

Abstract
The sex-ratio X-chromosome (SR) is a selfish chromosome that promotes its own transmission to the next generation by destroying Y-bearing sperm in the testes of carrier males. In some natural populations of the fly Drosophila neotestacea, up to 30% of the X-chromosomes are SR chromosomes. To investigate the molecular evolutionary history and consequences of SR, we sequenced SR and standard (ST) males at 11 X-linked loci that span the ST X-chromosome and at seven arbitrarily chosen autosomal loci from a sample of D. neotestacea males from throughout the species range. We found that the evolutionary relationship between ST and SR varies among individual markers, but genetic differentiation between SR and ST is chromosome-wide and likely due to large chromosomal inversions that suppress recombination. However, SR does not consist of a single multi-locus haplotype: we find evidence for gene flow between ST and SR at every locus assayed. Furthermore, we do not find long-distance linkage disequilibrium within SR chromosomes, suggesting that recombination occurs in females homozygous for SR. Finally, polymorphism on SR is reduced compared to ST, and loci displaying signatures of selection on ST do not show similar patterns on SR. Thus, even if selection is less effective on SR, our results suggest that gene flow with ST and recombination between SR chromosomes may prevent the accumulation of deleterious mutations and allow its long-term persistence at relatively high frequencies.
Keywords: genetic conflict, sex-ratio drive, meiotic drive, molecular evolution, sex chromosome evolution, Drosophila neotestacea
Introduction
Genetic conflict occurs when one part of the genome promotes its own transmission to the detriment of the rest of the genome. Conflict is ubiquitous across the tree of life, and selfish genetic elements can range in size from gene-sized transposable elements to entire chromosomes (Burt & Trivers, 2006; Rice, 2013). Selection against selfish activity is hypothesized to have contributed to the evolution of fundamental processes and structures such as meiotic recombination and centromeres (Werren, 2011; Rice, 2013). Meiotic drivers are selfish elements that cause genetic conflict by manipulating gametogenesis to favor their own transmission to the next generation (Lindholm et al., 2016). One example is sex-ratio (SR) drive, which occurs when an X-chromosome kills Y-bearing sperm in carrier males, causing them to sire only daughters that also carry SR (Jaenike, 2001). X-chromosomes may be predisposed to evolve selfish behavior because of the inherent conflict among the autosomes, X-chromosome, and Y-chromosome over the optimal proportion of male and female offspring (Hurst & Pomiankowski, 1991; Jaenike, 2001; Meiklejohn & Tao, 2010). The resulting bias in the offspring sex ratio may also alter the population-level sex ratio, which can favor the evolution of suppressors of drive located in other parts of the genome (Hamilton, 1967; Hall, 2004; Meiklejohn & Tao, 2010).
If the selfish genetic system escapes suppression and restores drive, but now requires two or more components on the driving chromosome to act, selection for genetic linkage may favor suppressed recombination between them (Prout et al., 1973; Charlesworth & Hartl, 1978). Empirically, most drive systems are found in heterochromatic regions or are located within chromosomal inversions, and thus experience reduced recombination between the driving and wild-type, or Standard (ST), arrangements (Jaenike, 2001; Lindholm et al., 2016). This suppressed recombination can have important consequences for patterns of molecular evolution (Navarro & Barton, 2003; Hoffmann & Rieseberg, 2008). For instance, in addition to high divergence between inverted and noninverted arrangements, selection on variants within an inversion can lead to low polymorphism due to Hill-Robertson effects (e.g. Andolfatto et al., 2001; Cheng et al., 2012; Fabian et al., 2012). The increase in frequency of the driving haplotype due to the selfish behavior of the driving loci can also leave a signature similar to that caused by an increase in frequency due to positive natural selection. For example, on the X-chromosome in Drosophila mauritiana, striking polymorphism patterns congruent with strong selective sweeps were detected around loci involved in SR drive in the closely related species D. simulans (Nolte et al., 2013).
SR chromosomes are common in Diptera and have evolved independently many times (Jaenike, 2001). Neither of the two well-studied SR systems in D. simulans is associated with inversions, but both show evidence of selective-sweep like patterns of low polymorphism at the causal driving loci and at very closely linked loci (Derome et al., 2008; Kingan et al., 2010; Helleu et al., 2016). The SR chromosome in D. pseudoobscura is associated with several inversions, but in a study of five X-linked loci, a reduction in polymorphism on SR was only observed at one locus (Babcock & Anderson, 1996; Kovacevic & Schaeffer, 2000). This could be due to the small size of the inversion, the location of this locus near the breakpoint of an inversion, or proximity to a causal driving locus (Kovacevic & Schaeffer, 2000). At the other extreme, the SR chromosome of D. recens is completely bound in overlapping inversions and shows extremely high interlocus linkage disequilibrium (LD) and a dramatic reduction in polymorphism at all sampled loci compared to the standard X-chromosome (Dyer et al., 2007). Similar patterns are observed in the SR chromosome of the stalk-eyed fly, Teleopsis dalmanni (Christianson et al., 2011). In both of these cases, the SR inversions carry pleiotropic deleterious alleles; they cause female sterility when homozygous in D. recens and decreased eyestalk size in T. dalmanni (Dyer et al., 2007; Cotton et al., 2014). Overall, it appears that the rapid increase in frequency of the SR chromosome and restricted recombination with ST tend to result in decreased polymorphism on SR chromosomes. Because SR systems have arisen many times and thus have different evolutionary histories and characteristics, we can compare them to understand how a SR system’s age, chromosomal inversions, and pleiotropic effects alter the molecular evolutionary consequences it has for the X-chromosome.
In this study we investigate SR drive in Drosophila neotestacea, which is a North American boreal and temperate forest mushroom-feeding fruit fly that harbors an SR chromosome (James & Jaenike, 1990). In some populations of D. neotestacea, SR is found at frequencies as high as 30%, which appear to be stable on the order of decades (James & Jaenike, 1990; Dyer, 2012; Pinzone & Dyer, 2013). In D. neotestacea, there is also no evidence of active SR suppressors, and SR homozygote females are fully fertile (Dyer, 2012). Previous genetic work is limited to microsatellite repeat markers, which showed strong genetic differentiation between SR and ST suggesting the presence of one or more inversions on SR (Dyer, 2012; Dyer et al., 2013). This work also identified an excess of microsatellite polymorphism on SR that could be due to occasional recombination or gene conversion between SR and ST in heterozygote females (Dyer, 2012; Dyer et al., 2013). High, stable frequencies of SR in some populations, a lack of phenotypic suppressors, and highly differentiated inverted regions potentially protected from mutation accumulation by occasional recombination suggest that SR may have had a large effect on X-chromosome evolution in this species.
We examine the molecular evolutionary consequences of SR and determine the evolutionary relationship between ST and SR. Using a sample of D. neotestacea males from throughout the species range, we sequenced SR and ST males at 11 randomly chosen X-linked loci that span the ST X-chromosome. For comparison we also sequenced a subset of these individuals at seven randomly chosen autosomal loci. We analyzed patterns of genetic differentiation between the ST and SR chromosomes, performed a phylogenetic analysis to infer the evolutionary history of SR, and compared levels of recombination, linkage disequilibrium, and nucleotide polymorphism between ST and SR. We confirmed the presence of inversions and decreased recombination between ST and SR. Our results indicate that while SR and ST are strongly differentiated and selection may be acting differently on SR, SR is not segregating as a single extended haplotype. We suggest that SR may be able to persist long-term at relatively high frequencies due to occasional gene flow with ST and among SR chromosomes, which prevents SR from degrading via mutation accumulation.
