

Summary
Transendothelial migration (TEM) of PMN involves a carefully orchestrated dialogue of adhesion and signaling events between leukocyte and endothelial cell. This chapter will focus on the endothelial cells’ contribution to transmigration. The initiation of TEM itself generally requires interaction of PECAM on the leukocyte with PECAM at the endothelial cell border. This is responsible for the transient elevation of cytosolic free calcium ions in endothelium that is required for TEM and for recruitment of membrane from the lateral border recycling compartment (LBRC). TEM requires LBRC to move to the site at which TEM will take place and for VE-cadherin to move away. Targeting of the LBRC to this site likely precedes movement of VE-cadherin, and may play a role in clearing VE-cadherin from the site of TEM.
The process of TEM can be dissected into steps mediated by distinct pairs of PMN/endothelial interacting molecules. CD99 regulates a step at or close to the end of TEM. CD99 signals through soluble adenylyl cyclase to activate PKA to trigger ongoing targeted recycling of the LBRC. Paracellular transmigration predominates (≥ 90% of events) in the cremaster muscle circulation, but transcellular migration may be more important at sites such as the blood brain barrier. Both processes involve many of the same molecules as well as recruitment of the LBRC.
Keywords: transmigration, endothelial cell junctions, platelet/endothelial cell adhesion molecule 1 (PECAM), lateral border recycling compartment (LBRC), CD99
Introduction
This chapter will cover the process of transendothelial migration (TEM) or diapedesis, the stage in leukocyte extravasation in which leukocytes squeeze across the endothelial lining of blood vessels en route to the site of inflammation. There are many comprehensive reviews of transmigration (1–6) to which the reader is referred; our knowledge of this process has increased remarkably in the past decade. In keeping with the spirit of Immunological Reviews, we will “review our own work within the context of the field…” and point out where controversies lie and where important information still awaits discovery. Since there are other chapters in this volume on interactions of neutrophils (PMN) with epithelial cells and even transmigration of PMN, this chapter will discuss transendothelial migration from the point of view of the endothelial cells. The focus will be on TEM of neutrophils. Many of the same molecules and mechanisms mediate TEM of monocytes and lymphocytes. However, where there are known differences, these will be pointed out.
The molecules and signaling pathways that are most important for TEM vary depending on the leukocyte type, the vascular bed, the inflammatory stimulus, and the chronicity of the inflammatory response. PMN generally participate in the acute inflammatory response, but are otherwise subjected to the same caveats. The molecules and signaling pathways described here have been found to be generally relevant and robust using both in vitro and in vivo models. However, there will undoubtedly be exceptions, since blocking any single molecule or pathway never inhibits leukocyte rolling, adhesion, or TEM completely.
Most transmigration in response to inflammation takes place at postcapillary venules. At these sites transmigration follows the steps of capture, rolling, activation, adhesion, and intraluminal crawling in the extravasation cascade (3, 7, 8). Exceptions occur in the lung in which most TEM takes place through capillaries thinner than the diameter of PMN so this elaborate sequence of adhesive events is not necessary to arrest them at the site (9–11). These steps are reversible, and most of the neutrophils (PMN) that enter the postcapillary venule feeding the site of inflammation do not roll; most of those that roll do not adhere, and even most of the adherent PMN do not extravasate (12). However, once a PMN commits to diapedesis, with rare exceptions (13), it traverses the endothelium and underlying basal lamina to enter the inflamed tissue. Most of the benefits as well as the unintended consequences of inflammation take place after leukocytes cross the blood vessels. Thus, we have felt that TEM is the “point of no return” of the inflammatory response and will focus on the molecules and mechanisms regulating it.
There are two pathways for TEM. In paracellular migration leukocytes move between apposing endothelial cells; in transcellular migration they move through the body of the endothelial cell (1, 14–16). Transcellular migration was controversial for a long time, since evidence to support it was limited to static thin section electron micrographs in which TEM at a tricellular junction (a preferred site for PMN transmigration (17)) could resemble transcellular migration close to an endothelial cell junction (18). However, studies employing extensive serial sectioning (15) as well as high resolution intravital microscopy (13) demonstrate that transcellular migration can and does occur in vivo. Based in intravital microscopic observations few PMN (< 10%) transmigrate transcellularly in vivo (13). Nevertheless, there is evidence that a greater proportion of lymphocytes transmigrate transcellularly at special sites such as the blood-brain barrier, and in some in vitro models (19, 20). Paracellular and transcellular migration seem at first glance to be very different pathways. However, as we learn more about them, we find more similarities than differences (1, 14, 21, 22).
While increased vascular permeability at the site of inflammation and leukocyte transmigration occur in spatial and temporal proximity, they are distinct events (23). Plasma leakage takes place more proximally in smaller (20 – 40 μm) postcapillary venules, while PMN exit from larger (40 – 60 μm) venules (24, 25). This makes sense, since leakage of plasma provides the hemoconcentration and slower flow that allows leukocytes to make productive contacts with the endothelial wall. These processes can even be dissected genetically. Cortactin-deficient mice have constitutively elevated vascular permeability but defective TEM (26). Many of the same signaling molecules and pathways regulate vascular permeability as well as TEM. What likely contributes to the maintenance of the endothelial barrier during leukocyte diapedesis is that these signals are localized to the portion of the endothelial cell interacting with the leukocyte during TEM, while they are more diffusely activated in response to soluble mediators that trigger vascular permeability (23).
Preludes to transendothelial migration
As PMN approach the site of transmigration (whether it is paracellular or transcellular) intercellular cell adhesion molecule-1 (ICAM-1) on the endothelial cell becomes concentrated below the leukocyte (14, 27, 28) due to interactions with leukocyte function associated antigen (LFA-1, CD11a/CD11b) on the leukocyte (27). Vascular cell adhesion molecule-1 (VCAM-1) shows a similar concentration under and around lymphocytes and monocytes (29). Contemporaneously, tetraspanin molecules on the endothelial cell become co-localized with these molecules (30). Engagement of ICAM-1 activates the actin-bundling molecule cortactin, which helps bring f-actin into this site and also recruits more of the cytoplasmic tail of ICAM-1 to continue the process in what may be a positive feedback loop (31, 32).
Cortactin-deficient mice have defective neutrophil TEM due to defective ICAM-1 clustering. They also have inherently leaky venules due to reduced levels of Ras-related protein 1 (Rap1) the absence of activation by cortactin (26). In addition to confirming a role for cortactin in both of these phenomena, this paper demonstrated genetically what many in the field had recognized from much indirect evidence: That TEM and opening of endothelial junctions are distinct and separable events.
When tracked with fluorescent molecules or stained post hoc with fluorescent antibodies, the co-localization of ICAM-1 and LFA-1 appears to continue during the course of transmigration, with LFA-1 redistributing to the uropod (lagging edge) of the PMN to keep in contact as the PMN migrates below the endothelium (27). As diapedesis begins ICAM-1, VCAM-1, tetraspanins, and actin become highly concentrated along the endothelial cell membrane surrounding the leukocyte as it transmigrates, giving the appearance of a ring by immunofluorescent staining (27, 29, 30). Orthogonal views of confocal immunofluorescence images of these molecules show intense staining surrounding the transmigrating leukocyte (Figure 1) whether it is passing paracellularly or transcellularly (14, 33). In addition to these rings, fingers of fluorescence stretching above the apical surface of the endothelial and partially surrounding the leukocytes have been reported by some (28–30, 33, 34) but not others (14, 27, 35–37). The differences are likely due to differences in the in vitro assays used and the time it took for TEM to be completed (38). These have been interpreted as apical membrane forming “docking structures” (29) or “transmigratory cups” (33) or artifacts due to of out-of-focus light over the rings in the X-Y plane being reconstructed in the X-Z plane (1, 38). Their presence or absence does not seem to affect the rate or extent of TEM (28, 33).
Figure 1. Enrichment of endothelial cell adhesion molecules around leukocytes migrating transcellularly.

Representative confocal or deconvolved images of monocytes or PMN caught in the act of transcellular migration are shown stained with mAb for the indicated cell adhesion molecule (CAM, left panel, green in merge), phalloidin to mark actin (middle panel, red in merge), and DAPI to mark nuclei (blue in merge). Rings of enrichment of ICAM-1, PECAM, CD99, and JAM-A were consistently seen surrounding the site at which leukocytes migrated through the endothelial cell (arrows in left panel). Each event was verified by scanning through the monolayer. Orthogonal (X-Z) projections along the plane indicated by the white line (XZ) shown below the merged images demonstrate that the leukocyte was indeed crossing the endothelial cell (arrows in right panel). Thin white line underlies the basal surface of the endothlelial cell. The JAM-A panels show a monocyte (upper panel) and a PMN (lower panel). Note in the PECAM panel and upper JAM-A panel that several leukocytes are present on the endothelial cell, but only the site of transcellular migration stains for PECAM or JAM-A, respectively, demonstrating that the PECAM and JAM-A stained around the leukocyte belongs to the endothelial cell. (*) indicates leukocytes not in the act of transmigration and not stained for PECAM or JAM-A. Long arrow in PECAM and VE-cadherin panels point to the site of transcellular migration where endothelial cell actin is pushed aside. Arrowheads mark cell borders. Scale bar = 10 μm. Reproduced from (14) with permission.
Initiating transmigration: Major membrane movements
At least two events in the endothelial cell are critical for efficient transendothelial migration: Mobilization of VE-cadherin away from the site of TEM and influx of membrane from the lateral border recycling compartment (LBRC) to the site of TEM (36, 37, 39, 40). Both occur early in the process of transmigration and continue until TEM is complete (36, 41). Blocking either process reduces TEM by up to 90%, depending on the inflammatory model.
Mobilization of vascular endothelial cell cadherin (VE-cadherin)
VE-cadherin (cadherin-5, CD144) is a type I transmembrane protein that (after birth) is exclusively expressed in endothelial cells. It is concentrated at adherens junctions where it forms calcium dependent homophilic interactions and participates in barrier function and transendothelial migration (42–44). VE-cadherin surface expression is stabilized by its association with the cytoplasmic protein p120 catenin, and it can link to the actin cytoskeleton via its interaction with β-catenin and plakoglobin, members of the armadillo gene family (39, 45–47).
Adhesion of leukocytes to the endothelial cell activates downstream signaling pathways that induce VE-cadherin to be cleared from the site of transmigration to produce what appears as a gap in VE-cadherin staining along the junction (41, 48). VE-cadherin is internalized into clathrin coated vesicles under a variety of conditions (49–51). However, in the setting of TEM, VE-cadherin may not be internalized but rather pushed aside along the plane of the membrane and diffuse back to refill the junction once transmigration is complete (39, 41).
