

Abstract
Both siRNA and miRNA can serve as powerful gene-silencing reagents but their specific delivery to cancer cells in vivo without collateral damage to healthy cells remains challenging. We report here the application of RNA nanotechnology for specific and efficient delivery of anti-miRNA seed-targeting sequence to block the growth of prostate cancer in mouse models. Utilizing the thermodynamically ultra-stable three-way junction of the pRNA of phi29 DNA packaging motor, RNA nanoparticles were constructed by bottom-up self-assembly containing the anti-prostate-specific membrane antigen (PSMA) RNA aptamer as a targeting ligand and anti-miR17 or anti-miR21 as therapeutic modules. The 16 nm RNase-resistant and thermodynamically stable RNA nanoparticles remained intact after systemic injection in mice and strongly bound to tumors with little or no accumulation in healthy organs 8 hours postinjection, and subsequently repressed tumor growth at low doses with high efficiency.
Introduction
Functionality of living organisms is made up by a wide variety of molecules consisting of DNA, RNA, and proteins. In the past, DNA has been used for its simplistic and defined structure in biomaterials,1,2,3,4 while proteins have extensively been utilized for their diverse structure and functionalities.5 RNA brings together the characteristics of the simplicity of DNA and the wide variety of folding structures and functions of proteins, making it an attractive candidate for use in nanobiotechnology.6,7 Utilizing RNA in therapies provides several advantages over other technologies, including: known stoichiometry and defined folding of nanoparticles, thereby decreasing the possibility of side effects and toxicity.8,9,10,11 Multiple RNA moieties such as receptor-binding aptamer,12,13 siRNA,14,15,16 ribozyme,17,18,19 miRNA,20,21,22 and riboswitch23,24 can be incorporated through the use of bottom up self-assembly.8,14 RNA nanoparticles normally are in the 10–50 nm size range, which is the optimal to pass through cell membranes through cell surface receptor mediated endocytosis while still being retained by the body but with little accumulation in organs.25,26,27 Finally, RNA nanoparticles can prevent antibody detection by being protein free, but maintain their selectivity using receptor-binding aptamers.28,29,30 The previous problem of instability of RNA nanoparticles once hindered the field of RNA nanotechnology31; but 2'-Fluoro (2'-F) or -OMethyl modifications to uracil and cytosine backbones not only improves the thermodynamic stability but makes the resulting RNA resistant to RNase degradation.32,33 With these benefits, a stable RNA nanoparticle would prove to be a beneficial therapeutic delivery vehicle that could be used in the specific targeting and treatment of cancers and viral diseases.
Affecting one in every six men and being the second deadliest cancer in men (behind lung cancer), prostate cancer is a severe disease that affects a wide population. There are roughly 250,000 new cases of prostate cancer diagnosed each year with 30,000 deaths (American Cancer Society statistics). LNCaP cells are a class of prostate cancer cells known for being hormone dependent and androgen sensitive for growth.34,35,36 Extensive work on prostate cancer therapies has previously been done using LNCaP cells as they are normally considered a less aggressive cancer.37,38,39,40 It is beneficial to use the early stage cancer cells as a target for therapies, as the tumor has not had time to develop into an aggressive state, is still hormonal dependent, and overall easier to manage than an hormone independent tumor. This allows for a contained treatment and a higher chance of successful removal of the disease. Furthermore, it has been shown that LNCaP tumor cells over express prostate-specific membrane antigen (PSMA),36,41,42,43 making it an even more attractive target as it can be easily identified from healthy prostate cells allowing for a specific targeting and delivery of therapeutics while having low toxicity effects on normally functioning prostate cells.
Within cancers, it has been found that many microRNA (miRNA) are either downregulated or overexpressed.44,45,46 The differences in expression from healthy tissues have shown adverse downstream effects on many protein expressions, most notably leading to downregulation of tumor suppressors and increased antiapoptotic genes. LNCaP-FGC prostate cancer cells are known to have miR17 and miR21 play important roles,47,48 two common oncomirs that are commonly seen as important players in cancers. These two microRNAs lead to the downregulation of tumor suppressors such as PTEN and PDCD4 and upregulation of antiapoptotic genes.48,49,50,51,52,53 The regulation of these important oncogenes could correct the hindered tumor suppression and sensitize the cells to apoptosis. Recently anti-miRNA LNA sequences have been developed for miR17 and miR21, resulting in the silencing of their respective miRNAs.54
Previously, a thermodynamically and chemically stable RNA three-way junction (3WJ) was discovered within the packaging RNA (pRNA) of the phi29 bacteriophage DNA packaging motor.10 pRNA forms into a hexameric ring on the packaging connector and consists of two domains, the helical domain and central domain containing two interlocking loops.55,56,57,58 The two domains are connected together through a 3WJ that has been shown to form from three short RNA oligo strands in the absence of metal ions.10 The junction also has the ability to harbor RNA moieties off each branch while still retaining the central folding structure of the 3WJ as well as the functionality of the RNA extensions.10 This ultra-stable platform has proven to be a viable scaffold to carry RNA functionalities for the use in therapeutics in cancers and viral diseases.10,59,60,61
Using the pRNA-3WJ, we propose to create a RNA molecule with specific targeting ability to prostate cancer cells to carry miRNA LNAs.54 Here, we report the use of the pRNA-3WJ for the construction of RNA nanoparticles for the specific targeting of prostate cancer tumor cells. LNCaP-FGC cells were used as in vitro cell model and the overexpression of the PSMA in LNCaP-FGC cells was used as a specific target. Targeting was achieved through the use of the PSMA A9g RNA aptamer that has been previously developed,62,63 and conjugated onto the pRNA-3WJ through bottom-up construction. Furthermore, we have conjugated anti-miRNA LNAs as well as fluorescent tags onto the remaining two branches of the three way junction for monitoring the binding and entry into the tumor cells. Nanoparticles were successfully constructed with defined stoichiometry and size that maintained the folding structures of both the pRNA-3WJ and A9g PSMA aptamer, keeping the functionality of the aptamer. The molecules further showed specific targeting to LNCaP-FGC cells in vitro through flow cytometry, cell confocal microscopy, and dual luciferase assays. Dual luciferase assays and qRT-PCR (quantitative Real Time Polymerase Chain Reaction) reported specific knockdown of miR21 and miR17 in LNCaP cells. Through these studies, we have proven that the combination of the A9g PSMA aptamer with anti-miRNA LNAs through the pRNA-3WJ into therapeutic nanoparticles, selective targeting to prostate cancer, specifically LNCaP-FGC cells, can be accomplished, providing a vehicle for therapeutical elements like siRNAs, miRNAs, or chemotherapies for tumor treatments.
Results
Construction of pRNA-3WJ nanoparticles harboring PSMA binding aptamer
The truncated PSMA A9g aptamer was placed onto the pRNA-3WJ, creating a branched RNA motif to specifically target prostate cancer cells. RNA 3WJs were created with the PSMA aptamer attached to the 3WJa/3WJc branch in three different orientations (Figures 1 and 3) in order to test if there is any variation in the aptamer folding once placed on the pRNA-3WJ. Each of the three variants of the A9g-3WJ displayed proper folding and sizing on polyacrylamide gels ran in native conditions.
Figure 1.

Design and construction of pRNA-3WJ nanoparticles harboring prostate-specific membrane antigen (PSMA) binding aptamer and anti-miRNA LNA. (a) The sequence and secondary structure of bacteriophage phi29 packaging RNA (pRNA). (b) 3WJ core of pRNA. (c) Design of pRNA-3WJ nanoparticles harboring PSMA binding aptamer and anti-miRNA LNA. (d) 10% native TBM PAGE of A9g-3WJ-anti-miR21 nanoparticle.
