

Abstract
Background
The described species from the Metarhizium genus are cosmopolitan fungi that infect arthropod hosts. Interestingly, while some species infect a wide range of hosts (host-generalists), other species infect only a few arthropods (host-specialists). This singular evolutionary trait permits unique comparisons to determine how pathogens and virulence determinants emerge. Among the several virulence determinants that have been described, secondary metabolites (SMs) are suggested to play essential roles during fungal infection. Despite progress in the study of pathogen-host relationships, the majority of genes related to SM production in Metarhizium spp. are uncharacterized, and little is known about their genomic organization, expression and regulation. To better understand how infection conditions may affect SM production in Metarhizium anisopliae, we have performed a deep survey and description of SM biosynthetic gene clusters (BGCs) in M. anisopliae, analyzed RNA-seq data from fungi grown on cattle-tick cuticles, evaluated the differential expression of BGCs, and assessed conservation among the Metarhizium genus. Furthermore, our analysis extended to the construction of a phylogeny for the following three BGCs: a tropolone/citrinin-related compound (MaPKS1), a pseurotin-related compound (MaNRPS-PKS2), and a putative helvolic acid (MaTERP1).
Results
Among 73 BGCs identified in M. anisopliae, 20 % were up-regulated during initial tick cuticle infection and presumably possess virulence-related roles. These up-regulated BGCs include known clusters, such as destruxin, NG39x and ferricrocin, together with putative helvolic acid and, pseurotin and tropolone/citrinin-related compound clusters as well as uncharacterized clusters. Furthermore, several previously characterized and putative BGCs were silent or down-regulated in initial infection conditions, indicating minor participation over the course of infection.
Interestingly, several up-regulated BGCs were not conserved in host-specialist species from the Metarhizium genus, indicating differences in the metabolic strategies employed by generalist and specialist species to overcome and kill their host. These differences in metabolic potential may have been partially shaped by horizontal gene transfer (HGT) events, as our phylogenetic analysis provided evidence that the putative helvolic acid cluster in Metarhizium spp. originated from an HGT event.
Conclusions
Several unknown BGCs are described, and aspects of their organization, regulation and origin are discussed, providing further support for the impact of SM on the Metarhizium genus lifestyle and infection process.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-016-3067-6) contains supplementary material, which is available to authorized users.
Keywords: Metarhizium spp, Secondary metabolite biosynthetic gene clusters, Infection process, Transcriptome analysis, Biological control, Cattle tick
Background
The genus Metarhizium comprises entomopathogenic fungi that have been employed for the biological control of crop plagues and vector-borne diseases since these species were first described [1]. The wide range of arthropod hosts infected by Metarhizium spp. has resulted in a need to better understand the infection process and to improve its modulation for biocontrol. Metarhizium spp. are models for host-pathogen interaction studies and virulence factor discovery [2–4] as well as for the development of potential novel applications [5–7]. Additionally, this genus comprises unique evolutionary traits, harboring well-characterized transitional species with varying degrees of host specificity. Some species are host-specialists (M. acridum and M. album), some demonstrate an intermediate host range (M. guizhouense and M. majus) and some are host-generalists (M. anisopliae, M. robertsii, and M. brunneum) (Table 1) [8]. Comparative genomic analyses have suggested that generalists evolved from specialists via transitional species with intermediate host ranges [8].
Table 1.
Host range of Metarhizium species
| Metarhizium species | Host range | Hosts |
|---|---|---|
| Metarhizium album | Specialist | Hemiptera; |
| Metarhizium acridum | Specialist | Orthoptera; |
| Metarhizium majus | Intermediate | Coleoptera and Lepidoptera; |
| Metarhizium guizhouense | Intermediate | Coleoptera and Lepidoptera; |
| Metarhizium brunneum | Generalist | More than seven orders of insects, as well as arachnids; |
| Metarhizium robertsii | Generalist | More than seven orders of insects, as well as arachnids; |
| Metarhizium anisopliae | Generalist | More than seven orders of insects, as well as arachnids; |
Metarhizium spp. infection begins when fungal conidia adhere to the surface of a suitable host. Host cuticle composition and fungal characteristics determine the host specificity [8, 9]. Under appropriate humidity and temperature conditions, conidia germination gives rise to the germ-tube and to a specialized infection structure, the appressorium. This structure assists the fungus in breaching the host cuticle to reach the hemocoel, where host colonization and sepsis commence, ultimately resulting in host death. During infection, several hydrolytic enzymes, such as chitinases, proteases and lipases, act as important virulence determinants [2]. These enzymes not only facilitate nutritional processes but also morphogenesis and autolytic processes in fungal development [10]. In addition to hydrolytic enzymes, secondary metabolites (SMs) are also produced by Metarhizium to overcome and kill the host [11].
SMs are small molecules with diverse biological activities and applications. Numerous SMs of interest have been isolated from entomopathogenic fungi in recent years (reviewed by [11]), such as beauvericin from Beauveria bassiana, which possesses insecticidal, antifungal, antibacterial and potent cytotoxic activities against human cells [11]. Cordycepin, an SM product from Cordyceps militaris, exhibits apoptotic and anti-proliferative activities against cancer cells [12], and hirsutellic acid A from Hirsutella spp. demonstrates activity against the malarial parasite Plasmodium falciparum [13]. Additionally, many subclasses of destruxins, which exert insecticide, antiviral and cytotoxic effects, have been isolated from Metarhizium spp. [11]. The various biotechnological applications of such compounds have aroused great interest in Metarhizium spp. as sources of novel control drugs [14, 15].
In fungi, genes for the biosynthesis of SMs are often arranged in clusters and are co-regulated. These biosynthetic gene clusters (BGCs) usually contain backbone genes such as polyketide synthases (PKS), non-ribosomal peptide synthetases (NRPS), hybrids (PKS-NRPS), terpene cyclases (TCs) and prenyltransferases (PTs) as well as adjacent genes that assist in regulation, transport and metabolite trimming [15, 16]. Massive sequence data availability, combined with tools to predict BGCs, have revealed that fungal genomes encode far greater numbers of SMs than previously estimated. This diversity of silent metabolites, which are not accessible under normal laboratory culture conditions, reflects habitat complexity [17] and represents great scientific and commercial opportunities [14]. Furthermore, these BGCs are also evolutionarily interesting. It has been proposed that clustering favors the survival of SM genes, and BGCs partially depend on horizontal gene transfer (HGT) for their dispersal [18]. In fact, several horizontally transferred BGCs have been described. For example, the sterigmatocystin cluster was transferred from Aspergillus spp. to Podospora anserina [19], and the homologous ACE1 gene cluster in Aspergillus clavatus originated via HGT from a donor related to Magnaporthe spp. [20]. HGT events for BGCs have also been linked with the success of emergent pathogens, such as Mycosphaerella populorum, which acquired a chaetoglobosin-like cluster from an unknown donor that is potentially involved in poplar tree infection [21].
Although a vast array of SM compounds has been isolated from Metarhizium species [11, 22–25], few BGCs have been examined at the gene level employing functional mutants [26–30]. The deletion of five genes, specifically a gene from the serinocyclin BGC (synthesis of cyclic peptides in conidia), a gene from the NG39x BGC (synthesis of mutagenic fusarin-like compounds NG391 and NG393), a gene from metachelin BGC (synthesis of siderophore) and two PKS genes (MrPKs1 and MrPKs2), did not significantly affect virulence. Until now, only the deletion of the destruxin and siderophore ferricrocin synthesis genes has been shown to affect virulence. An M. robertsii mutant lacking destruxin demonstrated reduced infection efficiency against Bombyx mori and Locusta migratoria [28], and an M. robertsii mutant lacking the siderophore ferricrocin exhibited reduced virulence in Spodoptera exigua [30]. Furthermore, it has been suggested that the retention of the destruxin BGC is evolutionarily related to the host range, given that host-specialists do not possesses a fully functional destruxin synthesis cluster [28]. This was also suggested in reports predicting BGCs using bioinformatics tools, which indicated that host-specialist species of Metarhizium have a different set of BGCs than host-generalist species [8, 31]. In general, these results suggest that the presence of a different range of SMs may be related to the narrowed virulence and specialization of host-specialist species.
Therefore, it is reasonable to assume that many BGCs, including clusters that are not expressed under normal laboratory conditions, participate in the Metarhizium spp. infection process. However, the activation of silent clusters, and functional gene analysis methods are laborious and time-consuming. Alternatively, to investigate genes related to infection in a genome-wide strategy, in this work we have deepened the existing knowledge of SMs in the genus Metarhizium. We have performed an exhaustive survey and description of BGCs in M. anisopliae and assessed the conservation of BGCs and related genes within the Metarhizium genus. To validate some of these BGCs, we analyzed RNA-seq data from M. anisopliae grown on cattle-tick (Rhipicephalus microplus) cuticles to evaluate their differential expression. In addition, we selected three up-regulated BGCs (Ma-PKS1, MaNRPS-PKS2, and MaTERP1) and applied phylogeny and comparative genomic analyses to predict their metabolic pathways and evolutionary history.
Methods
Genomes and RNA-seq data
All fungal genomes were downloaded from the NCBI Genome Database, and the descriptions and accession numbers are displayed in Additional file 1. For RNA-seq experiments, briefly, cattle tick R. microplus cuticles were sterilized and used as the sole carbon source for M. anisopliae E6 growth. Spore suspensions (5 × 106 spores per ml) were used to inoculate the cuticles by immersion for 30 s. The inoculated cuticles were dispersed over 1 % water agar plates and maintained for 48 h (48hI) and 144 h (144hI) at 28 °C. As a control, the fungus was cultivated in 100 mL of liquid Cove’s complete medium (MCc) for 48 h (48hC) at 28 °C. The detailed RNA-seq experimental procedure, sequencing and data management have been previously described [32], and sequencing data are available under accession number PRJNA257269.
Normalization and expression analysis
For expression analysis, we considered RPKM values > = 2 to indicate detectable expression. Genes were considered differentially expressed if the corresponding log2-fold change ratios were > = 1 or = < −1, with a 5 % false discovery rate (FDR < =0.05) [32].
BGCs and related gene predictions
Putative BGCs in the M. anisopliae genome were identified with the antiSMASH 3.0 [33], SMURF [34], and SMIPS [35] algorithms and previous results from the literature were also examined [31, 36, 37]. The borders of each cluster were initially detected based on the antiSMASH 3.0 prediction [33] and subsequently confirmed with CASSIS, which assumes the existence of common regulatory patterns in cluster promoters for cluster delimitation [35]. The conservation of predicted clusters among Metarhizium spp. was assessed with MultiGeneBlast [38], based primarily on backbone gene conservation (e-value < 1 × 10−5, query coverage > 60 % and identity > 60 %). Afterward, BLASTP (non-redundant protein sequences database, recovering the best 500 hits) was used to search and curate orthologous clusters among other filamentous fungi genomes, and to select putative orthologous backbone genes for the phylogenetic analysis of MaPKS1, MaNRPS-PKS2, and MaTERP1 (e-value < 1 × 10−5, query coverage > 50 and identity > 45 %, ignoring more than one sequence under the same species) [39]. To further confirm that the collected genes were truly orthologous, the backbone genes of MaPKS1, MaNRPS-PKS2, and MaTERP1 were blasted against the MetaPhOrs database [40], and complete genomes were subjected to OrthoMCL curation, a Markov-based algorithm (clustering thresholds: e-value < 1e-05 and identity > = 30 %) (Additional file 2) [41]. Forty species with complete annotated genomes representing each taxon shown in this study were selected for OrthoMCL analysis (Additional file 1). Additionally, several fungal genomes from the Clavicipitaceae family were deposited at NCBI as raw or incomplete assemblies from projects that generally employed whole genome shotgun (WGS) strategies [42–44]. The contents of several unannotated genomes (Epichlöe festucae, Balasia obtecta, Epichlöe baconii, Pochonia chlamydosporia, Periglandula ipomoeae, Claviceps fusiformis, Aciculosporium take, Epichlöe sylvatica, Neotyphodium gansuense, Hypocrella siamensis and Atkinsonella hypoxylon) were accessed using BLASTN against the WGS database and MultiGeneBlast. The putative orthologous genes were annotated with FGENESH (gene-finding parameters for Metarhizium spp. or Claviceps spp.) and aligned with the backbone genes from M. anisopliae [45]. Genes that satisfied the previously fixed cutoffs were added to the phylogenetic analysis and cluster curation. Moreover, known global regulators that affect SM biosynthesis were also identified in M. anisopliae and their expression was evaluated.
Phylogenetic analysis
A special procedure was adopted for the MaPKS1 phylogeny: given that the phylogeny of PKS genes can be chaotic, particularly for ortholog definition as it is difficult to differentiate orthologous from non-orthologous genes, we generated a tree that included collected entries comprising putative orthologs of MaPKS1, all PKS from M. anisopliae E6, and all characterized PKS from MIBiG, a database of characterized biosynthetic gene clusters [46]. An amino acid alignment was built using PRANK, and the evolutionary history was inferred using the Maximum Parsimony method with 1,000 bootstrap replicates and MrBayes [47–49] for 107 generations (sampled every 100 steps), applying an average standard deviation of split frequencies < 0.01 as the convergence criterion. Parameters and trees obtained through the Bayesian approach were summarized by applying a 25 % burn-in (these trees are contained in Additional file 3) [50, 51].