Under resting conditions, the vascular endothelial specific protein tyrosine phosphatase (VE-PTP) is associated with VE-cadherin via plakoglobin, maintaining it in a hypophosphorylated state. Interaction of activated endothelial cells with leukocytes triggers dissociation of VE-PTP from VE-cadherin, allowing the latter to be phosphorylated (52, 53). Engagement of endothelial ICAM-1 and VCAM-1 by leukocyte integrins transduces signals that phosphorylate VE-cadherin and its associated catenins (54–57). Phosphorylation of VE-cadherin at its cytoplasmic tail results in the weakening of the junctions as the cytoplasmic tail is uncoupled from the actin cytoskeleton. Two key tyrosine residues, Y658 and Y731 on the cytoplasmic tail of VE-cadherin have been implicated in this process (40, 58). (Note that Y658 is distinct from the Y685 implicated to have a role in permeability in response to soluble mediators (58).) The Vestweber group recently showed that mice in which VE-cadherin was replaced with the Y731F mutant to mimic the dephosphorylated state of VE-cadherin had a specific defect in leukocyte transmigration (58).
Targeted recycling of membrane from the LBRC
When a non-blocking monoclonal antibody against platelet/endothelial cell adhesion molecule 1 (PECAM, CD31) is added to endothelial cells at 37°C, it is seen by immunoelectron microscopy not only along the endothelial border but also in a subjunctional structure of interconnected 50 nm vesicles and tubules (36). PECAM from this compartment exchanges with the surface membrane at the cell border with a half-time of approximately 10 minutes. Therefore this compartment was called the Lateral Border Recycling Compartment or LBRC (36). The LBRC is contiguous with the plasma membrane, suggesting that this compartment is a complex invagination of the junctional membrane that extends several hundred nanometers into the cell. Interestingly, the compartment is labeled well at 37°C, but incubating endothelial cells with the same antibodies at 4°C only labeled the junction (36, 59). Electron micrographs clearly show that the compartment still exists under this condition, indicating that PECAM in the LBRC is protected from large molecules at 4°C. This is not because the vesicles have pinched off, as protons and small molecules such as biotin can still enter (36, 60). Under resting conditions roughly 30% of the total PECAM is in the LBRC (36).
The LBRC is distinct from other endothelial vesicular organelles such as the vesiculo- vacuolar organelle (VVO) and caveolae. VVOs are much larger (>150 nm) in size, more heterogeneous in shape, and typically communicate between the apical and basal surfaces (61). VVOs open and close in response to VEGF whereas the LBRC is completely and continuously accessible to the exterior of the cell (62). VVOs have not been observed in vitro. Likewise, caveolae are rare in endothelial cells in vitro and are typically observed as single vesicles or single invaginations on the apical and basal membranes, seldom present at the junction. By immunofluorescence, caveolin-1 (a marker of both caveolae and VVOs (63, 64)) does not co-localize with PECAM at the junction or during TEM (14, 36). In addition, biochemical analysis of PECAM and caveolin-1 shows that the two localize to different membrane microdomains (36). This was examined using sucrose gradients to purify cholesterol-rich membrane microdomains from endothelial cells solubilized with cold non-ionic detergent (36). In this analysis, caveolin-1 was predominantly recovered in the buoyant fractions that correspond to lipid rafts whereas PECAM was recovered from the dense fractions which contained the solubilized proteins that are excluded from membrane microdomains. (This is in distinction to monocytes, where activation of PECAM led to its phosphorylation and partition into cholesterol-rich microdomains (65) and in platelets where a small fraction of palmitoylated PECAM can be recovered in such domains (66).)
The LBRC also contains other molecules reported to positively regulate TEM: junctional adhesion molecule A (JAM-A) (14), CD99 (14), PVR (59), and nepmucin (67), which regulates lymphocyte TEM in high-endothelial venules. The LBRC is also observed in vivo as EM examination blood vessels from mice injected intravenously with anti-mouse PECAM-HRP shows an identical sub-junctional staining pattern (unpublished results).
The same immunoelectron microscopy experiments show that VE-cadherin, a molecule that negatively regulates TEM, is not in the LBRC (14, 59). We have recently developed a way to isolate the LBRC from endothelial cells (68). A proteomic approach was taken to identify proteins unique to or enriched in the LBRC. These studies confirmed that caveolin-1 is not a component of the LBRC and revealed that in addition to VE-cadherin, its associated catenins were also excluded from the LBRC. We were unable to identify any unique proteins in the LBRC; however, both IQ motif GTPase-activating protein 1(IQGAP1) and vimentin were specifically enriched in the LBRC fraction (69).
We recently used chimeric molecules of CD25, PECAM, and VE-cadherin to study the signals for inclusion of membrane proteins into the LBRC (60). Inclusion of proteins from the lateral border of the endothelial cell plasma membrane into the LBRC seems to be the default pathway. VE-cadherin is actively excluded by its homophilic interaction motif (RVDAE). Removal of this domain from VE-cadherin or competition with a soluble RVDAE peptide allowed its entry into the LBRC. This explains why no unique proteins were found in the isolated LBRC and enriched proteins were associated with the cytoplasmic side (69). This study also demonstrated that the constituents of the LBRC move in concert during TEM; there is no separation of membrane enriched in PECAM or CD99 (60).
During TEM, the LBRC moves along microtubules to engage the transmigrating leukocyte in a process called ‘targeted recycling.’ This is a critical step in diapedesis. Disrupting PECAM-PECAM homophilic interactions ablates both TEM and targeted recycling (36). The enrichment of the LBRC at the interface with the leukocyte is similarly blocked when the endothelial cells are treated with reagents demecholcine or taxol, which depolymerize microtubules or cause microtubule bundling, respectively (37). Neither treatment affected the size or the constitutive recycling of the compartment suggesting instead that microtubules are essential for LBRC movement during TEM. This is supported by the finding that LBRC targeted recycling is also impaired upon microinjection of function blocking antibodies against the conserved motor domain of kinesin, a molecular motor that facilitates intracellular traffic along microtubules toward the plus end (37).
There are 45 kinesin heavy chains that comprise 15 kinesin families. We recently found that knockdown of kinesin-1 with shRNA or blockade by intraendothelial cell injection of a blocking monoclonal antibody specific for kinesin-1 arrested targeted recycling of the LBRC and TEM of monocytes to 10–15% of control levels (70). Kinesin-1 associates with several known light chains, each of which has multiple isoforms generated by alternative splicing. Light chains assist in binding of cargo to the kinesin heavy chain and the alternatively spliced carboxy terminal ends help define the cargo specificity (71–74). We found that kinesin light chain 1 (KLC1) was the light chain responsible for targeted recycling of the LBRC and surprisingly, a single splice form of KLC1, KLC1 variant 1 (also known as KLC1C) was the major—perhaps the only—isoform involved: Knockdown of KLC1C alone blocked targeted recycling of the LBRC and TEM maximally; re-expression of KLC1C restored both. Other splice variants had no effect (70). These results were true for PMN as well as for monocytes.
Available data suggest that the molecules in the LBRC, not those on the surface of the junction are the ones that are required for TEM. When endothelial cells were treated with blocking antibodies against PECAM, PVR, or CD99 at 4°C, which leaves the protected LBRC pool of these molecules untouched, TEM remained unaffected, while the same antibodies given at 37°C (conditions under which they enter the LBRC) blocked TEM (59, 75).
Relationship of VE-cadherin movement to LBRC targeted recycling
Removal of VE-cadherin from the endothelial junction at the site of TEM and targeted recycling of the LBRC to the site of TEM are required for efficient TEM. Are these processes independent, cooperative, or sequential? Does one depend on the other? We recently investigated this question for transmigration of both PMN and monocytes (76). By reversibly depolymerizing microtubules in endothelial cells or injecting a function-blocking kinesin antibody, we were able to inhibit targeted recycling and TEM, as previously published (37). Under these conditions VE-cadherin was not cleared from the membrane underneath the adherent leukocytes (76). We then performed the converse experiment, transfecting endothelial cells to express a form of VE-cadherin with tyrosines 658 and 731 mutated to phenylalanine so they could not leave the junction (40). Under these conditions, TEM was blocked; however, targeted recycling of LBRC still took place (76). Thus, targeted recycling of the LBRC is triggered and can occur in the absence of movement of the LBRC.
This suggests that LBRC movement occurs first in response to leukocytes positioned at the endothelial border and may actually participate in moving VE-cadherin-containing membrane out of the way (68, 76). Recall that the LBRC does not contain VE-cadherin; in fact it is actively excluded from the LBRC (60). The influx of LBRC membrane to surround the leukocyte could either dilute or push aside VE-cadherin and its associated catenins. When the leukocyte has passed across the endothelium, retrieval of this membrane back into the LBRC would allow VE-cadherin to become enriched at the junction again by exclusion from the LBRC or by lateral movement along the plasma membrane. This would allow junctions to re-form rapidly without having to reassemble their complex three dimensional structure (1, 60, 68, 76). In fact, not only adherens junctions but also tight junctions reform very rapidly after transmigration (77).
The presence within the LBRC of the molecules that positively regulate TEM suggests that it could act as a reservoir of these molecules for TEM as well as a reservoir of junctional membrane. By simple geometry, about 2/3 of the membrane surface area necessary to surround the transmigrating leukocyte could come from the junctional plasma membrane at the cell borders immediately adjacent to the leukocyte (68). The additional 1/3 could come from the LBRC; this is precisely the fraction of membrane that resides in the LBRC (36). In this way, transmigration could proceed without retraction of the endothelial cells.
Initiating transmigration: Major signaling events
When leukocytes arrive at what will become the site of transmigration on the apical surface of the endothelial cell, at least two critical signaling events occur: 1) PECAM on the leukocyte engages PECAM at the endothelial cell border and 2) there is a transient increase in cytosolic free calcium ion concentration (↑[Ca+2]i ) in the endothelial cells (1, 3, 78, 79). Interfering with either process results in a similar phenotype: Neutrophils (80) or monocytes (81) arrested over the endothelial cell borders tightly adherent, but unable to transmigrate (75). Live imaging (82) confirms what was suspected from transmission and scanning electron micrographs (75, 83): that the leukocytes are actively moving along the endothelial borders probing with lamellapodia, as if looking for a spot to transmigrate. There is no change in the ability of neutrophils, monocytes, or lymphocytes to adhere to the endothelial cells or move to the junctions; only TEM is inhibited (37, 75, 84).
The initial reports of these phenomena were published the same year (75, 80). Despite the similarity of these phenotypes, in vitro studies did not show a connection between PECAM ligation and ↑ [Ca+2]i in endothelial cells (85)(and unpublished results from our lab) that could be detected by calcium sensitive fluorescent dyes in vitro. However, recently a link has been established.
PECAM-PECAM engagement
PECAM is comprised of six extracellular immunoglobulin-like domains, a transmembrane domain, and a cytoplasmic tail containing two immunotyrosine inhibitory motifs (ITIM) (86, 87). Interaction of the homophilic interaction domain (the amino-terminal immunoglobulin family domain 1) (83, 88) of PECAM on the leukocyte with PECAM domain 1 on the endothelial cell is required for TEM (75, 83). Monoclonal antibodies recognizing this domain (75) or PECAM domain 1-IgG fusion proteins (83) block TEM of PMN and monocytes in vitro and in vivo (89–91). Incubating either leukocytes or endothelial cells with anti-PECAM antibody blocks equally efficiently, and as well as blocking both together, consistent with a homophilic mechanism (75).
