

Abstract
The fluorination of unactivated Csp3–H bonds remains a desirable, challenging transformation for pharmaceutical, agricultural, and materials scientists. Previous methods for this transformation have used bench-stable fluorine atom sources; however, many still rely on the use of UV-active photocatalysts for the requisite high-energy hydrogen atom abstraction event. Herein, we describe the use of uranyl nitrate hexahydrate as a convenient, hydrogen atom abstraction catalyst that can mediate fluorinations of certain alkanes upon activation with visible light.
Keywords: Uranium, photocatalysis, hydrogen atom transfer, C–H activation, fluorination
Graphical abstract
U can do it: Uranyl cation (UO22+) is able to effect the catalytic fluorination of unactivated Csp3–H bonds under visible light irradiation. This report highlights uranyl nitrate as a convenient, molecular C–H abstraction catalyst that exhibits selectivity distinct from previously reported catalytic systems.

Fluorine has gained a privileged position in the fields of medicinal,1 agricultural,2 and materials chemistry3 for the desirable characteristics that it can confer on the constituent matter of each field. Isosteric (but certainly not electronically similar) with hydrogen, the fluorine atom permits modulation of myriad molecular properties, including partitioning behavior, acidity of neighboring groups, and metabolic stability.4 Fluorine incorporation has traditionally been achieved through use of pre-fluorinated building blocks, limiting the possible sources of fluorine to commercially available compounds. Even deoxyfluorination, one of the more robust techniques for the targeted incorporation of fluorine, requires the pre-existence of oxygenated functionality. For this reason, direct, late-stage fluorinations of unactivated Csp3–H bonds presents an enticing platform for accessing compounds beyond the confines of fine chemicals catalogues.5
While the fluorination of Csp3–H bonds using elemental fluorine has been common since the Second World War,6 the low selectivity of this transformation, combined with the operationally non-trivial nature of handling elemental fluorine, has fueled the recent interest5 in selective Csp3–H fluorination using safe, bench stable reagents. Indeed, the recent reports from Lectka,7 Britton,8 Chen,9 and Tan10 suggests that, despite progress, this transformation is not yet a solved problem. With some exceptions,11,12,13 recent strategies for selectively fluorinating Csp3–H bonds have largely recruited photo-hydrogen atom abstraction (HAT) catalysts (denoted [cat]), such as acetophenone, anthraquinone, 1,2,4,5-tetracyanobenzene, and tetra-n-butylammonium decatungstate (TBADT, Figure 1), to generate a carbon-centered radical. This radical can react with a fluorine atom source (RnN–F), typically the electrophilic N-F fluorination reagents N-fluorobenzenesulfonimide (NFSI, Figure 1) or Selectfluor (figure 1), in a mode first recognized by Sammis,14 to furnish the desired product. These processes are rendered catalytic through the oxidizing nature of the generated aminyl radical (denoted RnN•) (NFSI) or radical cation (Selectfluor), which can return the photoreduced HAT catalyst to its initial state. These methods, while enabling, share one drawback: the need for ultraviolet (UV, or hν 200–400 nm) irradiation. While not all UV photoreactions require specialized equipment, many use low efficiency light sources and can induce side reactions. Therefore, it would be desirable to modulate the light requirement of the photo-HAT catalyst into the visible (hν 400–750 nm) range.15 Toward realizing this aim, we sought a HAT catalyst that could be activated with low energy light.
Figure 1.