After this confirmation, amino acid (BLAST collected entries for MaPKS1 and MaNRPS-PKS2) and nucleotide alignments (tef1 gene, detailed below) were built and trimmed with GUIDANCE using PRANK as an MSA algorithm with default parameters [50, 52] to generate the phylogenetic analysis (these alignments are contained in Additional files 4, 5, 6). The best-fit evolutionary model for each alignment was assessed using Prottest 3.4 [53] for proteins and jmodeltest-2.1.9 [54] for nucleotides (Additional file 7). Phylogenetic trees were constructed using PhyML 3.1 [55] with 100 bootstrap replicates, and MrBayes with the same parameters as described above.
To generate the phylogeny of the putative helvolic acid cluster (MaTERP1; possibly obtained via HGT), protein sequences for each orthologous gene (excluding genes involved in fusidic acid biosynthesis; for a detailed explanation see Additional file 8) that belong to this cluster were recovered and processed as described above. An matrix representation parsimony (MRP) supertree was constructed based on the inferred trees with CLANN 3.1.3 with 100 bootstrap replicates [56]. Alternatively, the alignments were concatenated into a supermatrix using SeaView (supermatrix alignment is contained in Additional file 9) [57]. Evolutionary model analysis and phylogenetic inference for this supermatrix followed the procedures described above, and the tree was inferred using PhyML 3.1 (run with 1,000 bootstrap replicates). The cluster trees were rooted at species from the Aspergillus genus, and the topology was similar when the trees were unrooted or rooted at the midpoint. Both the supertree and the supermatrix tree were compared with the species tree to highlight possible HGT events implicated in the evolutionary history of this BGC [21, 58]. The species tree was based on the translation elongation factor 1-alpha (tef1) barcode and rooted at fungal species that did not belong to the Pezizomycotina class. The tef1 gene is the current barcode pattern for species delimitation and classification in the Metarhizium genus and Clavicipitaceae family [59, 60]. The inferred species tree was analyzed manually for conflicts and incongruities with the current fungi and Clavicipitaceae evolutionary history [61, 62].
Results
BGC predictions and boundaries delimitation
The genome survey predicted 73 putative BGCs, comprising to twenty-two PKS, thirteen NRPS, nine terpenes (TERP), seven NRPS-PKS, three indoles (IND), two IND-NRPS, 1 IND-TERP, 1 TERP-PKS, 1 siderophore (SID), and fourteen BGCs, classified by antiSMASH as “OTHER”, a generic class of clusters encoding unusual BGCs (Additional file 10). Our survey found more BGCs than any other survey previously published for Metarhizium spp. [8, 31].
To refine the BGC boundaries, the predicted backbone genes were subjected to CASSIS, which assumes the presence of common regulatory patterns among genes from the same cluster. CASSIS was chosen because other tools only predict backbone genes (e.g., SMIPS), ignoring accessory genes, or overrating cluster boundaries (e.g., SMURF and antiSMASH). Based on the CASSIS prediction, 49 BGC boundaries were reassigned when compared to the previous antiSMASH prediction (Additional file 11; BGCs with reassigned boundaries are marked). However, the CASSIS results must be carefully analyzed, because other layers of regulation may be present, and some accessory genes may not exhibit the same regulation patterns found in the rest of the cluster [35].
Conservation of BGCs in the Metarhizium genus and host range
The majority of BGCs (> 83 %) found in M. anisopliae are well conserved in host-generalists (M. robertsii and M. brunneum) and intermediate-host-range species (M. guizhouense and M. majus); including M. robertsii ARSEF23 (69 conserved clusters [cc]), M. brunneum ARSEF3297 (70 cc), M. guizhouense ARSEF977 (64 cc) and M. majus ARSEF297 (61 cc) (Additional file 10). Some SM clusters were also found to be conserved in host-specialist species, such as M. acridum CQMa102 (35 cc) and M. album ARSEF1941 (30 cc); however, this conservation was present to a lesser degree (Additional file 10).
Comparative genomic analysis of BGCs and phylogeny
Comparative genomic analysis was employed to clarify the predictable final products of the assigned BGCs, integrating these data with BGCs previously characterized in Metarhizium spp. These comparisons revealed certain interesting clusters, which are listed in Additional file 10. MaPKS2 (MANI_004781) was predicted to be responsible for the biosynthesis of aurovertins, which are metabolites that have already been isolated from Metarhizium spp. cultures [63] but lack a characterized BGC. MaPKS2 exhibited 42–77 % identity with the BGC responsible for aurovertin biosynthesis in C. arbuscula (Additional file 12) [64]. MaTERP2 (MANI_002110) was assigned as a lanosterol cyclase, exhibiting 79 % identity with the partially characterized lanosterol cyclase from Trichoderma harzianum [65]. The final product of MaIND-NRPS1 (MANI_029655) was predicted to be an elymoclavine/ergovaline-related compound. This generic classification took into account the conservation between MaIND-NRPS1 and both elymoclavine and ergovaline characterized clusters (Additional file 12). MaIND-NRPS1 (MANI_029655) exhibited 57–77 % identity with a portion of the elymoclavine BGC from C. fusiformis [66]. Furthermore, the NRPS gene (MANI_029666) internal to the cluster exhibited 59 % identity with the biosynthetic ergovaline NRPS from Neotyphodium lolii [67] (Additional file 12). A generic classification was also applied for MaIND-TERP1 (MANI_011022) and MaNRPS-PKS3 (MANI_023437), which were designated as a terpendole E/lolitrem-related compound, and xenolozoyenone-related compound, respectively. MaIND-TERP1 exhibited 60–75 % identity with the biosynthetic terpendole E BGC characterized in Chaunopycnis (Tolypocladium) alba [68], and 59–77 % identity with the biosynthetic lolitrem BGC characterized in E. festucae [69, 70] (Additional file 12). However, MaIND-TERP1 cluster contains additional genes that are not conserved in the terpendole E and lolitrem clusters; these genes could potentially participate in the biosynthesis of the resulting terpendole E/lolitrem-related compound. MaNRPS-PKS3 exhibited 31–50 % identity with the xenolozoyenone BGC characterized in Glarea lozoyensis [71] (Additional file 12). Despite the low identity, both clusters were phylogenetically related as determined by Yue and coworkers (2015), further supporting our proposed assignment.
For the MaNRPS-PKS2 (MANI_018878), MaTERP1 (MANI_010527/MANI_010530/MANI_010531/MANI_010532), and MaPKS1 (MANI_014762) clusters, we deepened the comparative genomic analysis by performing a phylogeny. This phylogeny was performed because these three clusters are up-regulated during early infection (48hC x 48hI; following section), have a narrow cluster distribution among fungi (differing from MaTERP2, for example, which is also up-regulated in early infection, but is ubiquitous among Ascomycota), may have originated in Metarhizium spp. via HGT events and are located in singular genomic regions.
MaNRPS-PKS2 (MANI_018878) matched the characterized pseurotin BGC from A. fumigatus with considerable identity (63–81 %) (Fig. 1a and b) [72]. Furthermore, the search for orthologs and phylogenetic analysis revealed a restricted cluster distribution among filamentous fungi, with conservation observed only in host-generalist Metarhizium spp. (Fig. 1a). In A. fumigatus, this BGC is located in a singular genomic region with intertwined biosynthetic genes involved in the formation of fumitremorgin, fumagillin, and pseurotin. This region is under the control of the global regulator LaeA, and fumagillin and pseurotin are co-regulated by a supercluster-embedded regulatory gene [36]. In the genus Metarhizium, this BGC appears to have been horizontally acquired from an unknown donor, and fumitremorgin and fumagillin backbone genes are absent, although it is likely that some fumagillin accessory genes were also horizontally acquired (Fig. 2a). These accessory genes are strongly up-regulated, similar to the remaining pseurotin cluster (Table 2), indicating their likely participation in compound biosynthesis and leading us to classify the final product of this cluster as a pseurotin-related compound. Although CASSIS was unable to detect similar regulatory regions in the promoters of the pseurotin-related compound BGC, the embedded Zn(II) 2-Cys(6) transcription factor highlighted in A. fumigatus is conserved in M. anisopliae (MANI_018928; 34 % identity). This transcription factor can regulate the entire cluster and genes in the vicinity, analogous to the regulation that occurs in A. fumigatus. In addition, there is one other up-regulated backbone gene (MaPKS14; MANI_018879; Additional file 11) that is located near the pseurotin-related compound in Metarhizium, indicating possible co-regulation (Fig. 2a). Similarly, a pseurotin-related cluster located near an orthologous for MaPKS1 (although MaPKS1 is located in another genomic region in Metarhizium spp. genomes) was detected in Tolypocladium ophioglossoides (Fig. 2a). These results suggest that pseurotin and pseurotin-related compound clusters can be embedded in superclusters in different vicinities and configurations. Furthermore, these differences in pseurotin cluster location favor the proposed explanation that this cluster is located in highly variable regions in different genomes.
Fig. 1.
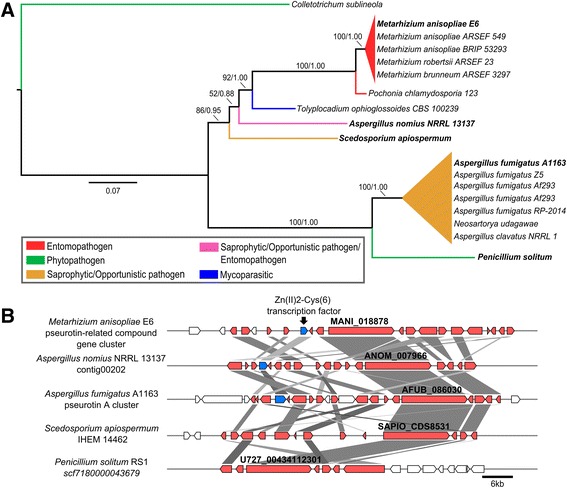
Pseurotin-related compound BGC (MaNRPS-PKS2). a Phylogenetic analysis was performed using Maximum-likelihood and Bayesian methods, based on the pseurotin-related backbone gene and orthologous sequences exhibited by several fungi. The orthologous sequences were classified according to fungal lifestyle trait, represented by different colors. The Bayesian tree is displayed, and branch support values (bootstrap proportions and Bayesian posterior probability) are associated with nodes. The Bayesian inference ran for 9,997,000 generations. Species in bold in (a) were used for the cluster conservation analysis presented in (b). b Some genes from M. anisopliae MaNRPS-PKS2 BGC resembled the characterized pseurotin BGC from A. fumigatus (34–81 % identity) and putative BGCs from A. nomius (49–85 % identity), S. apiospermum (63–84 % identity) and P. solitum (59–81 % identity). The M. anisopliae Zn(II) 2-Cys(6) transcription factor resembles the embedded transcription factor found in A. fumigatus (34 % identity), and the putative transcription factor from A. nomius (49 % identity). Interestingly, S. apiospermum and P. solitum do not have orthologs for this transcription factor. Orthologous genes were assigned the same color; white boxes represent genes that were not predicted to be part of M. anisopliae cluster, and blue boxes represent the conserved Zn(II)2-Cys(6) transcription factor
Fig. 2.
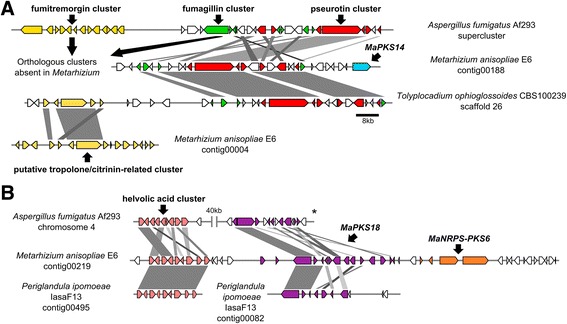
Conservation of supercluster regions in several species. a Comparison of the fumagillin/pseurotin supercluster region among M. anisopliae, A. fumigatus and T. ophioglossoides. The backbone gene from fumagillin (green) is absent in M. anisopliae and T. ophioglossoides, but some accessory genes are present and intertwined with the well-conserved pseurotin BGC (red). These accessory genes appear to participate in metabolite biosynthesis; therefore, the final product of this cluster was speculated to be a pseurotin-related compound. Upstream of the pseurotin-related BGC, the fumitremorgin cluster (yellow) is present in A. fumigatus, but absent in M. anisopliae, and there is a putative tropolone/citrinin-related BGC at this location in T. ophioglossoides. The tropolone/citrinin-related BGC has orthologous sequences (MaPKS1) in M. anisopliae, although they are located in a different genomic region. The MaPKS14 (light-blue) BGC is located downstream the pseurotin-related BGC only in M. anisopliae. b Comparison of a putative supercluster region in M. anisopliae, A. fumigatus, A. niger, and P. ipomoeae. Three BGCs (helvolic acid, MaPKS18, and MaNRPS-PKS6) were assigned to this M. anisopliae region. The helvolic acid (pink) and MaPKS18 (purple) clusters appear to be co-regulated. Additionally, both are conserved in A. fumigatus and P. ipomoeae, although the BGCs are distantly located in chromosome 4 in A. fumigatus. * This locus was inverted to fit in the figure
Table 2.
