

Abstract
Glioblastoma (GBM) contains a population of stem-like cells that promote tumor invasion and resistance to therapy. Identifying and targeting stem cell factors in GBM may lead to the development of more effective therapies. High Mobility Group AT-hook 2 (HMGA2) is a transcriptional modulator that mediates motility and self-renewal in normal and cancer stem cells. We identified increased expression of HMGA2 in the majority of primary human GBM tumors and cell lines compared to normal brain. Additionally, HMGA2 expression was increased in CD133+ GBM neurosphere cells compared to CD133- cells. Targeting HMGA2 with lentiviral short hairpin RNA (shRNA) led to decreased GBM stemness, invasion, and tumorigenicity. Ectopic expression of HMGA2 in GBM cell lines promoted stemness, invasion, and tumorigenicity. Our data suggests that targeting HMGA2 in GBM may be therapeutically beneficial.
Keywords: glioma, motility, clonogenicity, GSC, HMGI-C
1. Introduction
Glioblastoma (GBM) is the most aggressive and incurable form of astrocytoma with over 10,000 new cases estimated to be diagnosed in 2015 [1]. The average survival time after diagnosis is 12-14 months [2-4]. The standard of care for GBM is maximal surgical resection followed by radiation and chemotherapy [5]. GBM tumors are heterogeneous and contain a hierarchy of cell types, a small percentage of which are GBM stem-like cells [6] that are highly invasive and resistant to current therapies [2-4]. Failure of current GBM therapeutics warrants identification of novel targets and pathways regulating stemness, invasion and tumor formation.
HMGA2 (also called HMGI-C) is a DNA binding protein that acts as an architectural transcription regulator that can assemble and maintain enhanceosomes [7]. HMGA2 binds to the AT-rich minor grooves of DNA through three conserved sequences called AT-hooks [8]. HMGA2 can regulate the expression of both oncogenes and tumor suppressors [9]. HMGA2 is important during embryonic morphogenesis [10] and is aberrantly expressed in cancer [7, 10-32]. High levels of HMGA2 in cancer are associated with increased invasiveness, stemness and poor prognosis [12, 14, 18, 22, 24-36] However, the functional importance of HMGA2 in regulating stemness and tumorigenicity in GBM are poorly understood.
HMGA2 can be regulated by different mechanisms including the LIN28 pathway [7, 17, 37-39]. LIN28A and LIN28B negatively regulate the let-7 tumor suppressor family of microRNAs [40-43]. HMGA2 has seven conserved let-7 binding sites in the 3’-UTR region [44], enabling negative regulation by let-7 in GBM [17]. We previously identified LIN28A and LIN28B as being expressed in GBM, and showed that knockdown of LIN28A or overexpression of let-7 could decrease GBM invasion [17]. We further found that manipulation of LIN28A and let-7 levels in GBM led to changes in HMGA2 expression, suggesting that the LIN28/let-7/HMGA2 pathway was active in GBM[17]. We here further explore this pathway by directly investigating the function of HMGA2 in GBM.
2. Materials and Methods
2.1. Cell culture
The human adherent GBM cell lines, U-87 MG and LN-229, were obtained from the American Type Culture Collection (ATCC) and were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; Invitrogen™, Life technologies, New York) supplemented with fetal bovine serum (FBS; 10% for U-87 MG and 5% for LN-229; GE Healthcare Life Sciences, Utah). The adult human GBM neurosphere cell lines, GBM14, GBM10, JHH520, and HSR-GBM1 were cultured in neurosphere media containing human epidermal growth factor (EGF; PeproTech, New Jersey), fibroblast growth factor (FGF; PeproTech, New Jersey) and heparin sulfate (Sigma-Aldrich, Missouri) as previously described [45, 46]. HEK293T and HEK293GP cells were used for producing viral particles and were cultured in DMEM containing 10% FBS. All these cell lines were cultured in a humidified atmosphere of 5% CO2 at 37°C.
2.2. DNA constructs
Two lentiviral shRNA constructs TRCN0000342673 (shHMGA2#1) and TRCN0000342671 (shHMGA2#2) targeting the 3’-UTR region and the coding region, respectively (Sigma-Aldrich, Missouri), of human HMGA2 mRNA were used to knockdown HMGA2. MISSION® pLKO.1-puro Empty Vector Control (does not contain shRNA construct) and MISSION® pLKO.1-puro Non-Target shRNA Control Plasmid DNA (SCRAMBLE shRNA control that does not target any known sequence from any species) were used as controls for the two shRNA constructs. pBABE zeo HMGA2 [47] was a gift from Tyler Jacks (Addgene plasmid # 17411) that was used to retrovirally express HMGA2. pBABE zeo (Addgene plasmid # 1766) was used as a control for the retroviral HMGA2-expressing plasmid.
2.3. Virus preparation, titration and infection
Viral particles were produced and titered as previously described [48]. For infection, GBM cells were treated with trypsin (Sigma-Aldrich, Missouri; for U-87 MG and LN-229) or Accutase (Sigma-Aldrich, Missouri; for neurosphere cell lines) and gently triturated and plated at low density. The concentrated virus (approximately 2.5 × 107 TU/ml) was added to the cells and incubated for 48 hours, after which the transduced cells were selected using puromycin (Sigma-Aldrich, Missouri; 1-2μg/ml) or Zeocin (Invitrogen™, life technologies, New York; 250µg/ml). After 7 days of selection, HMGA2 knockdown or expression was confirmed by western blotting.
2.4. Colony formation in soft agar and methyl cellulose
GBM1 cells expressing control or HMGA2 vector were triturated to single cells and plated in soft agar to form colonies for 14-18 days as previously described [17]. For serial clonogenic methylcellulose assays, HSR-GBM1 cells expressing control or HMGA2 vector were triturated into single cells and plated in methylcellulose as previously described [45] with the following modifications. The primary assay was plated into 6-well plates with 4 wells each for control and HMGA2 overexpressing cells. After 19-20 days of the primary assay, three wells were stained with p-Nitro Blue Tetrazolium Chloride (NBT; Affymetrix, Inc., Ohio) and from the fourth well, spheres greater than 100μm in diameter were pulled, triturated into single cells and replated into methylcellulose (secondary assay). After 17-19 days of secondary assay, the process described above was repeated again (tertiary assay). Stained colonies from primary, secondary and tertiary assays that were greater than 50 μm in diameter were imaged and counted using MCID Core software (Cambridge, UK). At least 3 images per well were counted and analyzed.