Next, the prostate targeting 3WJs were tested for nuclease and thermodynamic stability. First each of the 2'-F modified A9g-3WJs were incubated with 10% fetal bovine serum over 24 hours (Figure 2a). Samples were then run on polyacrylamide gels, where the band corresponding to the assembled RNA nanoparticles remained stable throughout all time points. This data shows that the RNA nanoparticles will remain stable during in vivo applications as RNases are not able to recognize and degrade the fluoro-modified nucleic acids. Furthermore, the thermodynamic stability of the PSMA targeting 3WJ was assayed using temperature-gradient gel electrophoresis. Temperature gradient was applied perpendicular to the electrical current in order to find the melting temperature of the 3WJ harboring the aptamer (Figure 2b). Previously, the melting temperature (TM) of 2'-F-modified pRNA-3WJ core was found to be 69.8 °C.64 From the temperature-gradient gel electrophoresis melting curve, the melting temperature of the A9g-3WJ was found to be 61.2 °C. Although the TM of the aptamer-3WJ complex is less than the 3WJ itself, the still rather high melting temperature of the nanoparticle indicates the PSMA A9 aptamer does not significantly hinder the stability of the pRNA-3WJ core, and in fact the 3WJ actually provides added stability to the PSMA aptamer.
Figure 2.

Stability and characterization of assembled pRNA-3WJ nanoparticles harboring prostate-specific membrane antigen binding aptamer and anti-miRNA LNA. (a) Assessment of chemical stability of A9g-3WJ-anti-miRNA LNA treated with 10% serum contained cell culture medium in 8% TBM native PAGE. (b) Assessment of thermodynamic stability of A9g-3WJ-anti-miRNA LNA by temperature-gradient gel electrophoresis assay. Melting profile of nanoparticle derived from PAGE with sigmoidal fitting of data to find melting temperature. (c) Hydrodynamic sizing and zeta potential measurements of the A9g-3WJ-anti-miR21 nanoparticle using a Zetasizer nano-ZS.
After establishing that A9g-3WJ displayed the high stability previously seen in the pRNA-3WJ, the targeting 3WJ was expanded to harbor anti-miRNA LNAs for testing the delivery of therapeutics to LNCaP-FGC cells (Figure 1c). The extension of the unoccupied branch of the 3WJ posed no problem in folding of the RNA strands, as all completed nanoparticles were assayed on polyacrylamide gels ran in native conditions (Figure 1d). Assembly gels showed high yield of the folded RNA nanoparticle, with any side product bands being contributed to mismatch in RNA concentrations during assembly. Within the assembly gel, two bands were seen from the A9g-3WJ strand and the A9g-3WJ-anti-miRNA samples; this was attributed to formation of a self-dimer of the A9g aptamer. Furthermore, assembled nanoparticles, A9g-3WJ and A9g-3WJ-anti-miR21 LNA, were tested for zeta potential and hydrodynamic size (Figure 2c). Here, it was found that the size of the nanoparticles were 3.77 ± 0.59 and 4.27 ± 0.32 nm, with a zeta potential of −18.0 ± 1.45 and −22.87 ± 2.40 mV, respectively. These values are in line with predictions and previously published results of other RNA nanoparticles.65,66,67
Targeting of pRNA-3WJ nanoparticles to LNCaP-FGC cells
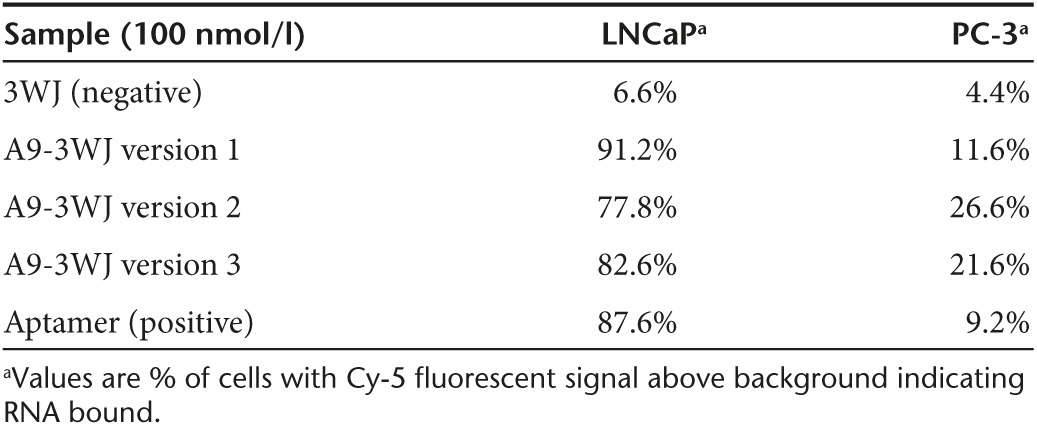
In order to test the targeting of the pRNA-3WJ, either a fluorescent Alexa647 or whole chain labeling with Cy5 fluorophore was placed on the 3WJb strand to allow for tracking of the RNA nanoparticles created and described above. The A9g-3WJ nanoparticles were incubated with LNCaP-FGC cells, as well as PC-3 cells (PSMA−) as a negative control. Following the RNA incubation and washing steps cells were analyzed by flow cytometry to confirm the binding of each of the three A9g-3WJs. FACS data from each of the designs showed strong binding (>75%) to LNCaP-FGC cells while the aptamer-negative pRNA-3WJ displayed only 6.59% binding (Figure 3). However, with this high binding, it was found that the two designs with hinging aptamers produced an increased and undesired nonspecific binding to PC-3 cells. Therefore connecting the A9g PSMA aptamer to the pRNA-3WJ directly to the two helical regions of the 3WJa/3WJc branch provided the highest binding to LNCaP-FGC (91.22%), while having low non-specific binding to PSMA- PC-3 cell line (11.60%) (Table 1). Flow cytometry data displayed that the addition of the pRNA-3WJ to the end of the PSMA A9g aptamer did not interrupt cell binding, and provided a branched scaffold for the addition of therapeutic elements such as siRNAs, miRNAs, and anti-miRNAs.
Figure 3.

Flow cytometry assay for studying specific binding of pRNA-3WJ nanoparticles on prostate cancer cells. (a) Illustration of various design of conjugating prostate-specific membrane antigen (PSMA) binding A9g aptamer onto the pRNA-3WJ core. Alexa647 labelled A9g-3WJ RNA nanoparticles were incubated with (b) PSMA+ LNCaP-FGC cells and (c) PSMA- PC-3 cells. Nanoparticles indicated positive binding of nanoparticles to LNCaP cells while avoiding nonspecific binding to PC-3 cells.
Table 1. Summary of flow cytometery binding data.
Next, endocytosis entry of the RNA nanoparticles harboring the PSMA A9g aptamer into LNCaP-FGC cells was examined. In order for proper delivery of therapeutics and RNA nanoparticles to act as anticancer agents, entry into the cells are required for proper release of therapeutic agents. Alexa647-labeled A9g-3WJ nanoparticles were incubated with LNCaP-FGC and PC-3 cells, cells were then fixed and the nuclei and cytoplasm were stained. Confocal microscopy images shows proper binding of the A9g aptamer to LNCaP cells with very little signal seen on PC-3 cells. Furthermore, confocal imaging along with Z-axis stacking imaging (Figure 4) confirmed Alexa647 signal within LNCaP-FGC cells on A9g-positive nanoparticles. Additionally, little Alexa647 signal was seen around cells samples without the PSMA aptamer and the signal was only seen accumulating around the LNCaP cells and not within. Additionally, cell binding assays were completed using VCaP (PSMA+) and Jurkat (PSMA−) cell lines to further test the specificity of the A9g-3WJ nanoparticles (data not shown).
Figure 4.

