RS, Goldhaber JI & Philipson KD (2002). The Na+‐Ca2+ exchanger is essential for the action of cardiac glycosides. Circ Res 90, 305–308. [DOI] [PubMed] [Google Scholar]
- Reuter H & Schwinger RH (2012). Calcium handling in human heart failure – abnormalities and target for therapy. Wien Med Wochenschr 162, 297–301. [DOI] [PubMed] [Google Scholar]
- Rindler TN, Dostanic I, Lasko VM, Nieman ML, Neumann JC, Lorenz JN & Lingrel JB (2011). Knockout of the Na,K‐ATPase α2‐isoform in the cardiovascular system does not alter basal blood pressure but prevents ACTH‐induced hypertension. Am J Physiol Heart Circ Physiol 301, H1396–H1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rindler TN, Lasko VM, Nieman ML, Okada M, Lorenz JN & Lingrel JB (2013). Knockout of the Na,K‐ATPase α2‐isoform in cardiac myocytes delays pressure overload‐induced cardiac dysfunction. Am J Physiol Heart Circ Physiol 304, H1147–H1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez JS, Velez Rueda JO, Salas M, Becerra R, Di Carlo MN, Said M, Vittone L, Rinaldi G, Portiansky EL, Mundina‐Weilenmann C, Palomeque J & Mattiazzi A (2014). Increased Na+/Ca2+ exchanger expression/activity constitutes a point of inflection in the progression to heart failure of hypertensive rats. PLoS One 9, e96400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos KP, Jordan MC, Fishbein MC, Ritter MR, Friedlander M, Chang HC, Rahgozar P, Han T, Garcia AJ, Maclellan WR, Ross RS & Philipson KD (2007). Hypertrophy and heart failure in mice overexpressing the cardiac sodium‐calcium exchanger. J Card Fail 13, 318–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schatzmann HJ (1953). [Cardiac glycosides as inhibitors of active potassium and sodium transport by erythrocyte membrane.]. Helv Physiol Pharmacol Acta 11, 346–354. [PubMed] [Google Scholar]
- Schneider R, Wray V, Nimtz M, Lehmann WD, Kirch U, Antolovic R & Schoner W (1998). Bovine adrenals contain, in addition to ouabain, a second inhibitor of the sodium pump. J Biol Chem 273, 784–792. [DOI] [PubMed] [Google Scholar]
- Schoner W & Scheiner‐Bobis G (2007). Endogenous and exogenous cardiac glycosides: their roles in hypertension, salt metabolism, and cell growth. Am J Physiol Cell Physiol 293, C509–C536. [DOI] [PubMed] [Google Scholar]
- Scriven DR, Dan P & Moore ED (2000). Distribution of proteins implicated in excitation‐contraction coupling in rat ventricular myocytes. Biophys J 79, 2682–2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semb SO, Lunde PK, Holt E, Tønnessen T, Christensen G & Sejersted OM (1998). Reduced myocardial Na+, K+‐pump capacity in congestive heart failure following myocardial infarction in rats. J Mol Cell Cardiol 30, 1311–1328. [DOI] [PubMed] [Google Scholar]
- Shah JR, Laredo J, Hamilton BP & Hamlyn JM (1998). Different signaling pathways mediate stimulated secretions of endogenous ouabain and aldosterone from bovine adrenocortical cells. Hypertension 31, 463–468. [DOI] [PubMed] [Google Scholar]
- Sharma K & Kass DA (2014). Heart failure with preserved ejection fraction: mechanisms, clinical features, and therapies. Circ Res 115, 79–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shattock MJ, Ottolia M, Bers DM, Blaustein MP, Boguslavskyi A, Bossuyt J, Bridge JH, Chen‐Izu Y, Clancy CE, Edwards A, Goldhaber J, Kaplan J, Lingrel JB, Pavlovic D, Philipson K, Sipido KR & Xie ZJ (2015). Na+/Ca2+ exchange and Na+/K+‐ATPase in the heart. J Physiol 593, 1361–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelly DA, He S, Moseley A, Weber C, Stegemeyer M, Lynch RM, Lingrel J & Paul RJ (2004). Na+ pump α2‐isoform specifically couples to contractility in vascular smooth muscle: evidence from gene‐targeted neonatal mice. Am J Physiol Cell Physiol 286, C813–C820. [DOI] [PubMed] [Google Scholar]
