

Abstract
Purpose
Transforming growth factor-β–induced epithelial–mesenchymal transition (EMT) is one of the main causes of posterior capsular opacification (PCO) or secondary cataract; however, the signaling events involved in TGF-β–induced PCO have not been fully characterized. Here, we focus on examining the role of β-catenin/cyclic AMP response element–binding protein (CREB)-binding protein (CBP) and β-catenin/T-cell factor (TCF)-dependent signaling in regulating cytoskeletal dynamics during TGF-β–induced EMT in lens epithelial explants.
Methods
Rat lens epithelial explants were cultured in medium M199 in the absence of serum. Explants were treated with TGF-β2 in the presence or absence of the β-catenin/CBP interaction inhibitor, ICG-001, or the β-catenin/TCF interaction inhibitor, PNU-74654. Western blot and immunofluorescence experiments were carried out and analyzed.
Results
An increase in the expression of fascin, an actin-bundling protein, was observed in the lens explants upon stimulation with TGF-β, and colocalized with F-actin filaments. Inhibition of β-catenin/CBP interactions, but not β-catenin/TCF interactions, led to a decrease in TGF-β–induced fascin and stress fiber formation, as well as a decrease in the expression of known markers of EMT, α-smooth muscle actin (α-SMA) and matrix metalloproteinase 9 (MMP9). In addition, inhibition of β-catenin/CBP–dependent signaling also prevented TGF-β–induced downregulation of epithelial cadherin (E-cadherin) in lens explants.
Conclusions
We show that β-catenin/CBP–dependent signaling regulates fascin, MMP9, and α-SMA expression during TGF-β–induced EMT. We demonstrate that β-catenin/CBP–dependent signaling is crucial for TGF-β–induced EMT in the lens.
Keywords: TGFβ, EMT, β-catenin signaling
Fibrosis of the ocular lens or fibrotic cataract is caused by epithelial–mesenchymal transition (EMT) of lens epithelial cells, which ultimately results in lens opacification and loss of vision. Treatment for primary cataract is surgical replacement of the cataractous lens with a synthetic intraocular lens; however, a major postoperative complication of this procedure is the development of secondary cataract or posterior capsular opacification (PCO).1 Posterior capsular opacification results from remnant lens epithelial cells remaining on the anterior lens capsule following cataract surgery that show enhanced capabilities of migration, evasion of apoptosis, production and deposition of extracellular matrix (ECM) components, and capsular wrinkling.2–4 Features of transformed lens epithelial cells include extensive cytoskeletal remodeling, including downregulation of epithelial cadherin (E-cadherin), F-actin polymerization, and upregulation of α-smooth muscle actin (α-SMA), the latter of which is a known marker of EMT.3,5–7 Many of these features have been shown to be induced by transforming growth factor-β (TGF-β).3
Transforming growth factor-β is the major cytokine that promotes EMT-driven fibrosis in many systems and diseases.8–10 Importantly, both active and latent forms of TGF-β have been detected in the aqueous humor of the eye and have been correlated with EMT-mediated lens fibrotic disorders, including both anterior subcapsular cataract (ASC) and PCO.11 Numerous experimental models, including those in rodents, have further shown that TGF-β can induce EMT in the lens. For example, incubation of whole rat lenses in vitro with active TGF-β disrupted normal lens morphology and led to the conversion of lens epithelial cells (LECs) into a myofibroblast phenotype indicative of ASC formation.12–14 Stimulation of ex vivo rat and mouse lens explants with TGF-β generated spindle-shaped myofibroblasts that exhibited increased accumulation of ECM, increased expression of α-SMA, and decreased E-cadherin.15–17 Similarly, transgenic mice overexpressing TGF-β, specifically in the lens, as well as adenoviral-mediated gene transfer of TGF-β to the lens, resulted in EMT of LECs and ASC formation, resembling that observed in humans.18,19
Transforming growth factor-β–induced EMT in the lens has been shown to be mediated through the receptor Smads (Smad2 or Smad3), which associate with Smad4 in the nucleus to control transcription.20 However, Smad-independent TGF-β–activated pathways have also been shown to regulate EMT, and have been linked with fibrotic lens pathologies.15 Transforming growth factor-β–induced EMT has also been shown to occur through cross-talk with components of other signaling pathways, such as the Wnt/β–catenin pathway.21,22 In particular, during cataractogenesis, TGF-β was found to promote expression of Wnts along with its receptor, Frizzled, as well as facilitate translocation of β-catenin from the cell margin to the nucleus.23 However, the nature of the β-catenin signaling involved in the formation of these cataracts remains unclear. A number of recent reports have demonstrated that the interaction between β-catenin and Smad3,24–26 as opposed to β-catenin/T-cell factor (TCF) interaction,26 is critical for TGF-β–induced EMT. The formation of the β-catenin/Smad3 complex was further shown to be dependent on cyclic AMP response element–binding protein (CREB)-binding protein (CBP), a β-catenin transcriptional coactivator.25 For example, when disruption of the β-catenin/CBP complex was caused in alveolar epithelial cells by treatment with ICG-001, a small-molecule inhibitor that specifically inhibits β-catenin/CBP interaction and thereby prevents the interaction between β-catenin and Smad3, TGF-β–induced EMT was prevented.25,26 These findings demonstrate the importance of CBP in β-catenin/Smad3 complex formation.25 Further use of lithium chloride, a stimulant of β-catenin/TCF–dependent signaling, and ICG-001 in Smad reporter assay and TOP flash reporter assay demonstrated that β-catenin/CBP/Smad–dependent signaling, and not β-catenin/TCF/LEF–dependent signaling, was responsible for EMT and profibrotic effects in murine renal tubular epithelial cells.26
Despite the growing evidence for the importance of β-catenin/CBP–dependent signaling during TGF-β–induced EMT in other systems, there are no reports of β-catenin/CBP–dependent signaling during TGF-β–induced EMT in the lens. The current study explores this using ICG-001, an inhibitor that specifically inhibits β-catenin/CBP–dependent signaling, in epithelial cells of the rat lens explant. We further examine the role of β-catenin/CBP–dependent signaling in the induction of fascin, an actin-bundling protein known to be involved in EMT27–30 and shown to be regulated by β-catenin–dependent signaling.31 Fascin, induced by TGF-β–mediated signaling,32 has been shown to facilitate cell migration through its specific role in the formation and turnover of focal adhesions, mainly in cellular projections such as filopodia.33–35 Our results demonstrate that treatment of lens explants with ICG-001 prevented TGF-β–induced expression of α-SMA as well as fascin and stress fiber formation. In contrast, inhibition of the β-catenin/TCF interaction using PNU-74654, an inhibitor that inhibits nuclear β-catenin/TCF interaction, failed to prevent TGF-β–induced EMT as evidenced by the retention in expression of known markers of EMT, such as α-SMA and fascin. Together, our data support the requirement for β-catenin signaling in EMT in the lens and further demonstrate that modulation of β-catenin/CBP interaction via small-molecule inhibitors such as ICG-001 may provide a novel approach to therapeutic intervention in ocular fibrosis.