2.5. Boyden chamber transwell invasion assay
Transwell invasion assays were performed as previously described [17] with the following modifications. Transduced adherent cells were triturated to single cells and plated onto Matrigel-coated transwell inserts in the upper chamber containing serum-deprived medium while the lower chamber contained DMEM supplemented with either 5% (for LN-229) or 10% (for U-87 MG) FBS as a chemoattractant. Transduced neurosphere cells were plated in neurosphere media lacking growth factors while the lower chamber contained media supplemented with EGF and FGF. The cells were allowed to invade through Matrigel for 24-26 hours after which the invading cells collected in the bottom part of the filter were stained with hematoxylin for quantitation. At least 2 transwells per condition and 6 high power fields (HPF) per transwell were analyzed.
2.6. RNA extraction and quantitative PCR
RNA was extracted and purified using RNeasy kit (Qiagen, California). Reverse transcription and quantitative PCR was performed using SYBR Green reagents (Applied Biosystems, California) on an I-Cycler IQ Real-Time detection system (Bio-Rad, California) according to the manufacturer’s instructions. The ΔΔCT method was used to quantify RNA expression levels and all values were normalized to 18S rRNA. Expression levels were analyzed from at least three experimental and 2 technical replicates. The following primer sets (for all human sequences) were used: HMGA2 forward 5’-AAAGCAGCTCAAAAGAAAGCA -3’, reverse 5’-TGTTGTGGCCATTTCCTAGGT-3’, and 18S forward 5’-GTAACCCGTTGAACCCCATT-3’, reverse 5’-CCATCCAATCGGTAGTAGCG-3’.
2.7. Western Blotting
Cells and primary tumor tissues were lysed, separated on SDS-polyacrylamide gels and subjected to Western blotting as described before [17] using the following antibodies: HMGA2 (1:1000 dilution, Cell Signaling Technology, Inc., Massachusetts), glyceraldehyde-3-dehydrogenase (GAPDH) (1:3000 dilution, RDI, New York), and β-actin (1:5000 dilution, Santa Cruz Biotechnology, Inc., Texas). Goat anti-mouse and goat anti-rabbit secondary antibodies conjugated to horse-radish peroxidase (1:3000 to 1:5000 dilution, KPL, Maryland) were used to detect the primary antibodies. Western Lightning Plus ECL (PerkinElmer, Massachusetts) was used to detect the secondary antibodies. Densitometry analysis was performed using Image J software [49].
2.8. Intracranial xenograft tumors and bioluminescence imaging
Transduced cells were xenografted intracranially into immunocompromised mice as described previously [17] with the following modifications. Stereotactic technique [50] was used for orthotopic xenografting into the striatum with the coordinates -3 from bregma, +2 from the sagittal suture and 3 mm deep. We implanted 1 × 104 transduced tumor cells overexpressing HMGA2 and 1 × 105 transduced cells wherein HMGA2 expression was reduced using shRNA. Tumor growth was monitored periodically using bioluminescence imaging by the Xenogen IVIS ® Spectrum in vivo imaging system (PerkinElmer, Inc., Massachusetts) as described previously [46]. Animals were monitored regularly for signs of distress (cachexia, neurologic deficits, weight loss, hunching) suggestive of increasing tumor mass and were sacrificed at that time. Resected brains with intracranial tumors were embedded in paraffin and processed for immunohistochemistry. 3/10 animals from the shHMGA2 group were censored and not included in the survival analysis due to the following reasons: one died post-operation, one died from lymphoma, and the other was lost during cage maintenance. All procedures performed on animals were in accordance with approved protocols from the Institutional Animal Care and Use Committee.
2.9. Immunohistochemistry
Human glioma tissue microarrays (TMAs) containing tumor cores from 45 adult and 35 pediatric primary GBM tumors were constructed at Johns Hopkins as described in [17]. The TMAs and deparaffinized intracranial xenografted tissue sections were subjected to immunohistochemistry as described before [17] at the Johns Hopkins Histopathology Core. The tissue sections were stained with hematoxylin and eosin (H&E) to visualize tumor cells. For HMGA2 immunohistochemistry on the TMAs, human specific HMGA2 primary antibody (1:7 dilution, Abcam, Massachusetts) was used and nuclear staining was scored by a neuropathologist (F.R.) as high (3), moderate (2), low (1) or negative (0, if no immunoreactivity or only a blush/trace was observed).
2.10. Human tumor tissue processing
Primary human brain tumor and normal samples were obtained by surgical resections performed at the Johns Hopkins Hospital following approved Institutional Review Board protocols and after informed consent from patients. The tissue was cryopreserved immediately post resection. For preparation of tumor lysates, the cryopreserved tumor and normal samples were lysed in RIPA buffer and processed for Western blotting as described previously [17].
2.11. Cell proliferation and apoptosis assays
Cell proliferation was measured by treating the transduced cells with bromodeoxyuridine (BrdU) (Sigma-Aldrich, St. Louis, Missouri, USA) for 3 hours and the samples were then processed for immunofluorescence as described previously [51]. For assessing apoptosis, transduced cells were cytospun and processed for cleaved caspase-3 staining as described in [51] using cleaved caspase-3 antibody (Cell Signaling Technology, Massachusetts).
2.12. Flow cytometric analysis
Transduced cells overexpressing HMGA2 were triturated to single cells and processed for CD133 flow sorting as described previously [45].
2.13. Statistical methods and survival analysis
Data was analyzed from each experiment performed at least twice with concordant results and representative results are shown. Statistical tests were performed using Excel (Microsoft, Washington). Error bars denote standard error of the mean (SEM). Statistical significance was determined using an unpaired, two-sided Student’s t-test, where P<0.05 was considered to be statistically significant. For in vivo experiments, data was collected from at least 5 animals injected per condition and statistical significance was determined by log-rank test using GraphPad Prism (GraphPad software, Inc., California).