Activating PECAM in platelets, lymphocytes, and endothelial cells results in phosphorylation of the ITIM domain tyrosines 663 and 686 (87, 92–94). PECAM has no endogenous tyrosine kinase activity, but is phosphorylated by src and fer family kinases (95–97) to recruit src homology region 2 domain-containing tyrosine phosphatase 2 (SHP-2) (98–100). In T cells and platelets, it has an inhibitory role (92, 101, 102). In most of these cases, tyrosine 686 is the critical phosphorylation site with 663 playing a secondary role.
It therefore came as a surprise when we found that when interacting with monocytes, phosphorylation of tyrosine 663 on endothelial cell PECAM was critical for TEM; tyrosine 686 phosphorylation was not necessary (103). Gain of function experiments showed that expression of PECAM containing tyrosine 663 in ECV-304 cells could impart the ability to support TEM even if tyrosine 686 was mutated to phenylalanine (Y686F). On the other hand, expression of PECAM with Y663F could not support TEM. In this case, TEM was rescued by transfection of PECAM in which Y663 was not mutated (103). Mechanistically, wild-type PECAM and PECAM Y686F could enter the LBRC and undergo targeted recycling. On the other hand, PECAM Y663F had difficulty entering and leaving the LBRC and was not targeted efficiently to the site of monocyte interaction with the endothelial junction (103).
The mechanism by which phosphorylation of tyrosine 663 on the cytoplasmic tail of PECAM affects targeted recycling of the LBRC is not known. To be honest, we do not even know that phosphorylation is necessary. We know that mutation of tyrosine 663 to phenylalanine inhibits the process (103). It is possible that an unmodified tyrosine residue at that position is required for efficient targeted recycling. In a somewhat related manner, de-phosphorylation of mouse VE-cadherin tyrosine 731 is thought to be critical for VE-cadherin movement during TEM (58).
Transient increase in endothelial cell cytosolic free calcium ion concentration (↑ [Ca2+]i )
As mentioned above, pharmacological chelation of endothelial Ca2+ during TEM results in a phenotype in which leukocytes adhere normally to the apical surface of endothelial cells, but are unable to transmigrate across (33, 80, 81, 104, 105) similar to blocking PECAM homophilic interactions with primary antibody (75), suggesting that these two processes might be related.
Further evidence came when we found that chelation of endothelial Ca2+ inhibited targeted recycling of the LBRC to the same extent that it inhibited TEM (84). Transient receptor potential canonical family member six (TRPC6) is a non-selective cation channel that is five-fold more permeable to Ca2+ than Na+ (106–108). It is the most abundant TRPC channel in endothelial cells (109, 110) and is concentrated along endothelial cell borders, where it seems to be further enriched in the endothelial membrane surrounding transmigrating PMN (84).
Expression of a dominant negative TRPC6 or knockdown of endogenous TRPC6 in human umbilical vein endothelial cells dramatically reduced targeted recycling of the LBRC and TEM of PMN and monocytes (84). Both processes were restored in endothelial cells by expression of a rescue construct expressing TRPC6 resistant to the shRNA (84). In TRPC6-deficient mice reconstituted with wild-type bone marrow, PMN were still recruited to a site of inflammation in the skin, but there was a selective defect in transmigration (84). PMN were arrested on the luminal surface of postcapillary venules, similar to the phenotype seen when PECAM was blocked (89, 91, 111, 112). The block was qualitatively and quantitatively similar to that achieved in the global TRPC6 knockout, demonstrating that absence of TRPC6 in endothelial cells was primarily responsible for the effect (84). TRPC6-deficient mice have previously been shown to have defects in the inflammatory response (106, 113–122). This work provides a mechanism to explain the failure to recruit leukocytes in these models.
Transendothelial migration as a multistep process
After the LBRC has been recruited to the site of TEM via PECAM-PECAM interactions, and VE-cadherin has left the site of TEM, migration still requires additional molecular interactions. This is demonstrable because one can selectively and reversibly block the transmigration step using antibodies against specific molecules. In a sequential transmigration assay (Fig. 2), one can block TEM using monoclonal antibody (mAb) against molecule A for an appropriate time, then wash out the block, add a mAb against molecule B, and resume the TEM assay. If molecule B is involved in a process downstream of A, mAb B will block any further TEM. If molecule B is involved in a step of TEM before molecule A, it will not (59, 68, 123). This was first shown for the relationship of PECAM and CD99 (123). CD99 is expressed at the borders of EC and is in the LBRC (14). If TEM is blocked by anti-CD99 monoclonal antibody, the initial round of PECAM-mediated targeted recycling takes place (124), and PMN (124, 125) or monocytes (123, 124) begin TEM, but cannot complete it. They are stuck partway through, with their leading edge below the endothelial monolayer, but their uropod remaining on the apical surface (123–125). Recent studies showed that after the initial round of targeted recycling, further targeted recycling does not occur under these conditions (124).
Figure 2. Schematic diagram of the sequential block assay.
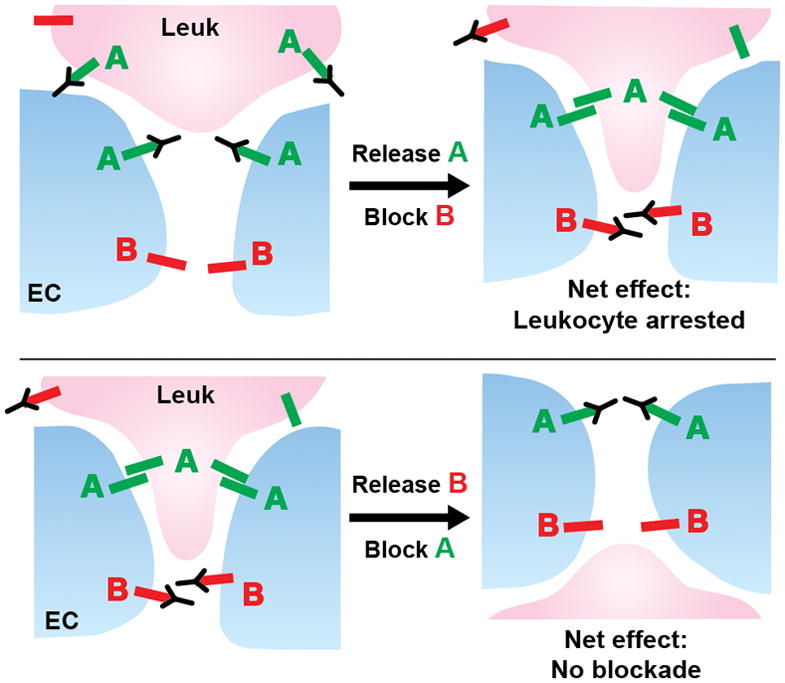
Molecules A and B on the EC regulate TEM in that order. (Top) If TEM is blocked by antibody against molecule A, and that block is released, antibody against B can still arrest TEM. (Bottom) If TEM is blocked by antibody against molecule B, and that block is released, TEM can no longer be blocked by antibody against A.
The fact that two distinct molecules regulate different steps in TEM begs the question of whether there are other molecules that mediate additional steps in the process. In fact, poliovirus receptor (CD155, PVR), expressed at EC borders, is involved in TEM of monocytes (126). Using the sequential TEM assay, we found that PVR, interacting with the leukocyte ligand DNAX accessory molecule-1, controls a step in TEM that is between those regulated by PECAM and CD99 (59). PVR is also in the LBRC and, as expected, PVR mAb delivered to the LBRC blocked TEM (59).
Completing transmigration: The role of CD99
CD99 is a type 1 membrane protein concentrated at the cell borders of endothelial cells and expressed diffusely on the surfaces of circulating blood cells to varying degrees. It is highly glycosylated by O-linked carbohydrates, which account for 44% of its apparent molecular weight of 32 kD. It is a fairly unique molecule with only one paralog in the genome (127) and no signaling motifs on its cytoplasmic tail. As mentioned above, inhibition of homophilic interaction between CD99 on the leukocyte and CD99 on the endothelial cell arrests TEM with the leukocyte partway across the endothelial junction with its leading lamellipodium under the EC and a trailing uropod on the apical surface (123, 125), in contrast to the appearance of leukocytes blocked at the PECAM-dependent step, which appear entirely on the apical surface hovering over the junctions (75).
CD99 is constitutively in a complex at the endothelial cell border with three intracellular molecules: the A-kinase anchoring protein ezrin, the unique soluble adenylate cyclase (sAC), and protein kinase A (PKA). Engagement of CD99 activates sAC to produce cAMP locally to activate PKA and orchestrate the next wave of LBRC recruitment (124). An evolutionarily conserved basic region in the juxtamembrane region of the cytoplasmic tail is critical for maintaining CD99 in this complex. Mutation of these lysine residues to glutamate abolished the ability of CD99 to form a complex with ezrin and abolished the ability of the endothelial cells to support TEM (124).
In vivo as well as in vitro a small molecule inhibitor of sAC (KH7) and endothelial cell specific knockout or knockdown of sAC inhibited PMN and monocyte TEM in a model of acute inflammatory dermatitis (124). The phenotype of the block appeared identical to the blockade imparted by anti-CD99 mAb (123–125).
Completing transmigration: Membrane and cytoskeletal movements
Following TEM, the junctions must reseal and tight apposition of adjacent endothelial cells must be re-established. Unfortunately, relatively little is known about how adherens and tight junctions rapidly reform following TEM (77). Nothing is known about retrieval of the LBRC; is it continuously recycled during the leukocyte passage or is it retrieved in one large membrane movement at the end?
How do the endothelial transmigration pores close? The increase in isometric tension in endothelial cells that has been reported to follow ↑ [Ca2+]i due to phosphorylation of myosin light chain kinase was thought to help open the endothelial borders in preparation for TEM (128). Recently, this tension was shown to come during the second half of TEM when the transmigration pore was decreasing in size (129). This corresponded to contraction of f-actin fibers surrounding the transmigration pore and an increase in RhoA activity as registered by RhoA activity sensors (129). The authors hypothesize that during the initial phase of TEM, vascular leakage across the transmigration pore is minimized because the leukocyte is actively pushing against the endothelial cells to open a passage for TEM (129). In addition, continuous interaction of adhesion molecules from the LBRC with ligands on the leukocyte would help maintain tight apposition (68).
What about movement of membrane after pore closure? As a result of the loss of isometric tension in the endothelial monolayer, lamellipodia form on the ventral surface of the endothelial cells and move across the substratum to contact the neighboring endothelial cell and seal the “wound.” This process was effective for both paracellular and transcellular migration and required several Rac1 effectors as well as localized production of hydrogen peroxide (130).