Methods
Sampling and DNA sequencing
X-chromosomes were sampled from 14 geographic populations that span the D. neotestacea species range (Table S1, See Table 1 of Dyer, 2012). These males are the same as used in Dyer (2012), in which wild-caught males were identified as carrying ST or SR X-chromosomes by the proportion of female offspring they produced. From each population where SR is present, at least three ST and three SR males were sequenced at each of 11 randomly chosen X-linked loci, which included six protein-coding genes (marf, mof, pgd, rpl, spk, and sxl), and the flanking regions of six microsatellite loci (neo5261, neo6002, neo7029, neo7040, neo8377, and neo8385)(Table S2)(Dyer, 2007). In total, each male was sequenced at 4,510 base pairs on the X-chromosome. In populations without SR present, only ST males were sampled. At least 53 ST and 41 SR males were included for every marker, though the total sample number is variable for each individual locus. From each population, at least three males were chosen randomly with respect to their X-chromosome type using a random number generator (random.org). These males were sequenced at seven arbitrary autosomal protein coding loci located on all of the other Muller elements except F, which included esc, gl, mago, ntid, sia, tpi, and wee for a total of 2,657 bp per individual (Table S2). All these loci were also sequenced in one or two individuals of D. orientacea and D. putrida, which are two of the other members of the testacea species group. From the final member of the testacea species group, D. testacea, one female from each of 24 isofemale lines collected in Munich, Germany were chosen for sequencing.
DNA extractions were performed with Qiagen Puregene Core Kit A. Fragments were amplified using standard PCR protocols (Table S3) and sequenced on an Applied Biosystems 3730xl DNA Analyzer at the Georgia Genomics Facility. Base calls were confirmed using Geneious (Kearse et al., 2012), and heterozygous SNPs in diploid loci were phased using PHASE (Scheet & Stephens, 2006). Sequences were aligned by hand in Geneious, and microsatellite repeats were removed manually, leaving only the flanking regions for use in analyses (Kearse et al., 2012). Open reading frames were assigned using annotated D. melanogaster orthologs as a guide (flybase.org).
Recombination mapping
We used recombination mapping to determine the order of the loci on the ST chromosome. We performed single pair crosses with flies from isofemale lines originally collected in Seattle, WA (Sea) and Coeur d’Alene, ID (ID-1). Sea females were crossed to ID-1 males, and the F1 females collected and crossed to males from an inbred lab stock. The F2 males (carrying a maternally-derived X-chromosome) were collected and frozen for genotyping. All flies were reared on Instant Drosophila Medium (Carolina Biological Supply, Burlington, NC) supplemented with commercial mushroom (Agaricus bisporus) at 20°C with 60% relative humidity on a 12-hour light/dark cycle. DNA from the parents and 92 F2 males was extracted as described above. Parents were genotyped using repeat number at X-linked microsatellite markers (as described in Pinzone & Dyer, 2013), and sequenced at X-linked protein coding loci to identify SNPs at restriction enzyme cut sites. BsaWI (New England Biolabs, Ipswich, MA) was used for restriction fragment analysis of spk. F2 males were genotyped at the microsatellite loci and spk, and were sequenced at marf and pgd to identify which parental allele they carried. The remaining two loci (rpl and mof) contained no polymorphisms in the parental cross and were thus unable to be mapped. The most likely map order and distances were calculated using the Kosambi mapping function in MapDisto with 1000 bootstrap replicates (Kosambi, 1943; Lorieux, 2012).
As SR/SR females are fertile we attempted to use two independently collected and maintained SR lines for recombination mapping of the SR chromosome. These lines were collected in Eugene, OR in 2001 (SR-Par) and in Rochester New York in 1990 (SR-Lab). However, based on sequencing each locus from each line there was not enough variation between them to determine anything except that neo6002 and neo7029 are at least 50 cM apart on SR, the same as on ST.
The ST lab stock was also originally collected in New York in 1990, and the SR-Lab stock is maintained in the same genetic background. Every generation, ST/Y males are crossed to SR/SR females to generate SR/Y males, and SR/Y males are also crossed to SR/SR females to produce more SR/SR females. We tested for recombination between ST and SR by crossing ST/SR heterozygous females with ST/Y males, and then genotyping 95 male offspring at the six X-linked microsatellites described above. Parental ST and SR genotypes were identified using four SR/SR females and two ST/Y males from these highly inbred SR and ST stocks.
Phylogenetic analysis
Multi-locus phylogenetic species trees were constructed using *BEAST with the HKY nucleotide substitution model and a chain length of 100 million, with 20% burn-in removed (Heled & Drummond, 2010; Bouckaert et al., 2014). Orthologs from D. testacea, D. orientacea, and D. putrida were included in the analysis, and based on previous phylogenetic work D. putrida was used as an outgroup to root the trees (Perlman & Jaenike, 2003; Dyer et al., 2011). Trees for X-chromosome and autosomal loci were constructed separately. For the X-linked species tree, D. neotestacea samples were divided into ST and SR. The X-linked tree also included the marker sxl, which was dropped from further analyses due to extremely low polymorphism. The autosomal tree used all seven autosomal markers. The individual gene trees generated as a part of these *BEAST analyses were used to infer the relationship between D. neotestacea ST and SR and D. testacea at the individual locus level.
Relationships between individual samples were inferred using a multi-locus phylogenetic tree constructed with all the X-linked makers except neo5261, which lacked a sequence from D. putrida used to root the tree. This tree was also built in *BEAST using the HKY substitution model and a chain length of 2 billion with 20% burn-in removed.
Patterns of genetic differentiation and recombination
To estimate patterns of genetic differentiation we calculated KST and Snn (Hudson et al., 1992; Hudson, 2000) in DnaSP (Librado & Rozas, 2009) using both geographic sampling location and ST and SR as groupings (Librado & Rozas, 2009). Significance of individual KST and Snn values was determined using 1,000 random permutations according to the method of (Hudson et al., 1992; Librado & Rozas, 2009). Statistical analyses were carried out in RStudio (2014).
To estimate patterns of linkage disequilibrium (LD) within and across loci, all X-linked markers were concatenated in the order of the ST genetic map and the pairwise correlation coefficient (R2) between each pair of parsimony informative sites was calculated according to the method of Hill & Robertson (1968). Significance of each association was calculated in DnaSP using a Fisher’s Exact Test with a Bonferroni correction for multiple testing using α = 0.05 (Librado & Rozas, 2009). LD was inferred using all samples (SR and ST) and separately for SR and ST chromosomes. ZnS for each locus was calculated according to the method of Kelly (1997). The population recombination rate (ρ) was estimated for each locus using the composite-likelihood method of Hudson (2001) in the program LDHat (McVean et al., 2004). For the autosomes, ρ = (1/2)*4Ner = 2Ner (as males do not recombine in D. neotesteacea), where Ne is the effective population size and r is the per-generation recombination rate. For the X-chromosome, ρ = (2/3)*3Ner = 2Ner. ρ was calculated for ST and SR separately for each marker, and then scaled by the number of sites. Genetic exchange between SR and ST was detected in the concatenated alignment of all X-linked markers using the method of Betran et al. (1997) as implemented in DnaSP (Librado & Rozas, 2009).
Patterns of nucleotide polymorphism
For each marker, nucleotide polymorphism was analyzed separately for ST and SR sample groups. In markers that contained open reading frames, segregating sites were split into silent site polymorphisms (synonymous changes and changes outside the open reading frame) and nonsynonymous polymorphisms. Microsatellite flanking regions were assumed to be silent sites. To evaluate patterns of nucleotide polymorphism, average pairwise nucleotide differences (π) (Nei, 1987), Watterson’s θ (Watterson, 1975), and Tajima’s D (Tajima, 1989) were calculated in DnaSP using only silent sites (Librado & Rozas, 2009). π was also calculated using only nonsynonymous variation for the protein coding genes. Net nucleotide substitutions per site (Da) between ST and SR was calculated for a combined set of all X-linked markers (Nei, 1987; Librado & Rozas, 2009). Parsimony informative sites were identified for ST and SR individually in DnaSP and used to identify private alleles. Expected Tajima’s D values were generated using 10,000 coalescent simulations in the program HKA (https://bio.cst.temple.edu/~hey/software/software.htm#HKA). Ka/Ks and πa/πs were calculated for all protein coding loci in DnaSP (Librado & Rozas, 2009), with orthologs from D. putrida used to identify substitutions.