3. Results
3.1. HMGA2 is expressed in human GBM tumors and cell lines
We analyzed HMGA2 protein expression in primary GBM tumors resected from human patients by western blotting and immunohistochemistry. High levels of HMGA2 protein compared to normal brain were detected in the majority (5 out of 7, 71%) of primary GBM tumors (Fig. 1A) by western blotting. We performed immunohistochemistry to determine the expression levels of HMGA2 protein in 45 adult GBM and 35 pediatric GBM tumors on a tissue microarray (TMA) (Fig. 1B). We found moderate to high nuclear expression of HMGA2 in 29 out of 45 (67%) adult GBM tumors and 24 out of 35 (70%) pediatric GBM tumors (Fig. 1B).
Figure 1.
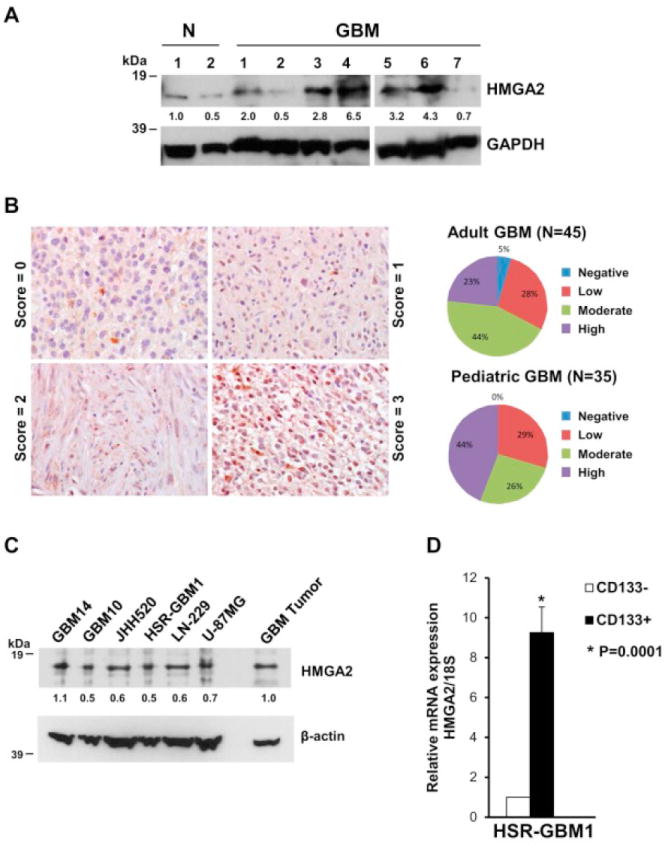
The majority of GBM primary tumors and cell lines show increased expression of HMGA2. (A) Western blot showing increased expression of HMGA2 protein in 71% (5/7) primary tumor samples from GBM patients compared to normal (N) brain tissue (cortex). GAPDH was used as a loading control. HMGA2 and GAPDH protein levels were quantitated using densitometry. Values normalized to sample#1 of normal brain tissue (N) are depicted below the blots. (B) Immunohistochemical analysis showing varying expression of HMGA2 in primary GBM samples on a tissue microarray (Scores: 0 or negative =no staining, 1=low staining, 2=moderate staining, 3=high staining). Quantitation of the percentage of tumor samples showing the staining is depicted at right. (C) Western blot showing HMGA2 expression in GBM cell lines (GBM14, GBM10, JHH520, HSR-GBM1, LN-229 and U-87 MG). A primary tumor sample is also shown for comparison of HMGA2 expression. β-actin was used as a loading control. HMGA2 and β-actin protein levels were quantitated on the western blot using densitometry and values normalized to the primary tumor sample for each cell line are depicted below the blots. (D) Increased expression of HMGA2 mRNA by quantitative PCR in CD133+ HSR-GBM1 neurospheres compared to CD133- cells. Data were normalized to 18S ribosomal RNA and are shown as relative expression (relative to CD133- cells) (*P=0.0001 by Student’s t-test, N=2).
We next tested the expression of HMGA2 protein in multiple patient-derived adherent (U-87 MG and LN-229) and neurosphere (GBM14, GBM10, JHH520, and HSR-GBM1 [52, 53]) cell lines by western blotting. All GBM cell lines analyzed showed some degree of HMGA2 protein expression (Fig. 1C).
HMGA2 promotes self-renewal capacity in embryonic and cancer stem cells [10]. CD133 is used as a marker to differentiate GBM stem-like cells from the other cell types in the heterogeneous tumor cell population [53, 54]. We used CD133 as a surrogate marker for GBM stem-like cells and flow-sorted HSR-GBM1 neurospheres for cells expressing CD133. Analysis of HMGA2 expression by quantitative PCR revealed a 9.3-fold higher expression of HMGA2 in HSR-GBM1 neurosphere cells expressing CD133 compared to CD133- cells (Fig. 1D, P<0.0001, N=2). This suggests that HMGA2 may play a role in promoting stemness or clonogenicity of GBM cells.
3.2. Reduction of HMGA2 expression inhibits GBM cell clonogenicity and invasion
To functionally dissect the role of HMGA2 in GBM, we used two GBM adherent (U-87 MG and LN-229) cell lines and a neurosphere (HSR-GBM1) cell line [46]. U-87 MG and LN-229 express levels of HMGA2 (Fig. 1C) comparable to the high levels of HMGA2 we detected in a subset of primary tumors, making them ideal for loss of function experiments. HSR-GBM1 expresses lower levels of HMGA2 (Fig. 1C), making it appropriate for gain and loss of function experiments. We used two separate lentiviral shRNA constructs as controls (pLKO.1 and SCRAMBLE) and two lentiviral HMGA2 shRNA constructs (shHMGA2#1 and shHMGA2#2) to decrease HMGA2 expression in GBM cell lines (Fig. 2A). Densitometric analysis of the bands in the western blots showed a reduction of HMGA2 protein by respectively 53% and 57% with shHMGA2#1 and shHMGA2#2 for HSR-GBM1, 28% and 42% with shHMGA2#1 and shHMGA2#2 for LN-229 compared to control (pLKO.1) infected cells (Fig. 2A). In U-87 MG cells, we found that shHMGA2#1 decreased HMGA2 protein expression by 14% compared to the cells expressing control shRNA (pLKO.1) (Supplementary Fig. S1-A). Although we have used shHMGA2#1 and shHMGA2#2 for suppressing HMGA2 expression up to 70% compared to the empty vector control shRNA in pediatric tumor cell lines [22], we were unable to achieve more than 57% knockdown of HMGA2 in GBM cells even after multiple attempts. This suggests that there is a greater requirement for HMGA2 in GBM tumor cells. To test the hypothesis that HMGA2 promotes clonogenicity in GBM, we performed clonogenic soft agar assays with HSR-GBM1 cells expressing either pLKO.1 or HMGA2 shRNA. HSR-GBM1 cells infected with pLKO.1 formed 79 ± 11 colonies, whereas the number of colonies formed by HSR-GBM1 cells expressing HMGA2 shRNA#2 were reduced to an average of 16 ± 2 (Fig. 2B, P<0.0001, N=2).