Confocal microscopy for assaying the binding and internalization of pRNA-3WJ nanoparticles via prostate-specific membrane antigen binding aptamer A9g. RNA nanoparticles were incubated with cell groups then fixed and stained for fluorescent imaging. (a) A9g-3WJ to LNCaP cells. (b) A9g-3WJ to PC-3 cells. (c) 3WJ control to LNCaP cells. (d) A9g aptamer control to LNCaP cells. Blue: DAPI-stained nucleus. Green: Phalloidin-Alexa 488-stained cytoplasm. Red: Alexa 647-labeled RNA nanoparticles. Overlay of three signals with z-axis scanning.
Delivery of anti-miRNA LNA to LNCaP-FGC cells
Anti-miRNA LNA targeting miR21 and miR17 were placed onto the PSMA A9g-3WJ by a DNA linker strand for delivery into LNCaP-FGC cells.65 Incubation studies of the RNA nanoparticles harboring the PSMA RNA aptamer with the miR17 and miR21 were then tested for the delivery using Promega's psi-check2 dual luciferase plasmid. The transfected plasmid contained sequences for Firefly and Renilla luciferase plasmids with each plasmid containing the microRNA targeting sequences at the 3'-UTR region of Renilla luciferase, respectively. Therefore in the native cells, the Renilla expression is knocked-out, as the miRNA binds to plasmid, preventing the translation of Renilla luciferase proteins, but Firefly expression remains unaffected. As anti-miRNA LNA sequences are delivered to the cells, miR17 and miR21 are knocked-down, thus resulting in an increased expression of Renilla in the dual luciferase assay.
By incubating RNA nanoparticles with LNCaP and PC-3 cells, miR17 and miR21 knock-down was assayed by the described dual luciferase assay. Data in LNCaP-FGC cells shows significant increase in Renilla expression in the A9g-3WJ-anti-miRNA LNA and anti-miRNA LNA transfected samples; however, little effect seen from PSMA aptamer negative samples for both miR17 and miR21 (Figure 5b). This shows the anti-miRNA sequences are being properly delivered to LNCaP-FGC cells by the PSMA A9g aptamer and processed by RISC. Additionally, studies completed in PC-3 cells showed significantly less increase in Renilla expression from the A9g-3WJ-anti-miRNA LNA samples, indicating RNA nanoparticles were selectively targeting LNCaP cells through PSMA binding, indicating a low toxicity to PSMA- cell lines. Furthermore, due to the fact that the aptamer sequence is on a different strand from the anti-miRNA sequences, these studies indicate that the RNA nanoparticles are remaining stable and intact through the pRNA-3WJ core throughout the studies.
Figure 5.

Assay for miRNA knockdown and downstream gene regulation effects of pRNA-3WJ nanoparticles harboring prostate-specific membrane antigen binding aptamer and anti-oncogenic miRNA LNA. (a) Dual-luciferase assay for evaluating delivered anti-miR21 LNA and (b) anti-miR17 LNA effects on prostate cancer cells from incubation of RNA nanoparticles. Knockdown of miRNAs led to spiked increase of reporter Renilla expression. (c) qPCR results of downstream genes PTEN and PDCD4 expression as a result of miR21 knockdown in LNCaP cells 72 hours postincubation with A9-3WJ-anti-miR21 LNA nanoparticles. (d) Design of A9g-3WJ-anti miR21 LNA and -anti-miR17 LNA nanoparticles. In all plots, *P < 0.1, **P < 0.01, ***P < 0.001.
To further confirm the delivery of anti-miRNA LNA sequences into LNCaP-FGC cells and the ability for the LNAs to effect downstream gene expressions, quantitative real-time PCR was completed, examining the expression of PTEN and PDCD4, known downstream genes of both miR17 and miR21. 18S was used as an internal control during these experiments to standardize the expressions of PTEN and PDCD4 mRNA. q-rtPCR results show significant increase of PTEN at only a 10 nmol/l concentration of RNA nanoparticles in the A9g-3WJ-anti-miR21 samples over the negative controls (Figure 5c). Additionally, it was seen that the RNA nanoparticles were able to slightly increase the PDCD4 expression over negative controls and the cell only samples. These data further prove the delivery of anti-miRNA LNAs to LNCaP-FGC cells through the A9g-3WJ complex. Furthermore, the increase of tumor suppressor genes not only shows a true knockdown of the miRNAs, but displays the RNA nanoparticles' ability to correct errors in gene expressions caused by the prostate cancer.
From the confirmed upregulation of tumor suppressor genes by delivery of miRNA LNAs by the 3WJ, testing of apoptotic effects were completed through assaying Caspase III signaling on LNCaP-FGC and PC-3 cells after incubation with A9g-3WJ-anti-miRNAs along with negative nanoparticle controls described before (Figure 6). The PC-3 cells and negative RNA nanoparticles with LNCaP cells were used to show the RNA nanoparticles were not toxic to the cells and apoptosis only occurred through specific delivery of the anti-miRNA LNAs. A spike in Caspase III signaling was seen in LNCaP cells treated with A9g-3WJ-anti-miR17 and transfected by RNAi Max anti-miRNA LNAs (Figure 6). This indicates that the silencing of miRNA was able to successfully lead to the death of LNCaP cells. Here, apoptosis caused by the decreased expression of miR17 shows the important role of the miRNA in LNCaP cells and the ability to control the cell growth and death through its expressions.
Figure 6.

Caspase III signaling assay for apoptosis effects of pRNA-3WJ nanoparticles harboring prostate-specific membrane antigen binding aptamer and antioncogenic miRNA LNA. RNA nanoparticles with anti-miR21 and -miR17 LNA were incubated with (a) LNCaP and (b) PC-3 cells cells for 24 hours. Caspase III signaling was assayed by fluorescent reporter with peak fluorescent of 440 nm as a reporter of cell apoptosis as a result of RNA nanoparticle delivery of anti-miRNA sequences. 5 μmol/l Camptothecin was used as a positive control and benchmark of apoptosis (**P < 0.01, ***P < 0.001).
In vivo delivery of anti-miRNA LNA to LNCaP
Subcutaneous xenograft tumors were developed in male nude mice with LNCaP C4-2 cells. Once tumors were fully developed (2 weeks) with cardio vasculature, 100 μl of 20 μmol/l solution of pRNA-3WJ nanoparticle harboring the PSMA A9g aptamer along with an Alexa647 fluorescent tag were administered to the mice through tail-vein injection. For all in vivo studies, nanoparticles were injected and delivered as bare oligos without the aid of any additives. Through a series of time points, mice were whole body imaged to examine the biodistribution of nanoparticles, along with the accumulation in tumors as well as healthy organs. Initially Alexa647 was detected throughout the whole body of the mice indicating nanoparticles successfully circulated through the mice. Through early time points, Alexa647 concentrated in the tumor, liver, and bladder of the mice; indicating the excess nanoparticles were excreted through the urine. After 8 hours, fluorescence signal was undetectable in all healthy organs, while remaining strong in the xenograft tumor (Figure 7a). Additionally, tumors were sectioned and imaged for fluorescent imaging (Figure 7c). Confocal imaging shows specific targeting and accumulation of the A9g-3WJ nanoparticles to the LNCaP xenograft tumor. This indicates nanoparticles have no lasting accumulation in the liver or kidneys, thus having low-toxicity profiles.
Figure 7.

In vivo delivery of pRNA-3WJ nanoparticles harboring prostate-specific membrane antigen binding aptamer and anti-miRNA LNA. RNA nanoparticles were delivered as bare oligos to LNCaP C4-2 subcutaneous xenografts in nude mice. (a) Biodistribution of A9g-3WJ-Alexa647 through nude mice. B: Brain, L: Lung, S: Spleen, Lv: Liver, K: Kidney, T: Tumor. (b) Tumor-bearing nude mice were administered nanoparticles through a series of five injections through the tail vein of 20 μmol/l solution at 100 μl (indicated by arrows), while tumor volume (mm3) and total mouse weight (g) were monitored (*P < 0.05, **P < 0.01, ***P < 0.0001). (c) Fluorescent confocal microscopy of tumor samples 8 hours post-i.v. administration of RNA nanoparticles. DAPI (Blue) is cell nucleus; Alexa647 (red) is RNA nanoparticle. (d) Western blot examining downstream expression of PTEN and PDCD4 from silencing of miR17 and miR21 using β-Actin as an internal control.