Polytene chromosome squashes
Inversions on SR were confirmed using squashes of polytene chromosomes from the salivary glands of D. neotestacea third instar larvae. Salivary glands were dissected out in phosphate-buffered saline, fixed in 45% acetic acid, stained with orcein, and then physically squashed on a microscope slide to separate the chromosomes (Sullivan et al., 2000). Chromosomes spreads were examined under 400X magnification using phase contrast. The X-chromosome was identified in ST males from the inbred lab stock because it showed no synapses or chromosomal inversions, and then ST/SR heterozygote females were generated and used to identify inversions between ST and SR.
Results
Genetic map
Recombination mapping of the ST X-chromosome revealed that the nine X-linked loci span a genetic distance of 113 cM (Figure 1). This distance likely encompasses the majority of the X-chromosome given the typical size of Muller element A of other species in the subgenus Drosophila (e.g. Gubenko & Evgenev, 1984; Schafer et al., 1993; Staten et al., 2004; Dyer et al., 2007). However, it is probable that the map order of the markers is rearranged on SR relative to ST. Not a single recombination event was detected between SR and ST chromosomes in the lab crosses; all 95 genotyped sons of ST/SR females inherited either the SR or ST haplotype at all six microsatellite loci.
Figure 1.

Linkage disequilibrium between SR and ST chromosomes. Recombination mapping was performed to determine location of the markers across the standard X-chromosome (ST) in centimorgans (cM). The matrix shows sites with significant R2 values between pairs of SNPs within and between each marker calculated using only parsimony informative sites. Only sites with significant associations are pictured, with the total numbers of sites considered for each marker noted in parentheses. All ST and SR samples were included in the analysis. Significance was determined using a Fisher’s Exact Test with a Bonferroni correction for multiple testing. Light grey squares indicate nonsignificant associations (p > 0.05) and blue squares indicate associations significant after correcting for multiple testing. mof and rpl were unable to be mapped but had no significant associations. See Figure S3 for R2 values when ST and SR are considered separately.
Differentiation among populations and between sex-ratio carrier status
We find little geographic differentiation among populations, similar to previous studies of D. neotestacea (Dyer, 2012; Dyer et al., 2013). KST and Snn based on geographic origin shows that average differentiation between geographic populations is low for both the autosomal markers (KST = 0.030, sd = 0.323) and the X-linked markers (0.020, sd = 0.037)(Figure 2a, Table S4). There is no significant difference in geographic-based population differentiation between the autosomes and the X-chromosome (t14 = 0.617, p = 0.547, two-tailed t-test). Therefore, for the remainder of the analyses we pool geographic samples together and focus on comparisons between SR and ST chromosomes.
Figure 2.
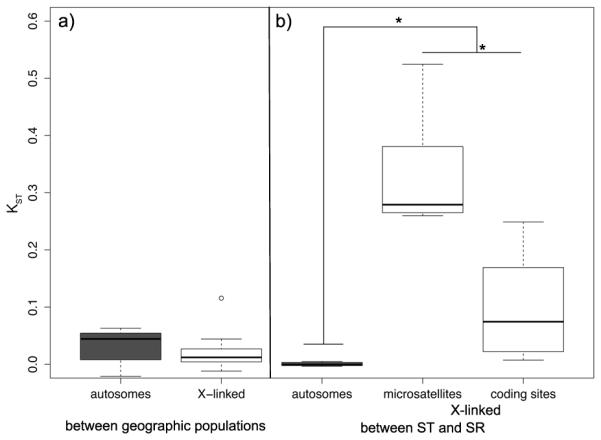
Pairwise KST per marker (a) between geographic populations and (b) between ST and SR males. White boxes are X-linked markers, and grey boxes are autosomal markers. The edges of the boxes are the first and third quartiles, and the dark grey line is the median. The whiskers extend to the last data point before 1.5x the interquartile range.
There is substantial genetic differentiation between the SR and ST X-chromosomes at every locus we surveyed (Table S4), indicating reduced gene flow between SR and ST on a chromosome-wide scale. The average KST between ST and SR for the protein coding genes was 0.1044 (sd = 0.1027) and for the microsatellite flanking regions was 0.3314 (sd = 0.1047; Figure 2b). In contrast, the average KST of autosomal markers between ST and SR males is extremely low (mean = 0.0005, sd = 0.0034; Figure 2b, Table S4), as expected given independent assortment of the autosomes and X-chromosome. Hudson’s Snn supports the results of the KST analysis (Table S4). An analysis of variance (ANOVA) comparing KST of the autosomal loci and the X-linked loci with locus type (microsatellite flanking region or protein coding gene) nested within X-chromosome (ST or SR) showed a significant effect of both chromosome and locus type, with the X-chromosome having significantly higher KST than the autosomes (F1,15 = 34.3, p < 0.0001) and microsatellite flanking regions on the X-chromosome having significantly higher KST than the protein coding genes (F1,15 = 21.72, p < 0.0001) (Figure 2b).
The differentiation between ST and SR can be seen in Figure 3, which depicts the X-linked haplotype of every individual sample at all parsimony informative segregating sites. With the exception of one locus (neo8385), all of the X-linked loci have segregating polymorphisms that are unique to ST or SR (Table S4). At eight of the eleven markers there are shared polymorphisms, and there are no fixed differences between ST and SR in any of the markers sampled (Table S4).
Figure 3.

X-chromosome haplotype structure of individual samples. Each row is an individual, with ST phenotype males above the dashed black line and SR phenotype males below. Each column is a single parsimony informative site in the concatenated alignment of all the X-linked markers in the order found on the ST map. The boundaries of the markers are noted on the x-axis. Dark grey represents the individual carries the major allele, and light grey is the minor allele. Some sites have a third segregating allele, which is represented by white. Gene conversion tracts that fall within a single marker are outlined in blue. Sites that mark the beginning and end of algorithmically detected gene conversion tracts that span two or more markers are highlighted in red, as are any sites within those tracts that appear characteristic of the opposite chromosome (e.g. SR-common alleles on ST).
Phylogenetic analysis
The genetic differentiation between ST and SR is also apparent in the multi-locus phylogeny of individual samples (Figure 4), where the only clade with any substantial support is the one containing all of the SR samples. It is likely that the very low branch support in the rest of the tree is due to the high amount of free recombination among ST chromosomes. Consistent with this, the species tree made from the X-linked markers suggests the origin of SR occurred after the split with D. testacea (Figure S1).
Figure 4.

Multi-locus phylogeny of individual samples, using only X-linked markers. The tree is rooted with D. putrida and includes the sister species D. testacea. Branches with a posterior probability support higher than 0.5 are indicated. Branch tips are marked with population of origin as indicated with the state or province abbreviation and X-chromosome type. Blue text denotes outgroups, red is SR, and black is ST.
The individual gene trees of the X-linked loci show such a much more variable relationship between ST and SR (Figure S2). Many markers include large polytomies that involve both ST and SR samples, and many which samples cluster with ST or SR varies from locus to locus (Figure S2). For no single marker is the association between SR status and the SR clade perfect; there are always SR individuals that cluster with ST, or vice versa. At a few markers, including marf and neo8377, SR is basal to ST (Figure S2). Consistent with the KST analyses, in the individual gene trees there is no phylogenetic sorting of ST and SR sequences at autosomal loci and no evidence of geographic clustering of samples at any locus (Figure S2).