Figure 2.

shRNA mediated knockdown of HMGA2 reduces GBM cell invasion and colony formation in vitro. (A) Western blots showing reduction of HMGA2 expression with two different shRNA (shHMGA2#1 and shHMGA2#2) compared to control vector (pLKO.1 and SCRAMBLE) infected cells in HSR-GBM1 and LN-229 cell lines. GAPDH was used as a loading control. HMGA2 and GAPDH protein levels were quantitated on the western blot using densitometry and values normalized to pLKO.1 for each cell line are depicted below the blots. (B) Left, HSR-GBM1 cells showing 79% reduction in colony formation with shHMGA2#2 in soft agar compared to control infected cells (*P<0.0001 by Student’s t-test, N=2). Right, Quantitation of the number of colonies is shown on the right. (C) Left, Photomicrographs (20X) showing reduced invasion of HSR-GBM1 and LN-229 cells infected with shHMGA2 compared to pLKO.1 infected cells in Matrigel transwell assay. Right, Quantitation of photomicrographs shown in (C). For HSR-GBM1: 57% and 50% reduction with shHMGA2#1 and #2, respectively, *P<0.001 for shHMGA2#1 and *P=0.001 for shHMGA2#2 by Student’s t-test, N=2. For LN-229: 73% and 44% reduction with shHMGA2#1 and shHMGA2#2, respectively, compared to pLKO.1 infected cells, *P<0.001 by Student’s t-test, N=2.
We further hypothesized that reduced expression of HMGA2 in GBM will decrease the invasive capacity of GBM cells. We performed transwell invasion assays in HSR-GBM1, LN-229 and U-87 MG cells expressing either pLKO.1 control or HMGA2 shRNA. HSR-GBM1 cells expressing HMGA2 shRNA showed a significant decrease in the ability of cells to invade Matrigel (57% and 50% reduction with shHMGA2#1 and shHMGA2#2, respectively), compared to pLKO.1 infected cells (Fig. 2C, P<0.001 for shHMGA2#1 and P=0.001 for shHMGA2#2, N=2). LN-229 cells expressing HMGA2 shRNA had a reduction in invasion of 73% and 44% with shHMGA2#1 and #2, respectively, compared to pLKO.1 infected cells (Fig 2C, P<0.001 for both HMGA2 shRNAs, N=2). U-87 MG cells infected with shHMGA2#1 showed a significant decrease in invasion by 30% compared to pLKO.1 infected cells (Supplementary Fig. S1-B, P=0.006 N=3), suggesting that HMGA2 is required for GBM cell invasion.
3.3. Reduction of HMGA2 suppresses GBM tumorigenicity in vivo in orthotopic xenografts
To determine whether HMGA2 is required for tumor formation and survival of tumor cells in an in vivo environment, we injected HSR-GBM1 cells expressing either pLKO.1 or HMGA2 shRNA into the striatum of the brain of immunocompromised mice (100,000 cells/mouse). We have engineered HSR-GBM1 cells to express luciferase constitutively allowing bioluminescence imaging [46]. After five weeks, animals injected with HSR-GBM1 cells transduced with pLKO.1 showed more luciferase intensity (Fig. 3A and B, mean bioluminescence intensity = 7273) compared to animals harboring HSR-GBM1 orthotopic xenografts transduced with HMGA2 shRNA (mean = 1666) (Fig. 3A and B, P=0.035, N=5 for each condition). The experiment was continued, and animals were sacrificed when they showed clinical signs of harboring a significant brain tumor. Log rank analysis of Kaplan-Meier survival curves showed significant increase in survival after HMGA2 knockdown with median survival for pLKO.1 control being 67.5 days and median survival for shHMGA2#2 being 118 days, P<0.0001 by log-rank test (Fig. 3C, N=10 for pLKO.1 and N=7 for shHMGA2#2). Animals were monitored for symptoms for six months, at which time the remaining mice harboring shHMGA2 xenografts were sacrificed. None of these long-surviving animals had a brain tumor when examined histologically (Supplementary Fig. S2).
Figure 3.
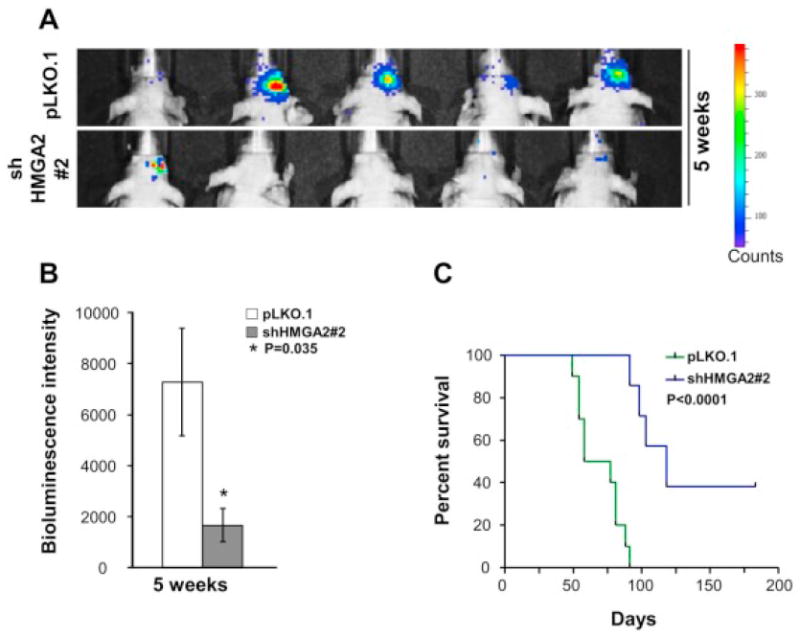
shRNA mediated knockdown of HMGA2 in GBM neurosphere cells inhibits intracranial tumor growth in immunocompromised mice. (A) Photographs of xenografted mice showing decreased bioluminescence intensity depicting reduced intracranial tumor growth with knockdown of HMGA2 (shHMGA2#2) in HSR-GBM1 cells at 5 weeks post injection compared to control mice (pLKO.1). (B) Quantitation of the intensity of bioluminescence showing 77% reduction with shHMGA2#2 xenografted mice at 5 weeks, respectively, compared to pLKO.1 mice, *P=0.035 by Student’s t-test, N=5. (C) Kaplan-Meier survival analysis depicting decreased survival of mice injected with HSR-GBM1 neurospheres expressing shHMGA2#2 compared to control infected cells (P<0.0001, N=10 for pLKO.1 and N=7 for shHMGA2#2).