Furthermore, the therapeutic effects of the A9g-3WJ-anti-miRNA nanoparticles were tested in vivo on LNCaP C4-2 subcutaneous xenografts. After tumors were developed to a volume of ~200 mm3, nanoparticles harboring the anti-miR17 and -miR21 LNAs were administered to the mice through a series of five tail-vein injections. Tumor volume and total weight of the mice were monitored throughout this experiment. From the tumor volume data (Figure 7b), it can clearly be seen that the A9-3WJ-anti-miRNA samples led to a stunted tumor growth and proliferation compared to negative controls, thus indicating the anti-miRNA oligos had a profound effect on the tumor environment and cell growth. Even after postadministration of the nanoparticles, a lack of growth in the tumors was seen for several days compared to nontreated tumors. Furthermore, there was no significant change in the mice weights over the observed RNA nanoparticle delivery indicating no toxicity in the mice and the tumor reduction was as a result of specific delivery to the tumors.
At the conclusion of in vivo delivery of the pRNA-3WJ nanoparticles, mice were sacrificed and tumors were harvested. PTEN and PDCD4 expressions were measured in the tumors, using β-Actin as an internal control. Western blots (Figure 7d) show an increase in PTEN and PDCD4 in the pRNA-3WJ nanoparticles harboring the anti-miRNA LNAs. These results display the same ability of the pRNA-3WJ nanoparticles as seen in vitro with the LNCaP cells, as the anti-miRNA sequences led to the increase of expression tumor suppressor genes as a result of decreased miR21 and miR17 expressions. On average an increase was seen in the A9g-3WJ sample over the control but less than A9g-3WJ-anti-miRNA groups. This may have been due to the repeated dosage of the RNA to the tumor cell in the animal trials, the larger volume of the nanoparticle may have had an effect on the gene expression within the tumor; however, the change in gene expression was not enough to create a change in tumor growth as shown in Figure 7b.
Discussion
An RNA aptamer using 2′-Fluoro-modified nucleotides was generated through SELEX for specific targeting of PSMA by Lupold et al.62 Furthermore, Rockey et al.63 rationally truncated the A9 PSMA aptamer to shorten the overall length while keeping the high specificity to PSMA. It is well known that prostate-specific membrane antigen is overexpressed in LNCaP-FGC prostate cancer cells.35,36,68–71,35,36,64–67 PSMA is an important target since it is overexpressed in primary prostate tumors as well as metastases in the lymph nodes.72,68 Furthermore, PSMA has been shown to be upregulated in tumors after patients have been treated with common androgen-deprivation therapies.70,73,66,69 The truncated PSMA A9g aptamer was placed onto the pRNA-3WJ, creating a branched RNA motif to specifically target prostate cancer cells.
Resulting nanoparticles display that PSMA A9g aptamer is not only specifically binding to PSMA+ cells, but entering through receptor mediated endocytosis as expected. Here, very similar binding profiles between flow cytometry and confocal microscopy were seen with LNCaP cells further confirming the targeting of the A9g aptamer. With positive detection of nanoparticles in the LNCaP-FGC cells, it is possible to deliver therapeutics in hopes of leading to apoptosis of LNCaP cells through RNA interference (RNAi). Upon confirmation of PSMA targeting nanoparticles entering into LNCaP-FGC cells, experiments were expanded to test the delivery of therapeutic components. LNCaP-FGC cells are known to overexpress miR17 and express miR21, two common oncomirs that are seen in cancers.47,48 These two microRNAs lead to the downregulation of tumor suppressors such as PTEN and PDCD4 and upregulation of antiapoptotic genes.48,49,50,51,52,53 Additionally, miR-21 has been shown to promote hormone independency prostate cancers74 and enhances the invasiveness of LNCaP cells75; while miR17 has been proven to promote tumor growth and invasiveness in prostate cancer cells.76 Therefore, silencing miR17 and miR21 could lead to the sensitization of the LNCaP-FGC cells leading to apoptosis. Anti-miRNA sequences have been developed to target, and bind miR17 and miR21, thus blocking their gene-silencing roles.54
The prostate cancer targeting RNA nanoparticles delivering the anti-miRNA LNAs shows the vast possibility of RNA nanoparticles. Here, we show constructed nanoparticles using the pRNA-3WJ were stable in fetal bovine serum, being resistant to RNases and stable at temperatures well above the 37 °C needed for in vivo applications. Furthermore, the 3WJ harboring the PSMA aptamer and anti-miRNA LNAs and positive functionality of each shows the versatility of the pRNA-3WJ to harbor RNA moieties while keeping the original functionality of each of the RNA groups, i.e., the targeting and binding of the PSMA aptamer and the silencing ability of the anti-miRNA LNAs. The LNCaP prostate cancer-specific nanoparticles produces excellent binding profiles, displaying binding at over 90% at 100 nmol/l RNA concentrations and later proved to provide specific delivery of anti-miRNA sequences for the knockdown of miR17 and miR21. Significant knockdown of miR17 and miR21 and upregulation of PTEN indicated the positive and specific delivery by the pRNA-3WJ RNA nanoparticles in in vitro and in vivo. We have demonstrated not only the ability of the RNA nanoparticles to accumulate and target the tumor specifically with high affinity, but deliver antioncogenic RNA sequences to regulate tumor growth and development of tumors.
Through these studies, RNA nanoparticles have been developed that have the capability to target tumors primary and metastatic tumors as well tumors after hormone deprivation treatments. Furthermore, anti-miRNA sequences have been shown to control tumor growth and proliferation, while having the possibility to prevent PSMA tumors from transitioning to hormonal independent states. Overall, stable RNA nanoparticles can be constructed for the specific targeting and treatment of cancers, and past hurdles that have held back the field of RNA nanotechnology are no longer a concern.
Conclusions
RNA nanoparticles were constructed using the pRNA-3WJ core from the phi29 packaging motor for specific targeting and treatment of prostate cancer cells. RNA nanoparticles were proven to remain chemically and thermodynamically stable after the addition of functional groups off each branch. The LNCaP prostate cancer-specific nanoparticles produces excellent binding profiles, displaying binding at over 90% at 100 nmol/l RNA concentrations and later proved to provide specific delivery of anti-miRNA sequences for the knockdown of miR17 and miR21, two common oncogenes. Significant knockdown of miR17 and miR21 and upregulation of PTEN indicated the positive and specific delivery by the pRNA-3WJ RNA nanoparticles in in vitro and in vivo.
Materials and Methods
Design and construction of pRNA-3WJ nanoparticles harboring PSMA binding aptamer and anti-miRNA LNA. RNA nanoparticles were constructed using a bottom-up approach, as previously described. Briefly, DNA oligos primers (Integrated DNA Technologies, Coralville, Iowa) were used to create dsDNA templates through polymerase chain reaction (PCR) for each RNA strand. RNA strands were then transcribed by T7 polymerase in vitro. Additionally, 2'-fluoro (2'-F)-modified cytosine and uracil were used in transcriptions along with an Y639F-mutant T7 polymerase, giving nuclease stability to the RNA strands. The anti-micro RNA LNAs were synthesized by Exiqon (Woburn, MA).