At a broader taxonomic level, the individual gene trees also show that the relationships among D. testacea, D. orientacea, and D. neotestacea are variable across markers. This is likely due to incomplete lineage sorting and/or hybridization among the testacea group species. We also note that while the species tree made from the X-linked markers places D. testacea sister to D. neotestacea, the species tree made from the autosomal markers places D. orientacea as most closely related to D. neotestacea (Figure S1).
Recombination and linkage disequilibrium
Compared to ST, the population recombination rate (ρ) for each marker on SR is reduced by a degree proportional to its average population frequency. A two-way ANOVA was used to test for significant differences on the X-chromosome, with locus type (silent sites in protein coding genes and microsatellite flanking regions) nested within X-chromosome type (ST or SR). ρ is significantly lower on SR (mean = 0.01, sd = 0.02) than on ST (mean = 0.17, sd = 0.14) (F1,16 = 11.703, p < 0.004), with no effect of marker type (F2,16 = 0.883, p = 0.43). Figure 5 depicts the ratio of ρ/(number of sites) between the X-chromosome and the autosomes; assuming an equal population sex ratio, the expectation for this ratio is 1. We scale this by the average frequency of ST and SR across all populations (0.85 and 0.15, respectively (Dyer, 2012)) to obtain an expectation for the estimate of ρ at each marker on ST and SR. On ST, there was no significant difference from the expectation for either the microsatellite flanking regions (mean = 1.28, sd = 0.97, t5 = 1.095, p = 0.32, two-tailed t-test) or the protein coding genes (mean = 0.77, sd = 0.67, t4 = −0.276, p = 0.80, two-tailed t-test). There was also no significant difference from the expectation on SR for either the microsatellites (mean = 0.07, sd = 0.13, t4 = −1.328, p = 0.25, two-tailed t-test) or the protein-coding genes (mean = 0.05, sd = 0.07, t3 = −3.00, p = 0.06, two-tailed t-test).
Figure 5.

Ratio of the population recombination rate scaled by number of sites (ρ/sites) on the X-chromosome to the average ρ/site on the autosomes for ST (grey boxes) and SR (white boxes). Total segregating sites were used to calculate ρ for each marker. The horizontal dashed lines represent the expected X/A ratio of 0.85 for ST and 0.15 for SR. The edges of the boxes are the first and third quartiles, and the dark grey line is the median. The whiskers extend to the last data point before 1.5x the interquartile range.
Long range LD across the entire X-chromosome is observed between loci when considering SR and ST chromosomes together (Figure 1). An R2 analysis identified 471 statistically significant SNP associations across all X-linked markers from a total of 14,535 comparisons between 171 parsimony informative sites. Associations within a locus were significantly overrepresented when considering their frequency (202 significant associations; 3,710 non-significant) compared to associations between SNPs in different loci (269 significant associations; 10,354 non-significant) (χ2 = 62.30, p < 0.0001) (Figure 1). While most of the LD is within loci, we do identify substantial long-range LD, even between sites that are over 100 cM apart on the ST chromosome.
In contrast, within each type of chromosome (SR or ST) nearly all of the significant pairs of associations are within rather than between loci, indicating the presence of recombination within each type (Figure S3). When considering only ST samples, 11,781 comparisons were made between 154 polymorphism informative sites, and 28 significant associations were identified, all of which were between SNPs located in the same marker. Despite the smaller number of sites considered for only the SR samples (2,080 comparisons between 65 parsimony informative sites), there were more significant SNP associations than on ST (186), and only three of these associations occurred between sites in different markers.
Evidence of gene flow between chromosome types is the detection of gene conversion tracts between ST and SR in both directions and in multiple individuals (Table S5, Figure 3). Tracts that begin and end in the same marker likely represent true instances of gene conversion, which typically only involve short stretches of nucleotides. However, many other algorithmically detected tracts crossed multiple markers in the concatenated alignment of all X-linked markers, with the longest tract covering 3,296 of the 4,510 bases in the alignment of concatenated X-linked markers. This tract had an ST donor and an SR recipient, and covered a total of eight markers, or roughly 100cM based on the ST map. Rather than a true gene conversion event, this very large tract and others like it represent ST-typical sequences found in a SR phenotype individual, at least at the endpoints of the identified tract (Figure 3). These are likely the result of gene flow between ST and SR due to other mechanisms such as double crossovers. Very long tracts may also represent two separate gene flow events that have been linked together due to a lack of informative sites between them. Out of the 33 total gene conversion events detected, 15 were from ST to SR, and 18 were from SR to ST (Fisher’s exact test, p = 0.8). However, the ST to SR conversion events are much larger generally, having an average tract length of 1571.4 bp (sd = 1096.07), significantly more than the average tract length of SR to ST conversion events (mean = 183.17, sd = 255.11) (t15 = 4.473, p < 0.001, two-tailed t-test). Of course, these lengths are only within the alignment of the sequence data used in this study, and the actual physical distance between the start and the end of the tract may be much larger. The gene order on SR is also likely highly rearranged relative to ST, so it is possible that loci that are very far apart on ST are actually much closer on SR.
Nucleotide polymorphism
At the autosomal markers, there is no significant difference between silent nucleotide diversity (πs) in flies that carried an ST (mean = 0.0245, sd = 0.0167) or SR chromosome (mean = 0.0222, sd = 0.0132; t6 = 0.744, p = 0.485, paired two-tailed t-test). This is expected given independent assortment of the autosomes and the X-chromosome during meiosis. Therefore all autosomal samples were grouped together and used as a single comparison for the X-linked markers. A set of general summary statistics for each locus can be found in Table S2.
Silent-site polymorphism on SR is less than half of what is found on ST X-chromosomes: on SR, the mean πs was 0.005 (sd = 0.003), and on ST it was 0.013 (sd = 0.012) (Figure S4). A two-way ANOVA with marker type (silent sites in protein coding genes and flanking regions of microsatellites) nested within X-chromosome type (ST or SR) was used to compare πs for the X-linked markers. There was a main effect of X-chromosome type, with SR having a significantly lower πs than the ST (F1,18 = 5.973, p = 0.025). There was also an effect of locus type, with the microsatellite markers having a significantly lower π than the silent sites of protein coding regions (F2,18 = 4.000, p = 0.037).
Nonsynonymous polymorphism (πa) was very low for every marker (Table S2), and a one-way ANOVA found no significant difference in πa between ST (mean = 0.001, sd = 0.002), SR (mean = 0.0005, sd = 0.0006), and the autosomes (mean = 0.001, sd = 0.001) (F2,14 = 0.67, p = 0.528). There was no significant difference between πa/πs values on ST (mean = 0.062, sd = 0.079) and SR (mean = 0.141, sd = 0.246) (t4 = −1.049, p = 0.35, two-tailed t-test), or between the X-chromosome (mean = 0.102, sd = 0.178) and the autosomes (mean = 0.057, sd = 0.050) (t10 = −0.753, p = 0.47, two-tailed t-test) (Table S2). There was also no significant different between Ka/Ks values on ST (mean = 0.116, sd = 0.175) and SR (mean = 0.087, sd = 0.113) (t4 = 1.009, p = 0.37, two-tailed t-test), or between the X-chromosome (mean = 0.102, sd = 0.139) and the autosomes (mean = 0.108, sd = 0.178) (t10 = 0.084, p = 0.93, two-tailed t-test) (Table S2a).