3.4. Overexpression of HMGA2 increases GBM clonogenicity and invasion
To test the hypothesis that increased expression of HMGA2 would promote GBM tumorigenicity, we expressed HMGA2 in our HSR-GBM1 neurosphere cell line (GBM1-HMGA2). Viral mediated expression of HMGA2 in HSR-GBM1 increased HMGA2 expression by 6.5 fold compared to the control virus infected cells (Fig. 4A).
Figure 4.
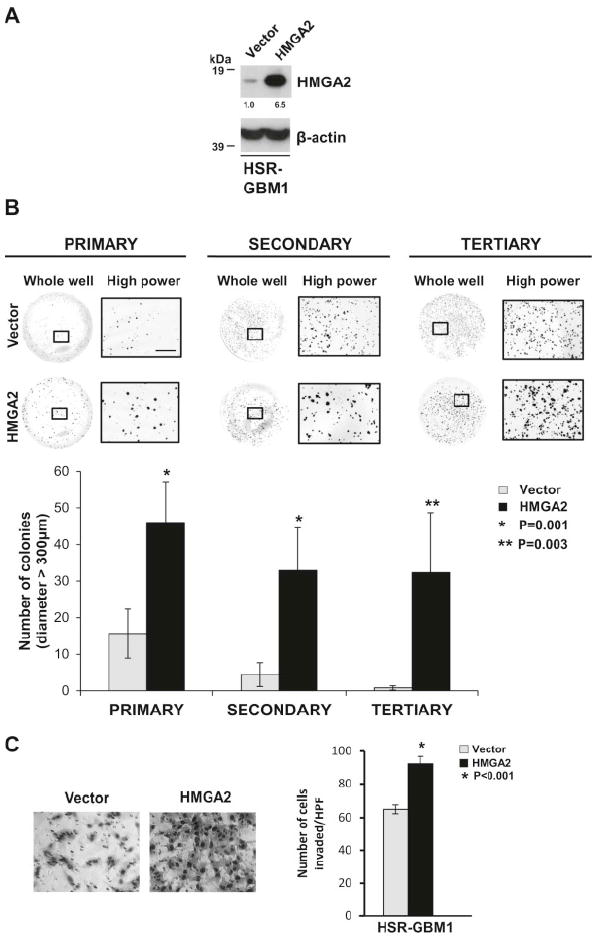
Overexpression of HMGA2 increases GBM cell invasion and clonogenicity in vitro. (A) Western blot showing increased expression of HMGA2 in HSR-GBM1 neurospheres infected with retroviral HMGA2 (HMGA2) compared to control vector (Vector). β-actin was used as a loading control. Densitometry values normalized to control vector are depicted below the blots. (B) Photos and quantification depicting increase in the number of larger colonies (diameter>300μm) formed by HSR-GBM1 neurospheres expressing retroviral HMGA2 compared to control vector cells in methylcellulose assays in vitro (*P=0.001 and 0.003 by Student’s t-test, N=3). Scale bar = 2500 micrometers on high power macrophotographs. (C) Photomicrographs (20X) depicting increased invasion of HSR-GBM1 neurospheres expressing HMGA2 compared to control vector infected cells in Matrigel-transwell invasion assays in vitro. Quantification depicting increase in invasion is shown on the right (*P<0.001 by Student’s t-test, N=3).
To confirm HMGA2’s role in promoting GBM clonogenicity, we performed clonogenic serial methylcellulose assays. We plated single cells of either control plasmid or HMGA2 transduced GBM1 cells in methylcellulose to form colonies (primary assay). We then extracted colonies that were greater than 100 micrometers (μm) in diameter and replated the assay twice (secondary and tertiary assays) as described in the Materials and Methods section. We have shown previously by clonogenic assays that HSR-GBM1 cells that form colonies greater than 100 μm in diameter are derived from a tumor stem-like cell and retain the ability to self-renew over multiple subsequent generations [45]. High expression of HMGA2 caused a significant increase in the number of larger colonies (diameter > 300μm) formed in primary (P=0.001, 2.9-fold increase), secondary (P=0.001, 7.4-fold increase) and tertiary assays (P=0.003, 40.4-fold increase) compared to control (Fig. 4B, N=3). Since increased HMGA2 expression in HSR-GBM1 does not lead to enhanced proliferation (Supplementary Figure S3-A), the increased colony size suggests that HMGA2 promotes the self-renewal capacity of GBM cells, a property that is important for tumor initiation.
To confirm that HMGA2 promotes GBM invasiveness, we performed Matrigel-transwell assays with GBM1-HMGA2 and GBM1-control cells. GBM1-HMGA2 cells showed 1.4 fold increase in the number of invading cells in 24 hours compared to those infected with the empty vector control plasmid (Fig. 4C, P<0.001, N=3).