Expression profiling of the M. anisopliae cluster related to the biosynthesis of a pseurotin-related compound
| NCBI gene locus ID | Expression (RPKM) | Differential expression (log2-fold change) | Gene product | |||
|---|---|---|---|---|---|---|
| 48hC | 48hI | 144hI | 48hCx48hI | 48hIx144hI | ||
| MANI_029058 | 0.00 | 0.34 | 0.34 | NA | NA | Hypothetical protein |
| MANI_018942 | 1.75 | 0.00 | 5.75 | NA | NA | Hypothetical protein |
| MANI_029071 | 0.00 | 209.91 | 134.67 | 9.77 | ND | Integral membrane protein |
| MANI_018916 | 0.55 | 492.48 | 212.39 | 9.64 | −1.28 | Cytochrome P450 |
| MANI_018955 | 1.03 | 37.39 | 59.92 | 5.02 | ND | Hypothetical protein |
| MANI_018958 | 2.21 | 355.01 | 165.40 | 7.30 | −1.10 | Phytanoyl-CoA dioxygenase |
| MANI_018941 | 1.92 | 422.89 | 226.54 | 7.76 | ND | Phytanoyl-CoA dioxygenase |
| MANI_018928 | 0.00 | 63.24 | 30.65 | 8.67 | −1.06 | C6 finger transcription factor |
| MANI_018959 | 1.84 | 986.40 | 323.46 | 8.90 | −1.57 | Hypothetical protein |
| MANI_018934 | 0.65 | 268.46 | 62.03 | 8.51 | −2.11 | Alpha/beta hydrolase |
| MANI_018878 | 0.44 | 736.13 | 182.13 | 10.78 | −1.96 | Hybrid NRPS/PKS enzyme |
| MANI_018952 | 0.00 | 1129.61 | 347.42 | 12.26 | −1.68 | Methyltransferase |
| MANI_029062 | 0.00 | 286.33 | 99.09 | 11.23 | −1.50 | Cytochrome P450 |
| MANI_120428 | 31.50 | 117.64 | 50.96 | 2.02 | −1.25 | Methionine aminopeptidase |
| MANI_029068 | 109.68 | 117.21 | 86.21 | ND | ND | Methionine aminopeptidase |
| MANI_018894 | 0.00 | 131.20 | 48.31 | 10.56 | −1.38 | Acetate-CoA ligase |
| MANI_111428 | 0.98 | 599.02 | 205.75 | 9.29 | −1.51 | Steroid monooxygenase |
| MANI_018962 | 0.00 | 269.62 | 67.44 | 9.55 | −1.94 | Hypothetical protein |
| MANI_018945 | 0.00 | 15.06 | 4.25 | NA | NA | Methyltransferase |
| MANI_018943 | 0.00 | 296.14 | 80.09 | 10.19 | −1.81 | Glutathione S-transferase |
| MANI_018919 | 0.74 | 889.71 | 213.01 | 10.07 | −2.08 | O-methyltransferase |
The relative changes in expression levels were estimated at 48 h for the control condition (hC) and both 48 and 144 h for infection conditions (hI). NA Not Available, ND No Difference
Given that supercluster arrangements are misleading when performing BGC predictions using search algorithms [36], we identified another putative supercluster in intermediate- and generalist-host-range Metarhizium spp. This putative supercluster is located in M. anisopliae contig 219, and is comprised of three clusters: MaPKS18 (MANI_010451), MaTERP1 (putatively enrolled in helvolic acid biosynthesis, as detailed below) and MaNRPS-PKS6 (MANI_010456/ MANI_121659) (Fig. 2b). This sequence region misleads the antiSMASH prediction, being the BGCs delimited by CASSIS and previous results from the literature. Furthermore, there is an apparent co-regulation of both the MaPKS18 and putative helvolic acid BGCs, which are up-regulated in early infection (48hC x 48hI) and down-regulated in late infection (48hI x 144hI), supporting the supercluster hypothesis (Table 3; Additional file 11). The putative helvolic acid (MaTERP1) and MaPKS18 BGCs have orthologs in A. fumigatus and P. ipomoeae (Fig. 2b). In A. fumigatus Af293, both clusters are located in the same chromosome and are separated by 40 kb, but the quality of the P. ipomoeae IasaF13 genome did not permit a synteny comparison. This set of results reinforces the notion that BGCs are located in rapidly evolving genomic regions and suggests that superclusters can be widespread in multiple fungal genomes.
Table 3.
Expression profiling of the M. anisopliae cluster related to the biosynthesis of a helvolic acid compound
| NCBI gene locus ID | Expression (RPKM) | Differential expression (log2-fold change) | Gene product | |||
|---|---|---|---|---|---|---|
| 48hC | 48hI | 144hI | 48hCx48hI | 48hIx144hI | ||
| MANI_010527 | 0.54 | 13.59 | 2.67 | 4.51 | −2.27 | Cytochrome P450 |
| MANI_010536 | 8.44 | 36.91 | 1.48 | 2.22 | −4.50 | Transferase family protein |
| MANI_010512 | 4.59 | 25.08 | 0.42 | 2.56 | −5.58 | FAD binding domain-containing |
| MANI_010537 | 3.03 | 30.78 | 4.47 | 3.42 | −2.72 | Transferase family protein |
| MANI_010532 | 0.00 | 12.57 | 0.91 | 6.73 | −3.60 | Cytochrome P450 |
| MANI_010594 | 3.34 | 25.10 | 6.37 | 2.94 | −1.90 | 3-oxoacyl-reductase 1 |
| MANI_010495 | 9.06 | 87.20 | 9.34 | 3.44 | −3.17 | Squalene-hopene-cyclase |
| MANI_010530 | 11.58 | 48.40 | 11.28 | 2.21 | −2.09 | Cytochrome P450 |
| MANI_010531 | 84.51 | 57.83 | 17.62 | NA | −1.67 | Cytochrome P450 |
The relative changes in expression levels were estimated at 48 h for the control condition (hC) and both 48 and 144 h for infection conditions (hI). NA Not Available, ND No Difference
The putative helvolic acid BGC (MaTERP1) showed considerable identity (41–65 %) with the helvolic acid cluster from A. fumigatus [37] (Fig. 3; Table 3). The helvolic acid cluster is organized around the prostadienol synthase gene AFU4G14770 in A. fumigatus. This cluster is unique, containing four backbone paralogous genes. As already suggested, this cluster evolved by gene duplication and differentiation from an ancestral monooxygenase, a transferase and two dehydrogenases [37]. This configuration is also observed in M. anisopliae, with four backbone genes (MANI_010527/MANI_010530/MANI_010531/MANI_010532). Furthermore, in addition to the backbone genes, all accessory genes highlighted in A. fumigatus are conserved in M. anisopliae (Fig. 3). Additionally, the isolation of helvolic acid from Metarhizium cultures supports the suggestion that MaTERP1 is responsible for metabolic biosynthesis [25].
Fig. 3.
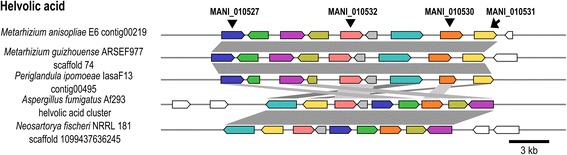
Putative helvolic acid (MaTERP1) conservation and synteny. The MaTERP1 cluster from M. anisopliae resembled the characterized helvolic acid cluster from A. fumigatus (41–65 % identity), and putative BGCs from N. fischeri (41–64 % identity) and P. ipomoeae (80–90 % identity). Notably, the BGC found in P. ipomoeae exhibits a strong synteny with clusters from the Metarhizium genus (e.g., M. anisopliae and M. guizhouense). The locus tags for the four backbone genes are given
Additionally, the phylogenetic analysis revealed a narrow cluster distribution (Fig. 4). This BGC is only found in intermediate- and generalist-host range Metarhizium spp., P. ipomoeae (Hypocreales order), and species from the Aspergillus genus (Eurotiales order), and is absent in Metarhizium host-specialist species and other members of the Clavicipitaceae family. The phylogenetic trees presented in this work, the strong gene conservation and uncommon cluster origin/formation, suggesting that this cluster may have been originated in Metarhizium species via an HGT event from a donor species closely related to the Eurotiales order (Fig. 4). This hypothesis is supported by the large evolutionary distance between Eurotiomycetes and Sordariomycetes (which diverged approximately 400 million years ago [MyA]) [73] and by the absence of a complete helvolic acid BGC in other species of the Hypocreales order (Fig. 3; Additional file 4).
Fig. 4.
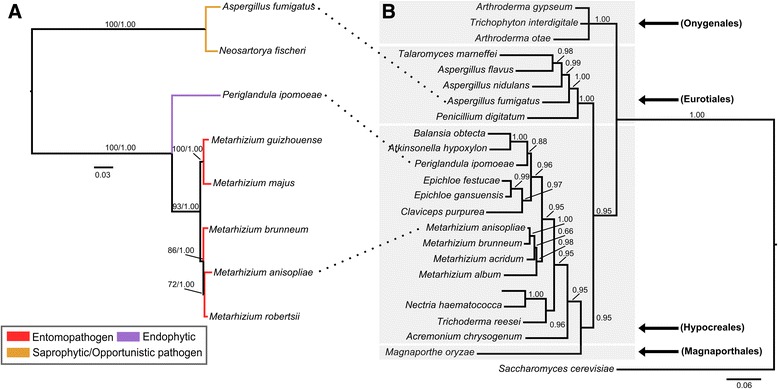
Species and helvolic acid BGC (MaTERP1) phylogeny. a Supermatrix tree of nine genes (MANI_010495/ MANI_010512/ MANI_010527/ MANI_010530/ MANI_010531/ MANI_010532/ MANI_010536/ MANI_010537/ MANI_010594) involved in helvolic acid biosynthesis. This supermatrix tree resembles the generated supertree (Additional file 13). The orthologous sequences were classified according to fungal lifestyle trait, represented by different colors. The Bayesian tree is displayed, and branch support values (bootstrap proportions and Bayesian posterior probability) are associated with nodes. The Bayesian inference ran for 1,000,000 generations. The cluster tree was compared with the species tree presented in (b). Note that the helvolic acid BGC is present in few Eurotiales and Hypocreales species. b The phylogeny of tef1, a barcode gene, showing established species relationships. Branch support values (Bayesian posterior probability) are associated with nodes. The Bayesian inference ran for 43,000 generations
Another contribution of functional genomics coupled with phylogenetic analysis was the partial elucidation of the first steps in the biosynthesis of up-regulated MaPKS1. The MaPKS1 backbone gene (MANI_014762) is orthologous to several characterized backbone genes: tropolone/stipitatic acid [74], citrinin [75], phomenoic acid [76] and azaphilone [77]. These orthologs suggest that a similar biosynthetic route is partially shared between MaPKS1 and these characterized metabolites (Fig. 5a e b). The conserved genes between MaPKS1 and the tropolone/stipitatic acid route are involved in the first steps of metabolic biosynthesis. The stipitatic acid backbone gene tropA exhibits 46 % identity with the MaPKS1 backbone gene (MANI_014762), tropB exhibits 47 % identity with MANI_014847, tropC exhibits 60 % identity with MANI_112407 and tropD exhibits 64 % identity with MANI_014887 (Fig. 5c). Similarly, two genes conserved between MaPKS1 and the citrinin biosynthetic route are also involved in the first steps of metabolic biosynthesis. The citrinin backbone gene CitS exhibits 45 % identity with the MaPKS1 backbone gene (MANI_014762), and mrl2 exhibits 37 % identity with MANI_014887, although mrl1 is absent (Fig. 5c). The potential conservation of these first metabolic steps supports the hypothesis that the final product of this BGC has, at minimum, the same biosynthetic origin and is related to tropolones and citrinins. However, the CASSIS prediction delineates a BGC comprised of 15 genes (several genes are not conserved in the tropolone or citrinin routes), leading us to classify the product of this cluster as a potential and generic tropolone/citrinin-related compound.
Fig. 5.