Materials and Methods
Reagents
Recombinant human TGF-β2 was obtained from R&D Systems (Minneapolis, MN, USA). Inhibitors ICG-001 and PNU-74654 were purchased from Sigma-Aldrich Corp. (St. Louis, MO, USA) and Calbiochem-EMD Millipore (Temecula, CA, USA), respectively. As to primary antibodies, fascin was from Millipore (Temecula, CA, USA), E-cadherin from BD Transduction Laboratories (Lexington, KY, USA), active β-catenin (clone 8E7) from Upstate (Lake Placid, NY, USA), MMP9 and GAPDH from Abcam (Cambridge, MA, USA), α-SMA fluorescein isothiocyanate (FITC) conjugated and unconjugated from Sigma-Aldrich Corp., Pan actin from Abcam. All secondary antibodies for immunofluorescence staining were purchased from Molecular Probes (Invitrogen, Eugene, OR, USA); secondary antibodies for Western blots were obtained from LI-COR Biosciences (Lincoln, NE, USA).
Ex Vivo Rat Lens Epithelium Explant Preparation and Treatment
Rats for this research were obtained and cared for in accordance with the Institutional Animal Care and Use Committee and Canadian Council of Animal Care rules. All animal procedures were carried out in accordance with the ARVO statement for the use of Animals in Ophthalmic and Vision research and were approved by the Ethics Committee on Animal Research of McMaster University (Hamilton, Ontario, Canada). Lens epithelial explants were obtained from 17- to 19-day-old Wistar rats (Charles River Laboratories, Montreal, Canada) as previously described.16 Briefly, rat lenses were isolated and placed in 35-mm culture dishes containing prewarmed, serum-free medium M199, supplemented with antibiotics (all from Invitrogen). The posterior suture of the lens was located and a small incision was made to remove the posterior capsule. This revealed the fiber mass, which was then gently peeled off the anterior epithelium. Once separated, the epithelium was then pinned to the culture dish with a blunt tool exposing LECs to culture medium. Twenty-four hours after explanting, confluent epithelial explants were left untreated in serum-free M199 or treated with TGF-β2 at 6 ng/mL for 48 hours. β-catenin inhibitors ICG-001 and PNU-74654 were dissolved in dimethyl sulphoxide (DMSO) at a final concentration of 17.6 and 10 mM, respectively. The working concentration for ICG-001 and PNU-74654 was 10 μM (1.14 μL/2 mL) and 20 μM (4 μL/2 mL), respectively. The working concentration of ICG-001 and PNU-74654 was determined based on dose–response experiments performed on lens epithelial explants. For β-catenin inhibitor studies, lens explants were pretreated with ICG-001 (5 μM) or PNU-74654 (20 μM) for 1 hour before coincubating with TGF-β2 (6 ng/mL) for 48 hours.
Immunofluorescence Staining
Lens explants were fixed with 10% neutral buffered formalin for 15 minutes and washed with phosphate-buffered saline (PBS; Invitrogen, Carlsbad, CA, USA). Explants were then detached from the culture dish and permeabilized at room temperature for 1 hour in 0.1% Triton X-100, 0.5% sodium dodecyl sulphate (SDS), and 5% donkey serum, followed by incubating overnight at 4°C with primary antibody at a dilution of 1:200 in PBS. Following further PBS washes, explants were incubated with corresponding secondary antibody, Alexa Fluor 488 or Alexa Fluor 568 (Invitrogen), at a dilution of 1:200 at room temperature for 1 hour, and washed three times with PBS (10 minutes for each wash). The explants were then mounted in Prolong Gold antifade reagent with 4′,6-diamidino-2-phenylindole (DAPI, Invitrogen) to visualize nuclei. Immunostained explants were analyzed using a Zeiss 510 Meta Confocal Microscope (Carl Zeiss Canada, Toronto, Canada) or Zeiss Apotome inverted fluorescence microscope (Carl Zeiss Canada) or Leica DMRA2 fluorescence microscope (Leica Microsystems Canada, Richmond Hill, Canada) equipped with a Q-Imaging RETIGA 1300i FAST digital camera (Q-Imaging, Surrey, BC, Canada). Minor adjustments were made to the captured images using Photoshop CS3 (Adobe Systems, Mountain View, CA, USA).
Western Blot Analysis
Lens explants (5–7 per sample) were lysed in Triton lysis buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 1% Triton X-100) with Complete Mini, EDTA-free Protease/phosphatase Inhibitor cocktail (Roche, Laval, QC, Canada). Equal amounts of total protein (5 μg) were loaded on a gel, as determined by DC Protein Assay (Bio-Rad, Mississauga, ON, Canada), and SDS-PAGE was performed. Resolved bands were transferred onto a nitrocellulose membrane (Pall Life Sciences, Pensacola, FL, USA). Membranes were blocked with Odyssey Blocking Buffer (LI-COR Biosciences) for 1 hour and incubated with primary antibody overnight at 4°C. Primary antibodies were diluted in Tris-buffered saline with 0.1% Tween-20 (TBST) at a dilution of 1:500 for fascin (Abcam), 1:1000 for E-cadherin (Millipore) and MMP9 (Abcam), and 1:10,000 for α-SMA (Sigma-Aldrich Corp.) and actin and GAPDH (Abcam). Blots were washed in TBST and probed with corresponding LI-COR near-infrared respective secondary antibodies (either 680- or 800-nm wavelengths) at a dilution of 1:10,000 for 1 hour. The blots were then washed with TBST and scanned using Odyssey near-infrared scanning system (LI-COR Biosciences). Densitometry on visualized bands was performed using Fiji image processing software (ImageJ, http://imagej.nih.gov/ij/; provided in the public domain by the National Institutes of Health, Bethesda, MD, USA). The peaks from the densitometric scans (ImageJ) were used to calculate the average density and the ratio between protein of interest and loading control. The average band density was further used to make graphs and calculate standard deviation using GraphPad Prism software (La Jolla, CA, USA). Unpaired Tukey's multiple comparison test was performed on the averages to calculate the P values using GraphPad Prism software.
Results
TGF-β2–Mediated EMT Leads to an Increase in the Expression of Fascin and Its Colocalization With Actin in Rat Lens Epithelial Explants
Transforming growth factor-β has been shown to induce fascin expression in gastric cancer cells.32 Additionally, increased expression of fascin and its actin binding activity have also been demonstrated to play a key role in cell adhesion and migration during EMT in cancer cells.36–38 To investigate the expression pattern of fascin during TGF-β–induced EMT in our lens system, primary rat lens epithelial explants were treated with TGF-β2, a known EMT inducer, followed by immunostaining for fascin. Immunostaining revealed a low-level, basal expression of fascin in untreated lens explants (Fig. 1A), along with an absence of F-actin staining, the latter of which was determined by rhodamine phalloidin. In comparison, a significant increase in expression of fascin was observed in lens explants following treatment with TGF-β2 for 48 hours, and this was correlated with the appearance of stress fibers (F-actin filaments) (Fig. 1A). Importantly, total actin protein levels remained similar between untreated and TGF-β2–treated lens explants (Supplementary Fig. S1). Interestingly, we observed a significant increase (∼13%, *P < 0.007, n = 4) in the colocalization of fascin with F-actin (Fig. 1B) in TGF-β2–treated explants, and this was specific to the filaments of fascin (arrowheads, Fig. 1A). Finally, to quantify the increased expression of fascin upon TGF-β2 stimulation, we performed Western blot analyses on lysates from untreated and TGF-β2–treated explants. A significant increase in the expression of fascin protein was observed in TGF-β2–treated explants when compared to the untreated controls (Fig. 1C, *P < 0.006, n = 4).