- Shi PP, Cao XR, Sweezer EM, Kinney TS, Williams NR, Husted RF, Nair R, Weiss RM, Williamson RA, Sigmund CD, Snyder PM, Staub O, Stokes JB & Yang B (2008). Salt‐sensitive hypertension and cardiac hypertrophy in mice deficient in the ubiquitin ligase Nedd4‐2. Am J Physiol Renal Physiol 295, F462–F470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shull GE, Lane LK & Lingrel JB (1986). Amino‐acid sequence of the β‐subunit of the (Na+ + K+)ATPase deduced from a cDNA. Nature 321, 429–431. [DOI] [PubMed] [Google Scholar]
- Shull GE, Schwartz A & Lingrel JB (1985). Amino‐acid sequence of the catalytic subunit of the (Na+ + K+)ATPase deduced from a complementary DNA. Nature 316, 691–695. [DOI] [PubMed] [Google Scholar]
- Shull MM & Lingrel JB (1987). Multiple genes encode the human Na+,K+‐ATPase catalytic subunit. Proc Natl Acad Sci USA 84, 4039–4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman BdZ, Warley A, Miller JI, James AF & Shattock MJ (2003). Is there a transient rise in sub‐sarcolemmal Na and activation of Na/K pump current following activation of I Na in ventricular myocardium? Cardiovasc Res 57, 1025–1034. [DOI] [PubMed] [Google Scholar]
- Sokolow S, Manto M, Gailly P, Molgo J, Vandebrouck C, Vanderwinden JM, Herchuelz A & Schurmans S (2004). Impaired neuromuscular transmission and skeletal muscle fiber necrosis in mice lacking Na/Ca exchanger 3. J Clin Invest 113, 265–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H, Karashima E, Hamlyn JM & Blaustein MP (2014). Ouabain–digoxin antagonism in rat arteries and neurones. J Physiol 592, 941–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H, Thompson SM & Blaustein MP (2013). Nanomolar ouabain augments Ca2+ signalling in rat hippocampal neurones and glia. J Physiol 591, 1671–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stella P, Manunta P, Mallamaci F, Melandri M, Spotti D, Tripepi G, Hamlyn JM, Malatino LS, Bianchi G & Zoccali C (2008). Endogenous ouabain and cardiomyopathy in dialysis patients. J Intern Med 263, 274–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocker SD, Lang SM, Simmonds SS, Wenner MM & Farquhar WB (2015). Cerebrospinal fluid hypernatremia elevates sympathetic nerve activity and blood pressure via the rostral ventrolateral medulla. Hypertension 66, 1184–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studer R, Reinecke H, Bilger J, Eschenhagen T, Bohm M, Hasenfuss G, Just H, Holtz J & Drexler H (1994). Gene expression of the cardiac Na+‐Ca2+ exchanger in end‐stage human heart failure. Circ Res 75, 443–453. [DOI] [PubMed] [Google Scholar]
- Su Z, Sugishita K, Ritter M, Li F, Spitzer KW & Barry WH (2001). The sodium pump modulates the influence of I Na on [Ca2+]i transients in mouse ventricular myocytes. Biophys J 80, 1230–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swift F, Tovsrud N, Enger UH, Sjaastad I & Sejersted OM (2007). The Na+/K+‐ATPase α2‐isoform regulates cardiac contractility in rat cardiomyocytes. Cardiovasc Res 75, 109–117. [DOI] [PubMed] [Google Scholar]
- Swift F, Tovsrud N, Sjaastad I, Sejersted OM, Niggli E & Egger M (2010). Functional coupling of α2‐isoform Na+/K+‐ATPase and Ca2+ extrusion through the Na+/Ca2+‐exchanger in cardiomyocyte. Cell Calcium 48, 54–60. [DOI] [PubMed] [Google Scholar]
- Szent‐Gyorgi A (1953). Chemical Physiology of Contraction in Body and Heart Muscle. Academic Press, New York, 135 pp. [Google Scholar]