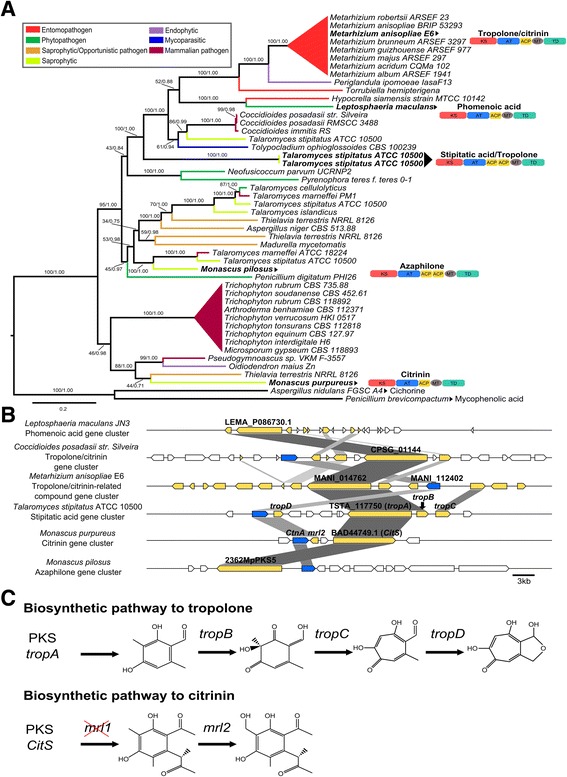
Tropolone/citrinin-related compound BGC (MaPKS1). a Phylogenetic analysis was performed using Maximum-likelihood and Bayesian methods, based on the tropolone/citrinin-related backbone gene and orthologous sequences in several fungi. Additionally, two PKS outgroup sequences were added: cichorine (Aspergillus nidulans FGSC A4) and mycophenolic acid (Penicillium brevicompactum). The orthologous sequences were classified according to fungal lifestyle trait, represented by different colors. The Bayesian tree is displayed, and branch support values (bootstrap proportions and Bayesian posterior probability) are associated with nodes. The Bayesian inference ran for 120,000 generations. Species in bold in (a) also have their domain organization shown with abbreviations (KS: Keto-synthase; AT: Acyltransferase; ACP: Acyl carrier protein; MT: Methyltransferase O- or C-; TD: Thioester reductase), and were used for the cluster conservation analysis presented in b. These clusters have characterized or partially characterized biosynthetic routes. b Some genes from M. anisopliae MaPKS1 BGC resembled the characterized stipitatic acid (tropolone) BGC from T. stipitatus and the citrinin BGC from M. purpureus. These conserved genes are involved in the first steps of the biosynthesis of their compound, as described in c. Note that the mrl1 gene of the citrinin biosynthetic pathway is absent in M. anisopliae. Additionally, the gene MANI_112402 resembles the ctnA citrinin regulator from M. purpureus (59 % identity), and putative transcription factors from C. posadasii, T. stipitatus, M. purpureus and M. pilosus as demonstrated in (b). Orthologous genes were assigned the same color; white boxes represent genes that are not predicted to be part of M. anisopliae cluster; and blue boxes represent the conserved transcription factor
Our analysis also suggested that the self-transcription factor (MANI_112402) is involved in MaPKS1 regulation (Table 4). This gene exhibits strong expression (>6-fold difference with P < 0.05) and 59 % identity with the transcription factor ctnA from the M. purpureus citrinin cluster (Fig. 5b). In M. purpureus, the deletion of ctnA caused a large decrease in citrinin production [78]. Moreover, a similar gene is also conserved in the Coccidioides posadasii putative tropolone/citrinin BGC, in the T. stipitatus tropolone BGC, and in the Monascus pilosus azaphilone BGC, suggesting a similar, widespread regulatory strategy among these BGCs (Fig. 5b; genes marked in blue). It would be simple to characterize this BGC in Metarhizium by constructing a knockout strain for this gene.
Table 4.
Expression profiling of the M. anisopliae cluster related to the biosynthesis of a tropolone/citrinin-related compound
| NCBI gene locus ID | Expression (RPKM) | Differential expression (log2-fold change) | Gene product | |||
|---|---|---|---|---|---|---|
| 48hC | 48hI | 144hI | 48hCx48hI | 48hIx144hI | ||
| MANI_014850 | 37.70 | 8.46 | 12.14 | −1.95 | ND | Mercuric reductase |
| MANI_014940 | 0.00 | 13.82 | 33.93 | ND | 1.36 | Hypothetical protein |
| MANI_014941 | 0.00 | 0.00 | 1.19 | NA | NA | Hypothetical protein |
| MANI_014846 | 0.55 | 3.11 | 0.00 | NA | NA | Major Facilitator Superfamily protein |
| MANI_014815 | 1.74 | 1.05 | 0.71 | NA | NA | Hypothetical protein |
| MANI_028157 | 23.92 | 1.45 | 0.98 | −3.76 | ND | Pantothenate transporter |
| MANI_014957 | 2.35 | 70.02 | 49.98 | 4.76 | ND | YCII-domain protein |
| MANI_014867 | 21.19 | 3.01 | 4.08 | −2.63 | ND | Major Facilitator Superfamily protein |
| MANI_014762 | 0.56 | 29.70 | 34.13 | 5.77 | ND | Polyketide synthase |
| MANI_014847 | 2.36 | 8.57 | 32.88 | ND | 1.92 | Salicylate 1-monooxygenase |
| MANI_014887 | 0.00 | 86.51 | 86.40 | 8.80 | ND | Leucoanthocyanidin Dioxygenase |
| MANI_112402 | 0.00 | 9.21 | 8.88 | 6.25 | ND | Citrinin biosynthesis transcriptional activator CtnR |
| MANI_112407 | 0.57 | 17.13 | 28.22 | 4.71 | ND | Cytochrome P450 |
| MANI_014818 | 1.86 | 23.25 | 28.56 | 3.68 | ND | Siderophore iron transporter |
| MANI_014903 | 0.00 | 56.20 | 81.77 | 8.09 | ND | Nucleoside-diphosphate-sugar epimerase |
The relative changes in expression levels were estimated at 48 h for the control condition (hC) and both 48 and 144 h for infection conditions (hI). NA Not Available, ND No Difference
Expression of BGCs
To validate some of our predictions regarding M. anisopliae BGCs, we conducted RNA-seq analysis using a system mimicking host infection. The fungus was cultured in Cove’s Complete Medium (C: Control condition) and in the presence of tick cuticles (I: Infection condition) for 48 or 144 h, as described in the Materials and Methods. Two pairwise comparisons were performed: 48hC x 48hI (early infection conditions) and 48hI x 144hI (late infection conditions). Nearly half of the predicted SM clusters, specifically 49 (36/73), were expressed (RPKM > = 2) under the analyzed culture conditions, and 20 % (15/73) were up-regulated under early infection conditions (48hI x 48hC), highlighting their potential relevance in the initial steps of infection. Conversely, 7 % (5/73) of the predicted BGCs were down-regulated when comparing 48hI versus 48hC. No up-regulated cluster was detected when 48hI was compared with 144hI; however, 14 BGCs were down-regulated. Moreover, of those 14 down-regulated BGCs, 9 were up-regulated under early infection conditions (Additional file 10).
The 15 BGCs up-regulated under early infection conditions included the already-cited tropolone/citrinin-related compound BGC (MaPKS1) (Table 4), the pseurotin-related compound BGC (MaNRPS-PKS2) (Table 2), the lanosterol cyclase BGC (MaTERP1), and the putative helvolic acid BGC (MaTERP2) (Table 3), in addition to the already-characterized destruxin (MaNRPS1) (Table 5), NG39x (MaNRPS-PKS1) (Table 6) and ferricrocin (MaNRPS9) (Additional file 11) clusters; Additionally, there were eight putative clusters of uncharacterized products (MaNRPS7; MaPKS14; MaPKS17; MaPKS18; MaIND1; MaOTHER8; MaOTHER12; MaOTHER13) (Additional file 11).
Table 5.
Expression profiling of the M. anisopliae cluster related to destruxin biosynthesis
| NCBI gene locus ID | Expression (RPKM) | Differential expression (log2-fold change) | Gene product | |||
|---|---|---|---|---|---|---|
| 48hC | 48hI | 144hI | 48hCx48hI | 48hIx144hI | ||
| MANI_024443 | 2.67 | 138.02 | 14.72 | 5.80 | −3.14 | ABC multidrug transporter |
| MANI_024450 | 2.78 | 88.43 | 8.54 | 5.06 | −3.34 | Hypothetical protein |
| MANI_131037 | 0.00 | 119.36 | 8.75 | 9.88 | −3.71 | Glutamate decarboxylase |
| MANI_130923 | 3.06 | 100.47 | 14.43 | 5.11 | −2.80 | Aldo-keto reductase |
| MANI_024448 | 4.58 | 480.60 | 52.47 | 6.81 | −3.08 | Cytochrome P450 |
| MANI_024437 | 9.41 | 261.50 | 15.65 | 4.92 | −3.96 | Destruxin synthetase |
The relative changes in expression levels were estimated at 48 h for the control condition (hC) and both 48 and 144 h for infection conditions (hI). NA Not Available, ND No Difference
Table 6.
Expression profiling of the M. anisopliae cluster related to NG39x biosynthesis
| NCBI gene locus ID | Expression (RPKM) | Differential expression (log2-fold change) | Gene product | |||
|---|---|---|---|---|---|---|
| 48hC | 48hI | 144hI | 48hCx48hI | 48hIx144hI | ||
| MANI_020814 | 6.73 | 6.92 | 7.65 | ND | ND | Monophenol monooxygenase |
| MANI_020801 | 0.48 | 0.00 | 0.00 | NA | NA | Ankyrin repeat protein |
| MANI_020903 | 0.00 | 1.63 | 0.00 | NA | NA | Eukaryotic aspartyl protease |
| MANI_020934 | 0.00 | 2.15 | 0.00 | NA | NA | eEF-1B gamma subunit-like protein |
| MANI_020906 | 0.00 | 2.23 | 0.00 | NA | NA | Alpha/beta hydrolase |
| MANI_020791 | 0.00 | 14.72 | 0.37 | 9.80 | −5.22 | Hybrid PKS-NRPS protein |
| MANI_020948 | 6.83 | 45.86 | 3.73 | ND | ND | Integral membrane protein |
| MANI_020870 | 4.31 | 19.98 | 2.65 | 2.24 | −2.81 | Major facilitator superfamily protein |
| MANI_121064 | 0.00 | 6.23 | 0.53 | NA | NA | Hypothetical protein |
| MANI_020865 | 2.30 | 13.40 | 1.88 | 2.58 | −2.72 | P450 monooxygenase |
| MANI_020911 | 5.66 | 7.82 | 5.96 | ND | ND | Carboxyl methyltransferase |
| MANI_121074 | 3.99 | 11.79 | 6.53 | 1.65 | ND | Hypothetical protein |
The relative changes in expression levels were estimated at 48 h for the control condition (hC) and both 48 and 144 h for infection conditions (hI). NA Not Available, ND No Difference
The five down-regulated BGCs under early infection conditions included: the xenolozoyenone-related compound (MaNRPS-PKS3) [71] and four putative clusters of uncharacterized products (MaNRPS8; MaNRPS10; MaOTHER1; MaOTHER11) (Additional file 11). The fourteen down-regulated BGCs under late infection conditions included the destruxin (MaNRPS1), serinocyclin (MaNRPS2), NG39x (MaNRPS-PKS1), helvolic acid (MaTERP1), xenolozoyenone-related compound (MaNRPS-PKS3), and pseurotin-related compound (MaNRPS-PKS2) BGCs, in addition to six putative clusters of uncharacterized products (MaNRPS11; MaPKS10; MaPKS14; MaPKS17; MaPKS18; MaTERP9; MaIND1; MaOTHER12) (Tables 2;3;5;6 and Additional file 11). Together, 30 % (22/73) of the predicted BGCs were differentially expressed in at least one of the pairwise comparisons.
Expression of global regulators of fungal traits
Many BGCs contain self-transcription factors integrated into the cluster organization [35, 79], but global regulators of fungal traits also influence the expression of BGCs [80, 81]. These global regulators extend from single transcription factors and histone-modifying enzymes (e.g., CreA, PacC, StuA, nscC, AreA, AreB, MeaB, GcnE and hdaA) to protein complexes (e.g., Velvet, and CCAAT-binding complexes) [80–85]. Our RNA-seq analysis detected the expression of all of the aforementioned global regulators at significant levels (RPKM > = 2) (Table 7).
Table 7.
Expression profiling of the M. anisopliae global regulators of fungal traits
| Global regulator | Metabolism | NCBI gene locus ID | Expression (RPKM) | Differential expression (log2-fold change) | |||
|---|---|---|---|---|---|---|---|
| 48hC | 48hI | 144hI | 48hCx48hI | 48hIx144hI | |||
| Velvet Complex | |||||||
| VeA | Light | MANI_008143 | 30.53 | 71.74 | 45.95 | 1.32 | ND |
| VelB | MANI_013601 | 25.91 | 29.21 | 18.58 | ND | ND | |
| LaeA | MANI_030399 | 3.64 | 6.84 | 4.97 | NA | NA | |
| CCAT-binding complex | |||||||
| HapB | Iron | MANI_001834 | 38.29 | 29.90 | 55.14 | ND | ND |
| HapC | MANI_119196 | 23.11 | 27.92 | 41.43 | ND | ND | |
| HapE | MANI_018323 | 41.24 | 22.68 | 30.49 | ND | ND | |
| HapX | MANI_009173 | 30.15 | 16.19 | 19.09 | ND | ND | |
| PacC | pH | MANI_008549 | 20.37 | 69.23 | 100.18 | 1.88 | ND |
| CreA | Carbon | MANI_015776 | 9.31 | 65.57 | 70.54 | 2.91 | ND |
| AreA | Nitrogen | MANI_016951 | 17.45 | 34.12 | 39.49 | 1.07 | ND |
| AreB | MANI_028387 | 22.46 | 15.89 | 28.44 | ND | ND | |
| MeaB | MANI_019405 | 40.46 | 54.10 | 50.67 | ND | ND | |
| StuA | Sporulation | MANI_024223 | 38.24 | 75.85 | 92.09 | 1.02 | ND |
| GcnE | Chromatin remodeling | MANI_000421 | 13.86 | 13.51 | 15.54 | ND | ND |
| hdaE | MANI_025732 | 17.80 | 17.52 | 14.26 | ND | ND | |
| nsdC | Asexual development | MANI_013461 | 9.74 | 20.40 | 18.76 | 1.19 | ND |
The relative changes in expression levels were estimated at 48 h for the control condition (hC) and both 48 and 144 h for infection conditions (hI). NA Not Available, ND No Difference
Under early infection condition, global regulators linked to carbon (CreA) and nitrogen (AreA) metabolism, pH (PacC), light stimuli (VeA), asexual development (nsdC) and sporulation (StuA) were up-regulated (Table 7). Conversely, the best characterized global regulator, the LaeA methyltransferase, demonstrated lower expression. Additionally, global regulators linked to iron (HapB; HapC; HapE; HapX) and nitrogen (AreB; MeaB) metabolism, light stimuli (VeB), the chromatic remodeling histone acetyltransferase (GcnE) and deacetylase (hdaA), did not exhibit significant changes in gene expression (Table 7). In addition to demonstrating that many of these global regulators are active (a significant amount remain unexplored in the Metarhizium genus), expression analysis provides clues regarding how infection conditions can be regulated and which genes may perform this regulation.