Figure 1.

TGF-β induces fascin expression. (A) Untreated and TGF-β2–treated lens explants were fixed and immunostained for fascin. Fascin-stained slides were costained for F-actin using rhodamine phalloidin. Stained slides were imaged using ×63 water lens of Zeiss LSM510 confocal microscope and analyzed using Zeiss LSM software. Scale bars: 10 μm. (B) A graph for colocalization of fascin with actin was made by scoring at least six different areas of the untreated and TGF-β2–treated lens explants (n = 4, *P < 0.007). (C) Western blot analysis for fascin and GAPDH was carried out using protein lysates extracted from untreated lens explants and TGF-β2–treated lens explants. Densitometric quantification (lower) shows fold increase in fascin normalized to untreated lens explants (n = 4, *P < 0.006).
Inhibition of β-Catenin/CBP–Dependent Signaling by ICG-001 Prevents TGF-β–Induced EMT and Fascin Induction in Rat LECs
Wnt/β–catenin signaling has been shown to play a critical role during TGF-β–induced EMT.21,22 In lens explants, β-catenin translocates to the nucleus from the cell margin following treatment with TGF-β.16,23 To corroborate our results with previous findings, we initially examined the status of β-catenin following TGF-β2 stimulation in the lens explants. Immunofluorescence staining showed membranous localization of β-catenin in untreated explants, whereas delocalization of β-catenin from the cell membrane was observed in the lens explants at 20 hours post TGF-β2 treatment (inset, panel 2, Fig. 2). Additionally, an increase in nuclear accumulation of β-catenin was observed in the explants incubated with TGF-β2 for 20 hours (inset, panel 2, Fig. 2). A complete delocalization of β-catenin from the cell membrane and concomitant increase in nuclear localization of β-catenin were observed upon incubation of explants with TGF-β2 at the later time point of 48 hours (inset, panel 3, Fig. 2).
Figure 2.

TGF-β disrupts membranous localization of β-catenin. Untreated and TGF-β2–treated lens explants were immunostained for β-catenin. Slides were imaged using ×40 lens of Leica DMRA2 fluorescent microscope. Scale bar: 100 μm. Untreated lens explant shows membranous localization of β-catenin (inset, merge image, upper) that is disrupted upon stimulation with TGF-β2 starting at 20 hours post treatment. TGF-β2 induces nuclear localization of β-catenin (inset, merge image, panels 2, 3).
During TGF-β–induced EMT, it has been proposed that α-SMA expression is regulated by the interaction between β-catenin and Smad3, which is further dependent on the transcriptional coactivator CBP.24,25 Inhibition of the β-catenin/CBP complex by the inhibitor ICG-00139 prevents β-catenin and Smad3 interaction and has been shown to inhibit TGF-β–induced EMT in other systems.25,26 We therefore utilized ICG-001 to determine whether disruption in β-catenin/CBP–dependent signaling in TGF-β2–treated explants can prevent fascin expression and EMT. Lens explants were treated with TGF-β2 in the presence or absence of ICG-001 (10 μM) for 48 hours. Upon stimulation with TGF-β2, LECs showed actin cytoskeleton remodeling, as demonstrated by a significant increase in both fascin and α-SMA expression (inset, Fig. 3A). Interestingly, TGF-β2–induced expression of fascin was prevented by cotreatment with ICG-001, and this suppression in fascin expression was further confirmed by Western blot analysis (Fig. 3B). A significant decrease in TGF-β2–induced expression of α-SMA was also observed upon disruption of β-catenin/CBP–dependent signaling by cotreatment with ICG-001 when compared to TGF-β-treated lens explants (inset, Fig. 3A; Figs. 3B–C, ***P < 0.0005). Untreated and ICG-001–treated explants did not exhibit any induction in either fascin or α-SMA expression. A previous report had shown that inhibition of Wnt/β–catenin signaling by ICG-001 induces cytotoxicity in multiple myeloma cells.40 However, we did not observe any significant increase in cell death in the lens explants treated with ICG-001 alone or in combination with TGF-β2.
Figure 3.

Effect of inhibition of β-catenin/CBP–dependent signaling by ICG-001 on TGF-β–induced EMT of lens explants. (A) Untreated, ICG-only, TGF-β2–only, and TGF-β2– and ICG-001-treated lens explants were immunostained for fascin and α-SMA. Slides were imaged using ×40 lens of Zeiss Apotome microscope and analyzed using Zeiss Zen software. Scale bar: 100 μm. (B) Western blot analysis for fascin, α-SMA, and GAPDH was carried out using protein lysates extracted from untreated, ICG-treated, TGF-β2–treated, and TGF-β2– and ICG-001-treated lens explants. (C) Densitometric quantification of α-SMA indicating fold reduction in α-SMA expression normalized to untreated lens explants (n = 3, **P < 0.005, ***P < 0.0005).
Since we had shown that fascin colocalizes with F-actin in TGF-β2–treated explants (Fig. 1), we further examined F-actin expression following treatment with TGF-β2 in the presence and absence of ICG-001. An increase in actin polymerization was observed in TGF-β2–treated explants, whereas coincubation with ICG-001 completely inhibited TGF-β2–induced F-actin polymerization (Fig. 4A).
Figure 4.

Inhibition of β-catenin/CBP–dependent signaling by ICG-001 prevents stress fiber formation and TGF-β–induced E-cadherin delocalization. LECs incubated with ICG only, TGF-β2 only, and TGF-β2 and ICG-001 immunostained for actin and E-cadherin. Untreated LECs were considered as controls. Stained slides were imaged using ×40 lens of Zeiss Apotome microscope and analyzed using Zeiss Zen software. Scale bar: 100 μm. (A) Actin staining reveals a decrease in TGF-β2–induced stress fiber formation (panel 3) with inhibition of β-catenin/CBP interaction by ICG-001 (panel 4). (B) The TGF-β2–induced loss of membranous E-cadherin (panel 3) was restored by ICG-001 (panel 4).
Our lab and others have shown that during TGF-β–induced EMT of LECs, E-cadherin is degraded, and its localization at the cell membrane is lost.14,41 Loss of E-cadherin leads to a dissociation of the E-cadherin/β–catenin complex that results in translocation of β-catenin to the nucleus and activation of its downstream targets.42 Thus, we examined the effects of TGF-β2 on E-cadherin expression in the presence of ICG-001. Untreated lens explants showed hexagonal, cobblestone arrangement with E-cadherin exhibiting membranous localization (inset, Fig. 4B), which was also observed in cells treated with ICG-001 alone. In comparison, explants treated with TGF-β2 exhibited a substantial decrease in the localization of E-cadherin at the cell membrane. In comparison, coincubation with TGF-β2 and ICG-001 was able to partially rescue the TGF-β–induced E-cadherin delocalization (Fig. 4B). These results suggest the involvement of β-catenin/CBP–dependent signaling in E-cadherin localization downstream43 of TGF-β signaling in lens explants.