Transcellular Migration
Although it is well accepted that leukocytes cross the endothelium at the cell borders (paracellular route) there is increasing evidence that leukocytes can also pass directly through endothelial cells (transcellular route). Much of the original evidence was indirect, based on single transmission electron micrographs that appeared to show leukocytes deeply indenting endothelial cells and/or passing across endothelial cells through a membrane-lined channel next to an intact junction (131–133). However, endothelial junctions are serpentine, and it was possible that the leukocyte was passing through a less structured junction (18). For a nice historical review of transcellular migration the reader is referred to reference (19). Transcellular migration was viewed with skepticism despite (or perhaps because of) the large amount of circumstantial evidence that supported it. The skeptics were to a great extent appeased by a paper published by the Dvorak lab in 1998 (15). This provided arguably the first indisputable evidence of neutrophil transcellular migration in vivo. In these studies neutrophil emigration was stimulated by direct injection of fMLF, a neutrophil chemoattractant and β2 integrin activator, into the skin of guinea pigs (15). The authors presented a collection of electron microscope serial sections in which neutrophils were shown to pass entirely across an endothelial cell without ever contacting a recognizable junction (15). Although the absolute frequency of transcellular migration was not addressed in this study, it demonstrated that transcellular migration was possible in vivo. High resolution intravital microscopy studies in which endothelial cell borders are labeled with non-blocking anti-PECAM antibody and PMN express eGFP have since demonstrated that transcellular migration does indeed occur under physiological conditions, but accounts for ≤ 10% of TEM events (13).
Molecules implicated in transcellular migration
Recently, several in vitro models were established that produced reliable transcellular migration (14, 33, 35, 134). These provided data on the molecules and mechanisms involved in this process. Many of the same molecules important for paracellular migration turn out to have a role in transcellular migration.
During transcellular TEM, ICAM-1 that is uniformly expressed on the luminal surface of HUVEC redistributes and is concentrated at the site of diapedesis. Furthermore, it is enriched in the membrane channel that surrounded the crossing leukocyte as it went through the EC body (14, 22, 33, 35, 134). However, for transcellular migration, as for paracellular migration, some investigators (27, 35, 37) do not observe the “docking structures” or “transmigratory cups” formed by projections of ICAM-1 above the plane of the endothelial cell membrane reported by others (29, 33).
In addition to ICAM-1, other molecules normally thought of to be restricted to the cell borders, including PECAM, JAM-A, and CD99 are seen around the leukocyte migrating transcellularly (14, 22, 33, 35, 134). In all cases, VE-cadherin is absent (See Fig. 1). These molecules are not only present, but appear to be functional. Somewhat surprisingly, transcellular migration was found to be dependent on PECAM and CD99. Blocking antibodies arrest transcellular migration (14). The reason for this may be explained by the mechanism, discussed below.
What determines the site of transmigration?
Whether the leukocyte migrates paracellularly or transcellularly may depend on the relative tightness of the endothelial junctions and the ability of the leukocyte to breach them. Carman and Springer (16) speculated that the leukocytes take the “path of least resistance” across the endothelium. If junctions are very tight, as for example, in the blood brain barrier, migrating across the cell at a thin point may be easier, as seen in cerebral inflammation (20, 135). However, that cannot be the only explanation since postcapillary venules, which are the sites of most inflammatory diapedesis (including transcellular migration (15)) have relatively leaky junctions. In fact, they are specialized for permeability, since this is the site of fluid re-uptake from the interstitium (136). Endothelial cells in culture, where transcellular migration has been best demonstrated, form a monolayer of low electrical resistance. When grown under conditions that result in ten-fold higher resistance, the frequency of transcellular TEM was no higher than under standard conditions (137).
Transcellular migration may also occur if the leukocyte has difficulty reaching the junction, such as cells in which CD11b is non-functional (82) or deficient (138). Phillipson, et al. (138) reported that neutrophils deficient in CD11b/CD18 adhered to the endothelium at a site of inflammation, but transmigrated poorly due to their inability to locomote along the endothelial surface. However, those that transmigrated tended to go transcellularly, perhaps because they achieved sufficient activation to transmigrate before they reached the cell border. Similarly, T cells deficient in the Rac activator Tiam1 are deficient in polarization and locomotion on endothelium, and tend to migrate transcellularly (139).
The role of leukocyte activation
We hypothesize that for low-resistance endothelia at least, transcellular migration may occur when leukocytes are highly and/or directly activated. The first indisputable evidence of neutrophil transcellular migration in vivo came from studies in which emigration was stimulated by direct injection of fMLF, a neutrophil chemoattractant and β2 integrin activator, into the skin of guinea pigs (15). The published in vitro studies of transcellular migration used MCP-1, platelet activating factor, or SDF-1 to stimulate migration of monocytes, neutrophils, and T cells, respectively (33) or employed mitogen-activated T lymphoblasts. (16, 134) Furthermore, these agents were added to the apical side of the endothelium where they would activate leukocytes, but not provide a chemotactic gradient. In the standard TEM assay system in our lab (140), paracellular migration predominates and transcellular migration is almost never seen across cytokine activated HUVEC. However, when we applied chemokine or chemoattractant to the apical side of endothelial monolayers, 10 to 30 % of leukocytes migrated transcellularly (14).
Leukocyte activation would promote polymerization of actin in their lamellipodia, which has been associated with transcellular migration in vivo (15) and shown to be necessary for transcellular migration in vitro (22). Leukocytes probe the apical surface of endothelial cells with “invasive podosomes” that contain polymerized actin. These deeply invaginated the surface in the areas of eventual transcellular migration (22).
The role of the Lateral Border Recycling Compartment
Membrane vesicles were seen accumulating in the region of transcellular migration (22). Millan et al. reported that caveolin-1 accumulated in the regions of transcellular migration implying that these might be caveolae (134). However, other groups did not see any enrichment in caveolin-1 at the site of diapedesis (14, 22, 33).
Mamdouh, et al. reported that the Lateral Border Recycling Compartment (LBRC) was critical for transcellular migration of leukocytes (14), similar to its involvement in paracellular migration (Fig. 3). The LBRC membrane components PECAM, CD99, and JAM-A moved in concert to surround neutrophil and monocytes migrating transcellularly. The source of the membrane was demonstrated to be the LBRC, and similar to paracellular migration (37), was dependent on functioning microtubules (14). Microtubule depolymerizing agents blocked targeted recycling and blocked transcellular as well as paracellular migration (14).
Figure 3. Targeted trafficking of membrane from the LBRC to surround leukocytes undergoing transcellular migration.

Staining of recycled PECAM as a surrogate for recycling LBRC (left panel, red in overlay) surrounding monocytes (upper panel) and a PMN (lower panel) demonstrates movement of membrane from the LBRC to the endothelial surface. Recycled LBRC is seen around the leukocytes migrating transcellularly (thin arrows) far from the endothelial border (arrowheads). Recycling LBRC enrichment is as great around the monocyte migrating transcellularly as it is around the monocyte migrating paracellularly (thick arrow). Actin staining (middle panel, green in merge) shows relative positions of the cells. Orthogonal (X-Z) projection demonstrates leukocytes in the act of transcellular migration. Arrows indicate recycled PECAM around leukocytes. Bar = 10 μm. Reproduced from (14) with permission.
Using transfected cell lines, Yang, et al. showed that overexpression of ICAM-1 promoted transcellular migration (35), implicating a role for ICAM-1 in this process. However, when LBRC recycling was inhibited, there was no effect on ICAM-1 enrichment around adherent leukocytes, even though both paracellular and transcellular migration were blocked (14). Therefore, while enrichment of ICAM-1 may help promote transcellular migration, and may even be a necessary prerequisite, it is not sufficient to promote transmigration in the absence of a functional LBRC.
The role of the LBRC in transcellular migration explains a number of observations made by several groups of investigators. The appearance in electron micrographs of membrane vesicles clustered around the leukocyte as it transmigrates (22) is consistent with recruitment of the LBRC. Many of the studies that provide evidence for transcellular migration in vivo often show electron micrographs of leukocytes passing through the cell within a micron or two of an intact endothelial junction (20, 135). This is exactly where the LBRC is situated and would put it in a prime location for its role in promoting transcellular as well as paracellular migration. Transcellular migration is also dependent on PECAM and CD99 (14). Since expression of PECAM and CD99 on the cell surface is essentially restricted to the cell borders, this was unexpected. However, once the LBRC is recruited and becomes part of the membrane forming the transmigration pore, interaction of leukocyte and endothelial cell PECAM and CD99 is required for transcellular passage.
Carman et al. (22) showed leukocytes appearing to probe the endothelial surface with lamellipodia (podosomes), often deeply invaginating the surface of the endothelial cell. They were highly enriched for actin filaments (15, 22), which were required for their function (22). Actin polymerization and lamellapodia formation are events triggered in leukocytes by integrin activation, so prominent podosomes in cells migrating transcellularly could be manifestations of high levels of leukocyte activation. The podosomes were hypothesized to initiate the formation of the transcellular channel. In previous reports such podosomes were observed invaginating the endothelial surface away from the junctions even under conditions where leukocytes crossed at the cell borders (141, 142) as well as under conditions where leukocytes are blocked in their attempts to cross at the junctions (Figure 4a in ref (83)). These lamellapodia may be a general mechanism for leukocytes to crawl across the endothelial surface. However, if they have a role in promoting transmigration, it is conceivable that they signal the recruitment of LBRC membrane to the leukocytes when they are at the cell borders as well as when they are not. In this case, the mechanisms of paracellular and transcellular migration would have even more in common.
At least one potential mechanistic difference between paracellular and transcellular migration remains. Since the LBRC is connected to the lateral endothelial cell surface at the cell borders, fusion of the LBRC membrane with the plasma membrane is not necessarily required to bring the LBRC in contact with the leukocyte for paracellular TEM. On the other hand, to bring this membrane compartment in contact with the leukocyte on the apical surface for transcellular migration would require membrane fusion. In fact, Carman et al. (22) provide evidence that membrane fusion is required for transcellular migration. However, since the LBRC vesicles are connected to each other (36), rather than multiple fusion events involving dozens or hundreds of vesicles, the entire surface area of the LBRC could be brought to surround the leukocyte with perhaps only two fusion events necessary: One at the apical surface and one at the basal surface of the endothelial cell.
The final frontier
Once the leukocyte has passed across the endothelial barrier, it still must cross the subendothelial basal lamina. This process generally requires far more time (13). Norshargh and colleagues have demonstrated that PMN (143) and monocytes (144) move between the abluminal surface of the endothelial cell and the basal lamina searching for areas where collagen IV and laminin are deposited at low density. These correspond to areas where there is a gap in pericyte coverage. Leukocytes exit to the interstitium in these zones. Migration to these sites is aided by interactions between LFA-1 on the leukocyte and ICAM-1 on pericytes (145). When modeled in vitro, there is a role for heterophilic interaction of domain 6 of leukocyte PECAM with some component of the basement membrane in this process, inasmuch as blockade of domain 6 selectively blocks migration across the basement membrane, but not across the endothelial cell (83). In vivo, PECAM-deficient leukocytes in the C57BL/6 strain show a delay in migration across the basal lamina (146), which is also associated with a failure to exteriorize α6β1 integrin, the receptor for laminin (147).
Unanswered Questions
As in other areas of science, the more we learn about neutrophil TEM, the more we realize there is to learn. Pursuing these questions will provide opportunities for future research and will ultimately lead to a more complete understanding of the inflammatory response.