Discussion
Although a vast number of interesting metabolites have been isolated from Metarhizium cultures, little is known regarding how BGCs are organized, expressed and regulated as well as their potential functions during host infection and their evolutionary history. To this end, a deep survey of BGCs was performed in the M. anisopliae genome and combined with a transcriptional profile analysis from an infection model in the economically important cattle-tick R. microplus, which is responsible for a variety of livestock infections and is a promising candidate for biological control by Metarhizium [86–89].
Our survey predicted and delimited 73 BGCs, of which 20 % were up-regulated under early infection conditions (48hC x 48hI), and a subset of these (9 out of 15) were down-regulated under late infection conditions (48hI x 144hI) (Additional file 10). These results point to a drastic change between the metabolic profiles of early and late infection. The Metarhizium infection process is dynamic and may end in arthropod or fungal death [90]. Once the fungus adheres to a host, rapid morphological and transcriptional changes occur, including the expression of several virulence factors [91]. Ment and coworkers (2012) showed that after 3 or 4 days of attachment to a suitable host, Metarhizium kills the host [90], switching to a saprophytic state, and virulence determinant expression is attenuated [2]. Conversely, in an unsuccessful infection scenario, the fungus will exhaust the endogenous spore nutrient reserves at 3 or 4 days post-cuticle adhesion while attempting to circumvent host defenses, resulting in the demise of the pathogen [90]. We suggest that some of the identified up-regulated BGCs participate in the first scenario (successful infection), given that this hypothesis corroborated with the observed strong expression of the destruxin cluster, a well-known virulence factor [92], and ferricrocin [30]. Additionally, we hypothesize that other BGCs are also expressed during early and late infection, such as BGCs for the production of antifungal and antibacterial compounds that help the fungus to circumvent competition with opportunistic and symbiotic microorganisms. These BGCs may be induced by direct interactions between M. anisopliae and other microorganisms, independent of the host interaction. Moreover, our experimental design employing tick cuticles may have blocked the expression of these antibiotic BGCs, as the natural tick gut microbiota was excluded [32]. Indeed, the antibacterial viridicatumtoxin BGC (MaPKS9) proposed by Gibson and coworkers (2014) was silent under our mimicked infection conditions, supporting this notion (Additional file 11) [31].
Some BGCs that were previously characterized in Metarhizium were silent or did not alter their expression under at least one of the tested conditions. This was the case for MaNRPS2 BGC (serinoclycin; MANI_020119), which was down-regulated under late infection conditions, and MaNRPS8 (metachelin; MANI_003049), which was down-regulated under early infection conditions. The MaPKS8 (MrPKS2; MANI_028434), already-cited MaPKS9, (viridicatumtoxin; MANI_003768) and MaPKS20 (MrPKS1; MANI_122426) BGCs did not demonstrate detectable expression. The reduced participation of these BGCs in the infection process is expected in accordance with previous literature [26, 29–31].
Similarly, the up-regulation of the destruxin cluster under early infection conditions was in accordance with the described insecticidal effects and phenotypic analysis of destruxin mutants. Similarly, the up-regulation of the ferricrocin cluster highlights the already-described importance of this siderophore in the Metarhizium lifestyle and infection process [30]. However, while destruxin metabolites directly affect the host defenses [92], the reduced infection resulting from the absence of ferricrocin is linked to delayed germination and alterations in endogenous fungal iron content [30].
Some putative clusters highlighted by the comparative genomic analysis do not appear to affect the virulence of Metarhizium species (Additional files 10 and 11). Such is the case for the putative BGCs for aurovertin (MaPKS2; MANI_004781), elymoclavine/ergovaline-related compound (MaIND-NRPS1; MANI_029655) and terpendole E/lolitrem-related compound (MaIND-TERP1; MANI_011022). Aurovertin metabolites have been isolated from M. anisopliae, P. chlamydosporia, and C. arbuscula [63, 64, 93]. These compounds exhibit potent inhibition of adenosine triphosphate synthase [64], and aurovertin D, which was isolated from P. chlamydosporia, induced the death of the free-living nematode Panagrellus redivivus [93]. The non-expression (RPKM < 2, under the three conditions) of this cluster is intriguing, particularly in light of the reports regarding P. chlamydosporia, a species closely related to the Metarhizium genus. Similarly, elymoclavine, ergovaline, terpendole E and lolitrem are ergot alkaloids produced by fungi from the Claviceps, Epichlöe and Tolypocladium genera, which are closely related to the Metarhizium genus [66, 67]. Ergot alkaloids are potent toxic alkaloids whose intake can lead to several effects ranging from poor weight gain to gangrene and death [67]. Similar to aurovertin, it could be postulated that these metabolites play a role in infection, although our results suggested the opposite. Certainly, the construction of gene knockouts for these BGCs will help to understand their importance in the Metarhizium lifestyle and interactions.
The xenolozoyenone-related compound (MaNRPS-PKS3; MANI_023437) was another cluster that was down-regulated, indicating decreased participation in the infection process. Xenolozoyenone is a pyrrolidinedione-containing compound isolated from G. lozoyensis. Although no biological activity or possible function has been linked to this compound, the backbone gene from this cluster (glpks3-glnrps7) was the first described classical fungal protein-coding operon [71]. The conservation of orthologs of these genes highlights the possibility that protein-coding operons are widespread among fungal genomes and BGCs and represent a new and virtually unexplored level of regulation.
The results of the transcriptomic analysis may also help to redefine the importance of NG39x (MaNRPS-PKS1; MANI_020791) in the infection process. The RNA-seq results suggested that the NG39x BGC might play a role in infection, as demonstrated by the differential expression (> 9.5-fold difference with P < 0.05 for the backbone gene MANI_020791) observed under early infection conditions (Table 6). Although NG39x cluster expression has been observed in vivo [27], a previous study performed RT-PCR analysis to evaluate the transcripts of the NG39x cluster while comparing in vitro fungal growth with growth in infected S. exigua larvae and did not revealed clear differences between the two conditions. It was postulated that the expression of the NG39x cluster is developmentally regulated, given that there was an increase in BGC expression related to biomass augmentation. Additionally, the knockout mutant for the backbone gene responsible for NG39x biosynthesis did not lead to diminished fungal virulence, indicating minor or no participation in the insect infection process [27]. NG39x compounds were recently reported to exert antiproliferative effects in human cell cultures, via a mechanism that involves impairment of the integrity of nucleic acid biosynthesis. However, it has not been established if these compounds directly interact with DNA or RNA or interact with some protein [94]. Therefore, it is possible that the toxicity of NG39x compounds varies according to the host and may exemplify cases of host specificity, which explains the increased expression of this cluster under early infection condition.
We expanded the comparative genomic analysis by constructing a phylogeny to investigate the metabolic pathways of three interesting up-regulated clusters: a pseurotin-related compound BGC (MaNRPS-PKS2), a putative helvolic acid BGC (MaTERP1) and a tropolone/citrinin-related compound BGC (MaPKS1).
Pseurotins are a group of compounds containing phenylalanine coupled to a polyketide with a spiro ring structure, and have been isolated from Aspergillus spp. and Pseudeurotium ovalis cultures [95]. Anti-angiogenic activities as well as IgE and chitin synthase inhibitory activities have been reported for pseurotins [96]. Although this metabolite has never been isolated from Metarhizium cultures, a recent report revealed a putative pseurotin cluster in M. robertsii [36] and served as the starting point for analysis. The expression and comparative genomic analysis suggested that the final product of this cluster is not pseurotin, but a related compound. Wiemann and coworkers (2013) speculate that the product of this BGC could be 12-hydroxy-ovalicin (Mer-f3). Mer-f3 exhibits inhibitory and immunosuppressive activities against certain tumor cell lineages, and ovalicin-related compounds isolated from M. anisopliae cultures have been tested for the treatment of atopic dermatitis in mice [97, 98].
The pseurotin-related compound BGC from M. anisopliae may also be embedded in a supercluster, analogous to the pseurotin BGC from Aspergillus species. This macro regulation exerted by a supercluster is an interesting and virtually unexplored area of research. Comparative genomic analysis and the transcription profile suggested the presence of another supercluster in M. anisopliae. In view of the apparent co-regulation of MaPKS18 and the putative helvolic acid BGC (MaTERP1) as well as the fact that the sequence region containing both clusters misled the antiSMASH prediction we suggest that this region is a supercluster. However, more investigation is needed to prove this hypothesis.
The aforementioned helvolic acid is another interesting secondary metabolite. Helvolic acid is a well-known fusadine triterpene antibiotic that is active against Gram-positive bacteria [11]. In A. fumigatus, this SM is suggested to play an important role in human pathogenesis, exerting inhibitory effects on macrophages and inducing epithelial damage [99]. This compound also exhibits antifungal activity against phytopathogens and demonstrates antifeedant properties in the armyworm Mythimna separata [100]. Our phylogenetic analysis supports the notion that this cluster was horizontally acquired from a species closely related to Eurotiales, given that this BGC is restricted to only a few species. Moreover, not only helvolic acid but its derivative (1,2-dihydrohelvolic acid) have been isolated from M. anisopliae cultures. Furthermore, 1,2-dihydrohelvolic acid was isolated from fungus grown in insect-derived material, suggesting that this cluster is active under infection conditions [25]. Conversely, purified helvolic acid from M. anisopliae cultures does not appear to be toxic to some insects [101]. Although expression analysis suggested a role in infection and antifeedant properties for this metabolite have been reported in the literature, the true importance of helvolic acid in the Metarhizium lifestyle must be assessed.
Our analysis also suggested that the final product of MaPKS1 is a tropolone/citrinin-related compound. Tropolones and citrinins are structurally very similar and are chemically grouped by the local suffering oxidation [74, 75]. Furthermore, MaPKS1 contains several genes that are not conserved in the characterized tropolone/stipitatic acid and citrinin biosynthetic routes. Thus, while some genes in the MaPKS1 BGC are conserved in the tropolone and citrinin routes, and these genes likely perform similar functions in M. anisopliae, the final metabolic function of this cluster is still unknown. Better classification of the final product of this BGC will emerge with the functional analysis of this cluster.
Our analysis also uncovered global regulators of fungal development, nutrition and niche adaptation that are up-regulated under early infection conditions and can govern the expression of both up- and down-regulated BGCs, as observed in other fungal species [82]. The up-regulation of PacC under early infection conditions (>1.88-fold difference with P < 0.05; MANI_008549) is in agreement with the ability of M. anisopliae to modulate pH during the infection process by alkalizing infected cuticles [102, 103]; under alkaline pH conditions, PacC serves as a positive regulator promoting the transcription of alkaline-expressed genes, and it has been suggested that PacC simultaneously represses acid-expressed genes [104]. The importance of PacC in Metarhizium infection and fungal growth has already been established via the construction of knockouts [105]. However, it has not been determined how this gene influences secondary metabolism in Metarhizium spp. PacC may potentially influence the BGC for destruxin production, as it was verified that destruxin production is favored by an alkaline pH [106]. In accordance with the central role of PacC in secondary metabolism regulation, there have been reports in Aspergillus spp., indicating that this gene regulates penicillin and sterigmatocystin BGCs [107, 108]. Another gene linked to secondary metabolism regulation is the carbon catabolite repressor (CreA), which is also up-regulated under early infection conditions (> 2.91-fold difference with P < 0.05; MANI_015776). CreA is a global repressor that ensures the utilization of preferred carbon sources, preventing the expression of genes linked to assimilatory traits of non-preferred carbon sources [109]. CreA is known to negatively influence the production of penicillin at high carbon concentrations and was suggested to act in ochratoxin A regulation [82, 110]; however, it still must be determined how this gene influences Metarhizium spp. secondary metabolism. Interestingly, in filamentous fungi, carbon and pH metabolisms appear to be related [111]. Recently, Bi and coworkers (2015) showed that many organisms acidify media under conditions of carbon excess, while alkalization occurs under carbon deprivation. Mutants for glutamate dehydrogenase 2 (gdh2), which catalyzes the deamination of non-preferred carbon sources, resulting in ammonia production, exhibited reduced virulence and alkalization potential, and gdh2 expression was negatively correlated with CreA in Colletotrichum gloeosporioides, Penicillium expansum, Aspergillus nidulans, and Fusarium oxysporum [111]. This similar regulation in distantly related species (e.g., Fusarium spp. and Aspergillus spp.) suggests a widespread form of regulation that can be present in Metarhizium species. However, our observation of both PacC and CreA up-regulation is contrary to this notion. The positive regulation of these global regulators under early infection conditions may indicate a switch from infection to a saprophytic state, which could lead to the down-regulation of BGCs involved in infection. Notably, the orthologs for the laeA gene in M. anisopliae exhibited lower expression (difference not available under both early and late infection conditions with P < 0.05; MANI_030399) (Table 7). LaeA demonstrates central importance in SM metabolism and virulence in Aspergillus spp. and Fusarium spp. [112, 113]. In Aspergillus carbonarius, the deletion of laeA led to a drastic decrease in ochratoxin A production [114], and the deletion of this gene in A. fumigatus blocks the expression of sterigmatocystin, penicillin, lovastatin and helvolic acid BGCs [115]. Future studies should be performed to assess the role of laeA in the Metarhizium lifestyle and virulence.