β-Catenin/TCF–Dependent Signaling Does Not Regulate Fascin Expression or EMT in TGF-β–Treated Rat Lens Explants
Previous studies have speculated that β-catenin/TCF–dependent signaling may regulate TGF-β–induced cataract formation and EMT of lens epithelial explants.23 To investigate this further, we inhibited the β-catenin/TCF–dependent signaling pathway utilizing PNU-74654 (PNU), an inhibitor that selectively inhibits the interaction between nuclear β-catenin and TCF4.44 We did not observe any change in the cellular morphology in the lens explants cotreated with TGF-β2 and PNU when compared to the cells of the explant incubated with TGF-β2. We first examined the change in expression of α-SMA following cotreatment of lens explants with TGFβ and PNU as compared to cells treated with TGFβ alone. We did not observe any change in expression of α-SMA–positive stress fibers (Fig. 5A) or levels of α-SMA protein (Fig. 5B) in explants cotreated with TGF-β2 and PNU (Fig. 5B). We further addressed the effect of PNU on E-cadherin expression. These findings revealed that while untreated explants showed membranous localization of E-cadherin (arrowhead, Fig. 6A), explants treated with TGF-β2 exhibited a decrease in the localization of E-cadherin at the cell membrane (arrowhead, Fig. 6A). As expected, we did not observe any change in the localization of E-cadherin in lens explants cotreated with TGF-β2 and PNU when compared to explants treated with TGF-β2 alone. These results were confirmed by Western blot analyses showing that cotreatment with PNU did not prevent the TGF-β–induced decrease in E-cadherin expression (Figs. 6A–C).
Figure 5.

Inhibition of β-catenin/TCF–dependent signaling fails to protect TGF-β–induced α-SMA expression and F-actin polymerization. (A) LECs incubated with TGF-β2 and TGF-β2 and PNU for 48 hours immunostained for α-SMA, and with rhodamine phalloidin for F-actin, and imaged using ×40 lens of Leica DMRA2 fluorescent microscope. Scale bar: 100 μm (B) Representative Western blot of α-SMA protein from untreated, TGF-β2–treated, PNU-treated, and TGF-β2– and PNU-treated LECs (n = 3).
Figure 6.

Effect on E-cadherin by inhibition of β-catenin/TCF interaction. (A) E-cadherin stained untreated, TGF-β2–treated, and TGF-β2– and PNU-cotreated LECs imaged using Zeiss LSM 510 confocal microscope. Scale bar: 10 μm. (B) Total protein from untreated, TGF-β2–treated, and TGF-β2– and PNU-cotreated LECs was subjected to Western blot analysis for E-cadherin and GAPDH (n = 3). (C) Graph showing average relative density ± SD of E-cadherin normalized to untreated LECs (n = 3, *P < 0.05, **P < 0.005).
Previous studies have shown that fascin gene expression is regulated by the Wnt/β–catenin signaling pathway, which promotes cancer cell migration and invasion.45 More specifically, the fascin promoter has been shown to contain binding sites for TCF/LEF, transcription factors that are activated specifically by β-catenin.31 To investigate the role of β-catenin/TCF interaction on fascin expression, rat lens epithelial explants were treated with TGF-β2 in the presence and absence of PNU. Immunofluorescence results revealed no change in the expression of fascin in lens explants cotreated with TGF-β2 and PNU when compared to explants treated with TGF-β2 alone (Fig. 7A). This finding was further confirmed using Western blot analysis, where the protein extracts from lens explants cotreated with TGF-β2 and PNU revealed no change in the levels of fascin protein when compared to explants treated with TGF-β2 alone (Figs. 7B, 7C).
Figure 7.

Effect of inhibition of β-catenin/TCF–dependent signaling on fascin. (A) Untreated, TGF-β2–treated, and TGF-β2– and PNU-cotreated LECs were stained for fascin and imaged using ×40 lens of Leica DMRA2 fluorescent microscope (n = 3). Scale bar: 100 μm. (B) Western blot analysis for fascin and GAPDH was carried out using protein lysates from untreated, TGF-β2–treated, PNU-treated, and TGF-β2– and PNU-cotreated LECs (n = 3). (C) Graph representing the relative density ± SD of fascin normalized to untreated LECs (n = 3, *P < 0.05).
Disruption of β-Catenin/CBP Interaction Inhibits MMP9 Expression
During EMT, cells undergoing transformation express mesenchymal markers, such as the matrix metalloproteinases (MMPs), as they lean toward a mesenchymal phenotype.46 Previous reports from our lab show an increase in MMP9 protein levels in the supernatants from TGF-β2–treated whole rat lenses.14 First, to show the mesenchymal phenotype of rat lens explants induced by TGF-β2 and to investigate whether β-catenin/CBP–dependent signaling has a role in the regulation of MMP9 expression, as observed in other systems,26 we examined MMP9 expression in TGF-β2–treated lens explants in the presence and absence of ICG-001. Western blot analyses revealed a significant 2.7 ± 0.8-fold reduction (P < 0.02) in TGF-β2–induced MMP9 protein levels upon inhibition of β-catenin/CBP–dependent signaling by ICG-001 compared to explants treated with TGF-β2 alone (Figs. 8A, 8B). Untreated and ICG-001–treated rat explants showed similar basal levels of MMP9 protein (Figs. 8A, 8B). Next, we investigated the ability of the PNU inhibitor to block TGF-β2–induced MMP9 expression and found that it was ineffective (data not shown). These results suggest a role for β-catenin/CBP–dependent signaling in regulating MMP9 expression during TGF-β–induced EMT in lens epithelial explants.
Figure 8.

Inhibition of β-catenin/CBP interaction by ICG-001 prevents MMP9 expression. (A) Untreated, ICG-001–treated, TGF-β2–treated, and TGF-β2– and ICG-001–cotreated lens explants were subjected to Western blot analysis for MMP9 and GAPDH (n = 3). (B) Graph representing the relative density ± SD of MMP9 normalized to untreated LECs (n = 3, *P < 0.02 across all samples).
Discussion
Understanding the mechanisms through which TGF-β induces EMT in the lens is important for developing novel therapeutic strategies for the prevention of cataract. In this study, we show that TGF-β–induced β-catenin/CBP–dependent signaling is critical during TGF-β–induced EMT in the lens. In particular, we show that TGF-β–induced β-catenin/CBP–dependent signaling is responsible for the upregulation of fascin, an actin-bundling protein known to be involved in EMT47 and migration and invasion in a number of different cancer cells.38 ICG-001, a specific inhibitor of β-catenin/CBP interactions, prevented TGF-β–induced fascin, α-SMA, and MMP9 induction, demonstrating the requirement of CBP-dependent β-catenin signaling in the regulation of proteins that facilitate TGF-β–induced EMT in the lens. These data add to the growing list of mechanisms regulated during TGF-β–induced ocular fibrosis, and identify novel signaling events and molecular markers that contribute to TGF-β–induced EMT, which could offer potential new therapeutic approaches to control fibrosis of the lens.