Transcribed RNA strands were purified on 8% polyacrylamide gels containing 8 mol/l Urea ran at 120 V for 1.5 hours on TBE running buffer (89 mmol/l Tris-borate, 2 mmol/l Ethylenediaminetetraacetic acid (EDTA)). RNA bands of interest were excised from the gel using UV shadow on thin layer chromatography plates and eluted from gel in elution buffer (0.5 M Ammonium Acetate, 0.1 mmol/l EDTA, 0.1% SDS) at 37 °C for a minimum of 3 hours and then precipitated by ethanol.
RNA nanoparticles were assembled by mixing strands at equal molar concentrations in TMS buffer (50 mmol/l Tris pH 8.0, 100 mmol/l NaCl, 10 mmol/l MgCl2) and heated to 80 °C for 5 minutes and slowly cooled over 40 minutes to 4 °C. Assembled RNA nanoparticles were then purified on 8% polyacrylamide gels at 100 V for 2 hours on TBM running buffer (89 mmol/l Tris, 200 mmol/l borate acid, and 5 mmol/l MgCl2) and at 4 °C. Samples were then excised and eluted as described above.
Temperature-gradient gel electrophoresis assay for studying thermodynamic stability of RNA nanoparticles. For temperature-gradient gel electrophoresis analysis, the experimental setup was adjusted to have a linear temperature gradient perpendicular to the electric field (BiometraHmbGh, Göttingen, Germany). The temperature gradient was set from 36.0 to 80.0 °C. RNA sample (500 nmol/l) was combined with 6× gel-loading buffer and run on 8% native polyacrylamide gel at 100 V for 1 hour. 10 mmol/l MgCl2 was present in both gel and electrophoresis buffer. The nanoparticles contained a Cy5 fluorophore on the 3WJb strand for monitoring melting and imaged by Typhoon FLA 7000 (GE Healthcare, Pittsburgh, PA). The pRNA-3WJ constructs within the total RNA was analyzed by ImageJ, and the melting curve of the construct was fitted using nonlinear Sigmoidal fitting. Melting temperatures (TM) were calculated where 50% of total RNA concentration was at ssRNA and 50% was in complexed 3WJ form.
Serum stability assay for studying chemical stability of RNA nanoparticles. Four hundred nanograms of pRNA-3WJ nanoparticle were incubated in TMS buffer containing fetal bovine serum (final concentration is 10%). The total volume was 10 µl. Samples were taken at multiple time points, including 0, 0.25, 0.5, 1, 2, 6, 12, and 24 hours after incubation at 37 °C. 8% Native TBM PAGE gel electrophoresis was applied to visualize RNA. After running for 1.5 hours at 4 °C, the gel was stained by ethidium bromide. Images were taken by Typhoon FLA 7000 (GE Healthcare).
Dynamic light scattering and zeta potential measurement of RNA nanoparticles. Apparent hydrodynamic sizes and zeta potential of pre-assembled A9g-3WJ-anti-miR21 LNA and A9g-3WJ nanoparticles were measured by a Zetasizer nano-ZS (Malvern Instrument, Malvern, UK). All RNA samples were measured at 2 μmol/l in Diethylpyrocarbonate H2O and PBS buffer (137 mmol/l NaCl, 2.7 mmol/l KCl, 100 mmol/l Na2HPO4, 2 mmol/l KH2PO4, pH 7.4) at 25 °C.
Cell culture. Human prostate cancer cell lines LNCaP-FGC, LNCaP C4-2 and PC-3 (American Type Culture Collection, Manassas, MA) were grown and cultured in RPMI 1640 medium (Invitrogen, Grand Island, NY) containing both 10% fetal bovine serum and penicillin/streptomycin in a 37 °C incubator with a 5% CO2 and a humidified atmosphere.
Flow cytometry assay of PSMA pRNA-3WJ nanoparticles binding to LNCaP-FGC cells. LNCaP-FGC and PC-3 cells were trypsinized and rinsed with blank RPMI-1640 medium. 100 nmol/l Cy5-labeled A9g-3WJ and the control pRNA-3WJ were each incubated with 2 × 105 LNCaP-FGC or PC-3 cells at 37 °C for 2 hours. After washing with PBS (137 mmol/l NaCl, 2.7 mmol/l KCl, 100 mmol/l Na2HPO4, 2 mmol/l KH2PO4, pH 7.4), the cells were resuspended in PBS buffer. Flow cytometry was performed by the UK Flow Cytometry & Cell Sorting core facility to observe the cell binding efficacy of the Cy-5 A9g-3WJ nanoparticles. Analysis was completed using Flowing Software v2.5 (Turku Centre for Biotechnology, Turku, Finland).
Confocal microscopy imaging analyzing RNA nanoparticle binding and entry into LNCaP-FGC cells. LNCaP-FGC and PC-3 cells were grown on glass coverslides in RPMI-1640 medium overnight. 100 nmol/l concentration Alexa647-labeled A9g-3WJ and the control pRNA-3WJ were each incubated with the cells at 37 °C for 2 hours. After washing with PBS, the cells were fixed by 4% paraformaldehyde and stained by Alexa Fluor 488 phalloidin (Invitrogen) for cytoskeleton and TO-PRO-3 iodide (642⁄661) (Invitrogen) for nucleus. The cells were then assayed for binding and cell entry by Zeiss LSM 510 laser scanning confocal microscope (Thornwood, New York).
Dual luciferase assay to analyze delivery of anti-miRNA by pRNA-3WJ nanoparticles. LNCaP-FGC and PC-3 cells were grown on 24-well plates in RPMI-1640 medium until reaching ~80–90% confluency. 100 and 50 nmol/l A9g-3WJ-anti miRNA LNA and the control RNAs including pRNA-3WJ/anti-miRNA-(17 or 21) LNA and A9g-3WJ were incubated with cells in opti-modified essential media (MEM) at 37 °C for 3 hours. As a positive control, anti-miRNA LNA transfected by RNAi MAX (following standard protocol by Invitrogen) into cells, while complete nanoparticles were merely incubated with cells. After incubation with the RNA, cells were washed once with blank RPMI-1640 medium and then transfected with psi-Check 2 plasmid (Promega, Madison, WI) which contains an oncogenic miRNA targeting sequences at the 3'-UTR region of Renilla Luciferase gene using Lipofectamine 2000 (Life Technologies, Carlsbad, CA). Dual-luciferase assay (Promega) was used to evaluate the anti-miRNA LNA effects 24 hours post-transfection upon manufacture's instruction. Briefly, cells were washed once with PBS and lysed with passive lysis buffer. The plates were shaken for 30 minutes at room temperature. 20 μl of the lysate were added to 50 μl of luciferase assay reagent (LAR II) in a luminometer tube and firefly luciferase activity was measured. Upon addition of 50 μl of Stop & Glo Reagent, control measurements of Renilla luciferase activity were then obtained. The Renilla luciferase activity obtained was then normalized with respect to the Firefly luciferase activity for determining the average ratio of Renilla to Firefly luciferase activity over several trials. Results were statistically analyzed by an analysis of variance two-way test in comparing Renilla expression to the cell only control and between PC-3 and LNCaP cell lines among each concentration.
qRT-PCR assay. Two A9g-pRNA-anti-miRNA LNA constructs were assayed for the subsequent gene upregulation effects: one harboring anti-miR21 LNA, and the other harboring anti-miR17 LNA.
LNCaP-FGC cells were incubated with 10 and 100 nmol/l of the individual A9g-3WJ-anti-miRNA LNA and control RNAs as described above. After 72-hour treatment, cells were collected and target gene upregulation effects were assessed by qRT-PCR. PC-3 cells were used as negative control cell line.