3.5. Overexpression of HMGA2 promotes GBM tumor formation in orthotopic xenografts
We hypothesized that increased HMGA2 would enhance GBM tumor formation. We injected 10,000 GBM1-HMGA2 or GBM-vector control cells into the striatum of immunocompromised mice. Luciferase imaging showed that all the animals xenografted with HMGA2-transduced GBM1 cells (5/5) showed large tumors (mean bioluminescence intensity = 24,834) by 4 weeks post injection (Fig. 5A and B). In comparison, at 4 weeks post injection, 4/5 control animals had tumors that were significantly smaller in bioluminescence intensity (mean = 3556) compared to the tumors formed by HMGA2 xenografted mice, and 1 animal did not have evidence of any tumor (Fig. 5A and B, P=0.0209 by Student’s t-test, N=5). The experiment was continued, and animals were sacrificed when they showed clinical signs of harboring a significant brain tumor. Log rank analysis of Kaplan-Meier curves showed a trend towards a decrease in median survival of high HMGA2-expressing GBM1 xenografted mice (86 days) compared to control vector mice (median survival =94.5 days, P=0.08 by log-rank test, N=5, Fig. 5C).
Figure 5.
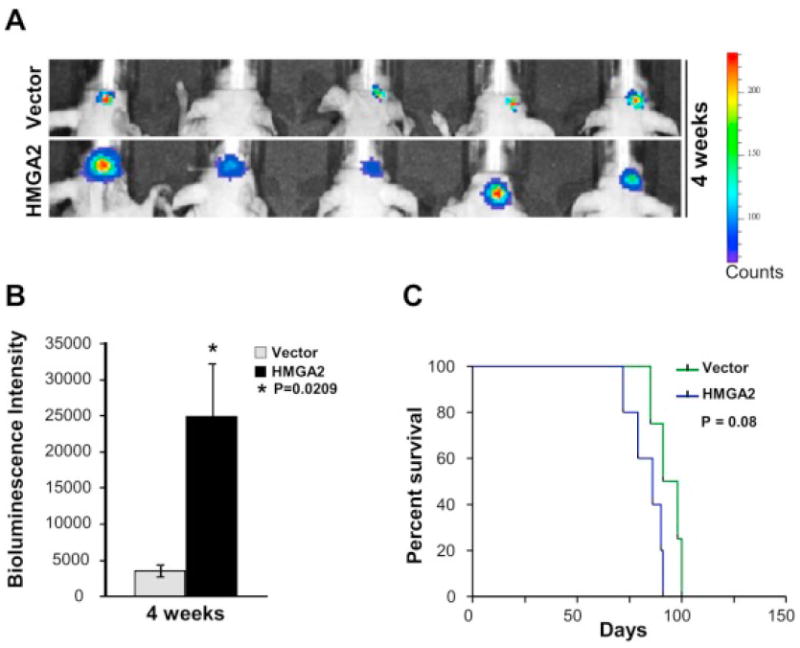
Overexpression of HMGA2 increases intracranial tumor growth in vivo (A) Increased bioluminescence intensity depicting increased intracranial tumor growth at 4 weeks post injection with overexpression of HMGA2 compared to control vector mice. (B) Quantitation of the increased bioluminescence intensity observed in (A) depicting 7.0 fold increase in HMGA2 overexpressing xenografted mice compared to controls (*P=0.0209 by Student’s t-test, N=5). (C) Kaplan Meier survival curves show a decrease in median survival with overexpression of HMGA2 in GBM1 neurospheres (P=0.08, N=5).
4. Discussion
The High Mobility Group AT-hook 2 (HMGA2) protein upregulates a pro-invasive program in diverse cancer types [7, 10, 12, 18-23, 34, 36]. We found increased expression of HMGA2 protein in the majority of GBM tumors by western blot analysis and immunohistochemistry using a cohort of 80 pediatric and adult GBM samples. Prior studies have investigated HMGA2’s role in GBM invasion and tumor formation in heterotopic flank tumors using adherent GBM cell lines [17, 55-57]. We have confirmed these findings using adherent cell lines and extended our interrogation of HMGA2 in stemness and tumorigenicity using GBM neurosphere lines and orthotopic intracranial mouse models. Neurospheres grow in a serum-free medium and are known to simulate GBM in culture and in vivo better than adherent cells [58, 59]. Since brain microenvironment plays profound roles in regulating tumor progression [60], we have confirmed the role of HMGA2 in promoting GBM progression using intracranial xenograft models.
We hypothesized that HMGA2 would promote tumorigenicity in GBM, and we found that HMGA2 enhances GBM clonogenicity and invasion. Supporting a role for HMGA2 in tumor initiation, the majority of GBM cells bearing shRNA against HMGA2 do not form colonies when plated as single cells in soft agar. The reduction in colony formation from single cells suggests that HMGA2 is required for clonogenicity and could affect survival of GBM cells. These cells also have reduced tumorigenicity in intracranial xenografts. However, it is difficult to assess invasiveness of these cells in vivo since the animals were sacrificed when they become symptomatic and the tumor growth is at an advanced stage. Since HSR-GBM1 cells are invasive at baseline, the tumors in both the animal groups (control and HMGA2-expressing) occupy a significant portion of the brain. We also did not observe significant changes in GBM cell proliferation in vitro (measured by BrdU cell growth assays) with perturbation of HMGA2 (Supplementary Fig. S3-A) suggesting that the reduced tumor formation in vivo upon knockdown of HMGA2 may be due to decreased tumor initiation and/or increased apoptosis. Consistent with prior studies [25, 56, 61, 62], we found increased apoptosis upon knockdown of HMGA2 in HSR-GBM1 cells (Supplementary Fig. S3-B). Our in vitro studies indicate HMGA2 promotes both increased invasion and clonogenicity, supporting the in vivo findings that modulation of HMGA2 levels affect tumorigenicity. Even though we could knockdown HMGA2 by 53-57% in our GBM cell lines, we observed prominent phenotypic effects. The inability of GBM cells to tolerate more than 57% reduction in HMGA2 supports the importance and greater dependence of GBM cells on HMGA2 for their survival.
We and others have demonstrated the importance of LIN28A or LIN28B in multiple brain tumor types [17, 51, 63-65]. We found that HMGA2 was altered coordinately after perturbation of LIN28A levels in GBM [17]. However, we identify LIN28A or LIN28B expression in approximately 40 percent of GBM [17]. In contrast, we find increased HMGA2 by immunohistochemistry and western blot in 70 percent of GBM, suggesting that multiple factors may regulate HMGA2. In other tumor types, HMGA2 levels are controlled by Transforming Growth Factor-β (TGF-β) [34], Raf-1 kinase inhibitory protein [66-68], RUNX1 [37], and several microRNAs [35, 39, 69-73].