The ratio of the effective population size (Ne) of X-chromosomes to autosomes in a population with an even sex ratio is expected to be 0.75, and thus the ratio of neutral polymorphism on the X-chromosome and the autosomes should also be 0.75. However, since a substantial proportion of X-chromosomes in this species are SR (15% species wide (Dyer, 2012)), the expected X/A polymorphism ratio for ST is (0.75*0.85) = 0.64, assuming SR frequency is at a long-term equilibrium. Silent polymorphism in protein-coding genes on ST trends higher than this expectation, but does not differ significantly (mean X/A ratio = 0.878, sd =0.641, t4 = 0.837, p = 0.45 two-tailed t-test) (Figure 6). Interestingly, the flanking regions of microsatellites on the ST X-chromosome have a significantly lower average level of polymorphism than expected (mean X/A ratio = 0.309, sd = 0.171, t5 = −4.696, p = 0.005, two tailed t-test) (Figure 6). The expected amount of polymorphism on SR compared to the autosomes relative to its frequency is (0.15*0.75) = 0.1125, again assuming that SR frequency is at a long-term equilibrium. In contrast to ST, the observed amount of silent polymorphism at both protein-coding genes and microsatellite loci on SR is higher than this expected value but not significantly different from it (mean X/A ratio: protein coding mean = 0.246, sd = 0.191, t4 = 1.564, p = 0.19, two tailed t-test; microsatellite flanking mean = 0.199, sd = 0.106, t5 = 1.991, p = 0.103 two tailed t-test) (Figure 6).
Figure 6.
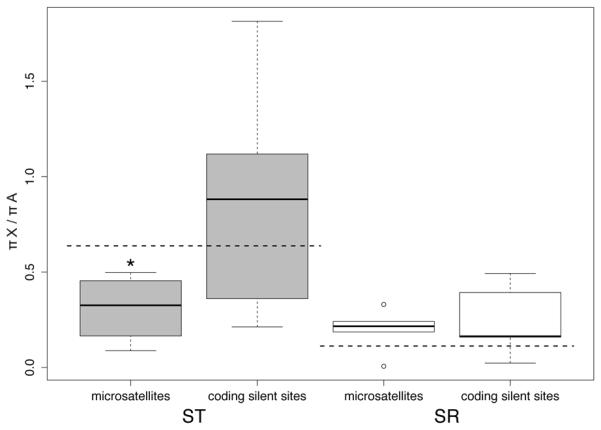
Ratio of average π (pairwise nucleotide differences) on the X-chromosome to average π on the autosomes for both ST (grey boxes) and SR (white boxes). Only silent sites were used for the protein coding genes. The horizontal dashed lines represent the expected X/A ratio of (0.75 * 0.85 = 0.64) for ST and (0.75 * 0.15 = 0.11) for SR. The edges of the boxes are the first and third quartiles, and the dark grey line is the median. The whiskers extend to the last data point before 1.5x the interquartile range.
Silent sites at autosomal loci have a generally negative Tajima’s D (mean = −1.74, sd = 0.407; Figure S4c and Table S2), with the observed D significantly lower than the neutral expectation for six of the seven autosomal loci (p < 0.05 for each). This suggests that this skew in the frequency spectrum is a demographic effect and these values can be taken as the genomic background. The average D is not significantly different between the autosomes and all sequences on the X-chromosome (mean = −1.617, sd = 0.597, t15 = −0.614, p = 0.55, two-tailed t-test) (Figure 7). On the X-chromosome, a two-way ANOVA with locus type (silent sites in protein coding genes and microsatellite flanking regions) nested within X-chromosome type (ST and SR) was used to determine significant differences between Tajima’s D values. There was there was no effect of maker type (F2,18 = 1.079, p = 0.36), but there was a significant effect of X-chromosome type, with markers on ST having a lower D than on SR (F1,18 = 5.99, p < 0.03) (Figure 7). However, this effect appears to be driven primarily by the microsatellite flanking regions, which have a very low D on ST (mean = −2.106, sd = 0.26) compared to SR (mean = −1.37, sd = 0.69). This trend can be observed when comparing D on an individual locus basis (Figure S4c, Table S2a).
Figure 7.
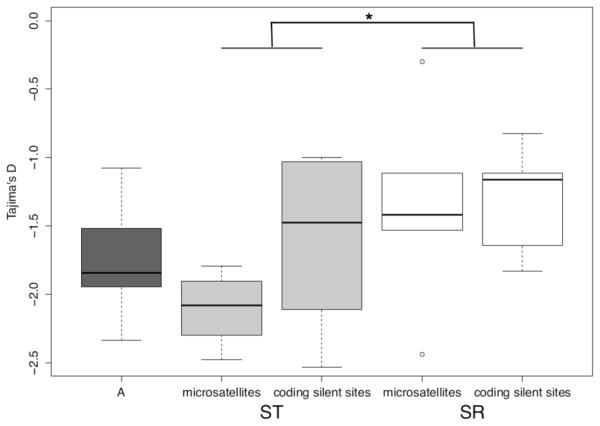
Tajima’s D values for microsatellite flanking regions on ST and SR and silent sites in protein coding genes on ST (light grey), SR (white), and the autosomes (A, in dark grey). The edges of the boxes are the first and third quartiles, and the dark grey line is the median. The whiskers extend to the last data point before 1.5x the interquartile range.
The X-chromosome in the sister species D. testacea harbors a similar level of polymorphism as the ST X-chromosome in D. neotestacea (mean π = 0.008, sd = 0.005, t13 −1.238, p = 0.238, two-tailed t-test)(Table S2b). However, polymorphism in D. testacea at the X-linked microsatellites is significantly higher than the expected value of 0.75 of the diversity on the autosomes (mean π = 0.011, sd = 0.005, t5 = 2.667, p = 0.045, two-tailed t-test), but matches that expectation at the X-linked protein coding loci (mean = 0.005, sd = 0.002, t5 = 0.128, p = 0.903, two-tailed t-test)(Table S2b). Overall, Tajima’s D values on the X-chromosome and autosomes in D. testacea are much closer to zero than in D. neotestacea, with only one locus being significantly different from the simulated expectation (Table S2b), and with no significant differences between the average D values of the X-linked microsatellites (mean = −1.282, sd = 0.631), the X-linked protein coding genes (mean = 1.501, sd = 0.278), and the autosomal loci (mean = −1.260, sd = 0.555)(F2,15 = 0.349, p = 0.711, three-way ANOVA)(Table S2b).
Confirmation of SR inversions
We confirmed chromosomal inversions between ST and SR by examining polytene chromosome squashes from ST/SR larvae (Figure 8). D. neotestacea has five visible chromosomes: the X-chromosome, one double-length telocentric autosome, two other autosomes, and the dot chromosome (Patterson & Stone, 1952). The X-chromosome was identified in ST males by its lack of inversions compared to the two similarly sized autosomes. Based on banding patterns, we believe that the X-chromosome described in James & Jaenike (1990) is actually one of these other autosomes. We then identified the X-chromosome in ST/SR females using shape and banding pattern. Several large inversions are clearly visible (Figure 8). Despite the use of an inbred lab stock for this analysis, chromosomal inversions occurred on every chromosome except the dot.
Figure 8.