Cells were processed for total RNA using Trizol RNA extraction reagent following manufacture's instruction (Life Technologies). The first cDNA strand was synthesized on total RNA (1 μg) from cells with the various RNAs treatment using SuperScript III First-Strand Synthesis System (Invitrogen). Real-time PCR was performed using Taqman assay. All reactions were carried out in a final volume of 20 μl using Taqman Fast Universal PCR Master Mix and assayed in triplicate. Primers/probe set for human PTEN and 18S were purchased from Life Technologies. PCR was performed on Step-One Plus real time PCR system (Applied Biosystem, Foster City, CA). The data were analyzed by the comparative CT Method (ΔΔCT Method). Data were statistically analyzed by an analysis of variance one-way test in comparing values with the cell only control in each concentration and gene.
Caspase III assay for studying cell viability upon pRNA-3WJ nanoparticle treatment. LNCaP-FGC cells were plated in 24-well plate and incubated with 100 nmol/l A9g-3WJ-anti miRNA LNA constructs and control RNAs as described above at 37 °C for 24 hours. A positive control of incubating cells with 5 μmol/l Camptothecin for 4 hours at 37 °C was used. Caspase III signaling was then assayed using Caspase III Assay Kit (BD Pharmigen, San Jose, CA) following the standard protocol. Cells were washed in 1× PBS buffer followed by incubation in 200 μl Cell Lysate Buffer for 30 minutes on ice. Hundred microliters of cell lysate were then incubated with 1 ml of 1× HEPES (4-(2-hydroxethyl)-1-piperazineethanesulfonic acid) buffer and 15 μl Ac-DEVD-AMC fluorescent substrate and incubated at 37 °C for 1 hour. Fluorescence was then measured from 420 to 460 nm with an excitation of 380 nm on fluorospectrometer (Horiba Jobin Yvon; SPEX Fluolog-3, Edison, NJ). Peak fluorescence at 440 nm was used to analyze Caspase III signaling.
In vivo biodistribution and tumor targeting of RNA nanoparticles. LNCaP C4-2 cells were cultured in vitro and subcutaneously injected under the skin of 4-week-old male nude mice. A total of 2 × 106 cells were injected in solution with Matrigel (Corning; Corning, New York) as a 50/50 % blend. Tumors were grown for 4 weeks until tumors reached a volume of 200 mm3. Mice were then administered PBS (blank control), pRNA-3WJ (negative control), and A9g-3WJ each with Alexa647 labels as naked oligos at a dose of 2 μmol/l at 100 μl through the tail vein. Mice were imaged for whole body fluorescence at time points 0, 1, 2, 3, 4, and 8 hours with an In Vivo Imaging System (IVIS) imager (Caliper Life Sciences, Waltham, MA). Upon the completion of the study, mice were sacrificed, and tumors, hearts, kidneys, livers, and brains were collected and imaged by the whole body imager for Alexa647 signal.
Furthermore, tumors were fixed in 4% paraformaldehyde with 10% sucrose in 1× PBS buffer at 4 °C overnight. Tumor samples were then placed in Tissue-Tek Optimum Cutting Temperature compound (Sakura Finetek USA, Torrance, CA) for frozen sectioning (10 μm thick). Sectioned tissue were then stained with DAPI (4′,6-diamidino-2-phenylindolem dihydrochloride) and mounted with ProLong Gold Anti-fade Reagent (Life Technologies) overnight. Slides were then fluorescently imaged by FluoView FV1000-Filter Confocal Microscope System (Olympus, Pittsburgh, PA).
In vivo tumor reduction by anti-miRNA LNA pRNA-3WJ nanoparticles. LNCaP C4-2 cells were grown and subcutaneously injected into 4-week-old male nude mice. 2 × 106 cells were injected as a 50% cell, 50% Matrigel (Corning, Corning, NY) blend into the flank of the mice with a total of five mice hosting two tumors each. Xenograft tumors were monitored until they reached roughly 200 mm3 in volume. Naked RNA nanoparticles harboring the PSMA A9g aptamer and anti-miRNA LNAs were injected through the tail vein of mice at a concentration of 20 μmol/l at 100 μl on days 7, 10, 13, 17, and 20 while monitoring tumor volume and total mouse weight up to day 29.
Western blot of miRNA downstream genes of xenograft tumors. Upon the completion of tumor reduction experiments, mice were sacrificed and tumors were harvested. LNCaP C4-2 tumors were homogenized in RIPA buffer containing Protease (ThermoFisher, Waltham, MA). Total protein concentration was measured by BCA protein assay (ThermoFisher) against protein standards. Hundred micrograms of total protein from the tumor were loaded onto TDX FastCast SDS PAGE gels (BioRad, Hercules, CA) and ran at 100 V for 2 hours. Gels were transferred onto membranes and stained with PTEN, PDCD4, and β-Actin antibodies (Cell Signaling, Danvers, MA) and imaged. β-Actin was used as an internal control and PTEN and PDCD4 are expressed as a ratio with β-Actin.
Acknowledgments
This research was supported by NIH grants R01EB003730 and U01CA151648 to P.G., R01CA186100 to B.G., and NCI CNTC training grant R25CA153954 to B.A. The UK Flow Cytometry & Cell Sorting core facility is supported in part by the Office of the Vice President for Research, the Markey Cancer Center and an NCI Center Core Support Grant (P30 CA177558) to the University of Kentucky Markey Cancer Center. Funding to Peixuan Guo's Endowed Chair position in Nanobiotechnology position is by the William Fairish Endowment Fund. Peixuan Guo's Sylvan G. Frank Endowed Chair position is funded by the CM Chen Foundation. P.G. is a consultant of Oxford Nanopore. His inventions at the University of Kentucky have been licensed to the Matt Holding and RNA Nanobio, Ltd.
References
- Seeman, NC (1998). DNA nanotechnology: novel DNA constructions. Annu Rev Biophys Biomol Struct 27: 225–248. [DOI] [PubMed] [Google Scholar]
- Seeman, NC (2010). Nanomaterials based on DNA. Annu Rev Biochem 79: 65–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeman, NC (2001) DNA nicks and nodes and nanotechnology. Nano Letters, 1: 22–26. [Google Scholar]
- Pinheiro, AV, Han, D, Shih, WM and Yan, H (2011). Challenges and opportunities for structural DNA nanotechnology. Nat Nanotechnol 6: 763–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellini, M, Mazzucchelli, S, Galbiati, E, Sommaruga, S, Fiandra, L, Truffi, M et al. (2014). Protein nanocages for self-triggered nuclear delivery of DNA-targeted chemotherapeutics in cancer cells. J Control Release 196: 184–196. [DOI] [PubMed] [Google Scholar]
- Guo, P (2010). The emerging field of RNA nanotechnology. Nat Nanotechnol 5: 833–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu, Y, Pi, F, Sharma, A, Rajabi, M, Haque, F, Shu, D et al. (2014). Stable RNA nanoparticles as potential new generation drugs for cancer therapy. Adv Drug Deliv Rev 66: 74–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu, D, Moll, WD, Deng, Z, Mao, C and Guo, P (2004). Bottom-up assembly of RNA arrays and superstructures as potential parts in nanotechnology. Nano Lett 4: 1717–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, P, Zhang, C, Chen, C, Garver, K and Trottier, M (1998). Inter-RNA interaction of phage phi29 pRNA to form a hexameric complex for viral DNA transportation. Mol Cell 2: 149–155. [DOI] [PubMed] [Google Scholar]
- Shu, D, Shu, Y, Haque, F, Abdelmawla, S and Guo, P (2011). Thermodynamically stable RNA three-way junction for constructing multifunctional nanoparticles for delivery of therapeutics. Nat Nanotechnol 6: 658–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonin, KA, Bindewald, E, Yaghoubian, AJ, Voss, N, Jacovetty, E, Shapiro, BA et al. (2010). In vitro assembly of cubic RNA-based scaffolds designed in silico. Nat Nanotechnol 5: 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellington, AD and Szostak, JW (1992). Selection in vitro of single-stranded DNA molecules that fold into specific ligand-binding structures. Nature 355: 850–852. [DOI] [PubMed] [Google Scholar]