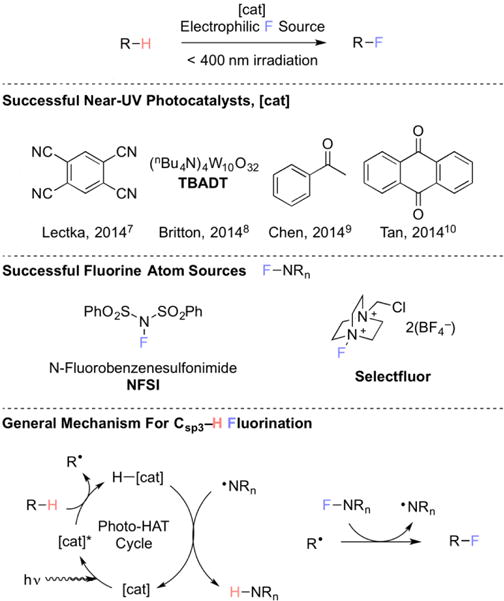
Several near-UV light HAT catalysts and electrophilic fluorine sources used for fluorinations of unactivated C–H bonds and a general mechanism illustrating their function
Uranium is an element that has become inextricably linked to the applications of its fissile isotope, 235U, toward power generation and nuclear weaponry. Surprisingly, 99.3% of natural uranium is made up of the non-fissile isotope 238U, a species whose main application is filling storage bins near enrichment facilities after removal of 235U (approximately 95% of all depleted uranium produced to date is stored this way).16 The removal of 235U makes handling depleted uranium no more arduous than that of other heavy metals.17 This depleted uranium represents a substantial untapped resource, as its crustal abundance exceeds that of molybdenum,18 an element common enough to be used as an enzymatic cofactor. Indeed, despite this glut of depleted uranium, much of its fundamental, structural chemistry is only recently coming to light,19 with its applications in catalysis lagging even further behind.20,21
One aspect of uranium chemistry that has been well studied, however, is the photochemistry of the uranyl cation (UO22+).22–24 Studies by multiple groups have revealed several intriguing characteristics, notably that a highly-oxidizing excited state, [UO2]2+* (+2.6 V vs SHE, almost equal to the oxidizing power of elemental fluorine!25) is accessible under blue light (hν 450–495 nm) irradiation. This excited state is sufficiently reactive to abstract hydrogen atoms from unactivated (BDE > 100 kcal/mol) C–H bonds to generate carbon-centered radicals. Furthermore, pioneering studies by Bakac and coworkers showed that this reactivity could be rendered catalytic for aerobic oxidation of alkanes (Figure 2); with some substrates, the quantum yield approaches unity.26,27 Despite this promising reactivity and abundance of desirable characteristics, the applications of the uranyl cation in catalysis remain largely underdeveloped.
Figure 2.
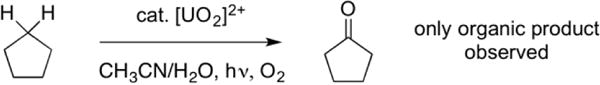
The catalytic aerobic oxidation of alkanes using uranyl cation has been reported
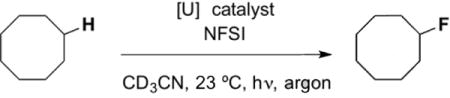
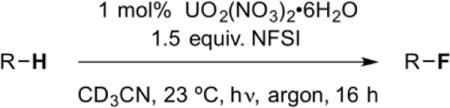
Based on this known photochemistry and initial report of catalytic activity, we hypothesized that the uranyl cation would be an ideal photo-HAT catalyst for the fluorination of unactivated Csp3–H bonds via the mechanism outlined in Figure 1. Putting this supposition to the test, we subjected cyclooctane, NFSI, and 1 mol% uranyl acetate dihydrate to blue LED irradiation to form fluorocyclooctane in a modest, catalytic 8% yield (Entry 1, Table 1). Replacing the uranyl source with uranyl nitrate hexahydrate, led to a greatly improved yield of 52%, corresponding to 52 turnovers (Entry 2, Table 1). Use of a higher intensity blue light source further improved the reaction efficiency, with the near quantitative formation of fluorocyclooctane possible after 16 hours of irradiation (>95%/>95 turnovers, Entry 3, Table 1), exceeding the efficiencies of the acetophenone9 and 1,2,4,5-tetracyanobenzene7 methods (yields of 82%/16 turnovers and 62%/6.2 turnovers, respectively). Both light and catalyst are required for the reaction, and reduction of the NFSI loading below 1.5 equivalents or the uranyl nitrate loading below 1 mol% reduced the efficiency (Table 1, Entries 4–7). Finally, acetone could be substituted as the reaction solvent with comparable efficiencies (Table 1, Entry 8).
Table 1.