- Takahashi H (2012). Upregulation of the renin‐angiotensin‐aldosterone‐ouabain system in the brain is the core mechanism in the genesis of all types of hypertension. Int J Hypertens 2012, 242786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takimoto E, Yao A, Toko H, Takano H, Shimoyama M, Sonoda M, Wakimoto K, Takahashi T, Akazawa H, Mizukami M, Nagai T, Nagai R & Komuro I (2002). Sodium calcium exchanger plays a key role in alteration of cardiac function in response to pressure overload. FASEB J 16, 373–378. [DOI] [PubMed] [Google Scholar]
- Tashko G, Rogowski R, Tilva V, Hamilton BP & Hamlyn JM (2010). Endogenous ouabain from bovine adrenal glands versus plant ouabain: implications for the biosynthesis of cardiotonic steroids by mammals and plants. Endocr Rev 31, (3) suppl., III p3–35. [Google Scholar]
- Therien AG & Blostein R (2000). Mechanisms of sodium pump regulation. Am J Physiol Cell Physiol 279, C541–C566. [DOI] [PubMed] [Google Scholar]
- Thomassen M, Christensen PM, Gunnarsson TP, Nybo L & Bangsbo J (2010). Effect of 2‐wk intensified training and inactivity on muscle Na+‐K+ pump expression, phospholemman (FXYD1) phosphorylation, and performance in soccer players. J Appl Physiol (1985) 108, 898–905. [DOI] [PubMed] [Google Scholar]
- Thomassen M, Murphy RM & Bangsbo J (2013). Fibre type‐specific change in FXYD1 phosphorylation during acute intense exercise in humans. J Physiol 591, 1523–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian J, Cai T, Yuan Z, Wang H, Liu L, Haas M, Maksimova E, Huang XY & Xie ZJ (2006). Binding of Src to Na+/K+‐ATPase forms a functional signaling complex. Mol Biol Cell 17, 317–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian J & Xie ZJ (2008). The Na‐K‐ATPase and calcium‐signaling microdomains. Physiology (Bethesda) 23, 205–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG (2008). The self‐tuning neuron: synaptic scaling of excitatory synapses. Cell 135, 422–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzanis G, Dimopoulos S, Agapitou V & Nanas S (2014). Exercise intolerance in chronic heart failure: the role of cortisol and the catabolic state. Curr Heart Fail Rep 11, 70–79. [DOI] [PubMed] [Google Scholar]
- Ufnal M & Skrzypecki J (2014). Blood borne hormones in a cross‐talk between peripheral and brain mechanisms regulating blood pressure, the role of circumventricular organs. Neuropeptides 48, 65–73. [DOI] [PubMed] [Google Scholar]
- Van Huysse JW, Amin MS, Yang B & Leenen FH (2012). Salt‐induced hypertension in a mouse model of Liddle syndrome is mediated by epithelial sodium channels in the brain. Hypertension 60, 691–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Huysse JW, Dostanic I, Lingrel JB, Hou X & Wu H (2011). Hypertension from chronic central sodium chloride in mice is mediated by the ouabain‐binding site on the Na,K‐ATPase α2‐isoform. Am J Physiol Heart Circ Physiol 301, H2147–H2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Huysse JW & Hou X (2004). Pressor response to CSF sodium in mice: mediation by a ouabain‐like substance and renin‐angiotensin system in the brain. Brain Res 1021, 219–231. [DOI] [PubMed] [Google Scholar]
- Vemuri R, Longoni S & Philipson KD (1989). Ouabain treatment of cardiac cells induces enhanced Na+‐Ca2+ exchange activity. Am J Physiol 256, C1273–C1276. [DOI] [PubMed] [Google Scholar]
- Verdonck F, Mubagwa K & Sipido KR (2004). [Na+] in the subsarcolemmal ‘fuzzy’ space and modulation of [Ca2+]i and contraction in cardiac myocytes. Cell Calcium 35, 603–612. [DOI] [PubMed] [Google Scholar]