HMGA2 is a stem cell factor primarily expressed during fetal development with almost no expression in adult human tissues [7, 10]. HMGA2 is aberrantly expressed at high levels in cancer [7] including GBM, which we show in this manuscript corroborating earlier reports [55, 56]. We demonstrate that CD133-expressing GBM cells (which are thought to represent GBM stem-like cells) show 9-fold higher expression of HMGA2 compared to CD133- cells. Since GBM stem-like cells are believed to be the root cause of self-renewal, invasion, tumor recurrence and therapy resistance, identifying targets in these stem-like cells is needed to improve outcomes. Our experiments show that depletion of HMGA2 in GBM neurospheres (which contain GBM stem-like cells) reduced invasion, increased apoptosis, decreased clonogenicity, and suppressed in vivo tumor-forming ability of GBM cells, suggesting that GBM-stem like cells need HMGA2 to maintain malignant properties. Therefore targeting HMGA2 in GBM stem-like cells may be a therapeutically beneficial strategy.
In summary, our studies demonstrate that HMGA2 plays an oncogenic role in GBM and can promote GBM cell clonogenicity, invasion, and tumorigenicity. This work validates HMGA2 as a potential therapeutic target in GBM. Developing anti-HMGA2 therapeutics may lead to improved outcomes for patients suffering from this deadly brain tumor.
Supplementary Material
Highlights.
HMGA2 is validated as a potential therapeutic target in GBM.
High levels of HMGA2 are present in GBM tumors, cell lines and stem-like cells.
HMGA2 promotes GBM stemness, invasion and in vivo tumor formation.
Acknowledgments
We thank Dr. Angelo Vescovi for sharing HSR-GBM1 cells and Dr. Gregory Riggins for kindly providing us with JHH520 cells. We also thank Ann Price and Jessica Hicks for assistance with immunohistochemistry. This work is dedicated to the memory of Haley House, whose family’s benefaction supports research in the Raabe laboratory. This work was supported by NCI Core Grant to the Johns Hopkins SKCCC P30 CA006973; The Children’s Brain Tumor Foundation; The Giant Food Pediatric Cancer Research Fund; a gift from the House family in memory of Haley House. E.H.R. is a St. Baldrick’s Scholar.
Abbreviations
- HMGA2
High Mobility Group AT-hook 2
- GBM
Glioblastoma
- CD133
Cluster-of-Differentiation-133
Footnotes
Conflicts of interest: None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ostrom QT, Gittleman H, Liao P, Rouse C, Chen Y, Dowling J, Wolinsky Y, Kruchko C, Barnholtz-Sloan J. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2007-2011. Neuro-oncology. 2014;16(Suppl 4):iv1–63. doi: 10.1093/neuonc/nou223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Agnihotri S, Burrell KE, Wolf A, Jalali S, Hawkins C, Rutka JT, Zadeh G. Glioblastoma, a brief review of history, molecular genetics, animal models and novel therapeutic strategies. Archivum immunologiae et therapiae experimentalis. 2013;61:25–41. doi: 10.1007/s00005-012-0203-0. [DOI] [PubMed] [Google Scholar]
- 3.Dunn GP, Rinne ML, Wykosky J, Genovese G, Quayle SN, Dunn IF, Agarwalla PK, Chheda MG, Campos B, Wang A, Brennan C, Ligon KL, Furnari F, Cavenee WK, Depinho RA, Chin L, Hahn WC. Emerging insights into the molecular and cellular basis of glioblastoma. Genes & development. 2012;26:756–784. doi: 10.1101/gad.187922.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Furnari FB, Fenton T, Bachoo RM, Mukasa A, Stommel JM, Stegh A, Hahn WC, Ligon KL, Louis DN, Brennan C, Chin L, DePinho RA, Cavenee WK. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes & development. 2007;21:2683–2710. doi: 10.1101/gad.1596707. [DOI] [PubMed] [Google Scholar]
- 5.Anton K, Baehring JM, Mayer T. Glioblastoma multiforme: overview of current treatment and future perspectives. Hematology/oncology clinics of North America. 2012;26:825–853. doi: 10.1016/j.hoc.2012.04.006. [DOI] [PubMed] [Google Scholar]
- 6.Lathia JD, Mack SC, Mulkearns-Hubert EE, Valentim CL, Rich JN. Cancer stem cells in glioblastoma. Genes & development. 2015;29:1203–1217. doi: 10.1101/gad.261982.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fedele M, Fusco A. HMGA and cancer. Biochimica et biophysica acta. 2010;1799:48–54. doi: 10.1016/j.bbagrm.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 8.Ozturk N, Singh I, Mehta A, Braun T, Barreto G. HMGA proteins as modulators of chromatin structure during transcriptional activation. Frontiers in cell and developmental biology. 2014;2:5. doi: 10.3389/fcell.2014.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Narita M, Narita M, Krizhanovsky V, Nunez S, Chicas A, Hearn SA, Myers MP, Lowe SW. A novel role for high-mobility group a proteins in cellular senescence and heterochromatin formation. Cell. 2006;126:503–514. doi: 10.1016/j.cell.2006.05.052. [DOI] [PubMed] [Google Scholar]
- 10.Pfannkuche K, Summer H, Li O, Hescheler J, Droge P. The high mobility group protein HMGA2: a co-regulator of chromatin structure and pluripotency in stem cells? Stem cell reviews. 2009;5:224–230. doi: 10.1007/s12015-009-9078-9. [DOI] [PubMed] [Google Scholar]
- 11.Chieffi P, Battista S, Barchi M, Di Agostino S, Pierantoni GM, Fedele M, Chiariotti L, Tramontano D, Fusco A. HMGA1 and HMGA2 protein expression in mouse spermatogenesis. Oncogene. 2002;21:3644–3650. doi: 10.1038/sj.onc.1205501. [DOI] [PubMed] [Google Scholar]