Epithelial–mesenchymal transition plays a major role in the pathogenesis of numerous fibrotic diseases including fibrotic cataract. Transforming growth factor-β is the growth factor involved in EMT of LECs following cataract surgery that results in the development of secondary cataract or PCO.10 In addition, EMT and the formation of anterior subcapsular plaques containing α-SMA–expressing, spindle-shaped cells can be induced by TGF-β.13,14 Transforming growth factor-β–induced EMT in LEC explants involves a loss of epithelial characteristics with downregulation/degradation of epithelial markers, such as E-cadherin, and a transition to a mesenchymal/fibroblast phenotype characterized by an upregulation of myofibroblast markers, namely, α-SMA incorporated into F-actin stress fibers.17,48 Formation of stress fibers requires the coordinated interaction between monomeric actin and its accessory proteins, such as fascin. Fascin has been shown to facilitate formation of peripheral actin-rich protrusions through its actin-bundling activity that results in increased cell migration.38 Recent reports have demonstrated more specific roles for fascin in the formation and turnover of adhesive structures of the cell.34,35 For example, fascin promotes cell migration by restricting the number and thickness of newly polymerized actin filaments into tight noncontractile bundles at the focal adhesions; depletion of fascin results in thicker and more contractile actin filaments that generate higher tensile forces and slow turnover of focal adhesions, ultimately causing decreased cell migration.35 Indeed, fascin along with other actin-modulating proteins helps cancer cells undergoing EMT to change their cellular morphology,49,50 aiding in tumor cell migration and invasion.51 Overexpression of fascin has also been shown to modulate EMT during progression of breast cancer52 and pancreatic tumors53; however, its fate during TGF-β–induced EMT in the lens had yet to be examined. In the current study, we demonstrate that stimulation of rat lens epithelial explants with active TGF-β led to a significant increase in the expression of fascin (Figs. 1A, 1C). In accordance with the role of fascin discussed above, this increase in expression of fascin in our lens explant model system following TGF-β treatment may contribute to the migratory phenotype of LECs often observed during PCO. Since our lens explant model often consists of a confluent culture of cells, migration is reserved only to the edges of the explant. Thus, future studies examining the role of fascin in the cellular protrusions of migratory lens explant cells will be important.
It is well known that TGF-β signaling in the lens is mediated through a canonical, Smad-dependent pathway20; however, Smad-independent pathways are also involved in regulating EMT. Our group has demonstrated that overexpression of TGF-β in the lens leads to the formation of ASC in the absence of Smad3.54 This finding indicates that Smad3 signaling alone is not required for TGF-β to induce a lens fibrotic response. Transforming growth factor-β–induced Smad signaling has also been shown to cross-talk with other pathways, such as Wnt/β-catenin. In other epithelial cell systems, β-catenin has been shown to form a complex with Smad3 that then translocates to the nucleus, and cooperatively promotes the transcription of EMT target genes.24–26 The translocation of β-catenin from cell margins to the nucleus in TGF-β–treated lens epithelial explants as shown in this study (Fig. 2), and as reported previously,16,23 suggests the role of β-catenin–dependent signaling during TGF-β–induced EMT in our lens explant model system. It is possible that TGF-β may be initiating β-catenin signaling through upregulation of Wnts and their receptors, frizzled (Fz), in our lens explant model system, as has been observed previously by others using a TGF-β transgenic mouse model.23 The formation of β-catenin/Smad3 complex is further dependent on the β-catenin transcriptional coactivator, CBP, which differentially regulates a subset of target genes.39 Inhibition of the interaction between β-catenin and CBP by ICG-001 prevents β-catenin/Smad3 complex formation and EMT.25,26 Here, we demonstrate that in the presence of ICG-001, TGF-β–induced α-SMA expression (Fig. 3) and stress fiber formation (Fig. 4A) were completely inhibited in lens epithelial explants, indicating the prevention of a contractile, myofibroblast phenotype. Further, the drastic decrease in TGF-β–induced expression of fascin in the presence of ICG-001 (Figs. 3A–C) indicates a novel role of β-catenin/CBP–dependent signaling in regulation of cofactors that are essential for cell migration and invasion. These observations show a complete inhibition of the TGF-β–induced expression of EMT-related proteins such as α-SMA by ICG-001 in LECs. The morphology of lens explant cells cotreated with TGF-β2 and ICG-001 showed a cobblestone arrangement of cells resembling that of untreated control explants, indicative of a prevention of TGF-β–induced transformation of LECs (Supplementary Fig. S2). As well, the known membrane delocalization of E-cadherin induced by TGF-β signaling55 was partially restored in the presence of ICG-001 (Fig. 4B), further indicating an inhibition of TGF-β–induced EMT in lens explants. However, since the inhibition was not complete, it is possible that TGF-β–induced loss of E-cadherin localization is also regulated by β-catenin–independent repressive activity of transcription factors such as slug on E-cadherin.56 Nonetheless, our findings suggest the importance of β-catenin/CBP–dependent signaling during TGF-β–induced EMT in the lens.
In addition to β-catenin/CBP–dependent signaling, β-catenin/TCF–dependent signaling has also been implicated in EMT in other model systems.57–59 In the lens, it is speculated that β-catenin/TCF signaling plays a role in regulating EMT/fibrosis.23 To further understand the role of β-catenin/TCF signaling during TGF-β–induced EMT in LECs, a specific inhibitor of β-catenin/TCF interaction, PNU-74654,44,60 was used. The morphology of lens explant cells cotreated with TGF-β2 and PNU resembles that of TGF-β2–treated LECs, indicating that PNU was unable to prevent TGF-β–induced transformation of LECs (Supplementary Fig. S3). The observed appearance of contractile, α-SMA–expressing fibers and presence of polymerized F-actin/stress fibers in our lens explant cells in the presence of TGF-β and PNU indicates that β-catenin/TCF–dependent signaling is not required in the regulation of TGF-β–induced α-SMA expression and stress fiber formation (Fig. 5). These observations are in line with those reported in other EMT model systems, in which β-catenin/CBP–dependent signaling as compared to β-catenin/TCF–dependent signaling is more critical during TGF-β–induced EMT.26 The ICG-001 inhibitor has, however, in some cases been shown to interfere with β-catenin/TCF–dependent signaling.61 Hence, we cannot rule out the possibility that β-catenin/TCF–dependent signaling may have partially contributed to TGF-β–induced EMT in LECs. However, β-catenin/CBP–dependent signaling, as opposed to β-catenin/TCF–dependent signaling, has been shown to be required for α-SMA expression and, importantly, the cis elements of the α-SMA promoter do not possess β-catenin/TCF–binding sites,62,63 but instead are regulated by transcription factors that are controlled by β-catenin/CBP/Smad3 signaling.24,25,64 Interestingly, inhibition of β-catenin/CBP–dependent signaling by ICG-001, and not inhibition β-catenin/TCF–dependent signaling by PNU, partially inhibited TGF-β–induced nuclear translocation of β-catenin (Supplementary Fig. S4). Moreover, neither ICG-001 nor PNU treatment alone resulted in the decrease in β-catenin expression (Supplementary Fig. S4). Finally, nuclear accumulation of β-catenin/Smad3, but not β-catenin/TCF complex, has been shown to coincide with α-SMA expression during TGF-β–induced EMT in other epithelial cells, such as tubular cells.26 Indeed, further investigation is needed to elucidate the role of the interaction between β-catenin/CBP and Smad3 in the regulation of α-SMA expression during TGF-β–induced EMT in LECs.