Neutrophil Activation
Anyone who has ever isolated PMN from peripheral blood knows how easy it is to activate them. It seems that just looking at them the wrong way will cause them to clump and adhere to the walls of the centrifuge tube. How, then, do they get activated enough to adhere to the endothelial surface, locomote to the junctions and transmigrate (which involves squeezing the entire cell through a pore that is maximally 4–6 μm wide (129)) but remain quiescent enough not to degranulate until they enter the inflamed tissue? One presumes that there are bidirectional activating and calming signals exchanged between the PMN and endothelial cell, but the nature of these is not known.
The LBRC
Much remains to be learned about the LBRC: What are the directional signals that recruit it to the site of TEM? Are these the same for PECAM, PVR/DNAM-1, and CD99 interactions? What happens to the membrane after targeted recycling; how is membrane retrieved for another round of targeted recycling? What is the purpose of constitutive recycling? We hypothesize that it may be a mechanism for endothelial cells to adapt to rapid and/or cyclical changes in junction surface area as vessels expand and contract during the cardiac cycle (arterial circulation) or in response to changes in tonicity or fluid volume (venous circulation) (60).
The presence in the LBRC of all the molecules that positively regulate TEM and the fact that they move together during TEM (60) begs the question of how these molecules can function sequentially in TEM? This is a focus of active research in our lab.
Mouse strain differences
It is well known that biological responses vary among mouse strains, which are variably resistant to many pathogens and disease models. Transendothelial migration is no exception. The vast majority of mouse strains respond to PECAM blockade or deficiency with a phenotype that resembles what is found with human cells—leukocytes arrested on the apical surface of the junctions, tightly adherent, but unable to transmigrate efficiently (111, 112, 148). However, the most frequently used mouse strain in immunology, C57Bl/6, has a different phenotype: Depending on the inflammatory model, PMN and monocytes are either not inhibited from transmigrating or are transiently arrested between the abluminal surface of the endothelium and the underlying basement membrane (112, 146, 148–151). A similar phenomenon is seen with CD99, although it has been less-well studied (123, 151–155).
The reason for the strain differences are not known, and the only reason to pursue understanding this discrepancy would be to gain insight into the regulation of TEM. In this regard, while the phenotype of PECAM or CD99 block is different among strains when comparing the effect of antibody blockade or genetic deletion of these molecules themselves, when interrupting the signaling pathways downstream of PECAM (TRPC6) and CD99 (sAC), the effect is consistent: The TRPC6-deficient mice in the C57Bl/6 strain showed a phenotype similar to other strains in which TEM was blocked by anti-PECAM antibody (84). Similarly, sAC knockout mice in the C57Bl/6 background showed a phenotype similar to other strains in which TEM was blocked by anti-CD99 antibody (124).
Great progress has been made in uncovering the molecules and mechanisms that govern TEM. It is likely that the pace of discovery will increase with better and faster systems for genetically manipulating mice and cells, better imaging techniques, and better reporter molecules for detecting signal transduction in living cells. Many of the unanswered questions above will be answered, although new ones will arise as we get closer to fully understanding the acute inflammatory response and the role of the neutrophil.
Acknowledgments
This work was supported by NIH Grants R01 HL046489 and R37 HL064774. I thank Dr. David Sullivan for drafting Figure 2.
Footnotes
DISCLOSURES
The author has no conflicts of interest to declare, financial or otherwise.
References
- 1.Muller WA. Mechanisms of leukocyte transendothelial migration. Annu Rev Pathol. 2011;6:323–344. doi: 10.1146/annurev-pathol-011110-130224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Muller WA. The regulation of transendothelial migration: new knowledge and new questions. Cardiovascular research. 2015;107:310–320. doi: 10.1093/cvr/cvv145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7:678–689. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- 4.van Buul JD, Kanters E, Hordijk PL. Endothelial signaling by Ig-like cell adhesion molecules. Arterioscler Thromb Vasc Biol. 2007;27:1870–1876. doi: 10.1161/ATVBAHA.107.145821. [DOI] [PubMed] [Google Scholar]
- 5.Fernandez-Borja M, van Buul JD, Hordijk PL. The regulation of leucocyte transendothelial migration by endothelial signalling events. Cardiovasc Res. 2010;86:202–210. doi: 10.1093/cvr/cvq003. [DOI] [PubMed] [Google Scholar]
- 6.Nourshargh S, Hordijk PL, Sixt M. Breaching multiple barriers: leukocyte motility through venular walls and the interstitium. Nat Rev Mol Cell Biol. 2010;11:366–378. doi: 10.1038/nrm2889. [DOI] [PubMed] [Google Scholar]
- 7.Butcher EC. Leukocyte-endothelial cell recognition: three (or more) steps to specificity and diversity. Cell. 1991;67:1033–1036. doi: 10.1016/0092-8674(91)90279-8. [DOI] [PubMed] [Google Scholar]
- 8.Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: The multistep paradigm. Cell. 1994;76:301–314. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 9.Hawkins HK, Heffelfinger SC, Anderson DC. Leukocyte adhesion deficiency: Clinical and postmortem observations. Pediatric Pathology. 1992;12:119–130. doi: 10.3109/15513819209023288. [DOI] [PubMed] [Google Scholar]
- 10.Doerschuk CM, Winn RK, Coxson HO, Harlan JM. CD18-dependent and -independent mechanisms of neutrophil emigration in the pulmonary and systemic microciruclation of rabbits. JImmunol. 1990;144:2327–2333. [PubMed] [Google Scholar]
- 11.Muller WA. Leukocyte-Endothelial Cell Adhesion Molecules in Transendothelial Migration. In: Gallin JI, Snyderman R, editors. Inflammation: Basic Principles and Clinical Correlates. 3. Philadelphia: Lippincott Williams & Wilkins; 1999. pp. 585–592. [Google Scholar]
- 12.Sumagin R, Prizant H, Lomakina E, Waugh RE, Sarelius IH. LFA-1 and Mac-1 define characteristically different intralumenal crawling and emigration patterns for monocytes and neutrophils in situ. J Immunol. 2010;185:7057–7066. doi: 10.4049/jimmunol.1001638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Woodfin A, et al. The junctional adhesion molecule JAM-C regulates polarized transendothelial migration of neutrophils in vivo. Nat Immunol. 2011;12:761–769. doi: 10.1038/ni.2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mamdouh Z, Mikhailov A, Muller WA. Transcellular migration of leukocytes is mediated by the endothelial lateral border recycling compartment. J Exp Med. 2009;206:2795–2808. doi: 10.1084/jem.20082745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feng D, Nagy JA, Pyne K, Dvorak HF, Dvorak AM. Neutrophils emigrate from venules by a transendothelial cell pathway in response to fMLF. Journal of Experimental Medicine. 1998;187:903–915. doi: 10.1084/jem.187.6.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carman CV, Springer TA. Trans-cellular migration: cell-cell contacts get intimate. Curr Opin Cell Biol. 2008;20:533–540. doi: 10.1016/j.ceb.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burns AR, et al. Neutrophil transendothelial migration is independent of tight junctions and occurs preferentially at tricellular corners. J Immunol. 1997;159:2893–2903. [PubMed] [Google Scholar]
- 18.Muller WA. Migration of leukocytes across endothelial junctions: Some concepts and controversies. Microcirculation. 2001;8:181–193. doi: 10.1038/sj/mn/7800078. [DOI] [PubMed] [Google Scholar]
- 19.Sage PT, Carman CV. Settings and mechanisms for trans-cellular diapedesis. Front Biosci. 2009;14:5066–5083. doi: 10.2741/3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wolburg H, Wolburg-Buchholz K, Engelhardt B. Diapedesis of mononuclear cells across cerebral venules during experimental autoimmune encephalomyelitis leaves tight junctions intact. Acta Neuropathol. 2005;109:181–190. doi: 10.1007/s00401-004-0928-x. [DOI] [PubMed] [Google Scholar]
- 21.Carman CV. Mechanisms for transcellular diapedesis: probing and pathfinding by ‘invadosome-like protrusions’. J Cell Sci. 2009;122:3025–3035. doi: 10.1242/jcs.047522. [DOI] [PubMed] [Google Scholar]
- 22.Carman CV, et al. Transcellular diapedesis is initiated by invasive podosomes. Immunity. 2007;26:784–797. doi: 10.1016/j.immuni.2007.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Muller WA. Localized signals that regulate transendothelial migration. Current opinion in immunology. 2016;38:24–29. doi: 10.1016/j.coi.2015.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baluk P, Bolton P, Hirata A, Thurston G, McDonald DM. Endothelial gaps and adherent leukocytes in allergen-induced early- and late-phase plasma leakage in rat airways. Am J Pathol. 1998;152:1463–1476. [PMC free article] [PubMed] [Google Scholar]
- 25.McDonald DM. Endothelial gaps and permeability of venules in rat tracheas exposed to inflammatory stimuli. The American journal of physiology. 1994;266:L61–83. doi: 10.1152/ajplung.1994.266.1.L61. [DOI] [PubMed] [Google Scholar]
- 26.Schnoor M, et al. Cortactin deficiency is associated with reduced neutrophil recruitment but increased vascular permeability in vivo. J Exp Med. 2011;208:1721–1735. doi: 10.1084/jem.20101920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shaw SK, et al. Coordinated redistribution of leukocyte LFA-1 and endothelial cell ICAM-1 accompany neutrophil transmigration. J Exp Med. 2004;200:1571–1580. doi: 10.1084/jem.20040965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carman CV, Jun CD, Salas A, Springer TA. Endothelial cells proactively form microvilli-like membrane projections upon intercellular adhesion molecule 1 engagement of leukocyte LFA-1. J Immunol. 2003;171:6135–6144. doi: 10.4049/jimmunol.171.11.6135. [DOI] [PubMed] [Google Scholar]
- 29.Barreiro O, et al. Dynamic interaction of VCAM-1 and ICAM-1 with moesin and ezrin in a novel endothelial docking structure for adherent leukocytes. J Cell Biol. 2002;157:1233–1245. doi: 10.1083/jcb.200112126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barreiro O, et al. Endothelial tetraspanin microdomains regulate leukocyte firm adhesion during extravasation. Blood. 2005;105:2852–2861. doi: 10.1182/blood-2004-09-3606. [DOI] [PubMed] [Google Scholar]