Optimization of cyclooctane fluorination
| ||||
|---|---|---|---|---|
| Entry | NFSI [equiv.] | Catalyst [mol%] | Light Source[a] | Yield [%][b] |
| 1 | 1.5 | UO2(OAc)2•4H2O (1) | A | 8 |
| 2 | 1.5 | UO2(NO3)2•6H2O (1) | A | 52 |
| 3 | 1.5 | UO2(NO3)2•6H2O (1) | B | >95 |
| 4 | 1.5 | UO2(NO3)2•6H2O (1) | none | not observed |
| 5 | 1.5 | none | B | not observed |
| 6 | 1.2 | UO2(NO3)2•6H2O (1) | B | 73 |
| 7 | 1.5 | UO2(NO3)2•6H2O (0.5) | B | 52 |
| 8[c] | 1.5 | UO2(NO3)2•6H2O (1) | B | >95 |
Light source A: High Density blue LED strip; Light source B: high intensity blue LED lamp
Yield determined by NMR through integration relative to a methyl acetate internal standard
Acetone-d6 used as solvent
With a rapid, efficient, visible light-mediated fluorination method in hand, we turned our attention to probing the substrate scope of the reaction. Initial testing found that cyclohexane and cyclopentane exhibited diminished, yet still moderate efficiencies at 42% and 32% yields, respectively (Table 2, Entries 2 and 3). The cause of this lower progress is currently unknown; however, a similar (though less pronounced) reduction in efficiency compared to cyclooctane was observed for the fluorination of cyclohexane using both the acetophenone9 and 1,2,4,5-tetracyanobenzene7 systems (yields of 59%/12 turnovers and 45%/4.5 turnovers, respectively). n-Alkanes provided a mixture of fluorinated products at methylene positions (Table 2, Entries 4 and 5).
Table 2.
Fluorination of hydrocarbons
| |||
|---|---|---|---|
| Entry | substrate | product | Yield [%][a] |
| 1 |
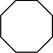
|
|
> 95 |
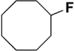
| 2 |
|
|
42 |
| 3 |
|
|
32 |
| 4 |
|
|
55 |
| 5 |
|
|
60 |
| 6 |
|
|
trace |
| 7 |
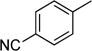
|
|
trace |
yield determined by NMR through integration relative to a methyl acetate internal standard
A fluorinated motif that occupies a privileged place in the fields of medicinal and agricultural chemistry is the trifluoromethylaryl28 functionality. Toward producing this functionality, toluene was subjected to the reaction conditions to furnish benzyl fluoride in trace yields, corresponding to a minimal consumption of starting material and NFSI; 4-cyanotoluene behaved similarly (Table 2, Entries 6 and 7).
Having achieved validation of the method on hydrocarbon substrates, our interests turned to the reaction of related oxygenates. Acetone, the simplest aliphatic ketone, displayed no detectable reactivity under our reaction conditions (Table 3, Entry 1). Increasing the chain length of the ketone substrate led to the production of some fluorinated products in trace yields (Table 3, Entries 2–4), with residual mass balance being unreacted starting material; cyclopentanone (Table 3, Entry 5) was similarly inert. As with ketones, esters were resistant to fluorination for short chain lengths (Table 3, Entries 6–8), but became suitable substrates as chain length increased, providing distal, internal functionalization (Table 3, Entry 9). Interestingly, ethyl isovalerate (Table 3, Entry 10) performed significantly better than its linear counterpart (Table 3, Entry 8), with the more complex natural product sclareolide performing yet better, furnishing 26% of the combined α- and β-fluorinated products (Table 3, Entry 11). While it appears that proximity to carbonyl functionalities is highly deactivating for the fluorination reaction, the low reactivity of carbonyl-containing substrates cannot be explained by electronic effects alone.
Table 3.