- Verdonck F, Volders PG, Vos MA & Sipido KR (2003). Intracellular Na+ and altered Na+ transport mechanisms in cardiac hypertrophy and failure. J Mol Cell Cardiol 35, 5–25. [DOI] [PubMed] [Google Scholar]
- Wang H, Haas M, Liang M, Cai T, Tian J, Li S & Xie Z (2004). Ouabain assembles signaling cascades through the caveolar Na+/K+‐ATPase. J Biol Chem 279, 17250–17259. [DOI] [PubMed] [Google Scholar]
- Wang J, Gao E, Rabinowitz J, Song J, Zhang XQ, Koch WJ, Tucker AL, Chan TO, Feldman AM & Cheung JY (2011). Regulation of in vivo cardiac contractility by phospholemman: role of Na+/Ca2+ exchange. Am J Physiol Heart Circ Physiol 300, H859–H868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Chen L, Li M, Cha H, Iwamoto T & Zhang J (2015). Conditional knockout of smooth muscle sodium calcium exchanger type‐1 lowers blood pressure and attenuates angiotensin II‐salt hypertension. Physiol Rep 3, e12273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Chen L, Wier WG & Zhang J (2013). Intravital Forster resonance energy transfer imaging reveals elevated [Ca2+]i and enhanced sympathetic tone in femoral arteries of angiotensin II‐infused hypertensive biosensor mice. J Physiol 591, 5321–5336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wansapura AN, Lasko V, Xie Z, Fedorova OV, Bagrov AY, Lingrel JB & Lorenz JN (2009). Marinobufagenin enhances cardiac contractility in mice with ouabain‐sensitive α1 Na+‐K+‐ATPase. Am J Physiol Heart Circ Physiol 296, H1833–H1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wansapura AN, Lasko VM, Lingrel JB & Lorenz JN (2011). Mice expressing ouabain‐sensitive α1‐Na,K‐ATPase have increased susceptibility to pressure overload‐induced cardiac hypertrophy. Am J Physiol Heart Circ Physiol 300, H347–H355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendt‐Gallitelli MF, Voigt T & Isenberg G (1993). Microheterogeneity of subsarcolemmal sodium gradients. Electron probe microanalysis in guinea‐pig ventricular myocytes. J Physiol 472, 33–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westcott KV, Huang BS & Leenen FH (2009). Brain renin‐angiotensin‐aldosterone system and ventricular remodeling after myocardial infarct: a review. Can J Physiol Pharmacol 87, 979–988. [DOI] [PubMed] [Google Scholar]
- Woo AL, James PF & Lingrel JB (1999). Characterization of the fourth alpha isoform of the Na,K‐ATPase. J Membr Biol 169, 39–44. [DOI] [PubMed] [Google Scholar]
- Wu J, Akkuratov EE, Bai Y, Gaskill CM, Askari A & Liu L (2013). Cell signaling associated with Na+/K+‐ATPase: activation of phosphatidylinositide 3‐kinase IA/Akt by ouabain is independent of Src. Biochemistry 52, 9059–9067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J, Ye Q, Cui X, Madan N, Yi Q, Pierre SV & Xie Z (2015). Expression of rat Na‐K‐ATPase α2 enables ion pumping but not ouabain‐induced signaling in α1‐deficient porcine renal epithelial cells. Am J Physiol Cell Physiol 309, C373–C382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z & Askari A (2002). Na+/K+‐ATPase as a signal transducer. Eur J Biochem 269, 2434–2439. [DOI] [PubMed] [Google Scholar]
- Yan Y, Shapiro AP, Haller S, Katragadda V, Liu L, Tian J, Basrur V, Malhotra D, Xie ZJ, Abraham NG, Shapiro JI & Liu J (2013). Involvement of reactive oxygen species in a feed‐forward mechanism of Na/K‐ATPase‐mediated signaling transduction. J Biol Chem 288, 34249–34258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Wei SG, Zhang ZH, Gomez‐Sanchez E, Weiss RM & Felder RB (2008). Does aldosterone upregulate the brain renin‐angiotensin system in rats with heart failure? Hypertension 51, 727–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan CM, Manunta P, Hamlyn JM, Chen S, Bohen E, Yeun J, Haddy FJ & Pamnani MB (1993). Long‐term ouabain administration produces hypertension in rats. Hypertension 22, 178–187. [DOI] [PubMed] [Google Scholar]