- 31.Yang L, et al. Endothelial cell cortactin coordinates intercellular adhesion molecule-1 clustering and actin cytoskeleton remodeling during polymorphonuclear leukocyte adhesion and transmigration. J Immunol. 2006;177:6440–6449. doi: 10.4049/jimmunol.177.9.6440. [DOI] [PubMed] [Google Scholar]
- 32.Yang L, Kowalski JR, Zhan X, Thomas SM, Luscinskas FW. Endothelial cell cortactin phosphorylation by Src contributes to polymorphonuclear leukocyte transmigration in vitro. Circ Res. 2006;98:394–402. doi: 10.1161/01.RES.0000201958.59020.1a. [DOI] [PubMed] [Google Scholar]
- 33.Carman CV, Springer TA. A transmigratory cup in leukocyte diapedesis both through individual vascular endothelial cells and between them. J Cell Biol. 2004;167:377–388. doi: 10.1083/jcb.200404129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van Buul JD, et al. RhoG regulates endothelial apical cup assembly downstream from ICAM1 engagement and is involved in leukocyte trans-endothelial migration. J Cell Biol. 2007;178:1279–1293. doi: 10.1083/jcb.200612053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang L, Froio RM, Sciuto TE, Dvorak AM, Alon R, Luscinskas FW. ICAM-1 regulates neutrophil adhesion and transcellular migration of TNF-alpha-activated vascular endothelium under flow. Blood. 2005;106:584–592. doi: 10.1182/blood-2004-12-4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mamdouh Z, Chen X, Pierini LM, Maxfield FR, Muller WA. Targeted recycling of PECAM from endothelial cell surface-connected compartments during diapedesis. Nature. 2003;421:748–753. doi: 10.1038/nature01300. [DOI] [PubMed] [Google Scholar]
- 37.Mamdouh Z, Kreitzer GE, Muller WA. Leukocyte transmigration requires kinesin-mediated microtubule-dependent membrane trafficking from the lateral border recycling compartment. J Exp Med. 2008;205:951–966. doi: 10.1084/jem.20072328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Muller WA. Transmigratory cups: Half-full or half-empty? Nature Immunology. 2008 [Google Scholar]
- 39.Alcaide P, et al. p120-Catenin regulates leukocyte transmigration through an effect on VE-cadherin phosphorylation. Blood. 2008;112:2770–2779. doi: 10.1182/blood-2008-03-147181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Allingham MJ, van Buul JD, Burridge K. ICAM-1-mediated, Src- and Pyk2-dependent vascular endothelial cadherin tyrosine phosphorylation is required for leukocyte transendothelial migration. J Immunol. 2007;179:4053–4064. doi: 10.4049/jimmunol.179.6.4053. [DOI] [PubMed] [Google Scholar]
- 41.Shaw SK, Bamba PS, Perkins BN, Luscinskas FW. Real-time imaging of vascular endothelial-cadherin during leukocyte transmigration across endothelium. J Immunol. 2001;167:2323–2330. doi: 10.4049/jimmunol.167.4.2323. [DOI] [PubMed] [Google Scholar]
- 42.Dejana E, Giampietro C. Vascular endothelial-cadherin and vascular stability. Curr Opin Hematol. 2012;19:218–223. doi: 10.1097/MOH.0b013e3283523e1c. [DOI] [PubMed] [Google Scholar]
- 43.Dejana E, Orsenigo F, Lampugnani MG. The role of adherens junctions and VE-cadherin in the control of vascular permeability. Journal of cell science. 2008;121:2115–2122. doi: 10.1242/jcs.017897. [DOI] [PubMed] [Google Scholar]
- 44.Vestweber D, Winderlich M, Cagna G, Nottebaum AF. Cell adhesion dynamics at endothelial junctions: VE-cadherin as a major player. Trends Cell Biol. 2009;19:8–15. doi: 10.1016/j.tcb.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 45.Dejana E, Tournier-Lasserve E, Weinstein BM. The control of vascular integrity by endothelial cell junctions: molecular basis and pathological implications. Dev Cell. 2009;16:209–221. doi: 10.1016/j.devcel.2009.01.004. [DOI] [PubMed] [Google Scholar]
- 46.Gumbiner BM. Regulation of cadherin adhesive activity. J Cell Biol. 2000;148:399–404. doi: 10.1083/jcb.148.3.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xiao K, et al. Cellular levels of p120 catenin function as a set point for cadherin expression levels in microvascular endothelial cells. J Cell Biol. 2003;163:535–545. doi: 10.1083/jcb.200306001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Allport JR, Muller WA, Luscinskas FW. Monocytes induce reversible focal changes in vascular endothelial cadherin complex during transendothelial migration under flow. J Cell Biol. 2000;148:203–216. doi: 10.1083/jcb.148.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nanes BA, et al. p120-catenin binding masks an endocytic signal conserved in classical cadherins. J Cell Biol. 2012;199:365–380. doi: 10.1083/jcb.201205029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chiasson CM, Wittich KB, Vincent PA, Faundez V, Kowalczyk AP. p120-catenin inhibits VE-cadherin internalization through a Rho-independent mechanism. Molecular biology of the cell. 2009;20:1970–1980. doi: 10.1091/mbc.E08-07-0735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xiao K, et al. p120-Catenin regulates clathrin-dependent endocytosis of VE-cadherin. Mol Biol Cell. 2005;16:5141–5151. doi: 10.1091/mbc.E05-05-0440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nottebaum AF, et al. VE-PTP maintains the endothelial barrier via plakoglobin and becomes dissociated from VE-cadherin by leukocytes and by VEGF. J Exp Med. 2008;205:2929–2945. doi: 10.1084/jem.20080406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vockel M, Vestweber D. How T cells trigger the dissociation of the endothelial receptor phosphatase VE-PTP from VE-cadherin. Blood. 2013;122:2512–2522. doi: 10.1182/blood-2013-04-499228. [DOI] [PubMed] [Google Scholar]
- 54.Turowski P, et al. Phosphorylation of vascular endothelial cadherin controls lymphocyte emigration. J Cell Sci. 2008;121:29–37. doi: 10.1242/jcs.022681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cook-Mills JM, Johnson JD, Deem TL, Ochi A, Wang L, Zheng Y. Calcium mobilization and Rac1 activation are required for VCAM-1 (vascular cell adhesion molecule-1) stimulation of NADPH oxidase activity. Biochem J. 2004;378:539–547. doi: 10.1042/BJ20030794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.van Wetering S, et al. Reactive oxygen species mediate Rac-induced loss of cell-cell adhesion in primary human endothelial cells. J Cell Sci. 2002;115:1837–1846. doi: 10.1242/jcs.115.9.1837. [DOI] [PubMed] [Google Scholar]
- 57.van Wetering S, et al. VCAM-1-mediated Rac signaling controls endothelial cell-cell contacts and leukocyte transmigration. Am J Physiol Cell Physiol. 2003;285:C343–352. doi: 10.1152/ajpcell.00048.2003. [DOI] [PubMed] [Google Scholar]
- 58.Wessel F, et al. Leukocyte extravasation and vascular permeability are each controlled in vivo by different tyrosine residues of VE-cadherin. Nature immunology. 2014;15:223–230. doi: 10.1038/ni.2824. [DOI] [PubMed] [Google Scholar]
- 59.Sullivan DP, Seidman MA, Muller WA. Poliovirus receptor (CD155) regulates a step in transendothelial migration between PECAM and CD99. Am J Pathol. 2013;182:1031–1042. doi: 10.1016/j.ajpath.2012.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Feng G, Sullivan DP, Han F, Muller WA. Segregation of VE-cadherin from the LBRC depends on the ectodomain sequence required for homophilic adhesion. J Cell Sci. 2015;128:576–588. doi: 10.1242/jcs.159053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dvorak AM, Kohn S, Morgan ES, Fox P, Nagy JA, Dvorak HF. The vesiculo-vacuolar organelle (VVO): a distinct endothelial cell structure that provides a transcellular pathway for macromolecular extravasation. J Leukoc Biol. 1996;59:100–115. [PubMed] [Google Scholar]
- 62.Feng D, Nagy JA, Hipp J, Dvorak HF, Dvorak AM. Vesiculo-vacuolar organelles and the regulation of venule permeability to macromolecules by vascular permeability factor, histamine, and serotonin. J Exp Med. 1996;183:1981–1986. doi: 10.1084/jem.183.5.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rothberg KG, Heuser JE, Donzell WC, Ying Y-S, Glenney JR, Anderson RGW. Caveolin, a protein component of caveolae membrane coats. Cell. 1992;68:673–682. doi: 10.1016/0092-8674(92)90143-z. [DOI] [PubMed] [Google Scholar]
- 64.Dvorak AM, Feng D. The vesiculo-vacuolar organelle (VVO). A new endothelial cell permeability organelle. J Histochem Cytochem. 2001;49:419–432. doi: 10.1177/002215540104900401. [DOI] [PubMed] [Google Scholar]
- 65.Florey O, Durgan J, Muller W. Phosphorylation of leukocyte PECAM and its association with detergent-resistant membranes regulate transendothelial migration. J Immunol. 2010;185:1878–1886. doi: 10.4049/jimmunol.1001305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sardjono CT, et al. Palmitoylation at Cys595 is essential for PECAM-1 localisation into membrane microdomains and for efficient PECAM-1-mediated cytoprotection. Thrombosis and haemostasis. 2006;96:756–766. [PubMed] [Google Scholar]
- 67.Jin S, et al. Nepmucin/CLM-9, an Ig domain-containing sialomucin in vascular endothelial cells, promotes lymphocyte transendothelial migration in vitro. FEBS Lett. 2008;582:3018–3024. doi: 10.1016/j.febslet.2008.07.041. [DOI] [PubMed] [Google Scholar]
- 68.Sullivan DP, Muller WA. Neutrophil and monocyte recruitment by PECAM, CD99, and other molecules via the LBRC. Seminars in immunopathology. 2014;36:193–209. doi: 10.1007/s00281-013-0412-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sullivan DP, Ruffer C, Muller WA. Isolation of the lateral border recycling compartment using a diaminobenzidine-induced density shift. Traffic. 2014;15:1016–1029. doi: 10.1111/tra.12184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cyrus BF, Muller WA. A Unique Role for Endothelial Cell Kinesin Light Chain 1, Variant 1 in Leukocyte Transendothelial Migration. The American journal of pathology. 2016 doi: 10.1016/j.ajpath.2016.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Khodjakov A, Lizunova EM, Minin AA, Koonce MP, Gyoeva FK. A specific light chain of kinesin associates with mitochondria in cultured cells. Mol Biol Cell. 1998;9:333–343. doi: 10.1091/mbc.9.2.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gyoeva FK, Bybikova EM, Minin AA. An isoform of kinesin light chain specific for the Golgi complex. Journal of cell science. 2000;113( Pt 11):2047–2054. doi: 10.1242/jcs.113.11.2047. [DOI] [PubMed] [Google Scholar]