- Gold, L (1995). The SELEX process: a surprising source of therapeutic and diagnostic compounds. Harvey Lect 91: 47–57. [PubMed] [Google Scholar]
- Khaled, A, Guo, S, Li, F and Guo, P (2005). Controllable self-assembly of nanoparticles for specific delivery of multiple therapeutic molecules to cancer cells using RNA nanotechnology. Nano Lett 5: 1797–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, S, Tschammer, N, Mohammed, S and Guo, P (2005). Specific delivery of therapeutic RNAs to cancer cells via the dimerization mechanism of phi29 motor pRNA. Hum Gene Ther 16: 1097–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, S, Huang, F and Guo, P (2006). Construction of folate-conjugated pRNA of bacteriophage phi29 DNA packaging motor for delivery of chimeric siRNA to nasopharyngeal carcinoma cells. Gene Ther 13: 814–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeprich, S, Zhou, Q, Guo, S, Shu, D, Qi, G, Wang, Y et al. (2003). Bacterial virus phi29 pRNA as a hammerhead ribozyme escort to destroy hepatitis B virus. Gene Ther 10: 1258–1267. [DOI] [PubMed] [Google Scholar]
- Sarver, N, Cantin, EM, Chang, PS, Zaia, JA, Ladne, PA, Stephens, DA et al. (1990). Ribozymes as potential anti-HIV-1 therapeutic agents. Science 247: 1222–1225. [DOI] [PubMed] [Google Scholar]
- Liu, H, Guo, S, Roll, R, Li, J, Diao, Z, Shao, N et al. (2007). Phi29 pRNA vector for efficient escort of hammerhead ribozyme targeting survivin in multiple cancer cells. Cancer Biol Ther 6: 697–704. [DOI] [PubMed] [Google Scholar]
- Pegtel, DM, Cosmopoulos, K, Thorley-Lawson, DA, van Eijndhoven, MA, Hopmans, ES, Lindenberg, JL et al. (2010). Functional delivery of viral miRNAs via exosomes. Proc Natl Acad Sci USA 107: 6328–6333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Y, Zhu, X, Zhang, X, Liu, B and Huang, L (2010). Nanoparticles modified with tumor-targeting scFv deliver siRNA and miRNA for cancer therapy. Mol Ther 18: 1650–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye, X, Liu, Z, Hemida, MG and Yang, D (2011). Targeted delivery of mutant tolerant anti-coxsackievirus artificial microRNAs using folate conjugated bacteriophage Phi29 pRNA. PLoS One 6: e21215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler, WC, Nahvi, A, Roth, A, Collins, JA and Breaker, RR (2004). Control of gene expression by a natural metabolite-responsive ribozyme. Nature 428: 281–286. [DOI] [PubMed] [Google Scholar]
- Mulhbacher, J, St-Pierre, P and Lafontaine, DA (2010). Therapeutic applications of ribozymes and riboswitches. Curr Opin Pharmacol 10: 551–556. [DOI] [PubMed] [Google Scholar]
- Gao, H, Shi, W and Freund, LB (2005). Mechanics of receptor-mediated endocytosis. Proc Natl Acad Sci USA 102: 9469–9474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain, KK (2005). The role of nanobiotechnology in drug discovery. Drug Discov Today 10: 1435–1442. [DOI] [PubMed] [Google Scholar]
- Li, W and Szoka, FC Jr (2007). Lipid-based nanoparticles for nucleic acid delivery. Pharm Res 24: 438–449. [DOI] [PubMed] [Google Scholar]
- Abdelmawla, S, Guo, S, Zhang, L, Pulukuri, SM, Patankar, P, Conley, P et al. (2011). Pharmacological characterization of chemically synthesized monomeric phi29 pRNA nanoparticles for systemic delivery. Mol Ther 19: 1312–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu, Y, Cinier, M, Shu, D and Guo, P (2011). Assembly of multifunctional phi29 pRNA nanoparticles for specific delivery of siRNA and other therapeutics to targeted cells. Methods 54: 204–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerchia, L, Giangrande, PH, McNamara, JO and de Franciscis, V (2009). Cell-specific aptamers for targeted therapies. Methods Mol Biol 535: 59–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, P, Haque, F, Hallahan, B, Reif, R and Li, H (2012). Uniqueness, advantages, challenges, solutions, and perspectives in therapeutics applying RNA nanotechnology. Nucleic Acid Ther 22: 226–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallan, PS, Greene, EM, Jicman, PA, Pandey, RK, Manoharan, M, Rozners, E et al. (2011). Unexpected origins of the enhanced pairing affinity of 2'-fluoro-modified RNA. Nucleic Acids Res 39: 3482–3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, J, Guo, S, Cinier, M, Shlyakhtenko, LS, Shu, Y, Chen, C et al. (2011). Fabrication of stable and RNase-resistant RNA nanoparticles active in gearing the nanomotors for viral DNA packaging. ACS Nano 5: 237–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horoszewicz, JS, Leong, SS, Kawinski, E, Karr, JP, Rosenthal, H, Chu, TM et al. (1983). LNCaP model of human prostatic carcinoma. Cancer Res 43: 1809–1818. [PubMed] [Google Scholar]
- Pettaway, CA, Pathak, S, Greene, G, Ramirez, E, Wilson, MR, Killion, JJ et al. (1996). Selection of highly metastatic variants of different human prostatic carcinomas using orthotopic implantation in nude mice. Clin Cancer Res 2: 1627–1636. [PubMed] [Google Scholar]
- Israeli, RS, Powell, CT, Corr, JG, Fair, WR and Heston, WD (1994). Expression of the prostate-specific membrane antigen. Cancer Res 54: 1807–1811. [PubMed] [Google Scholar]
- Chen, Z, Penet, MF, Nimmagadda, S, Li, C, Banerjee, SR, Winnard, PT Jr et al. (2012). PSMA-targeted theranostic nanoplex for prostate cancer therapy. ACS Nano 6: 7752–7762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrich, M, Raum, T, Lutterbuese, R, Voelkel, M, Deegen, P, Rau, D et al. (2012). Regression of human prostate cancer xenografts in mice by AMG 212/BAY2010112, a novel PSMA/CD3-Bispecific BiTE antibody cross-reactive with non-human primate antigens. Mol Cancer Ther 11: 2664–2673. [DOI] [PubMed] [Google Scholar]
- Wu, X, Ding, B, Gao, J, Wang, H, Fan, W, Wang, X et al. (2011). Second-generation aptamer-conjugated PSMA-targeted delivery system for prostate cancer therapy. Int J Nanomedicine 6: 1747–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dassie, JP, Liu, XY, Thomas, GS, Whitaker, RM, Thiel, KW, Stockdale, KR et al. (2009). Systemic administration of optimized aptamer-siRNA chimeras promotes regression of PSMA-expressing tumors. Nat Biotechnol 27: 839–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway, RE, Petrovic, N, Li, Z, Heston, W, Wu, D and Shapiro, LH (2006). Prostate-specific membrane antigen regulates angiogenesis by modulating integrin signal transduction. Mol Cell Biol 26: 5310–5324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao, V and Bacich, DJ (2006). Prostate specific membrane antigen (PSMA) expression gives prostate cancer cells a growth advantage in a physiologically relevant folate environment in vitro. Prostate 66: 867–875. [DOI] [PubMed] [Google Scholar]
- Silver, DA, Pellicer, I, Fair, WR, Heston, WD and Cordon-Cardo, C (1997). Prostate-specific membrane antigen expression in normal and malignant human tissues. Clin Cancer Res 3: 81–85. [PubMed] [Google Scholar]