Finally, the conservation of the BGCs found among Metarhizium species is yet another fundamental topic. The absence of the destruxin BGC in the host-specialists M. acridum and M. album led to the conclusion that this SM could play a pivotal role in the host-generalist lifestyle [28]. The comparative genomic analyses of BGCs in the Metarhizium genus indicate that only one half of these BGCs are conserved in host-specialist species. In addition to this, host-specialist species also encode BGCs that not conserved in host-generalist species, which represents an important difference in the metabolic profile and niche adaptation potential of these species. Entomopathogenic fungi may retain some BGCs related to the infection process, and SM acquisition can act as a driving force towards host generalization. Accordingly, our results showed that only 6 out of the 15 up-regulated clusters are conserved in M. acridum and M. album (Additional file 10). The results suggest that the plethora of SM produced by host-generalist and host-intermediate species exert an underestimated impact in the infection process, and destruxins as well as many other SMs may be essential for fungal adaptation to new hosts.
Conclusions
Although considerable progress has been made in understanding the Metarhizium infection process, unanswered questions remain, in particular those related to the definition of host specificity, which is a central and still not fully understood topic in the study of entomopathogenic fungi. Different fungal species, and even different strains, are able to synthesize different combinations of SM compounds that may result in distinct adaptation strategies [73]. In general, the results point to a fundamental role for SM in initial infection, with a notable difference in the BGCs present in host-specialist versus host-generalist Metarhizium species. Transcript analysis of samples obtained under conditions mimicking tick infection, showed the activation of several BGCs, with some up-regulated only during the early steps of infection. The conservation and expression of these genes can actively support M. anisopliae as a successful generalist pathogen. The origin and evolution of SM clusters is another interesting topic, and the results suggest that HGT events may have shaped the metabolic potential of generalist species, but this hypothesis still must be explored.
Several BGCs characterized by other groups, which were shown to participate in the infection process only to a minor degree, as well as several putative BGCs highlighted by this work were silent under mimicked conditions, reaffirming the hypothesis regarding their participation in the infection process. In this work, we took the first steps toward the characterization of several previously unexplored BGCs and examined their regulation, focusing on their potential participation in infection. These are important issues to investigate, not only to acquire basic knowledge regarding fungal lifestyles and adaptation, but also to explore for future biological control applications, and the identified genes and global regulators represent valuable targets for further experimental study.
Acknowledgments
We would like to thank the staff of LNCC for their support. Authors are indebted to the anonymous referee that provided a detailed and enthusiastic review that much improved the final manuscript.
Declarations
About this supplement
This article has been published as part of BMC Genomics Volume 17 Supplement 8: Selected articles from the Sixth International Conference of the Iberoamerican Society for Bioinformatics on Bioinformatics and Computational Biology for Innovative Genomics. The full contents of the supplement are available online at https://bmcgenomics.biomedcentral.com/articles/supplements/volume-17-supplement-8.
Funding
This study was supported by grants and fellowships from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Programa de Aperfeiçoamento Pessoal de Nível Superior (CAPES) [Grant: Biocomputacional Processo 23038.010041/2013¬13], Fundação de Amparo a Pesquisa do Estado do RS (FAPERGS) and Fundação de Amparo a Pesquisa do Estado do RJ (FAPERJ) and is part of the Advanced Network of Computational Biology (RABICÓ). The publication charges for this article were funded by CAPES (process no. 23038.010041/2013¬13).
Availability of data and materials
Not applicable.
Authors’ contribution
Conceived and designed the experiments: NS, RLMG, MHV, ATRV, and AS. Transcriptome analysis: NS, RLMG, and GLM. BGCs curation and comparative analyses: NS, AJ, and RLMG. Phylogenetic analyses: NS and FCA. Contributed reagents/materials/analysis tools: MHV, ATRV, and AS. Wrote the article: NS, AJ, RLMG, FCA, MHV and AS. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Additional files
Fungal genomes used in this work. (PDF 158 kb)
OrthoMCL clustering results. (PDF 157 kb)
Phylogenetic trees constructed with putative orthologs of MaPKS1, all PKS from M. anisopliae E6, and all characterized PKS from MIBiG. (PDF 600 kb)
Amino acid alignment for the pseurotin-related backbone gene and related orthologs. (FASTA 67 kb)
Amino acid alignment for the tropolone/citrinin-related backbone gene and related orthologs. (FASTA 125 kb)
Nucleotide alignment for the tef1 gene. (FASTA 40 kb)
Best-fit evolutionary models predicted with Prottest 3.4 or jmodeltest-2.1.9 for each alignment. (PDF 12 kb)
The partially conserved cluster found in A. chrysogenum and species from the Arthroderma genus is putatively linked to fusidic acid biosynthesis. (PDF 307 kb)
Amino acid alignment for helvolic acid supermatrix. (FASTA 40 kb)
Overview of the BGCs in M. anisopliae E6, their domain structures, and conservation among six Metarhizium species. (PDF 256 kb)
Expression profile of predicted BGCs. (PDF 934 kb)
Comparative genomic analysis and synteny for the aurovertin, elymoclavine/ergovaline-related compound, terpendole E/lolitrem-related compound, and xenolozoyenone-related compound BGCs. (PDF 161 kb)
Additional phylogenetic trees. (PDF 463 kb)
References
- 1.Zimmermann G. Review on safety of the entomopathogenic fungus Metarhizium anisopliae. Biocontrol Sci Tech. 2007;17(9):879–920. doi: 10.1080/09583150701593963. [DOI] [Google Scholar]
- 2.Schrank A, Vainstein MH. Metarhizium anisopliae enzymes and toxins. Toxicon. 2010;56(7):1267–1274. doi: 10.1016/j.toxicon.2010.03.008. [DOI] [PubMed] [Google Scholar]
- 3.Dubovskiy IM, Whitten MMA, Yaroslavtseva ON, Greig C, Kryukov VY, Grizanova EV, Mukherjee K, Vilcinskas A, Glupov VV, Butt TM. Can insects develop resistance to insect pathogenic fungi? Plos One. 2013;8(4):9. doi: 10.1371/journal.pone.0060248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Butt TM, Greenfield BPJ, Greig C, Maffeis TGG, Taylor JWD, Piasecka J, Dudley E, Abdulla A, Dubovskiy IM, Garrido-Jurado I, et al. Metarhizium anisopliae pathogenesis of mosquito larvae: a verdict of accidental death. Plos One. 2013;8(12):11. doi: 10.1371/journal.pone.0081686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang CS, St Leger RJ. A scorpion neurotoxin increases the potency of a fungal insecticide. Nat Biotechnol. 2007;25(12):1455–1456. doi: 10.1038/nbt1357. [DOI] [PubMed] [Google Scholar]
- 6.Ortiz-Urquiza A, Luo ZB, Keyhani NO. Improving mycoinsecticides for insect biological control. Appl Microbiol Biotechnol. 2015;99(3):1057–1068. doi: 10.1007/s00253-014-6270-x. [DOI] [PubMed] [Google Scholar]
- 7.Kern MF, Maraschin SD, Vom Endt D, Schrank A, Vainstein MH, Pasquali G. Expression of a chitinase gene from metarhizium anisopliae in tobacco plants confers resistance against rhizoctonia solani. Appl Biochem Biotechnol. 2010;160(7):1933–1946. doi: 10.1007/s12010-009-8701-1. [DOI] [PubMed] [Google Scholar]
- 8.Hu X, Xiao G, Zheng P, Shang Y, Su Y, Zhang X, Liu X, Zhan S, St Leger RJ, Wang C. Trajectory and genomic determinants of fungal-pathogen speciation and host adaptation. Proc Natl Acad Sci. 2014;111:16796. doi: 10.1073/pnas.1412662111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pedrini N, Crespo R, Juarez MP. Biochemistry of insect epicuticle degradation by entomopathogenic fungi. Comparative Biochemistry and Physiology C-Toxicology & Pharmacology. 2007;146(1–2):124–137. doi: 10.1016/j.cbpc.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 10.Junges A, Boldo JT, Souza BK, Guedes RLM, Sbaraini N, Kmetzsch L, Thompson CE, Staats CC, de Almeida LGP, de Vasconcelos ATR, et al. Genomic analyses and transcriptional profiles of the glycoside hydrolase family 18 genes of the entomopathogenic fungus metarhizium anisopliae. Plos One. 2014;9(9):16. doi: 10.1371/journal.pone.0107864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Molnar I, Gibson DM, Krasnoff SB. Secondary metabolites from entomopathogenic Hypocrealean fungi. Nat Prod Rep. 2010;27(9):1241–1275. doi: 10.1039/c001459c. [DOI] [PubMed] [Google Scholar]
- 12.Tian XW, Li YJ, Shen YY, Li QQ, Wang QL, Feng LS. Apoptosis and inhibition of proliferation of cancer cells induced by cordycepin (Review) Oncol Lett. 2015;10(2):595–599. doi: 10.3892/ol.2015.3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thongtan J, Saenboonrueng J, Rachtawee P, Isaka M. An antimalarial tetrapeptide from the entomopathogenic fungus Hirsutella sp BCC 1528. J Nat Prod. 2006;69(4):713–714. doi: 10.1021/np050549h. [DOI] [PubMed] [Google Scholar]
- 14.Yaegashi J, Oakley BR, Wang CCC. Recent advances in genome mining of secondary metabolite biosynthetic gene clusters and the development of heterologous expression systems in Aspergillus nidulans. Journal of Industrial Microbiology & Biotechnology. 2014;41(2):433–442. doi: 10.1007/s10295-013-1386-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lazarus CM, Williams K, Bailey AM. Reconstructing fungal natural product biosynthetic pathways. Nat Prod Rep. 2014;31(10):1339–1347. doi: 10.1039/C4NP00084F. [DOI] [PubMed] [Google Scholar]
- 16.Inglis DO, Binkley J, Skrzypek MS, Arnaud MB, Cerqueira GC, Shah P, Wymore F, Wortman JR, Sherlock G. Comprehensive annotation of secondary metabolite biosynthetic genes and gene clusters of aspergillus nidulans, a. Fumigatus, a. Niger and a. Oryzae. BMC Microbiol. 2013;13:23. doi: 10.1186/1471-2180-13-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Keller NP. Translating biosynthetic gene clusters into fungal armor and weaponry. Nat Chem Biol. 2015;11(9):671–677. doi: 10.1038/nchembio.1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Walton JD. Horizontal gene transfer and the evolution of secondary metabolite gene clusters in fungi: An hypothesis. Fungal Genet Biol. 2000;30(3):167–171. doi: 10.1006/fgbi.2000.1224. [DOI] [PubMed] [Google Scholar]
- 19.Slot JC, Rokas A. Horizontal transfer of a large and highly toxic secondary metabolic gene cluster between fungi. Curr Biol. 2011;21(2):134–139. doi: 10.1016/j.cub.2010.12.020. [DOI] [PubMed] [Google Scholar]
- 20.Khaldi N, Collemare J, Lebrun MH, Wolfe KH. Evidence for horizontal transfer of a secondary metabolite gene cluster between fungi. Genome Biol. 2008;9(1):10. doi: 10.1186/gb-2008-9-1-r18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dhillon B, Feau N, Aerts AL, Beauseigle S, Bernier L, Copeland A, Foster A, Gill N, Henrissat B, Herath P, et al. Horizontal gene transfer and gene dosage drives adaptation to wood colonization in a tree pathogen. Proc Natl Acad Sci U S A. 2015;112(11):3451–3456. doi: 10.1073/pnas.1424293112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Katou Y, Endo N, Suzuki T, Yu J, Kikuchi H, Oshima Y, Homma Y. Metarhizin a suppresses cell proliferation by inhibiting cytochrome c oxidase activity. Life Sci. 2014;103(1):1–7. doi: 10.1016/j.lfs.2014.03.023. [DOI] [PubMed] [Google Scholar]
- 23.Patrick MS, Adlard MW, Keshavarz T. Swainsonine production from batch cultures of Metarhizium anisopliae in stirred-tank reactors. Enzym Microb Technol. 1996;18(6):428–432. doi: 10.1016/0141-0229(95)00123-9. [DOI] [Google Scholar]