- 73.Liao GGG. Kinesin is a candidate for cross-bridging microtubules and intermediate filaments. Selective binding of kinesin to detyrosinated tubulin and vimentin. J Biol Chem. 1998;273:9797–9803. doi: 10.1074/jbc.273.16.9797. [DOI] [PubMed] [Google Scholar]
- 74.WoŸniak MJ, Allan VJ. Cargo selection by specific kinesin light chain 1 isoforms. The EMBO Journal. 2006;25:5457–5468. doi: 10.1038/sj.emboj.7601427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Muller WA, Weigl SA, Deng X, Phillips DM. PECAM-1 is required for transendothelial migration of leukocytes. J Exp Med. 1993;178:449–460. doi: 10.1084/jem.178.2.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gonzalez AM, Cyrus BF, Muller WA. Targeted Recycling of the Lateral Border Recycling Compartment Precedes Adherens Junction Dissociation during Transendothelial Migration. The American journal of pathology. 2016 doi: 10.1016/j.ajpath.2016.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Winger RC, Koblinski JE, Kanda T, Ransohoff RM, Muller WA. Rapid Remodeling of Tight Junctions during Paracellular Diapedesis in a Human Model of the Blood-Brain Barrier. Journal of immunology. 2014;193:2427–2437. doi: 10.4049/jimmunol.1400700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Muller WA. Mechanisms of transendothelial migration of leukocytes. Circ Res. 2009;105:223–230. doi: 10.1161/CIRCRESAHA.109.200717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Muller WA. PECAM: Regulating the start of diapedesis. In: Ley K, editor. Adhesion Molecules: Function and Inhibition. Basel: Birkhauser Verlag AG; 2007. pp. 201–220. [Google Scholar]
- 80.Huang AJ, Manning JE, Bandak TM, Ratau MC, Hanser KR, Silverstein SC. Endothelial cell cytosolic free calcium regulates neutrophil migration across monolayers of endothelial cells. JCell Biol. 1993;120:1371–1380. doi: 10.1083/jcb.120.6.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Su WH, Chen HI, Huang JP, Jen CJ. Endothelial [Ca(2+)](i) signaling during transmigration of polymorphonuclear leukocytes. Blood. 2000;96:3816–3822. [PubMed] [Google Scholar]
- 82.Schenkel AR, Mamdouh Z, Muller WA. Locomotion of monocytes on endothelium is a critical step during extravasation. Nat Immunol. 2004;5:393–400. doi: 10.1038/ni1051. [DOI] [PubMed] [Google Scholar]
- 83.Liao F, Huynh HK, Eiroa A, Greene T, Polizzi E, Muller WA. Migration of monocytes across endothelium and passage through extracellular matrix involve separate molecular domains of PECAM-1. J Exp Med. 1995;182:1337–1343. doi: 10.1084/jem.182.5.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Weber EW, Han F, Tauseef M, Birnbaumer L, Mehta D, Muller WA. TRPC6 is the endothelial calcium channel that regulates leukocyte transendothelial migration during the inflammatory response. The Journal of experimental medicine. 2015;212:1883–1899. doi: 10.1084/jem.20150353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lorenzon P, et al. Endothelial cell E-and P-selectin and vascular cell adhesion molecule-1 function as signaling receptors. J Cell Biol. 1998;142:1381–1391. doi: 10.1083/jcb.142.5.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Newman PJ, et al. PECAM-1 (CD31) cloning and relation to adhesion molecules of the immunoglobulin gene superfamily. Science. 1990;247:1219–1222. doi: 10.1126/science.1690453. [DOI] [PubMed] [Google Scholar]
- 87.Newman PJ. Switched at birth: A new family for PECAM-1. JClinInvest. 1999;103:5–9. doi: 10.1172/JCI5928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sun Q-H, Delisser HM, Zukowski MM, Paddock C, Albelda SM, Newman PJ. Individually distinct Ig homology domains in PECAM-1 regulate homophilic binding and modulate receptor affinity. J Biol Chem. 1996;271:11090–11098. doi: 10.1074/jbc.271.19.11090. [DOI] [PubMed] [Google Scholar]
- 89.Bogen S, Pak J, Garifallou M, Deng X, Muller WA. Monoclonal antibody to murine PECAM-1 [CD31] blocks acute inflammation in vivo. J Exp Med. 1994;179:1059–1064. doi: 10.1084/jem.179.3.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Vaporciyan AA, et al. Involvement of platelet-endothelial cell adhesion molecule-1 in neutrophil recruitment in vivo. Science. 1993;262:1580–1582. doi: 10.1126/science.8248808. [DOI] [PubMed] [Google Scholar]
- 91.Liao F, Ali J, Greene T, Muller WA. Soluble domain 1 of platelet-endothelial cell adhesion molecule (PECAM) is sufficient to block transendothelial migration in vitro and in vivo. J Exp Med. 1997;185:1349–1357. doi: 10.1084/jem.185.7.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Newman PJ. The biology of PECAM-1. JClinInvest. 1997;99:3–8. doi: 10.1172/JCI119129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ilan N, Madri JA. PECAM-1: old friend, new partners. Curr Opin Cell Biol. 2003;15:515–524. doi: 10.1016/s0955-0674(03)00100-5. [DOI] [PubMed] [Google Scholar]
- 94.Liu G, et al. ICAM-1-activated Src and eNOS signaling increase endothelial cell surface PECAM-1 adhesivity and neutrophil transmigration. Blood. 2012;120:1942–1952. doi: 10.1182/blood-2011-12-397430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dasgupta B, Muller WA. Endothelial Src kinase regulates membrane recycling from the lateral border recycling compartment during leukocyte transendothelial migration. Eur J Immunol. 2008;38:3499–3507. doi: 10.1002/eji.200838605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cicmil M, Thomas JM, Leduc M, Bon C, Gibbins JM. Platelet endothelial cell adhesion molecule-1 signaling inhibits the activation of human platelets. Blood. 2002;99:137–144. doi: 10.1182/blood.v99.1.137. [DOI] [PubMed] [Google Scholar]
- 97.Kogata N, et al. Identification of Fer Tyrosine Kinase Localized on Microtubules as a Platelet Endothelial Cell Adhesion Molecule-1 Phosphorylating Kinase in Vascular Endothelial Cells. Molecular biology of the cell. 2003;14:3553–3564. doi: 10.1091/mbc.E03-02-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gratzinger D, Barreuther M, Madri JA. Platelet-endothelial cell adhesion molecule-1 modulates endothelial migration through its immunoreceptor tyrosine-based inhibitory motif. Biochem Biophys Res Commun. 2003;301:243–249. doi: 10.1016/s0006-291x(02)02982-0. [DOI] [PubMed] [Google Scholar]
- 99.Masuda M, Osawa M, Shigematsu H, Harada N, Fujiwara K. Platelet endothelial cell adhesion molecule-1 is a major SH-PTP2 binding protein in vascular endothelial cells. FEBS Lett. 1997;408:331–336. doi: 10.1016/s0014-5793(97)00457-2. [DOI] [PubMed] [Google Scholar]
- 100.Newton-Nash DK, Newman PJ. A new role for platelet-endothelial cell adhesion molecule-1 (CD31): inhibition of TCR-mediated signal transduction. J Immunol. 1999;163:682–688. [PubMed] [Google Scholar]
- 101.Newman DK, Hamilton C, Newman PJ. Inhibition of antigen-receptor signaling by Platelet Endothelial Cell Adhesion Molecule-1 (CD31) requires functional ITIMs, SHP-2, and p56(lck) Blood. 2001;97:2351–2357. doi: 10.1182/blood.v97.8.2351. [DOI] [PubMed] [Google Scholar]
- 102.Newman PJ, Newman DK. Signal Transduction Pathways Mediated by PECAM-1: New Roles for an Old Molecule in Platelet and Vascular Cell Biology. Arterioscler Thromb Vasc Biol. 2003;23:953–964. doi: 10.1161/01.ATV.0000071347.69358.D9. [DOI] [PubMed] [Google Scholar]
- 103.Dasgupta B, Dufour E, Mamdouh Z, Muller W. A novel and critical role for tyrosine 663 in PECAM trafficking and transendothelial migration. J Immunol. 2009;182:5041–5051. doi: 10.4049/jimmunol.0803192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kielbassa-Schnepp K, Strey A, Janning A, Missiaen L, Nilius B, Gerke V. Endothelial intracellular Ca2+ release following monocyte adhesion is required for the transendothelial migration of monocytes. Cell Calcium. 2001;30:29–40. doi: 10.1054/ceca.2001.0210. [DOI] [PubMed] [Google Scholar]
- 105.Etienne-Manneville S, Manneville JB, Adamson P, Wilbourn B, Greenwood J, Couraud PO. ICAM-1-coupled cytoskeletal rearrangements and transendothelial lymphocyte migration involve intracellular calcium signaling in brain endothelial cell lines. J Immunol. 2000;165:3375–3383. doi: 10.4049/jimmunol.165.6.3375. [DOI] [PubMed] [Google Scholar]
- 106.Hofmann T, Obukhov AG, Schaefer M, Harteneck C, Gudermann T, Schultz G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature. 1999;397:259–263. doi: 10.1038/16711. [DOI] [PubMed] [Google Scholar]
- 107.Shi J, et al. Multiple regulation by calcium of murine homologues of transient receptor potential proteins TRPC6 and TRPC7 expressed in HEK293 cells. J Physiol. 2004;561:415–432. doi: 10.1113/jphysiol.2004.075051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Dietrich A, Mederos y Schnitzler M, Emmel J, Kalwa H, Hofmann T, Gudermann T. N-linked protein glycosylation is a major determinant for basal TRPC3 and TRPC6 channel activity. J Biol Chem. 2003;278:47842–47852. doi: 10.1074/jbc.M302983200. [DOI] [PubMed] [Google Scholar]
- 109.Paria BC, et al. Tumor necrosis factor-alpha-induced TRPC1 expression amplifies store-operated Ca2+ influx and endothelial permeability. Am J Physiol Lung Cell Mol Physiol. 2004;287:L1303–1313. doi: 10.1152/ajplung.00240.2004. [DOI] [PubMed] [Google Scholar]
- 110.Yip H, et al. Expression of TRPC homologs in endothelial cells and smooth muscle layers of human arteries. Histochemistry and cell biology. 2004;122:553–561. doi: 10.1007/s00418-004-0720-y. [DOI] [PubMed] [Google Scholar]
- 111.Liao F, Schenkel AR, Muller WA. Transgenic mice expressing different levels of soluble platelet/endothelial cell adhesion molecule-IgG display distinct inflammatory phenotypes. J Immunol. 1999;163:5640–5648. [PubMed] [Google Scholar]
- 112.Schenkel AR, Chew TW, Muller WA. Platelet endothelial cell adhesion molecule deficiency or blockade significantly reduces leukocyte emigration in a majority of mouse strains. J Immunol. 2004;173:6403–6408. doi: 10.4049/jimmunol.173.10.6403. [DOI] [PubMed] [Google Scholar]
- 113.Singh I, Knezevic N, Ahmmed GU, Kini V, Malik AB, Mehta D. Galphaq-TRPC6-mediated Ca2+ entry induces RhoA activation and resultant endothelial cell shape change in response to thrombin. J Biol Chem. 2007;282:7833–7843. doi: 10.1074/jbc.M608288200. [DOI] [PubMed] [Google Scholar]
- 114.Sel S, et al. Loss of classical transient receptor potential 6 channel reduces allergic airway response. Clin Exp Allergy. 2008;38:1548–1558. doi: 10.1111/j.1365-2222.2008.03043.x. [DOI] [PubMed] [Google Scholar]