The presence of fascin in the lens explants cells incubated with both TGF-β and PNU (Fig. 7) is in accordance with a previous report showing that CREB–AhR signaling, as opposed to β-catenin/TCF–dependent signaling, is responsible for the regulation of fascin.65 Furthermore, incubation of lens explants with PNU did not prevent TGF-β–induced downregulation of E-cadherin (Figs. 6A–C), a key component of adherens junction, which is in agreement with the previous report showing TGF-β–induced Smad-dependent signaling, but not β-catenin/TCF–dependent signaling, as the key mechanism involved in downregulation of E-cadherin during TGF-β–induced EMT.55 Overall, the presence of α-SMA, fascin, and E-cadherin shows that inhibition of β-catenin/TCF–dependent signaling by PNU was unable to prevent the TGF-β–induced contractile, myofibroblast phenotype and EMT in LECs.
During TGF-β–induced EMT, epithelial cells begin to express mesenchymal markers such as MMPs as they lean toward a more mesenchymal phenotype. Previously, we have shown that in the absence of MMP9 expression, TGF-β–induced EMT-like changes, such as induction of α-SMA in LECs, are prevented.16 In tubular epithelial cells, it has been demonstrated that the inhibition of β-catenin/CBP interactions by ICG-001 decreases active levels of MMP9, as assessed by gelatin zymography of supernatants, thereby completely preventing the mesenchymal transition.26 Here, we found that inhibition of β-catenin/TCF interactions by PNU was not able to prevent TGF-β–induced MMP9 expression, suggesting that β-catenin/TCF–dependent signaling may not be required in the regulation of MMP9 during TGF-β–induced EMT in LECs (data not shown). Interestingly, inhibition of β-catenin/CBP–dependent signaling by ICG-001 showed a significant decrease in TGF-β–induced MMP9 expression along with suppression of the mesenchymal transition of LECs (Fig. 8). A decrease in TGF-β–induced MMP9 expression upon inhibition of β-catenin/CBP interactions also shows the novel role of β-catenin/CBP–dependent signaling in regulation of MMP9 during TGF-β–induced EMT.
Taken together, our findings have revealed a β-catenin–mediated mechanism underlying the regulation of fascin, α-SMA, and MMP9 expression during TGF-β–induced EMT in the lens. We propose that TGF-β–mediated downregulation of E-cadherin results in disruption of E-cadherin/β–catenin complex that leads to increased cytosolic β-catenin, which interacts and forms a complex with CBP (Fig. 9). We demonstrate for the first time that β-catenin/CBP–dependent signaling is indispensable in TGF-β–mediated EMT in the lens. β-catenin/CBP complex may further interact with other regulators of EMT and regulate TGF-β–induced EMT of LECs. The interaction between β-catenin and Smad3 is known to promote transcription activity of myocardin-related transcription factor (MRTF), the master regulator of cytoskeletal genes, during TGF-β–induced EMT in tubular epithelial cells.24 Interestingly, our previous finding suggests translocation of MRTF to the nucleus during TGF-β–induced EMT in LECs.66 Hence, it is possible that the interplay between β-catenin, Smad3, and MRTF may be playing a crucial role in TGF-β–mediated EMT in LECs. Indeed, a detailed molecular investigation is needed to dissect out the role of β-catenin, Smad3, and MRTF interactions in TGF-β–induced EMT in the lens. In conclusion, the inhibition of TGF-β–induced EMT by ICG-001 may represent a new therapeutic approach to target ocular fibrosis of the lens.
Figure 9.
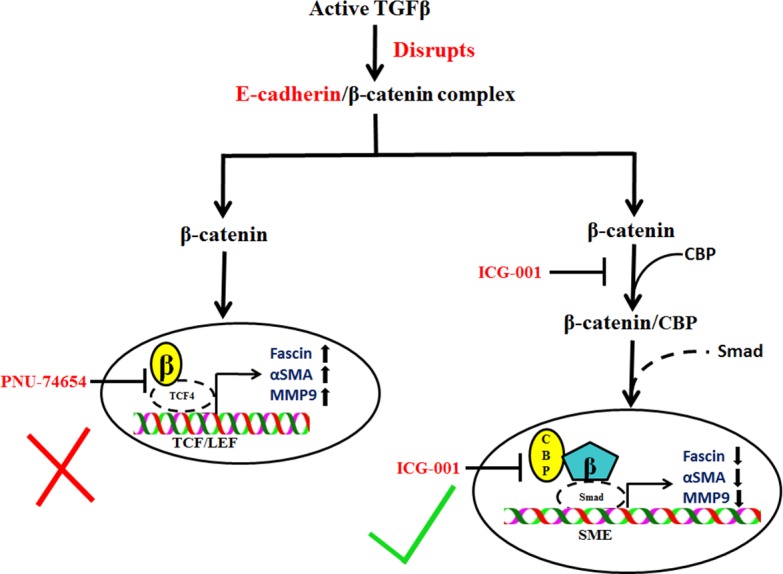
Schematic representation of the β-catenin–mediated mechanisms that regulate TGF-β–induced EMT in LECs. TGF-β–induced downregulation of E-cadherin leads to disruption of E-cadherin/β-catenin complex that results in interaction of β-catenin with CBP. Free β-catenin may interact with either Smad67 in a CBP-dependent manner or TCF, and regulate TGF-β–induced α-SMA, fascin, and MMP9 expression. Inhibition of interaction between β-catenin and TCF fails to inhibit TGF-β–induced EMT-like changes in LECs. However, inhibition of interaction between β-catenin and CBP prevents TGF-β–induced EMT in lens explants.
Supplementary Material
Acknowledgments
Supported by National Institutes of Health Grant EY017146 to JAW-M. AT is financially supported by a CREATE postdoctoral fellowship from Natural Sciences and Engineering Research Council (NSERC), Canada.