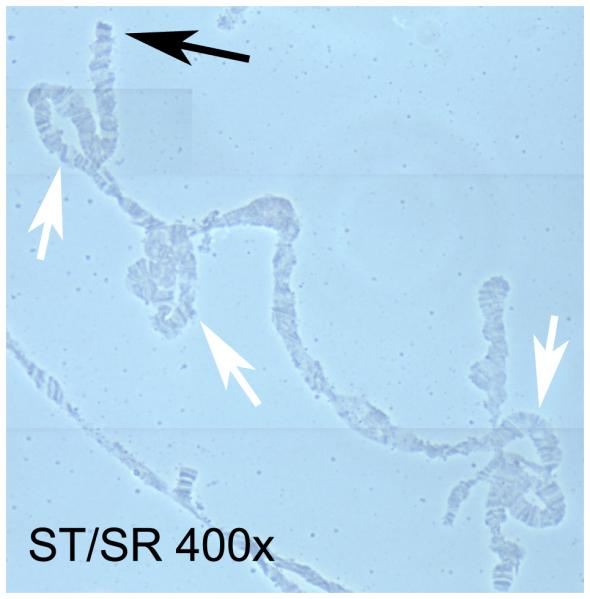
Polytene X-chromosomes of an ST/SR female. The black arrow marks the tip of the chromosome, and the white arrows mark inversions or more complex rearrangements.
Discussion
The effects of genetic conflict encompass the entire SR chromosome of D. neotestacea and affect all aspects of its evolution. Our findings of low genetic differentiation among geographic populations and high differentiation between ST and SR are consistent with previous work in this system that analyzed the repeat motifs of microsatellites (Figure 2) (Dyer, 2012; Dyer et al., 2013). We also used laboratory crosses and patterns of linkage disequilibrium to show that recombination is restricted between ST and SR along the entire length of the > 110 cM X-chromosome. These results suggest that most or all of SR is tied up in inversions, and we visually confirmed the presence of multiple, large rearrangements on SR. However, the amount of divergence between SR and ST varies from locus to locus, with no individual locus showing a perfect association with SR and no fixed differences between the two types of chromosomes (Figure S2, Table S4). ST individuals with SR haplotypes and vice versa can be seen within individual loci (Figure 3). This suggests ongoing gene flow in the form of gene conversion events or double crossovers, as well as incomplete lineage sorting of variation between ST and SR.
Such gene flow results in an imperfect association with SR over long genetic distances, so markers that are tightly linked to the driving locus can be expected to show the strongest differentiation between SR and ST. For instance, in the Winters and Paris SR systems of D. simulans only loci near the driver show clear evidence of selective sweeps (Derome et al., 2008; Kingan et al., 2010; Helleu et al., 2016). Likewise, decreased polymorphism is only found within inversions on the SR chromosome of D. pseudoobscura (Kovacevic & Schaeffer, 2000). The location of the driving loci is unknown in D. neotestacea, but previous work found microsatellite repeat genotypes at neo8385 and neo8377 that predict the SR phenotype at 94% accuracy (Dyer et al., 2013; Pinzone & Dyer, 2013). In our study these loci also have the highest values of KST between ST and SR (Table S4), and their gene trees show well-supported SR clades (Figure S2). As these markers are physically located only 3.5 cM from each other on the ST chromosome (Figure 1), of the loci we surveyed they may be physically closest to the driver(s). So while the presence of inversions precludes mapping of the SR gene(s), a history of occasional gene flow between SR and ST suggests that other approaches such as association mapping may identify loci or regions involved in the drive mechanism, because an SR causal locus would be perfectly associated with SR expression.
The lack of a single SR-associated haplotype in D. neotestacea is in contrast to the SR systems in D. recens and T. dalmanni (Dyer et al., 2007; Christianson et al., 2011), as well as the multiple segregating versions of the selfish autosomes SD in D. melanogaster and t-haplotype in mice (Lyon, 2003; Presgraves et al., 2009; Brand et al., 2015). In these systems, however, homozygous carriers of the driving chromosome usually have severe fitness costs, which have not been found in D. neotestacea. We suggest that SR has the capacity to purge deleterious mutations through occasional gene flow with ST as well as other SR chromosomes. While ST chromosomes appear to be highly recombining based on overall low levels of LD (Figures 5, S3), as a group SR chromosomes have substantial LD within loci but little LD between loci (Figure S3). This suggests that over longer distances recombination does still occur between SR chromosomes to break up genetic associations, despite reduced intra-locus population recombination rates on SR relative to ST (Figure 5). In D. neotestacea, females homozygous for SR are fully fertile and comprise about 2-3% of females in the wild (Dyer, 2012; Dyer et al., 2013). Even though these homozygous females are at a low prevalence, D. neotestacea is a common species with an overall large Ne, and thus there is likely substantial potential for recombination to occur between different SR chromosomes.
Nevertheless, SR’s lower frequency and restricted recombination could lead to a history of reduced Ne compared to ST, and consequently less effective selection. Ka/Ks and πa/πs values, which are often used as an estimate of selection in protein coding regions, show no difference between ST and SR. However, other data suggest selection may be acting differently on SR. Specifically, we found that the flanking regions of the microsatellites on ST had both reduced segregating polymorphism and a reduced Tajima's D compared to the expectation based on the autosomes (Figures 6, 7). This may indicate especially strong purifying selection at these loci. Some of the microsatellite markers may experience reduced recombination due to their location near the end of the ST X-chromosome (Figure 1)(Patterson & Stone, 1952), but the pattern remains even when the most distal locus with the lowest polymorphism on ST (neo5261) is dropped from the analysis (p = 0.001, two-tailed t-test).
The potential for selection in noncoding regions of the genome is well established (e.g., Andolfatto, 2005), though to our knowledge the flanking regions of microsatellites have never been shown to be the target of selection. Alternatively, the microsatellites themselves may be under selection for a particular repeat length (Haasl & Payseur, 2013). However, previous work using these same loci found repeat heterozygosity on ST is consistent with the expectation based on the autosomes (Dyer et al., 2013). Regardless of the cause of this putative selection on ST, our data indicate that it is not acting on equivalent loci on SR, as the pattern of lower than expected polymorphism and Tajima's D at these loci is not recovered on SR. This suggests that the selfish activity of SR may reduce the efficacy of selection and thus affect the evolution of linked sequences that are otherwise independent of the driving mechanism. More information is required to fully investigate this possibility.
Our species phylogeny combines the coalescent history of all X-linked loci to indicate that ST and SR diverged after the split with sister species D. testacea (Figure S2). We obtain a rough age estimate for SR of 330,000 years by using the net genetic divergence between ST and SR at all X-linked markers (Da = 0.568%) and a nucleotide substitution rate of 1.7% per million years (Table S2a)(Caccone et al., 1988). D. testacea is found Europe and Asia (Grimaldi et al., 1992), D. orientacea is found in Asia, and the range of D. neotestacea is thought to extend into Alaska (Patterson & Stone, 1952); it is possible that the last point of contact between D. neotestacea with the other species was across the Bering Land Bridge between 35,000 – 14,000 years ago (Meiri et al., 2014). However, the individual gene trees show that the relationship of SR and ST is variable across markers, and neo8377 shows a particularly interesting pattern where SR is basal to ST and D. testacea (Figure S2). As neo8377 is one of the markers most differentiated on SR and most closely associated with the selfish phenotype, it is possible that the relationship between SR, ST, and D. testacea at this locus represents the true evolutionary history of the driving loci, and at other sites on the X-chromosome recombination between ST and SR has reduced their genetic divergence since the split with D. testacea. Indeed, the age of SR is estimated to be much older when using only divergence between SR and ST at neo8377 (Da = 0.993%), roughly 580,000 years (Table S2a).
An old origin for parts of SR and variable evolutionary relationships across the X-chromosome could suggest a history of sex chromosome cycling in this species due to conflict, where genetic suppression of drive allowed inactivated SR chromosomes to remain in a population as ST chromosomes (Hall, 2004). A history of coevolution with suppressors would explain the barrier to recombination across the majority of the SR chromosome in D. neotestacea. No segregating suppressors have been identified on either the Y-chromosome or the autosomes in D. neotestacea (Dyer, 2012); the most recently evolved driving locus on SR may instead be held in check by population dynamic forces (Pinzone & Dyer, 2013). While no SR drive has been identified in North American populations of D. putrida or European populations of D. testacea, SR drive and suppressors of drive have been found segregating in populations of D. orientacea from Japan (K. Dyer, unpublished data). Given this and the estimated age of SR in D. neotestacea, it is plausible that the SR drive chromosomes in D. neotestacea and D. orientacea may share the same origin.