- Jansson, MD and Lund, AH (2012). MicroRNA and cancer. Mol Oncol 6: 590–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassen, S, Miska, EA and Caldas, C (2008). MicroRNA: implications for cancer. Virchows Arch 452: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, YS and Dutta, A (2009). MicroRNAs in cancer. Annu Rev Pathol 4: 199–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong, AY, Eischeid, AN, Xiao, J, Zhao, J, Chen, D, Wang, ZY et al. (2012). miR-17-5p targets the p300/CBP-associated factor and modulates androgen receptor transcriptional activity in cultured prostate cancer cells. BMC Cancer 12: 492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang, Y, Young, CY and Yuan, H (2010). MicroRNAs and prostate cancer. Acta Biochim Biophys Sin (Shanghai) 42: 363–369. [DOI] [PubMed] [Google Scholar]
- Li, T, Li, D, Sha, J, Sun, P and Huang, Y (2009). MicroRNA-21 directly targets MARCKS and promotes apoptosis resistance and invasion in prostate cancer cells. Biochem Biophys Res Commun 383: 280–285. [DOI] [PubMed] [Google Scholar]
- Meng, F, Henson, R, Wehbe-Janek, H, Ghoshal, K, Jacob, ST and Patel, T (2007). MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology 133: 647–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherr, M, Elder, A, Battmer, K, Barzan, D, Bomken, S, Ricke-Hoch, M et al. (2014). Differential expression of miR-17~92 identifies BCL2 as a therapeutic target in BCR-ABL-positive B-lineage acute lymphoblastic leukemia. Leukemia 28: 554–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriely, G, Wurdinger, T, Kesari, S, Esau, CC, Burchard, J, Linsley, PS et al. (2008). MicroRNA 21 promotes glioma invasion by targeting matrix metalloproteinase regulators. Mol Cell Biol 28: 5369–5380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickramasinghe, NS, Manavalan, TT, Dougherty, SM, Riggs, KA, Li, Y and Klinge, CM (2009). Estradiol downregulates miR-21 expression and increases miR-21 target gene expression in MCF-7 breast cancer cells. Nucleic Acids Res 37: 2584–2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obad, S, dos Santos, CO, Petri, A, Heidenblad, M, Broom, O, Ruse, C et al. (2011). Silencing of microRNA families by seed-targeting tiny LNAs. Nat Genet 43: 371–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz, C and Guo, P (2013). Ultrastable pRNA hexameric ring gearing hexameric phi29 DNA-packaging motor by revolving without rotating and coiling. Curr Opin Biotechnol 24: 581–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trottier,M, Garver,K, Zhang,C and Guo,P (1997) DNA-packaging pRNA as target for complete inhibition of viral assembly in vitro and in vivo. Nucleic Acids Symposium Series, 36: 187–189. [Google Scholar]
- Reid, RJ, Bodley, JW and Anderson, D (1994). Identification of bacteriophage phi 29 prohead RNA domains necessary for in vitro DNA-gp3 packaging. J Biol Chem 269: 9084–9089. [PubMed] [Google Scholar]
- Guo, P and Trottier,M (1994) Biological and biochemical properties of the small viral RNA (pRNA) essential for the packaging of the double-stranded DNA of phage phi29. Seminars in Virology, 5: 27–37. [Google Scholar]
- Lee, TJ, Haque, F, Shu, D, Yoo, JY, Li, H, Yokel, RA et al. (2015). RNA nanoparticle as a vector for targeted siRNA delivery into glioblastoma mouse model. Oncotarget 6: 14766–14776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rychahou, P, Haque, F, Shu, Y, Zaytseva, Y, Weiss, HL, Lee, EY et al. (2015). Delivery of RNA nanoparticles into colorectal cancer metastases following systemic administration. ACS Nano 9: 1108–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haque, F, Shu, D, Shu, Y, Shlyakhtenko, LS, Rychahou, PG, Evers, BM et al. (2012). Ultrastable synergistic tetravalent RNA nanoparticles for targeting to cancers. Nano Today 7: 245–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupold, SE, Hicke, BJ, Lin, Y and Coffey, DS (2002). Identification and characterization of nuclease-stabilized RNA molecules that bind human prostate cancer cells via the prostate-specific membrane antigen. Cancer Res 62: 4029–4033. [PubMed] [Google Scholar]
- Rockey, WM, Hernandez, FJ, Huang, SY, Cao, S, Howell, CA, Thomas, GS et al. (2011). Rational truncation of an RNA aptamer to prostate-specific membrane antigen using computational structural modeling. Nucleic Acid Ther 21: 299–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binzel, DW, Khisamutdinov, EF and Guo, P (2014). Entropy-driven one-step formation of Phi29 pRNA 3WJ from three RNA fragments. Biochemistry 53: 2221–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu, D, Li, H, Shu, Y, Xiong, G, Carson, WE 3rd, Haque, F et al. (2015). Systemic Delivery of Anti-miRNA for Suppression of Triple Negative Breast Cancer Utilizing RNA Nanotechnology. ACS Nano 9: 9731–9740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khisamutdinov, EF, Li, H, Jasinski, DL, Chen, J, Fu, J and Guo, P (2014). Enhancing immunomodulation on innate immunity by shape transition among RNA triangle, square and pentagon nanovehicles. Nucleic Acids Res 42: 9996–10004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasinski, DL, Khisamutdinov, EF, Lyubchenko, YL and Guo, P (2014). Physicochemically tunable polyfunctionalized RNA square architecture with fluorogenic and ribozymatic properties. ACS Nano 8: 7620–7629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregor, PD, Wolchok, JD, Turaga, V, Latouche, JB, Sadelain, M, Bacich, D et al. (2005). Induction of autoantibodies to syngeneic prostate-specific membrane antigen by xenogeneic vaccination. Int J Cancer 116: 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh, A, Wang, X, Klein, E and Heston, WD (2005). Novel role of prostate-specific membrane antigen in suppressing prostate cancer invasiveness. Cancer Res 65: 727–731. [PubMed] [Google Scholar]
- Meller, B, Bremmer, F, Sahlmann, CO, Hijazi, S, Bouter, C, Trojan, L et al. (2015). Alterations in androgen deprivation enhanced prostate-specific membrane antigen (PSMA) expression in prostate cancer cells as a target for diagnostics and therapy. EJNMMI Res 5: 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben Jemaa, A, Bouraoui, Y, Sallami, S, Banasr, A, Ben Rais, N, Ouertani, L et al. (2010). Co-expression and impact of prostate specific membrane antigen and prostate specific antigen in prostatic pathologies. J Exp Clin Cancer Res 29: 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweat, SD, Pacelli, A, Murphy, GP and Bostwick, DG (1998). Prostate-specific membrane antigen expression is greatest in prostate adenocarcinoma and lymph node metastases. Urology 52: 637–640. [DOI] [PubMed] [Google Scholar]
- Wright, GL Jr, Grob, BM, Haley, C, Grossman, K, Newhall, K, Petrylak, D et al. (1996). Upregulation of prostate-specific membrane antigen after androgen-deprivation therapy. Urology 48: 326–334. [DOI] [PubMed] [Google Scholar]
- Ribas, J, Ni, X, Haffner, M, Wentzel, EA, Salmasi, AH, Chowdhury, WH et al. (2009). miR-21: an androgen receptor-regulated microRNA that promotes hormone-dependent and hormone-independent prostate cancer growth. Cancer Res 69: 7165–7169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schramedei, K, Mörbt, N, Pfeifer, G, Läuter, J, Rosolowski, M, Tomm, JM et al. (2011). MicroRNA-21 targets tumor suppressor genes ANP32A and SMARCA4. Oncogene 30: 2975–2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, X, Du, WW, Li, H, Liu, F, Khorshidi, A, Rutnam, ZJ et al. (2013). Both mature miR-17-5p and passenger strand miR-17-3p target TIMP3 and induce prostate tumor growth and invasion. Nucleic Acids Res 41: 9688–9704. [DOI] [PMC free article] [PubMed] [Google Scholar]