Disclosure: A. Taiyab, None; A. Korol, None; P.A. Deschamps, None; J.A. West-Mays None
References
- 1. Spalton DJ. Posterior capsular opacification after cataract surgery. Eye (Lond). 1999; 13 (pt 3b): 489–492. [DOI] [PubMed] [Google Scholar]
- 2. Awasthi N,, Guo S,, Wagner BJ. Posterior capsular opacification: a problem reduced but not yet eradicated. Arch Ophthalmol. 2009; 127: 555–562. [DOI] [PubMed] [Google Scholar]
- 3. Kalluri R,, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009; 119: 1420–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Marcantonio JM,, Vrensen GF. Cell biology of posterior capsular opacification. Eye (Lond). 1999; 13 (pt 3b): 484–488. [DOI] [PubMed] [Google Scholar]
- 5. Lee JM,, Dedhar S,, Kalluri R,, Thompson EW. The epithelial-mesenchymal transition: new insights in signaling, development and disease. J Cell Biol. 2006; 172: 973–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Thiery JP,, Acloque H,, Huang RY,, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009; 139: 871–890. [DOI] [PubMed] [Google Scholar]
- 7. Yilmaz M,, Christofori G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 2009; 28: 15–33. [DOI] [PubMed] [Google Scholar]
- 8. Xu J,, Lamouille S,, Derynck R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009; 19: 156–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li M,, Krishnaveni MS,, Li C,, et al. Epithelium-specific deletion of TGF-beta receptor type II protects mice from bleomycin-induced pulmonary fibrosis. J Clin Invest. 2011; 121: 277–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Saika S. TGFbeta pathobiology in the eye. Lab Invest. 2006; 86: 106–115. [DOI] [PubMed] [Google Scholar]
- 11. Cousins SW,, McCabe MM,, Danielpour D,, Streilein JW. Identification of transforming growth factor-beta as an immunosuppressive factor in aqueous humor. Invest Ophthalmol Vis Sci. 1991; 32: 2201–2211. [PubMed] [Google Scholar]
- 12. Jampel HD,, Roche N,, Stark WJ,, Roberts AB. Transforming growth factor-beta in human aqueous humor. Curr Eye Res. 1990; 9: 963–969. [DOI] [PubMed] [Google Scholar]
- 13. Hales AM,, Chamberlain CG,, McAvoy JW. Cataract induction in lenses cultured with transforming growth factor-beta. Invest Ophthalmol Vis Sci. 1995; 36: 1709–1713. [PubMed] [Google Scholar]
- 14. Dwivedi DJ,, Pino G,, Banh A,, et al. Matrix metalloproteinase inhibitors suppress transforming growth factor-β-induced subcapsular cataract formation. Am J Pathol. 2006; 168: 69–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Banh A,, Deschamps PA,, Vijayan MM,, Sivak JG,, West-Mays JA. The role of Hsp70 and Hsp90 in TGF-beta-induced epithelial-to-mesenchymal transition in rat lens epithelial explants. Mol Vis. 2007; 13: 2248–2262. [PubMed] [Google Scholar]
- 16. Korol A,, Pino G,, Dwivedi D,, Robertson JV,, Deschamps PA,, West-Mays JA. Matrix metalloproteinase-9-null mice are resistant to TGF-beta-induced anterior subcapsular cataract formation. Am J Pathol. 2014; 184: 2001–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liu J,, Hales AM,, Chamberlain CG,, McAvoy JW. Induction of cataract-like changes in rat lens epithelial explants by transforming growth factor beta. Invest Ophthalmol Vis Sci. 1994; 35: 388–401. [PubMed] [Google Scholar]
- 18. Robertson JV,, Nathu Z,, Najjar A,, Dwivedi D,, Gauldie J,, West-Mays JA. Adenoviral gene transfer of bioactive TGFbeta1 to the rodent eye as a novel model for anterior subcapsular cataract. Mol Vis. 2007; 13: 457–469. [PMC free article] [PubMed] [Google Scholar]
- 19. Srinivasan Y,, Lovicu FJ,, Overbeek PA. Lens-specific expression of transforming growth factor beta1 in transgenic mice causes anterior subcapsular cataracts. J Clin Invest. 1998; 101: 625–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Saika S,, Miyamoto T,, Ishida I,, et al. TGFbeta-Smad signalling in postoperative human lens epithelial cells. Br J Ophthalmol. 2002; 86: 1428–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lamouille S,, Xu J,, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014; 15: 178–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nieto MA. The ins and outs of the epithelial to mesenchymal transition in health and disease. Annu Rev Cell Dev Biol. 2011; 27: 347–376. [DOI] [PubMed] [Google Scholar]
- 23. Chong CC,, Stump RJ,, Lovicu FJ,, McAvoy JW. TGFbeta promotes Wnt expression during cataract development. Exp Eye Res. 2009; 88: 307–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Charbonney E,, Speight P,, Masszi A,, Nakano H,, Kapus A. β-catenin and Smad3 regulate the activity and stability of myocardin-related transcription factor during epithelial-myofibroblast transition. Mol Biol Cell. 2011; 22: 4472–4485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhou B,, Liu Y,, Kahn M,, et al. Interactions between beta-catenin and transforming growth factor-beta signaling pathways mediate epithelial-mesenchymal transition and are dependent on the transcriptional co-activator cAMP-response element-binding protein (CREB)-binding protein (CBP). J Biol Chem. 2012; 287: 7026–7038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tian X,, Zhang J,, Tan TK,, et al. Association of beta-catenin with P-Smad3 but not LEF-1 dissociates in vitro profibrotic from anti-inflammatory effects of TGF-beta1. J Cell Sci. 2013; 126: 67–76. [DOI] [PubMed] [Google Scholar]
- 27. Hashimoto Y,, Skacel M,, Adams JC. Roles of fascin in human carcinoma motility and signaling: prospects for a novel biomarker? Int J Biochem Cell Biol. 2005; 37: 1787–1804. [DOI] [PubMed] [Google Scholar]
- 28. Hu W,, McCrea PD,, Deavers M,, Kavanagh JJ,, Kudelka AP,, Verschraegen CF. Increased expression of fascin, motility associated protein in cell cultures derived from ovarian cancer and in borderline and carcinomatous ovarian tumors. Clin Exp Metastasis. 2000; 18: 83–88. [DOI] [PubMed] [Google Scholar]
- 29. Grothey A,, Hashizume R,, Ji H,, et al. C-erbB-2/ HER-2 upregulates fascin, an actin-bundling protein associated with cell motility in human breast cancer cell lines. Oncogene. 2000; 19: 4864–4875. [DOI] [PubMed] [Google Scholar]
- 30. Grothey A,, Hashizume R,, Sahin AA,, McCrea PD. Fascin, an actin-bundling protein associated with cell motility is upregulated in hormone receptor negative breast cancer. Br J Cancer. 2000; 83: 870–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Vignjevic D,, Schoumacher M,, Gavert N,, et al. Fascin, a novel target of beta-catenin-TCF signaling is expressed at the invasive front of human colon cancer. Cancer Res. 2007; 67: 6844–6853. [DOI] [PubMed] [Google Scholar]
- 32. Li L,, Cao F,, Liu B,, Luo X,, Ma X,, Hu Z. TGF-beta induces fascin expression in gastric cancer via phosphorylation of smad3 linker area. Am J Cancer Res. 2015; 5: 1890–1896. [PMC free article] [PubMed] [Google Scholar]
- 33. Adams JC. Roles of fascin in cell adhesion and motility. Curr Opin Cell Biol. 2004; 16: 590–596. [DOI] [PubMed] [Google Scholar]
- 34. Hashimoto Y,, Parsons M,, Adams JC. Dual actin-bundling and protein kinase C-binding activities of fascin regulate carcinoma cell migration downstream of Rac and contribute to metastasis. Mol Biol Cell. 2007; 18: 4591–4602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Elkhatib N,, Neu MB,, Zensen C,, et al. Fascin plays a role in stress fiber organization and focal adhesion disassembly. Curr Biol. 2014; 24: 1492–1499. [DOI] [PubMed] [Google Scholar]