- 115.Tauseef M, et al. TLR4 activation of TRPC6-dependent calcium signaling mediates endotoxin-induced lung vascular permeability and inflammation. J Exp Med. 2012;209:1953–1968. doi: 10.1084/jem.20111355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lindemann O, et al. TRPC6 regulates CXCR2-mediated chemotaxis of murine neutrophils. J Immunol. 2013;190:5496–5505. doi: 10.4049/jimmunol.1201502. [DOI] [PubMed] [Google Scholar]
- 117.Wu QY, et al. Activation of calcium-sensing receptor increases TRPC3/6 expression in T lymphocyte in sepsis. Mol Immunol. 2015;64:18–25. doi: 10.1016/j.molimm.2014.10.018. [DOI] [PubMed] [Google Scholar]
- 118.Hamid R, Newman JH. Evidence for inflammatory signaling in idiopathic pulmonary artery hypertension: TRPC6 and nuclear factor-kappaB. Circulation. 2009;119:2297–2298. doi: 10.1161/CIRCULATIONAHA.109.855197. [DOI] [PubMed] [Google Scholar]
- 119.Kini V, Chavez A, Mehta D. A new role for PTEN in regulating transient receptor potential canonical channel 6-mediated Ca2+ entry, endothelial permeability, and angiogenesis. J Biol Chem. 2010;285:33082–33091. doi: 10.1074/jbc.M110.142034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Thilo F, et al. Pulsatile atheroprone shear stress affects the expression of transient receptor potential channels in human endothelial cells. Hypertension. 2012;59:1232–1240. doi: 10.1161/HYPERTENSIONAHA.111.183608. [DOI] [PubMed] [Google Scholar]
- 121.Damann N, Owsianik G, Li S, Poll C, Nilius B. The calcium-conducting ion channel transient receptor potential canonical 6 is involved in macrophage inflammatory protein-2-induced migration of mouse neutrophils. Acta physiologica. 2009;195:3–11. doi: 10.1111/j.1748-1716.2008.01918.x. [DOI] [PubMed] [Google Scholar]
- 122.Weissmann N, et al. Activation of TRPC6 channels is essential for lung ischaemia-reperfusion induced oedema in mice. Nat Commun. 2012;3:649. doi: 10.1038/ncomms1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Schenkel AR, Mamdouh Z, Chen X, Liebman RM, Muller WA. CD99 plays a major role in the migration of monocytes through endothelial junctions. Nat Immunol. 2002;3:143–150. doi: 10.1038/ni749. [DOI] [PubMed] [Google Scholar]
- 124.Watson RL, et al. Endothelial CD99 signals through soluble adenylyl cyclase and PKA to regulate leukocyte transendothelial migration. The Journal of experimental medicine. 2015;212:1021–1041. doi: 10.1084/jem.20150354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lou O, Alcaide P, Luscinskas FW, Muller WA. CD99 is a key mediator of the transendothelial migration of neutrophils. J Immunol. 2007;178:1136–1143. doi: 10.4049/jimmunol.178.2.1136. [DOI] [PubMed] [Google Scholar]
- 126.Reymond N, et al. DNAM-1 and PVR regulate monocyte migration through endothelial junctions. J Exp Med. 2004;199:1331–1341. doi: 10.1084/jem.20032206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Suh YH, et al. Cloning, genomic organization, alternative transcripts and expression analysis of CD99L2, a novel paralog of human CD99, and identification of evolutionary conserved motifs. Gene. 2003;307:63–76. doi: 10.1016/s0378-1119(03)00401-3. [DOI] [PubMed] [Google Scholar]
- 128.Hixenbaugh EA, Goeckeler ZM, Papaiya NN, Wysolmerski RB, Silverstein SC, Huang AJ. Chemoattractant-stimulated neutrophils induce regulatory myosin light chain phosphorylation and isometric tension development in endothelial cells. American Journal of Physiology. 1997;273:H981–H988. doi: 10.1152/ajpheart.1997.273.2.H981. [DOI] [PubMed] [Google Scholar]
- 129.Heemskerk N, et al. F-actin-rich contractile endothelial pores prevent vascular leakage during leukocyte diapedesis through local RhoA signalling. Nat Commun. 2016;7:10493. doi: 10.1038/ncomms10493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Martinelli R, et al. Release of cellular tension signals self-restorative ventral lamellipodia to heal barrier micro-wounds. The Journal of cell biology. 2013;201:449–465. doi: 10.1083/jcb.201209077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Williamson JR, Grisham JW. Electron microscopy of leukocytic margination and emigration in acute inflammation in dog pancreas. Am J Pathol. 1961;39:239–256. [PMC free article] [PubMed] [Google Scholar]
- 132.Marchesi VT, Gowans JL. The migration of lymphocytes through the endothelium of venules in lymph nodes: An electron microscope study. Proceedings of the Royal Society B. 1964;159:283–290. doi: 10.1098/rspb.1964.0002. [DOI] [PubMed] [Google Scholar]
- 133.Bamforth S, Lightman S, Greenwood J. Ultrastructural analysis of interleukin-1β-induced leukocyte recruitment to the rat retina. Investig Ophtalmol Vis Sci. 1997;38:25–35. [PubMed] [Google Scholar]
- 134.Millan J, Hewlett L, Glyn M, Toomre D, Clark P, Ridley AJ. Lymphocyte transcellular migration occurs through recruitment of endothelial ICAM-1 to caveola- and F-actin-rich domains. Nat Cell Biol. 2006;8:113–123. doi: 10.1038/ncb1356. [DOI] [PubMed] [Google Scholar]
- 135.Lossinsky AS, Shivers RR. Structural pathways for macromolecular and cellular transport across the blood-brain barrier during inflammatory conditions. Review. Histol Histopathol. 2004;19:535–564. doi: 10.14670/HH-19.535. [DOI] [PubMed] [Google Scholar]
- 136.Simionescu N, Simionescu M. The Cardiovascular System. In: Weiss L, editor. Histology Cell and Tissue Biology. 5. New York: Elseveier Science Publishing Co., Inc; 1983. [Google Scholar]
- 137.Furie MB, Cramer EB, Napstek BL, Silverstein SC. Cultured endothelial cell monolayers that restrict the transendothelial passage of macromolecules and electrical current. JCell Biol. 1984;98:1033–1041. doi: 10.1083/jcb.98.3.1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Phillipson M, Heit B, Colarusso P, Liu L, Ballantyne CM, Kubes P. Intraluminal crawling of neutrophils to emigration sites: a molecularly distinct process from adhesion in the recruitment cascade. J Exp Med. 2006;203:2569–2575. doi: 10.1084/jem.20060925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Gerard A, van der Kammen RA, Janssen H, Ellenbroek SI, Collard JG. The Rac activator Tiam1 controls efficient T-cell trafficking and route of trans-endothelial migration. Blood. 2009 doi: 10.1182/blood-2008-07-167668. blood-2008–2007–167668. [DOI] [PubMed] [Google Scholar]
- 140.Muller WA, Weigl S. Monocyte-selective transendothelial migration: Dissection of the binding and transmigration phases by an in vitro assay. J Exp Med. 1992;176:819–828. doi: 10.1084/jem.176.3.819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Furie MB, Naprstek BL, Silverstein SC. Migration of neutrophils across monolayers of cultured microvascular endothelial cells. An in vitro model of leucocyte extravasation. J Cell Sci. 1987;88( Pt 2):161–175. doi: 10.1242/jcs.88.2.161. [DOI] [PubMed] [Google Scholar]
- 142.Migliorisi G, Folkes E, Pawlowski N, Cramer EB. In vitro studies of human monocyte migration across endothelium in response to leukotriene B4 and f-Met-Leu-Phe. Am J Pathol. 1987;127:157–167. [PMC free article] [PubMed] [Google Scholar]
- 143.Wang S, et al. Venular basement membranes contain specific matrix protein low expression regions that act as exit points for emigrating neutrophils. J Exp Med. 2006;203:1519–1532. doi: 10.1084/jem.20051210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Voisin MB, Woodfin A, Nourshargh S. Monocytes and neutrophils exhibit both distinct and common mechanisms in penetrating the vascular basement membrane in vivo. Arterioscler Thromb Vasc Biol. 2009;29:1193–1199. doi: 10.1161/ATVBAHA.109.187450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Proebstl D, et al. Pericytes support neutrophil subendothelial cell crawling and breaching of venular walls in vivo. J Exp Med. 2012;209:1219–1234. doi: 10.1084/jem.20111622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Duncan GS, et al. Genetic evidence for functional redundancy of Platelet/Endothelial cell adhesion molecule-1 (PECAM-1): CD31-deficient mice reveal PECAM-1-dependent and PECAM-1-independent functions. J Immunol. 1999;162:3022–3030. [PubMed] [Google Scholar]
- 147.Wang S, Dangerfield JP, Young RE, Nourshargh S. PECAM-1, α6 integrins and neutrophil elastase cooperate in mediating neutrophil transmigration. J Cell Sci. 2005;118:2067–2076. doi: 10.1242/jcs.02340. [DOI] [PubMed] [Google Scholar]
- 148.Seidman MA, Chew TW, Schenkel AR, Muller WA. PECAM-independent thioglycollate peritonitis is associated with a locus on murine chromosome 2. PLoS ONE. 2009;4:e4316. doi: 10.1371/journal.pone.0004316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Thompson RD, et al. Platelet-endothelial cell adhesion molecule-1 (PECAM-1)-deficient mice demonstrate a transient and cytokine-specific role for PECAM-1 in leukocyte migration through the perivascular basement membrane. Blood. 2001;97:1854–1860. doi: 10.1182/blood.v97.6.1854. [DOI] [PubMed] [Google Scholar]
- 150.Schenkel AR, Chew TW, Chlipala E, Harbord MW, Muller WA. Different susceptibilities of PECAM-deficient mouse strains to spontaneous idiopathic pneumonitis. Exp Mol Pathol. 2006;81:23–30. doi: 10.1016/j.yexmp.2005.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Bixel MG, et al. CD99 and CD99L2 act at the same site as, but independently of, PECAM-1 during leukocyte diapedesis. Blood. 2010;116:1172–1184. doi: 10.1182/blood-2009-12-256388. [DOI] [PubMed] [Google Scholar]
- 152.Dufour EM, Deroche A, Bae Y, Muller WA. CD99 is essential for leukocyte diapedesis in vivo. Cell Commun Adhes. 2008;15:351–363. doi: 10.1080/15419060802442191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Bixel G, Kloep S, Butz S, Petri B, Engelhardt B, Vestweber D. Mouse CD99 participates in T cell recruitment into inflamed skin. Blood. 2004;104:3205–3213. doi: 10.1182/blood-2004-03-1184. [DOI] [PubMed] [Google Scholar]
- 154.Winger RC, et al. Cutting Edge: CD99 Is a Novel Therapeutic Target for Control of T Cell-Mediated Central Nervous System Autoimmune Disease. Journal of immunology. 2016;196:1443–1448. doi: 10.4049/jimmunol.1501634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Graesser D, et al. Altered vascular permeability and early onset of experimental autoimmune encephalomyelitis in PECAM-1-deficient mice. J Clin Invest. 2002;109:383–392. doi: 10.1172/JCI13595. [DOI] [PMC free article] [PubMed] [Google Scholar]