Fluorination of Oxygenated Molecules
| |||
|---|---|---|---|
| Entry | substrate | product | Yield [%][a] |
| 1 |
|
n/a | No fluorination |
| 2 |
|
|
Trace |
| 3 |
|
|
Trace |
| 4 |
|
|
Trace |
| 5 |
|
|
Trace |
| 6 |
|
n/a | No fluorination |
| 7 |
|
n/a | No fluorination |
| 8 |
|
|
Trace |
| 9 |
|
|
13 |
| 10 |
|
|
10 |
| 11 |
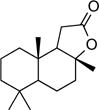
|
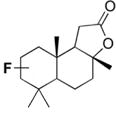
|
26[b] |
| 12 |
|
n/a | No fluorination |
| 13 |
|
n/a | No fluorination |
| 14 |
|
n/a | No fluorination |
Yield determined by NMR through integration relative to a methyl acetate internal standard;
1.6:1 C2/C3 fluorination
These results from carbonyl-containing substrates are in sharp contrast to those of previously reported photo-HAT fluorination methods, where high reactivity is observed despite sensitivity to electronic effects. An example is sclareolide: acetophenone9 (80%/16 turnovers), anthraquinone10 (77%/39 turnovers), 1,2,4,5-tetracyanobenzene7 (61%/6.1 turnovers), and TBADT8 (68%/34 turnovers) all produced mixtures of monofluorinated products in moderate to high yields as compared to the low result observed for the uranium system (26%/26 turnovers, Table 3, Entry 11). Similarly, acetal and ether functional groups are incompatible with the uranyl system, with the dioxolane derived from condensing cyclopentanone with ethylene glycol providing even less reactivity than the parent ketone (Table 3, Entry 12) and tert-butyl methyl ether not producing any fluorinated product. (Table 3, Entry 13). Taken together, the presence of a Lewis-basic oxygen site on the substrate, even the traditionally weak coordinator carbonyl, appears to be deleterious to the reaction.
An initial hypothesis combining these two observations posits that carbonyls might be quenching the uranyl excited state via a reversible, inner-sphere electron transfer. Indeed, it is known that ketones will readily coordinate the labile [UO2]2+ cation, with such intermediates having been observed experimentally.29 Bakac and coworkers27 previously observed this non-productive quenching via “exciplex decay” in the aerobic oxidation of toluene, finding that the vast majority of interactions between toluene and the uranyl excited state, [UO2]2+*, led to return to the ground-state with no HAT (figure 3). We encountered this same phenomenon when attempting to fluorinate toluene and, to an even greater extent, anisole, a substrate that would have been interesting as a model for the production of agrochemically valuable mono-, di-, and tri-fluoromethoxy30 ethers (Table 3, Entry 14).
Figure 3.
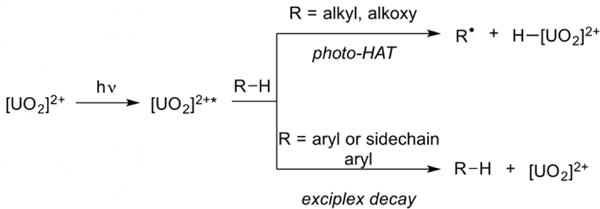
The uranyl excited state [UO2]2+* reacts with alkanes primarily through hydrogen atom transfer (HAT) and with arenes through unproductive exciplex decay
Compelling evidence for the exciplex decay of the arene substrates comes from a competitive quenching experiment, wherein an equimolar amount of toluene and cyclooctane was subjected to the fluorination and quantified. The low yields of both products (Figure 4) suggest that this is at least partially responsible for the low efficiency. Returning to the question of ketones, an analogous experiment using equimolar cyclopentanone and cyclooctane led to high yield of fluorocyclooctane (74%, Figure 4) and trace fluorinated cyclopentanone, suggesting that cyclopentanone is at best a weak quencher of the uranyl excited state, [UO2]2+*. If one imagines a slow, but competitive with HAT, intramolecular deactivation pathway for carbonyl compounds, the improved activity of ethyl isovalerate, long chain esters and sclareolide compared to cyclopentanone could be explained through several factors. These might include additional activation, and thus higher rate of reaction, of a methine proton (ethyl isovalerate); a greater number of potential reactive sites (longer chain substrates); and structural rigidity separating the reactive site from a potential quenching functionality (sclareolide).
Figure 4.
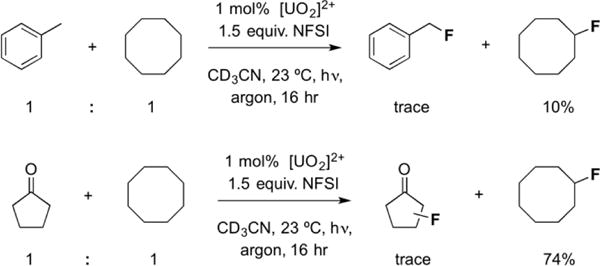
Reactions containing both cyclooctane and toluene (top) or cyclopentanone (bottom) behave differently with respect to reagent conversion. The toluene result suggests that it is an effective quencher of the uranyl excited state [UO2]2+*.