- 36. Mao X,, Chen D,, Wu J,, et al. Differential expression of fascin E-cadherin and vimentin: proteins associated with survival of cholangiocarcinoma patients. Am J Med Sci. 2013; 346: 261–268. [DOI] [PubMed] [Google Scholar]
- 37. Zou J,, Yang H,, Chen F,, et al. Prognostic significance of fascin-1 and E-cadherin expression in laryngeal squamous cell carcinoma. Eur J Cancer Prev. 2010; 19: 11–17. [DOI] [PubMed] [Google Scholar]
- 38. Jayo A,, Parsons M. Fascin: a key regulator of cytoskeletal dynamics. Int J Biochem Cell Biol. 2010; 42: 1614–1617. [DOI] [PubMed] [Google Scholar]
- 39. Ma H,, Nguyen C,, Lee KS,, Kahn M. Differential roles for the coactivators CBP and p300 on TCF/beta-catenin-mediated survivin gene expression. Oncogene. 2005; 24: 3619–3631. [DOI] [PubMed] [Google Scholar]
- 40. Grigson ER,, Ozerova M,, Pisklakova A,, Liu H,, Sullivan DM,, Nefedova Y. Canonical Wnt pathway inhibitor ICG-001 induces cytotoxicity of multiple myeloma cells in Wnt-independent manner. PLoS One. 2015; 10: e0117693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wormstone IM,, Tamiya S,, Anderson I,, Duncan G. TGF-beta2-induced matrix modification and cell transdifferentiation in the human lens capsular bag. Invest Ophthalmol Vis Sci. 2002; 43: 2301–2308. [PubMed] [Google Scholar]
- 42. Yamada S,, Pokutta S,, Drees F,, Weis WI,, Nelson WJ. Deconstructing the cadherin-catenin-actin complex. Cell. 2005; 123: 889–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Beyer C,, Schramm A,, Akhmetshina A,, et al. β-catenin is a central mediator of pro-fibrotic Wnt signaling in systemic sclerosis. Ann Rheum Dis. 2012; 71: 761–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Trosset JY,, Dalvit C,, Knapp S,, et al. Inhibition of protein-protein interactions: the discovery of druglike beta-catenin inhibitors by combining virtual and biophysical screening. Proteins. 2006; 64: 60–67. [DOI] [PubMed] [Google Scholar]
- 45. Gao W,, Zhang C,, Feng Y,, et al. Fascin-1 ezrin and paxillin contribute to the malignant progression and are predictors of clinical prognosis in laryngeal squamous cell carcinoma. PLoS One. 2012; 7: e50710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Savagner P. Leaving the neighborhood: molecular mechanisms involved during epithelial-mesenchymal transition. Bioessays. 2001; 23: 912–923. [DOI] [PubMed] [Google Scholar]
- 47. Hayashi Y,, Osanai M,, Lee GH. Fascin-1 expression correlates with repression of E-cadherin expression in hepatocellular carcinoma cells and augments their invasiveness in combination with matrix metalloproteinases. Cancer Sci. 2011; 102: 1228–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. West-Mays JA,, Pino G,, Lovicu FJ. Development and use of the lens epithelial explant system to study lens differentiation and cataractogenesis. Prog Retin Eye Res. 2010; 29: 135–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Li X,, Zheng H,, Hara T,, et al. Aberrant expression of cortactin and fascin are effective markers for pathogenesis, invasion metastasis and prognosis of gastric carcinomas. Int J Oncol. 2008; 33: 69–79. [PubMed] [Google Scholar]
- 50. Lin CK,, Su HY,, Tsai WC,, Sheu LF,, Jin JS. Association of cortactin fascin-1 and epidermal growth factor receptor (EGFR) expression in ovarian carcinomas: correlation with clinicopathological parameters. Dis Markers. 2008; 25: 17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Machesky LM,, Li A. Fascin: invasive filopodia promoting metastasis. Commun Integr Biol. 2010; 3: 263–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Xing P,, Li JG,, Jin F,, et al. Fascin an actin-bundling protein promotes breast cancer progression in vitro. Cell Biochem Funct. 2011; 29: 303–310. [DOI] [PubMed] [Google Scholar]
- 53. Li A,, Morton JP,, Ma Y,, et al. Fascin is regulated by slug, promotes progression of pancreatic cancer in mice, and is associated with patient outcomes. Gastroenterology. 2014; 146: 1386–1396.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Banh A,, Deschamps PA,, Gauldie J,, Overbeek PA,, Sivak JG,, West-Mays JA. Lens-specific expression of TGF-beta induces anterior subcapsular cataract formation in the absence of Smad3. Invest Ophthalmol Vis Sci. 2006; 47: 3450–3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Nawshad A,, Medici D,, Liu CC,, Hay ED. TGFbeta3 inhibits E-cadherin gene expression in palate medial-edge epithelial cells through a Smad2-Smad4-LEF1 transcription complex. J Cell Sci. 2007; 120: 1646–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Vincent T,, Neve EP,, Johnson JR,, et al. A SNAIL1-SMAD3/4 transcriptional repressor complex promotes TGF-beta mediated epithelial-mesenchymal transition. Nat Cell Biol. 2009; 11: 943–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. He W,, Dai C,, Li Y,, Zeng G,, Monga SP,, Liu Y. Wnt/beta-catenin signaling promotes renal interstitial fibrosis. J Am Soc Nephrol. 2009; 20: 765–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Akhmetshina A,, Palumbo K,, Dees C,, et al. Activation of canonical Wnt signalling is required for TGF-beta-mediated fibrosis. Nat Commun. 2012; 3: 735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wei J,, Melichian D,, Komura K,, et al. Canonical Wnt signaling induces skin fibrosis and subcutaneous lipoatrophy: a novel mouse model for scleroderma? Arthritis Rheum. 2011; 63: 1707–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Barker N,, Clevers H. Mining the Wnt pathway for cancer therapeutics. Nat Rev Drug Discov. 2006; 5: 997–1014. [DOI] [PubMed] [Google Scholar]
- 61. Henderson WR, Jr,, Chi EY,, Ye X,, et al. Inhibition of Wnt/beta-catenin/CREB binding protein (CBP) signaling reverses pulmonary fibrosis. Proc Natl Acad Sci U S A. 2010; 107: 14309–14314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Blank RS,, McQuinn TC,, Yin KC,, et al. Elements of the smooth muscle alpha-actin promoter required in cis for transcriptional activation in smooth muscle. Evidence for cell type-specific regulation. J Biol Chem. 1992; 267: 984–989. [PubMed] [Google Scholar]
- 63. Sun Q,, Chen G,, Streb JW,, et al. Defining the mammalian CArGome. Genome Res. 2006; 16: 197–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Velasquez LS,, Sutherland LB,, Liu Z,, et al. Activation of MRTF-A-dependent gene expression with a small molecule promotes myofibroblast differentiation and wound healing. Proc Natl Acad Sci U S A. 2013; 110: 16850–16855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hashimoto Y,, Loftis DW,, Adams JC. Fascin-1 promoter activity is regulated by CREB and the aryl hydrocarbon receptor in human carcinoma cells. PLoS One. 2009; 4: e5130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Gupta M,, Korol A,, West-Mays JA. Nuclear translocation of myocardin-related transcription factor-A during transforming growth factor beta-induced epithelial to mesenchymal transition of lens epithelial cells. Mol Vis. 2013; 19: 1017–1028. [PMC free article] [PubMed] [Google Scholar]
- 67. Tian YC,, Phillips AO. Interaction between the transforming growth factor-beta type II receptor/Smad pathway and beta-catenin during transforming growth factor-beta1-mediated adherens junction disassembly. Am J Pathol. 2002; 160: 1619–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.