- 24.Skrobek A, Butt TM. Toxicity testing of destruxins and crude extracts from the insect-pathogenic fungus Metarhizium anisopliae. Fems Microbiology Letters. 2005;251(1):23–28. doi: 10.1016/j.femsle.2005.07.029. [DOI] [PubMed] [Google Scholar]
- 25.Lee SY, Kinoshita H, Ihara F, Igarashi Y, Nihira T. Identification of novel derivative of helvolic acid from Metarhizium anisopliae grown in medium with insect component. J Biosci Bioeng. 2008;105(5):476–480. doi: 10.1263/jbb.105.476. [DOI] [PubMed] [Google Scholar]
- 26.Moon YS, Donzelli BGG, Krasnoff SB, McLane H, Griggs MH, Cooke P, Vandenberg JD, Gibson DM, Churchill ACL. Agrobacterium-mediated disruption of a nonribosomal peptide synthetase gene in the invertebrate pathogen Metarhizium anisopliae reveals a peptide spore factor. Appl Environ Microbiol. 2008;74(14):4366–4380. doi: 10.1128/AEM.00285-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Donzelli BGG, Krasnoff SB, Churchill ACL, Vandenberg JD, Gibson DM. Identification of a hybrid PKS-NRPS required for the biosynthesis of NG-391 in Metarhizium robertsii. Curr Genet. 2010;56(2):151–162. doi: 10.1007/s00294-010-0288-0. [DOI] [PubMed] [Google Scholar]
- 28.Wang B, Kang QJ, Lu YZ, Bai LQ, Wang CS. Unveiling the biosynthetic puzzle of destruxins in Metarhizium species. Proc Natl Acad Sci U S A. 2012;109(4):1287–1292. doi: 10.1073/pnas.1115983109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen Y, Feng P, Shang Y, Xu Y-J, Wang C. Biosynthesis of non-melanin pigment by a divergent polyketide synthase in Metarhizium robertsii. Fungal Genet Biol. 2016;0:142e149. doi: 10.1016/j.fgb.2014.10.018. [DOI] [PubMed] [Google Scholar]
- 30.Giuliano Garisto Donzelli B, Gibson DM, Krasnoff SB. Intracellular siderophore but not extracellular siderophore is required for full virulence in Metarhizium robertsii. Fungal Genet Biol. 2015;82:56–68. doi: 10.1016/j.fgb.2015.06.008. [DOI] [PubMed] [Google Scholar]
- 31.Gibson DM, Donzelli BGG, Krasnoff SB, Keyhani NO. Discovering the secondary metabolite potential encoded within entomopathogenic fungi. Nat Prod Rep. 2014;31(10):1287–1305. doi: 10.1039/C4NP00054D. [DOI] [PubMed] [Google Scholar]
- 32.Staats CC, Junges A, Guedes RL, Thompson CE, de Morais GL, Boldo JT, de Almeida LG, Andreis FC, Gerber AL, Sbaraini N, et al. Comparative genome analysis of entomopathogenic fungi reveals a complex set of secreted proteins. BMC Genomics. 2014;15(1):822. doi: 10.1186/1471-2164-15-822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weber T, Blin K, Duddela S, Krug D, Kim HU, Bruccoleri R, Lee SY, Fischbach MA, Muller R, Wohlleben W, et al. antiSMASH 3.0-a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 2015;43(W1):W237–W243. doi: 10.1093/nar/gkv437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Khaldi N, Seifuddin FT, Turner G, Haft D, Nierman WC, Wolfe KH, Fedorova ND. SMURF: genomic mapping of fungal secondary metabolite clusters. Fungal Genet Biol. 2010;47(9):736–741. doi: 10.1016/j.fgb.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wolf T, Shelest V, Nath N, Shelest E. CASSIS and SMIPS: promoter-based prediction of secondary metabolite gene clusters in eukaryotic genomes. Bioinformatics. 2015. [DOI] [PMC free article] [PubMed]
- 36.Wiemann P, Guo CJ, Palmer JM, Sekonyela R, Wang CCC, Keller NP. Prototype of an intertwined secondary-metabolite supercluster. Proc Natl Acad Sci U S A. 2013;110(42):17065–17070. doi: 10.1073/pnas.1313258110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lodeiro S, Xiong QB, Wilson WK, Ivanova Y, Smith ML, May GS, Matsuda SPT. Protostadienol biosynthesis and metabolism in the pathogenic fungus aspergillus fumigatus. Org Lett. 2009;11(6):1241–1244. doi: 10.1021/ol802696a. [DOI] [PubMed] [Google Scholar]
- 38.Medema MH, Takano E, Breitling R. Detecting sequence homology at the gene cluster level with MultiGeneBlast. Mol Biol Evol. 2013;30(5):1218–1223. doi: 10.1093/molbev/mst025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 40.Pryszcz LP, Huerta-Cepas J, Gabaldon T. MetaPhOrs: orthology and paralogy predictions from multiple phylogenetic evidence using a consistency-based confidence score. Nucleic Acids Res. 2011;39(5):8. doi: 10.1093/nar/gkq953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li L, Stoeckert CJ, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13(9):2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schardl CL, Young CA, Hesse U, Amyotte SG, Andreeva K, Calie PJ, Fleetwood DJ, Haws DC, Moore N, Oeser B, et al. Plant-symbiotic fungi as chemical engineers: multi-genome analysis of the clavicipitaceae reveals dynamics of alkaloid loci. Plos Genetics. 2013;9(2):26. doi: 10.1371/journal.pgen.1003323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schardl CL, Young CA, Moore N, Krom N, Dupont PY, Pan J, Florea S, Webb JS, Jaromczyk J, Jaromczyk JW, et al. Genomes of Plant-Associated Clavicipitaceae. In: Martin FM, et al., editors. Fungi. London: Academic Press Ltd-Elsevier Science Ltd; 2014. pp. 291–327. [Google Scholar]
- 44.Larriba E, Jaime M, Carbonell-Caballero J, Conesa A, Dopazo J, Nislow C, Martin-Nieto J, Lopez-Llorca L. Sequencing and functional analysis of the genome of a nematode egg-parasitic fungus, Pochonia chlamydosporia. Fungal Genet Biol. 2014;65:69–80. doi: 10.1016/j.fgb.2014.02.002. [DOI] [PubMed] [Google Scholar]
- 45.Solovyev V, Kosarev P, Seledsov I, Vorobyev D. Automatic annotation of eukaryotic genes, pseudogenes and promoters. Genome Biol. 2006;7:12. doi: 10.1186/gb-2006-7-s1-s10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Medema MH, Kottmann R, Yilmaz P, Cummings M, Biggins JB, Blin K, de Bruijn I, Chooi YH, Claesen J, Coates RC, et al. Minimum information about a biosynthetic gene cluster. Nat Chem Biol. 2015;11(9):625–631. doi: 10.1038/nchembio.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Altekar G, Dwarkadas S, Huelsenbeck JP, Ronquist F. Parallel metropolis coupled markov chain monte carlo for bayesian phylogenetic inference. Bioinformatics. 2004;20(3):407–415. doi: 10.1093/bioinformatics/btg427. [DOI] [PubMed] [Google Scholar]
- 48.Ayres DL, Darling A, Zwickl DJ, Beerli P, Holder MT, Lewis PO, Huelsenbeck JP, Ronquist F, Swofford DL, Cummings MP, et al. BEAGLE: an application programming interface and high-performance computing library for statistical phylogenetics. Syst Biol. 2012;61(1):170–173. doi: 10.1093/sysbio/syr100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Hohna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. MrBayes 3.2: efficient bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61(3):539–542. doi: 10.1093/sysbio/sys029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Loytynoja A, Goldman N. webPRANK: a phylogeny-aware multiple sequence aligner with interactive alignment browser. Bmc Bioinformatics. 2010;11:6. doi: 10.1186/1471-2105-11-579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30(12):2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sela I, Ashkenazy H, Katoh K, Pupko T. GUIDANCE2: accurate detection of unreliable alignment regions accounting for the uncertainty of multiple parameters. Nucleic Acids Res. 2015;43(W1):W7–W14. doi: 10.1093/nar/gkv318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Darriba D, Taboada GL, Doallo R, Posada D. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics. 2011;27(8):1164–1165. doi: 10.1093/bioinformatics/btr088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Darriba D, Taboada GL, Doallo R, Posada D. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 2012;9(8):772. doi: 10.1038/nmeth.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59(3):307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 56.Creevey CJ, McInerney JO. Clann: investigating phylogenetic information through supertree analyses. Bioinformatics. 2005;21(3):390–392. doi: 10.1093/bioinformatics/bti020. [DOI] [PubMed] [Google Scholar]
- 57.Gouy M, Guindon S, Gascuel O. SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol. 2010;27(2):221–224. doi: 10.1093/molbev/msp259. [DOI] [PubMed] [Google Scholar]
- 58.Campbell MA, Rokas A, Slot JC. Horizontal transfer and death of a fungal secondary metabolic gene cluster. Genome Biology and Evolution. 2012;4(3):289–293. doi: 10.1093/gbe/evs011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bischoff JF, Rehner SA, Humber RA. A multilocus phylogeny of the metarhizium anisopliae lineage. Mycologia. 2009;101(4):512–530. doi: 10.3852/07-202. [DOI] [PubMed] [Google Scholar]
- 60.Kepler RM, Humber RA, Bischoff JF, Rehner SA. Clarification of generic and species boundaries for Metarhizium and related fungi through multigene phylogenetics. Mycologia. 2014;106(4):811–829. doi: 10.3852/13-319. [DOI] [PubMed] [Google Scholar]
- 61.Sung GH, Sung JM, Hywel-Jones NL, Spatafora JW. A multi-gene phylogeny of clavicipitaceae (ascomycota, fungi): identification of localized incongruence using a combinational bootstrap approach. Mol Phylogenet Evol. 2007;44(3):1204–1223. doi: 10.1016/j.ympev.2007.03.011. [DOI] [PubMed] [Google Scholar]
- 62.Grigoriev IV, Nikitin R, Haridas S, Kuo A, Ohm R, Otillar R, Riley R, Salamov A, Zhao XL, Korzeniewski F, et al. MycoCosm portal: gearing up for 1000 fungal genomes. Nucleic Acids Res. 2014;42(D1):D699–D704. doi: 10.1093/nar/gkt1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Azumi M, Ishidoh K, Kinoshita H, Nihira T, Ihara F, Fujita T, Igarashi Y. Aurovertins F-H from the entomopathogenic fungus Metarhizium anisopliae. J Nat Prod. 2008;71(2):278–280. doi: 10.1021/np070494e. [DOI] [PubMed] [Google Scholar]
- 64.Mao XM, Zhan ZJ, Grayson MN, Tang MC, Xu W, Li YQ, Yin WB, Lin HC, Chooi YH, Houk KN, et al. Efficient biosynthesis of fungal polyketides containing the dioxabicyclo-octane ring system. J Am Chem Soc. 2015;137(37):11904–11907. doi: 10.1021/jacs.5b07816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cardoza RE, Vizcaino JA, Hermosa MR, Sousa S, Gonzalez FJ, Llobell A, Monte E, Gutierrez S. Cloning and characterization of the erg1 gene of trichoderma harzianum: effect of the erg1 silencing on ergosterol biosynthesis and resistance to terbinafine. Fungal Genet Biol. 2006;43(3):164–178. doi: 10.1016/j.fgb.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 66.Lorenz N, Wilson EV, Machado C, Schardl CL, Tudzynski P. Comparison of ergot alkaloid biosynthesis gene clusters in Claviceps species indicates loss of late pathway steps in evolution of C-fusiformis. Appl Environ Microbiol. 2007;73(22):7185–7191. doi: 10.1128/AEM.01040-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fleetwood DJ, Scott B, Lane GA, Tanaka A, Johnson RD. A complex ergovaline gene cluster in Epichloe endophytes of grasses. Appl Environ Microbiol. 2007;73(8):2571–2579. doi: 10.1128/AEM.00257-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Motoyama T, Hayashi T, Hirota H, Ueki M, Osada H. Terpendole E, a kinesin Eg5 inhibitor, is a Key biosynthetic intermediate of indole-diterpenes in the producing fungus chaunopycnis alba. Chemistry & Biology. 2012;19(12):1611–1619. doi: 10.1016/j.chembiol.2012.10.010. [DOI] [PubMed] [Google Scholar]
- 69.Young CA, Bryant MK, Christensen MJ, Tapper BA, Bryan GT, Scott B. Molecular cloning and genetic analysis of a symbiosis-expressed gene cluster for lolitrem biosynthesis from a mutualistic endophyte of perennial ryegrass. Mol Gen Genomics. 2005;274(1):13–29. doi: 10.1007/s00438-005-1130-0. [DOI] [PubMed] [Google Scholar]
- 70.Young CA, Felitti S, Shields K, Spangenberg G, Johnson RD, Bryan GT, Saikia S, Scott B. A complex gene cluster for indole-diterpene biosynthesis in the grass endophyte Neotyphodium lolii. Fungal Genet Biol. 2006;43(10):679–693. doi: 10.1016/j.fgb.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 71.Yue Q, Chen L, Li Y, Bills GF, Zhang X, Xiang M, Li S, Che Y, Wang C, Niu X, et al. Functional operons in secondary metabolic gene clusters in glarea lozoyensis (fungi, ascomycota, leotiomycetes) mBio. 2015;6:3. doi: 10.1128/mBio.00703-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Maiya S, Grundmann A, Li X, Li SM, Turner G. Identification of a hybrid PKS/NRPS required for pseurotin a biosynthesis in the human pathogen aspergillus fumigatus. Chembiochem. 2007;8(14):1736–1743. doi: 10.1002/cbic.200700202. [DOI] [PubMed] [Google Scholar]