Regardless of cause, the limited substrate scope of the uranyl fluorination, while initially disheartening, is not without benefits. Firstly, the (essentially) complete inertness of short-chain ketones and relative unreactivity of other carbonyl compounds contrasts with the TBADT8- and acetophenone9-mediated reactions, for which they are excellent substrates. In the substrate admixing experiments of Figure 4, the selective activation of cyclooctane (BDE 96 kcal•mol−1)32 over toluene (BDE 90 kcal•mol−1)33 is interesting and again diverges greatly from arylketone9,34- and TBADT35-catalyzed fluorination methods, wherein benzyllic positions are preferentially activated. This highly-discriminating nature of the uranyl catalyst opens the door for selective fluorination of electronically activated C–H bonds in the presence of others that would be a liability under previously-reported conditions.
A second upside to this observation is the contribution of a new data point to the sparsely populated chart of uranyl photocatalysis. The benchmarking of this promising complex against popular photo-HAT catalysts in an increasingly well-studied reaction class shows that while many comparisons can be made, so can many contrasts. The behavior of cyclooctane shows that the efficiency of uranyl photocatalysis can outstrip that of traditional near-UV photo-HAT catalysts while operating under visible light; however, the substrate must be chosen judiciously.
Ultimately, a new catalytic method to fluorinate certain unactivated Csp3–H bonds was developed; this method uses low-energy visible light to drive homolytic cleavages of strong C–H bonds by an activated uranyl catalyst and capitalizes on the reactivity of a putative organic radical. To the best of our knowledge, this chemistry constitutes the second catalytic transformation based on the HAT reactivity of a photo-activated uranyl catalyst. Our hope is that the research described herein will stimulate future efforts to expand the considerable potential of abundant, yet underutilized, uranyl complexes in catalysis.
Supplementary Material
Footnotes
This paper is dedicated with respect and admiration to Prof. Martin F. Semmelhack on the occasion of his 75th birthday.
This work was supported by NIGMS R01 GM065483 to E.J.S., NSF-GRFP DGE 1148900 to J.G.W., the NSF-CCI Center for Selective C–H Functionalization (CHE-1205646), and Princeton University.
Supporting information for this article is given via a link at the end of the document.
References
- 1.Kirk KL. Org Process Res Dev. 2008;12:305–321. [Google Scholar]
- 2.Fujiwara T, O’Hagan D. J Fluorine Chem. 2014;167:16–29. [Google Scholar]
- 3.Berger R, Resnati G, Metrangolo P, Weber E, Hulliger J. Chem Soc Rev. 2011;40:3496–3508. doi: 10.1039/c0cs00221f. [DOI] [PubMed] [Google Scholar]
- 4.Hagmann WG. J Med Chem. 2008;51:4359–4369. doi: 10.1021/jm800219f. [DOI] [PubMed] [Google Scholar]
- 5.Neumann CN, Ritter T. Angew Chem Int Ed. 2015;54:3216–3221. doi: 10.1002/anie.201410288. [DOI] [PubMed] [Google Scholar]
- 6.McBee ET. Ind Eng Chem. 1947;39:236–237. [Google Scholar]
- 7.Blum S, Knippel JL, Lectka T. Chem Sci. 2014;5:1175–1178. [Google Scholar]
- 8.Halperin SD, Fan H, Chang S, Martin RE, Britton R. Angew Chem Int Ed. 2014;53:4690–4693. doi: 10.1002/anie.201400420. [DOI] [PubMed] [Google Scholar]