We suggest that even with a potential decrease in the efficacy of selection on SR, the maintenance of variation through recombination with ST and other SR chromosomes has allowed the persistence of SR at high frequencies in natural populations. However, this may not be without negative effects for the ST chromosome, including a reduction in Ne if SR is at high prevalence and the acquisition of deleterious SR-linked alleles through recombination. The genetic conflict caused by SR drive may therefore affect the evolution of the X-chromosome regardless of SR carrier status. It is possible that this has already been the case. Ultimately, a whole-genome approach will indicate the extent to which recombination is suppressed across the entire SR chromosome, and the resulting consequences for genetic variation and the strength of selection for both types of chromosomes.
Supplementary Material
Acknowledgements
We are grateful to Brooke White, Jesse Lopez, and Michael Bray for technical assistance and to four anonymous reviewers for useful suggestions. Funding was provided by a Presidential Fellowship from the University of Georgia and by the National Institute of General Medical Sciences (NIGMS) of the National Institutes of Health (NIH) under award T32GM007103 to KEP, and by grants from the National Science Foundation (DEB-1149350), The Ellison Medical Research Foundation, and The University of Georgia Research Foundation to KAD.
Footnotes
References
- Andolfatto P, Depaulis F, Navarro A. Inversion polymorphisms and nucleotide variability in Drosophila. Genet. Res. 2001;77:1–8. doi: 10.1017/s0016672301004955. [DOI] [PubMed] [Google Scholar]
- Andolfatto P. Adaptive evolution of non-coding DNA in Drosophila. Nature. 2005;437:1149–1152. doi: 10.1038/nature04107. [DOI] [PubMed] [Google Scholar]
- Babcock CS, Anderson WW. Molecular evolution of the Sex-Ratio inversion complex in Drosophila pseudoobscura: analysis of the Esterase-5 gene region. Mol. Biol. Evol. 1996;13:297–308. doi: 10.1093/oxfordjournals.molbev.a025589. [DOI] [PubMed] [Google Scholar]
- Betran E, Rozas J, Navarro A, Barbadilla A. The estimation of the number and the length distribution of gene conversion tracts from population DNA sequence data. Genetics. 1997;146:89–99. doi: 10.1093/genetics/146.1.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouckaert R, Heled J, Kühnert D, Vaughan T, Wu C-H, Xie D, Suchard MA, Rambaut A, Drummond AJ. BEAST 2: A software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2014;10:e1003537. doi: 10.1371/journal.pcbi.1003537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand CL, Larracuente AM, Presgraves DC. Origin, evolution, and population genetics of the selfish Segregation Distorter gene duplication in European and African populations of Drosophila melanogaster. Evolution. 2015;69:1271–1283. doi: 10.1111/evo.12658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burt A, Trivers R. Genes in Conflict: The Biology of Selfish Genetic Elements. Belknap Press of Harvard University Press; Cambridge, Mass: 2006. [Google Scholar]
- Caccone A, Amato GD, Powell JR. Rates and patterns of scnDNA and mtDNA divergence within the Drosophila melanogaster subgroup. Genetics. 1988;118:671–683. doi: 10.1093/genetics/118.4.671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B, Hartl DL. Population dynamics of the Segregation Distorter polymorphism of Drosophila melanogaster. Genetics. 1978;89:171–192. doi: 10.1093/genetics/89.1.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng C, White BJ, Kamdem C, Mockaitis K, Costantini C, Hahn MW, Besansky NJ. Ecological genomics of Anopheles gambiae along a latitudinal cline: A population-resequencing approach. Genetics. 2012;190:1417–1432. doi: 10.1534/genetics.111.137794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson SJ, Brand CL, Wilkinson GS. Reduced polymorphism associated with X chromosome meiotic drive in the stalk-eyed fly Teleopsis dalmanni. PLoS ONE. 2011;6:1–7. doi: 10.1371/journal.pone.0027254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotton AJ, Foldvari M, Cotton S, Pomiankowski A. Male eyespan size is associated with meiotic drive in wild stalk-eyed flies (Teleopsis dalmanni) Heredity. 2014;112:363–369. doi: 10.1038/hdy.2013.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derome N, Baudry E, Ogereau D, Veuille M, Montchamp-Moreau C. Selective sweeps in a 2-locus model for sex-ratio meiotic drive in Drosophila simulans. Mol. Biol. Evol. 2008;25:409–416. doi: 10.1093/molbev/msm269. [DOI] [PubMed] [Google Scholar]
- Dyer KA. Identification and characterization of 21 polymorphic microsatellite loci from the mycophagous fly Drosophila neotestacea. Mol. Ecol. Notes. 2007;7:1120–1122. [Google Scholar]
- Dyer KA, Charlesworth B, Jaenike J. Chromosome-wide linkage disequilibrium as a consequence of meiotic drive. Proc. Natl. Acad. Sci. 2007;104:1587–1592. doi: 10.1073/pnas.0605578104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer KA, White BE, Bray MJ, Pique DG, Betancourt AJ. Molecular evolution of a Y chromosome to autosome gene duplication in Drosophila. Mol. Biol. Evol. 2011;28:1293–1306. doi: 10.1093/molbev/msq334. [DOI] [PubMed] [Google Scholar]
- Dyer KA. Local selection underlies the geographic distribution of sex-ratio drive in Drosophila neotestacea. Evolution. 2012;66:973–984. doi: 10.1111/j.1558-5646.2011.01497.x. [DOI] [PubMed] [Google Scholar]
- Dyer KA, Bray MJ, Lopez SJ. Genomic conflict drives patterns of X-linked population structure in Drosophila neotestacea. Mol. Ecol. 2013;22:157–169. doi: 10.1111/mec.12097. [DOI] [PubMed] [Google Scholar]
- Fabian DK, Kapun M, Nolte V, Kofler R, Schmidt PS, Schlotterer C, Flatt T. Genome-wide patterns of latitudinal differentiation among populations of Drosophila melanogaster from North America. Mol. Ecol. 2012;21:4748–4769. doi: 10.1111/j.1365-294X.2012.05731.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimaldi D, James AC, Jaenike J. Systematics and modes of reproductive isolation in the holarctic Drosophila testacea species group (Diptera: Drosophilidae) Ann. Entomol. Soc. Am. 1992;85:671–685. [Google Scholar]
- Gubenko IS, Evgenev MB. Cytological and linkage maps of Drosophila virilis chromosomes. Genetica. 1984;65:127–139. [Google Scholar]
- Haasl RJ, Payseur BA. Microsatellites as targets of natural selection. Mol. Biol. Evol. 2013;30:285–298. doi: 10.1093/molbev/mss247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall DW. Meiotic drive and sex chromosome cycling. Evolution. 2004;58:925–931. doi: 10.1111/j.0014-3820.2004.tb00426.x. [DOI] [PubMed] [Google Scholar]
- Hamilton WD. Extraordinary sex ratios. A sex-ratio theory for sex linkage and inbreeding has new implications in cytogenetics and entomology. Science. 1967;156:477–488. doi: 10.1126/science.156.3774.477. [DOI] [PubMed] [Google Scholar]
- Heled J, Drummond AJ. Bayesian inference of species trees from multilocus data. Mol. Biol. Evol. 2010;27:570–580. doi: 10.1093/molbev/msp274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helleu Q, Gérard PR, Dubruille R, Ogereau D, Prud’homme B, Loppin B, Montchamp-Moreau C. Rapid evolution of a Y-chromosome heterochromatin protein underlies sex chromosome meiotic drive. Proceedings of the National Academy of Sciences. 2016;113:4110–4115. doi: 10.1073/pnas.1519332113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill WG, Robertson A. Linkage disequilibrium in finite populations. Theor. Appl. Genet. 1968;38:226–231. doi: 10.1007/BF01245622. [DOI] [PubMed] [Google Scholar]