- 73.Marcet-Houben M, Gabaldon T. Horizontal acquisition of toxic alkaloid synthesis in a clade of plant associated fungi. Fungal Genet Biol. 2016;86:71–80. doi: 10.1016/j.fgb.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Davison J, Al Fahad A, Cai MH, Song ZS, Yehia SY, Lazarus CM, Bailey AM, Simpson TJ, Cox RJ. Genetic, molecular, and biochemical basis of fungal tropolone biosynthesis. Proc Natl Acad Sci U S A. 2012;109(20):7642–7647. doi: 10.1073/pnas.1201469109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.He Y, Cox RJ. The molecular steps of citrinin biosynthesis in fungi. Chem Sci. 2016;7(3):2119–2127. doi: 10.1039/C5SC04027B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Elliott CE, Callahan DL, Schwenk D, Nett M, Hoffmeister D, Howlett BJ. A gene cluster responsible for biosynthesis of phomenoic acid in the plant pathogenic fungus, Leptosphaeria maculans. Fungal Genet Biol. 2013;53:50–58. doi: 10.1016/j.fgb.2013.01.008. [DOI] [PubMed] [Google Scholar]
- 77.Balakrishnan B, Karki S, Chiu SH, Kim HJ, Suh JW, Nam B, Yoon YM, Chen CC, Kwon HJ. Genetic localization and in vivo characterization of a Monascus azaphilone pigment biosynthetic gene cluster. Appl Microbiol Biotechnol. 2013;97(14):6337–6345. doi: 10.1007/s00253-013-4745-9. [DOI] [PubMed] [Google Scholar]
- 78.Shimizu T, Kinoshita H, Nihira T. Identification and in vivo functional analysis by gene disruption of ctnA, an activator gene involved in citrinin biosynthesis in Monascus purpureus. Appl Environ Microbiol. 2007;73(16):5097–5103. doi: 10.1128/AEM.01979-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yin WB, Keller NP. Transcriptional regulatory elements in fungal secondary metabolism. J Microbiol. 2011;49(3):329–339. doi: 10.1007/s12275-011-1009-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bayram O, Krappmann S, Ni M, Bok JW, Helmstaedt K, Valerius O, Braus-Stromeyer S, Kwon NJ, Keller NP, Yu JH, et al. VelB/VeA/LaeA complex coordinates light signal with fungal development and secondary metabolism. Science. 2008;320(5882):1504–1506. doi: 10.1126/science.1155888. [DOI] [PubMed] [Google Scholar]
- 81.Lopez-Berges MS, Hera C, Sulyok M, Schafer K, Capilla J, Guarro J, Di Pietro A. The velvet complex governs mycotoxin production and virulence of Fusarium oxysporum on plant and mammalian hosts. Mol Microbiol. 2013;87(1):49–65. doi: 10.1111/mmi.12082. [DOI] [PubMed] [Google Scholar]
- 82.Brakhage AA. Regulation of fungal secondary metabolism. Nat Rev Microbiol. 2013;11(1):21–32. doi: 10.1038/nrmicro2916. [DOI] [PubMed] [Google Scholar]
- 83.Yu JH, Keller N. Annual review of phytopathology. Palo Alto: Annual Reviews; 2005. Regulation of secondary metabolism in filamentous fungi; pp. 437–458. [DOI] [PubMed] [Google Scholar]
- 84.Gilbert MK, Mack BM, Wei QJ, Bland JM, Bhatnagar D, Cary JW. RNA sequencing of an nsdC mutant reveals global regulation of secondary metabolic gene clusters in Aspergillus flavus. Microbiol Res. 2016;182:150–161. doi: 10.1016/j.micres.2015.08.007. [DOI] [PubMed] [Google Scholar]
- 85.Sheppard DC, Doedt T, Chiang LY, Kim HS, Chen D, Nierman WC, Filler SG. The Aspergillus fumigatus StuA protein governs the up-regulation of a discrete transcriptional program during the acquisition of developmental competence. Mol Biol Cell. 2005;16(12):5866–5879. doi: 10.1091/mbc.E05-07-0617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Frazzon AP, da Silva Vaz Junior I, Masuda A, Schrank A, Vainstein MH. In vitro assessment of Metarhizium anisopliae isolates to control the cattle tick Boophilus microplus. Vet Parasitol. 2000;94(1–2):117–125. doi: 10.1016/S0304-4017(00)00368-X. [DOI] [PubMed] [Google Scholar]
- 87.Da Silva WOB, Santi L, Schrank A, Vainstein MH. Metarhizium anisopliae lipolytic activity plays a pivotal role in Rhipicephalus (Boophilus) microplus infection. Fungal Biology. 2010;114(1):10–15. doi: 10.1016/j.mycres.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 88.Reck J, Klafke GM, Webster A, Dall’Agnol B, Scheffer R, Souza UA, Corassini VB, Vargas R, dos Santos JS, Martins JR. First report of fluazuron resistance in Rhipicephalus microplus: a field tick population resistant to six classes of acaricides. Vet Parasitol. 2014;201(1–2):128–136. doi: 10.1016/j.vetpar.2014.01.012. [DOI] [PubMed] [Google Scholar]
- 89.Webster A, Reck J, Santi L, Souza UA, Dall’Agnol B, Klafke GM, Beys-da-Silva WO, Martins JR, Schrank A. Integrated control of an acaricide-resistant strain of the cattle tick Rhipicephalus microplus by applying Metarhizium anisopliae associated with cypermethrin and chlorpyriphos under field conditions. Vet Parasitol. 2015;207(3–4):302–308. doi: 10.1016/j.vetpar.2014.11.021. [DOI] [PubMed] [Google Scholar]
- 90.Ment D, Churchill ACL, Gindin G, Belausov E, Glazer I, Rehner SA, Rot A, Donzelli BGG, Samish M. Resistant ticks inhibit Metarhizium infection prior to haemocoel invasion by reducing fungal viability on the cuticle surface. Environ Microbiol. 2012;14(6):1570–1583. doi: 10.1111/j.1462-2920.2012.02747.x. [DOI] [PubMed] [Google Scholar]
- 91.Ortiz-Urquiza A, Keyhani NO. Action on the surface: entomopathogenic fungi versus the insect cuticle. Insects. 2013;4(3):357–374. doi: 10.3390/insects4030357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Liu BL, Tzeng YM. Development and applications of destruxins: a review. Biotechnol Adv. 2012;30(6):1242–1254. doi: 10.1016/j.biotechadv.2011.10.006. [DOI] [PubMed] [Google Scholar]
- 93.Wang YL, Li LF, Li DX, Wang BL, Zhang KQ, Niu XM. Yellow pigment aurovertins mediate interactions between the pathogenic fungus pochonia chlamydosporia and its nematode host. J Agric Food Chem. 2015;63(29):6577–6587. doi: 10.1021/acs.jafc.5b02595. [DOI] [PubMed] [Google Scholar]
- 94.Bohnert M, Dahse HM, Gibson DM, Krasnoff SB, Hoffmeister D. The fusarin analog NG-391 impairs nucleic acid formation in K-562 leukemia cells. Phytochem Lett. 2013;6(2):189–192. doi: 10.1016/j.phytol.2013.01.001. [DOI] [Google Scholar]
- 95.Bladt TT, Frisvad JC, Knudsen PB, Larsen TO. Anticancer and antifungal compounds from aspergillus, penicillium and other filamentous fungi. Molecules. 2013;18(9):11338–11376. doi: 10.3390/molecules180911338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ishikawa M, Ninomiya T, Akabane H, Kushida N, Tsujiuchi G, Ohyama M, Gomi S, Shito K, Murata T. Pseurotin A and its analogues as inhibitors of immunoglobuline E production. Bioorg Med Chem Lett. 2009;19(5):1457–1460. doi: 10.1016/j.bmcl.2009.01.029. [DOI] [PubMed] [Google Scholar]
- 97.Kuboki H, Tsuchida T, Wakazono K, Isshiki K, Kumagai H, Yoshioka T. Mer-f3, 12-hydroxy-ovalicin, produced by Metarrhizium sp f3. J Antibiot. 1999;52(6):590–593. doi: 10.7164/antibiotics.52.590. [DOI] [PubMed] [Google Scholar]
- 98.Yoon CS, Nam SH, Jeon JY, Lee HS, Lee ML, Son HU, Lee SH. Ovalicin ameliorates compound 48/80-induced atopic dermatitis-related symptoms. Biol Pharm Bull. 2011;34(12):1881–1884. doi: 10.1248/bpb.34.1881. [DOI] [PubMed] [Google Scholar]
- 99.Abad A, Fernandez-Molina JV, Bikandi J, Ramirez A, Margareto J, Sendino J, Hernando FL, Ponton J, Garaizar J, Rementeria A. What makes Aspergillus fumigatus a successful pathogen? Genes and molecules involved in invasive aspergillosis. Revista Iberoamericana De Micologia. 2010;27(4):155–182. doi: 10.1016/j.riam.2010.10.003. [DOI] [PubMed] [Google Scholar]
- 100.Li XJ, Zhang Q, Zhang AL, Gao JM. Metabolites from aspergillus fumigatus, an endophytic fungus associated with melia azedarach, and their antifungal, antifeedant, and toxic activities. J Agric Food Chem. 2012;60(13):3424–3431. doi: 10.1021/jf300146n. [DOI] [PubMed] [Google Scholar]
- 101.Huxham IM, Lackie AM, McCorkindale NJ. Inhibitory effects of cyclodepsipeptides, destruxins, from the fungus metarhizium-anisopliae, on cellular-immunity in insects. J Insect Physiol. 1989;35(2):97–105. doi: 10.1016/0022-1910(89)90042-5. [DOI] [Google Scholar]
- 102.St Leger RJ, Nelson JO, Screen SE. The entomopathogenic fungus Metarhizium anisopliae alters ambient pH, allowing extracellular protease production and activity. Microbiology-Sgm. 1999;145:2691–2699. doi: 10.1099/00221287-145-10-2691. [DOI] [PubMed] [Google Scholar]
- 103.St Leger RJ, Joshi L, Roberts D. Ambient pH is a major determinant in the expression of cuticle-degrading enzymes and hydrophobin by Metarhizium anisopliae. Appl Environ Microbiol. 1998;64(2):709–713. doi: 10.1128/aem.64.2.709-713.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Alkan N, Meng XC, Friedlander G, Reuveni E, Sukno S, Sherman A, Thon M, Fluhr R, Prusky D. Global aspects of pacC regulation of pathogenicity genes in colletotrichum gloeosporioides as revealed by transcriptome analysis. Mol Plant-Microbe Interact. 2013;26(11):1345–1358. doi: 10.1094/MPMI-03-13-0080-R. [DOI] [PubMed] [Google Scholar]
- 105.Huang W, Shang Y, Chen P, Gao Q, Wang C. MrpacC regulates sporulation, insect cuticle penetration and immune evasion in Metarhizium robertsii. Environ Microbiol. 2014. [DOI] [PubMed]
- 106.Liu B-L, Rou T-M, Rao YK, Tzeng Y-M. Effect of pH and aeration rate on the production of destruxins A and B from metarhizium anisopliae. International Journal of Applied Science and Engineering. 2007;5(1):17–26. [Google Scholar]
- 107.Keller NP, Nesbitt C, Sarr B, Phillips TD, Burow GB. pH regulation of sterigmatocystin and aflatoxin biosynthesis in Aspergillus spp. Phytopathology. 1997;87(6):643–648. doi: 10.1094/PHYTO.1997.87.6.643. [DOI] [PubMed] [Google Scholar]
- 108.Bergh KT, Brakhage AA. Regulation of the Aspergillus nidulans penicillin biosynthesis gene acvA (pcbAB) by amino acids: Implication for involvement of transcription factor PACC. Appl Environ Microbiol. 1998;64(3):843–849. doi: 10.1128/aem.64.3.843-849.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Fernandez J, Wright JD, Hartline D, Quispe CF, Madayiputhiya N, Wilson RA. Principles of carbon catabolite repression in the rice blast fungus: Tps1, Nmr1–3, and a MATE-family pump regulate glucose metabolism during infection. Plos Genetics. 2012;8(5):29. doi: 10.1371/journal.pgen.1002673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Cepeda-Garcia C, Dominguez-Santos R, Garcia-Rico R, Garcia-Estrada C, Cajiao A, Fierro F, Martin J. Direct involvement of the CreA transcription factor in penicillin biosynthesis and expression of the pcbAB gene in Penicillium chrysogenum. Appl Microbiol Biotechnol. 2014;98(16):7113–7124. doi: 10.1007/s00253-014-5760-1. [DOI] [PubMed] [Google Scholar]
- 111.Bi F, Barad S, Ment D, Luria N, Dubey A, Casado V, Glam N, Mínguez JD, Espeso E, Fluhr R et al. Carbon regulation of environmental pH by secreted small molecules that modulate pathogenicity in phytopathogenic fungi. Molecular Plant Pathology 2015:1364–3703. doi: 10.1111/mpp.12355. [DOI] [PMC free article] [PubMed]
- 112.Bok JW, Keller NP. LaeA, a regulator of secondary metabolism in Aspergillus spp. Eukaryotic Cell. 2004;3(2):527–535. doi: 10.1128/EC.3.2.527-535.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Butchko RAE, Brown DW, Busman M, Tudzynski B, Wiemann P. Lae1 regulates expression of multiple secondary metabolite gene clusters in Fusarium verticillioides. Fungal Genet Biol. 2012;49(8):602–612. doi: 10.1016/j.fgb.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 114.Crespo-Sempere A, Marin S, Sanchis V, Ramos AJ. VeA and LaeA transcriptional factors regulate ochratoxin A biosynthesis in Aspergillus carbonarius. Int J Food Microbiol. 2013;166(3):479–486. doi: 10.1016/j.ijfoodmicro.2013.07.027. [DOI] [PubMed] [Google Scholar]
- 115.Keller N, Bok J, Chung D, Perrin RM, Shwab EK. LaeA, a global regulator of Aspergillus toxins. Med Mycol. 2006;44:S83–S85. doi: 10.1080/13693780600835773. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.