- 9.Xia JB, Zhu C, Chen C. Chem Commun. 2014;50:11701–11704. doi: 10.1039/c4cc05650g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kee CW, Chin KF, Wong MW, Tan CH. Chem Commun. 2014;50:8211–8214. doi: 10.1039/c4cc01848f. [DOI] [PubMed] [Google Scholar]
- 11.Chambers RD, Parsons M, Sanford G, Bowyden R. Chem Commun. 2000:959–960. [Google Scholar]
- 12.Liu W, Huang MJ, Nielsen RJ, Goddard W, Groves JT. Science. 2012;337:1322–1325. doi: 10.1126/science.1222327. [DOI] [PubMed] [Google Scholar]
- 13.Blum S, Pitts CR, Miller DC, Haselton N, Holl MG, Urheim E, Lectka T. Angew Chem Int Ed. 2012;51:10580–10583. doi: 10.1002/anie.201203642. [DOI] [PubMed] [Google Scholar]
- 14.Rueda-Becerril M, Sazepin CC, Leung JCT, Okbinoglu T, Kennepohl P, Paquin JF, Sammis GM. J Am Chem Soc. 2012;134:4026–4029. doi: 10.1021/ja211679v. [DOI] [PubMed] [Google Scholar]
- 15.Prier CK, Rankic DA, MacMillan DWC. Chem Rev. 2013;113:5322–5363. doi: 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hubbell MW. The Fundamentals of Nuclear Power Generation: Questions and Answers. Author House; Bloomington: 2011. [Google Scholar]
- 17.Princeton University. “Uranium and Thorium Use”, can be found under. 2015 https://ehs.princeton.edu/laboratory-research/radiation-safety/radioactive-materials/uranium-thorium-use.
- 18.Lide D. CRC Handbook of Chemistry and Physics, 2000–2001. CRC Press; Boca Raton: 2000. [Google Scholar]
- 19.Liddle ST. Angew Chem Int Ed. 2015;54:8604–8641. doi: 10.1002/anie.201412168. [DOI] [PubMed] [Google Scholar]
- 20.Fox AR, Bart SC, Meyer K, Cummins CC. Nature. 2008;455:341–349. doi: 10.1038/nature07372. [DOI] [PubMed] [Google Scholar]
- 21.Li Y, Su J, Mitchell E, Zhang G, Li J. Sci China Chem. 2013;56:1671–1681. [Google Scholar]
- 22.Burrows HD, Kemp TJ. Chem Soc Rev. 1974;3:139–165. [Google Scholar]
- 23.Syt’ko VV, Umreiko DS. J Appl Spectrosc. 1998;65:857–870. [Google Scholar]
- 24.Paine RT, Kite MS. Lanthanide and Actinide Chemistry and Spectroscopy. American Chemical Society; Washington D.C.: 2009. pp. 369–380. [Google Scholar]
- 25.Jørgensen CK, Reisfield R. Struct Bond. 1982;50:121–171. [Google Scholar]
- 26.Wang WD, Bakac A, Espensen JH. Inorg Chem. 1995;34:6034–6039. [Google Scholar]
- 27.Mao Y, Bakac A. J Phys Chem. 1996;100:4219–4223. [Google Scholar]
- 28.Barata-Vallejo S, Lantaño B, Postigo A. Chem Eur J. 2014;51:16806–16829. doi: 10.1002/chem.201404005. [DOI] [PubMed] [Google Scholar]
- 29.Rios D, Schoendorff G, Van Stipdonk MJ, Gordon MS, Windus TL, Gibson JK, de Jong WA. Inorg Chem. 2012;51:12768–12775. doi: 10.1021/ic3015964. [DOI] [PubMed] [Google Scholar]
- 30.Leroux FR, Manteax B, Vors JP, Pazenok S. Beilstein J Org Chem. 2008;4:13. doi: 10.3762/bjoc.4.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matsushima K. J Am Chem Soc. 1972;94:6010–6016. [Google Scholar]
- 32.Periyar A, Bose S, Biswas AN, Barman S, Bandyopadhyay P. Catal Sci Technol. 2014;4:3180–3185. [Google Scholar]
- 33.Nam PC, Nguyen MT. J Phys Chem A. 2005;109:10342–10347. doi: 10.1021/jp0534030. [DOI] [PubMed] [Google Scholar]
- 34.Xia JB, Zhu C, Chen C. J Am Chem Soc. 2013;135:17494–17500. doi: 10.1021/ja410815u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nodwell MB, Bagai A, Halperin SD, Martin RE, Knust H, Britton R. Chem Commun. 2015;51:11783–11786. doi: 10.1039/c5cc04058b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.