- Hoffmann AA, Rieseberg LH. Revisiting the impact of inversions in evolution: From population genetic markers to drivers of adaptive shifts and speciation? Annu. Rev. Ecol. Evol. Syst. 2008;39:21–42. doi: 10.1146/annurev.ecolsys.39.110707.173532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson RR, Boos DD, Kaplan NL. A statistical test for detecting geographic subdivision. Mol. Biol. Evol. 1992;9:138–151. doi: 10.1093/oxfordjournals.molbev.a040703. [DOI] [PubMed] [Google Scholar]
- Hudson RR. A new statistic for detecting genetic differentiation. Genetics. 2000;155:2011–2014. doi: 10.1093/genetics/155.4.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson RR. Two-locus sampling distributions and their application. Genetics. 2001;159:1805–1817. doi: 10.1093/genetics/159.4.1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst LD, Pomiankowski A. Causes of sex-ratio bias may account for unisexual sterility in hybrids - a new explanation of Haldane's rule and related phenomena. Genetics. 1991;128:841–858. doi: 10.1093/genetics/128.4.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaenike J. Sex chromosome meiotic drive. Annu. Rev. Ecol. Syst. 2001;32:25–49. [Google Scholar]
- James AC, Jaenike J. Sex-ratio meiotic drive in Drosophila testacea. Genetics. 1990;126:651–656. doi: 10.1093/genetics/126.3.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Meintjes P, Drummond A. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly JK. A test of neutrality based on interlocus associations. Genetics. 1997;146:1197–1206. doi: 10.1093/genetics/146.3.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingan SB, Garrigan D, Hartl DL. Recurrent selection on the Winters sex-ratio genes in Drosophila simulans. Genetics. 2010;184:253–265. doi: 10.1534/genetics.109.109587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosambi DD. The estimation of map distances from recombination values. Annals of Eugenics. 1943;12:172–175. [Google Scholar]
- Kovacevic M, Schaeffer SW. Molecular population genetics of X-linked genes in Drosophila pseudoobscura. Genetics. 2000;156:155–172. doi: 10.1093/genetics/156.1.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- Lindholm AK, Dyer KA, Firman RC, Fishman L, Forstmeier W, Holman L, Johannesson H, Knief U, Kokko H, Larracuente AM, Manser A, Montchamp-Moreau C, Petrosyan VG, Pomiankowski A, Presgraves DC, Safronova LD, Sutter A, Unckless RL, Verspoor RL, Wedell N, Wilkinson GS, Price TAR. The Ecology and Evolutionary Dynamics of Meiotic Drive. Trends in Ecology & Evolution. 2016;31:315–326. doi: 10.1016/j.tree.2016.02.001. [DOI] [PubMed] [Google Scholar]
- Lorieux M. MapDisto: Fast and efficient computation of genetic linkage maps. Molecular Breeding. 2012;30:1231–1235. [Google Scholar]
- Lyon MF. Transmission ratio distortion in mice. Annual Review of Genetics. 2003;37:393–408. doi: 10.1146/annurev.genet.37.110801.143030. [DOI] [PubMed] [Google Scholar]
- McVean GA, Myers SR, Hunt S, Deloukas P, Bentley DR, Donnelly P. The fine-scale structure of recombination rate variation in the human genome. Science. 2004;304:581–584. doi: 10.1126/science.1092500. [DOI] [PubMed] [Google Scholar]
- Meiklejohn CD, Tao Y. Genetic conflict and sex chromosome evolution. Trends. Ecol. Evol. 2010;25:215–223. doi: 10.1016/j.tree.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiri M, Lister AM, Collins MJ, Tuross N, Goebel T, Blockley S, Zazula GD, van Doorn N, Dale Guthrie R, Boeskorov GG, Baryshnikov GF, Sher A, Barnes I. Faunal record identifies Bering isthmus conditions as constraint to end-Pleistocene migration to the New World. Proc. R. Soc. B Biol. Sci. 2014;281 doi: 10.1098/rspb.2013.2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro A, Barton NH. Chromosomal speciation and molecular divergence--accelerated evolution in rearranged chromosomes. Science. 2003;300:321–324. doi: 10.1126/science.1080600. [DOI] [PubMed] [Google Scholar]
- Nei M. Molecular Evolutionary Genetics. Columbia University Press; New York: 1987. [Google Scholar]
- Nolte V, Pandey RV, Kofler R, Schlötterer C. Genome-wide patterns of natural variation reveal strong selective sweeps and ongoing genomic conflict in Drosophila mauritiana. Genome Research. 2013;23:99–110. doi: 10.1101/gr.139873.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson JT, Stone WS. Evolution in the Genus Drosophila. The Macmillan Company; New York: 1952. [Google Scholar]
- Perlman SJ, Jaenike J. Infection success in novel hosts: an experimental and phylogenetic study of Drosophila-parasitic nematodes. Evolution. 2003;57:544–557. doi: 10.1111/j.0014-3820.2003.tb01546.x. [DOI] [PubMed] [Google Scholar]
- Pinzone CA, Dyer KA. Association of polyandry and sex-ratio drive prevalence in natural populations of Drosophila neotestacea. Proc. R. Soc. B Biol. Sci. 2013;280:20131397. doi: 10.1098/rspb.2013.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presgraves DC, Gerard PR, Cherukuri A, Lyttle TW. Large-scale selective sweep among Segregation Distorter chromosomes in African populations of Drosophila melanogaster. PLoS Genet. 2009;5:e1000463. doi: 10.1371/journal.pgen.1000463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prout T, Bundgaard J, Bryant S. Population genetics of modifiers of meiotic drive I. The solution of a special case and some general implications. Theor. Popul. Bio. 1973;4:446–465. doi: 10.1016/0040-5809(73)90020-8. [DOI] [PubMed] [Google Scholar]
- RCoreTeam . R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; Vienna, Austria: 2014. [Google Scholar]
- Rice WR. Nothing in genetics makes sense except in light of genomic conflict. Annu. Rev. Ecol. Syst. 2013;44:217–237. [Google Scholar]
- Schafer DJ, Fredline DK, Knibb WR, Green MM, Barker JSF. Genetics and linkage mapping of Drosophila buzzatii. J. Heredity. 1993;84:188–194. doi: 10.1093/oxfordjournals.jhered.a111315. [DOI] [PubMed] [Google Scholar]
- Scheet P, Stephens M. A fast and flexible statistical model for large-scale population genotype data : Applications to inferring missing genotypes and haplotypic phase. Am. J. Hum. Genet. 2006;78:629–644. doi: 10.1086/502802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staten R, Schully SD, Noor MAF. A microsatellite linkage map of Drosophila mojavensis. BMC Genetics. 2004;5:8. doi: 10.1186/1471-2156-5-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan W, Ashburner M, Hawley RS. Drosophila Protocols. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, N.Y: 2000. [Google Scholar]
- Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watterson GA. On the number of segregating sites in genetical models without recombination. Theor. Popul. Biol. 1975;7:256–276. doi: 10.1016/0040-5809(75)90020-9. [DOI] [PubMed] [Google Scholar]
- Werren JH. Selfish genetic elements, genetic conflict, and evolutionary innovation. Proc. Natl. Acad. Sci. 2011;108:10863–10870. doi: 10.1073/pnas.1102343108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.