- Yuan X, Lin Z, Luo S, Ji G, Yuan C & Wu Y (2007). Effects of different magnitudes of cyclic stretch on Na+‐K+‐ATPase in skeletal muscle cells in vitro. J Cell Physiol 212, 509–518. [DOI] [PubMed] [Google Scholar]
- Zahler R, Sun W, Ardito T & Kashgarian M (1996). Na‐K‐ATPase alpha‐isoform expression in heart and vascular endothelia: cellular and developmental regulation. Am J Physiol 270, C361–C371. [DOI] [PubMed] [Google Scholar]
- Zelis R, Mason DT & Braunwald E (1968). A comparison of the effects of vasodilator stimuli on peripheral resistance vessels in normal subjects and in patients with congestive heart failure. J Clin Invest 47, 960–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Chen L, Raina H, Blaustein MP & Wier WG (2010. a). In vivo assessment of artery smooth muscle [Ca2+]i and MLCK activation in FRET‐based biosensor mice. Am J Physiol Heart Circ Physiol 299, H946–H956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Hamlyn JM, Karashima E, Raina H, Mauban JR, Izuka M, Berra‐Romani R, Zulian A, Wier WG & Blaustein MP (2009). Low‐dose ouabain constricts small arteries from ouabain‐hypertensive rats: implications for sustained elevation of vascular resistance. Am J Physiol Heart Circ Physiol 297, H1140–H1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Lee MY, Cavalli M, Chen L, Berra‐Romani R, Balke CW, Bianchi G, Ferrari P, Hamlyn JM, Iwamoto T, Lingrel JB, Matteson DR, Wier WG & Blaustein MP (2005). Sodium pump α2 subunits control myogenic tone and blood pressure in mice. J Physiol 569, 243–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Ren C, Chen L, Navedo MF, Antos LK, Kinsey SP, Iwamoto T, Philipson KD, Kotlikoff MI, Santana LF, Wier WG, Matteson DR & Blaustein MP (2010. b). Knockout of Na+/Ca2+ exchanger in smooth muscle attenuates vasoconstriction and L‐type Ca2+ channel current, and lowers blood pressure. Am J Physiol Heart Circ Physiol 298, H1472–H1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Dong H, Rubin LJ & Yuan JX (2007). Upregulation of Na+/Ca2+ exchanger contributes to the enhanced Ca2+ entry in pulmonary artery smooth muscle cells from patients with idiopathic pulmonary arterial hypertension. Am J Physiol Cell Physiol 292, C2297–C2305. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Yan X, Liu W & Li C (2012). Cyclic stretch stimulates recruitment of active Na+/K+‐ATPase subunits to the plasma membrane of skeletal muscle cells. Mol Cell Biochem 366, 299–308. [DOI] [PubMed] [Google Scholar]
- Zucker IH, Xiao L & Haack KK (2014). The central renin‐angiotensin system and sympathetic nerve activity in chronic heart failure. Clin Sci 126, 695–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zulian A, Linde CI, Pulina MV, Baryshnikov SG, Papparella I, Hamlyn JM & Golovina VA (2013). Activation of c‐SRC underlies the differential effects of ouabain and digoxin on Ca2+ signaling in arterial smooth muscle cells. Am J Physiol Cell Physiol 304, C324–C333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo L, Chuang CC, Hemmelgarn BT & Best TM (2015). Heart failure with preserved ejection fraction: Defining the function of ROS and NO. J Appl Physiol (1985) 119, 944–951. [DOI] [PubMed] [Google Scholar]