- 12.Di Cello F, Hillion J, Hristov A, Wood LJ, Mukherjee M, Schuldenfrei A, Kowalski J, Bhattacharya R, Ashfaq R, Resar LM. HMGA2 participates in transformation in human lung cancer. Molecular cancer research : MCR. 2008;6:743–750. doi: 10.1158/1541-7786.MCR-07-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fedele M, Pierantoni GM, Visone R, Fusco A. Critical role of the HMGA2 gene in pituitary adenomas. Cell cycle. 2006;5:2045–2048. doi: 10.4161/cc.5.18.3211. [DOI] [PubMed] [Google Scholar]
- 14.Fedele M, Visone R, De Martino I, Troncone G, Palmieri D, Battista S, Ciarmiello A, Pallante P, Arra C, Melillo RM, Helin K, Croce CM, Fusco A. HMGA2 induces pituitary tumorigenesis by enhancing E2F1 activity. Cancer cell. 2006;9:459–471. doi: 10.1016/j.ccr.2006.04.024. [DOI] [PubMed] [Google Scholar]
- 15.Mahajan A, Liu Z, Gellert L, Zou X, Yang G, Lee P, Yang X, Wei JJ. HMGA2: a biomarker significantly overexpressed in high-grade ovarian serous carcinoma. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2010;23:673–681. doi: 10.1038/modpathol.2010.49. [DOI] [PubMed] [Google Scholar]
- 16.Malek A, Bakhidze E, Noske A, Sers C, Aigner A, Schafer R, Tchernitsa O. HMGA2 gene is a promising target for ovarian cancer silencing therapy. International journal of cancer Journal international du cancer. 2008;123:348–356. doi: 10.1002/ijc.23491. [DOI] [PubMed] [Google Scholar]
- 17.Mao XG, Hutt-Cabezas M, Orr BA, Weingart M, Taylor I, Rajan AK, Odia Y, Kahlert U, Maciaczyk J, Nikkhah G, Eberhart CG, Raabe EH. LIN28A facilitates the transformation of human neural stem cells and promotes glioblastoma tumorigenesis through a pro-invasive genetic program. Oncotarget. 2013;4:1050–1064. doi: 10.18632/oncotarget.1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morishita A, Zaidi MR, Mitoro A, Sankarasharma D, Szabolcs M, Okada Y, D’Armiento J, Chada K. HMGA2 is a driver of tumor metastasis. Cancer research. 2013;73:4289–4299. doi: 10.1158/0008-5472.CAN-12-3848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morshedi A, Ren Z, Li J, Droge P. Probing into the Biological Processes Influenced by ESC Factor and Oncoprotein HMGA2 Using iPSCs. Stem cell reviews. 2012 doi: 10.1007/s12015-012-9373-8. [DOI] [PubMed] [Google Scholar]
- 20.Nishino J, Kim I, Chada K, Morrison SJ. Hmga2 promotes neural stem cell self-renewal in young but not old mice by reducing p16Ink4a and p19Arf Expression. Cell. 2008;135:227–239. doi: 10.1016/j.cell.2008.09.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang X, Liu X, Li AY, Chen L, Lai L, Lin HH, Hu S, Yao L, Peng J, Loera S, Xue L, Zhou B, Zhou L, Zheng S, Chu P, Zhang S, Ann DK, Yen Y. Overexpression of HMGA2 promotes metastasis and impacts survival of colorectal cancers. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011;17:2570–2580. doi: 10.1158/1078-0432.CCR-10-2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaur H, Hutt-Cabezas M, Weingart MF, Xu J, Kuwahara Y, Erdreich-Epstein A, Weissman BE, Eberhart CG, Raabe EH. The Chromatin-Modifying Protein HMGA2 Promotes Atypical Teratoid/Rhabdoid Cell Tumorigenicity. Journal of neuropathology and experimental neurology. 2015;74:177–185. doi: 10.1097/NEN.0000000000000161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang K, Gao H, Wu X, Wang J, Zhou W, Sun G, Wang Y, Mu B, Kim C, Chu P, Ho DM, Ann DK, Wong TT, Yen Y. Frequent overexpression of HMGA2 in human atypical teratoid/rhabdoid tumor and its correlation with let-7a3/let-7b miRNA. Clinical cancer research : an official journal of the American Association for Cancer Research. 2014;20:1179–1189. doi: 10.1158/1078-0432.CCR-13-1452. [DOI] [PubMed] [Google Scholar]
- 24.Madison BB, Jeganathan AN, Mizuno R, Winslow MM, Castells A, Cuatrecasas M, Rustgi AK. Let-7 Represses Carcinogenesis and a Stem Cell Phenotype in the Intestine via Regulation of Hmga2. PLoS genetics. 2015;11:e1005408. doi: 10.1371/journal.pgen.1005408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cai J, Shen G, Liu S, Meng Q. Downregulation of HMGA2 inhibits cellular proliferation and invasion, improves cellular apoptosis in prostate cancer. Tumour biology : the journal of the International Society for Oncodevelopmental Biology and Medicine. 2015 doi: 10.1007/s13277-015-3853-9. [DOI] [PubMed] [Google Scholar]
- 26.Liu Z, Wu K, Yang Z, Wu A. High-mobility group A2 overexpression is an unfavorable prognostic biomarker for nasopharyngeal carcinoma patients. Molecular and cellular biochemistry. 2015 doi: 10.1007/s11010-015-2521-0. [DOI] [PubMed] [Google Scholar]
- 27.Chang KP, Lin SJ, Liu SC, Yi JS, Chien KY, Chi LM, Kao HK, Liang Y, Lin YT, Chang YS, Yu JS. Low-molecular-mass secretome profiling identifies HMGA2 and MIF as prognostic biomarkers for oral cavity squamous cell carcinoma. Scientific reports. 2015;5:11689. doi: 10.1038/srep11689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xia YY, Yin L, Jiang N, Guo WJ, Tian H, Jiang XS, Wu J, Chen M, Wu JZ, He X. Downregulating HMGA2 attenuates epithelial-mesenchymal transition-induced invasion and migration in nasopharyngeal cancer cells. Biochem Biophys Res Commun. 2015;463:357–363. doi: 10.1016/j.bbrc.2015.05.068. [DOI] [PubMed] [Google Scholar]
- 29.Panagopoulos I, Bjerkehagen B, Gorunova L, Taksdal I, Heim S. Rearrangement of chromosome bands 12q14~15 causing HMGA2-SOX5 gene fusion and HMGA2 expression in extraskeletal osteochondroma. Oncol Rep. 2015;34:577–584. doi: 10.3892/or.2015.4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xia YY, Yin L, Tian H, Guo WJ, Jiang N, Jiang XS, Wu J, Chen M, Wu JZ, He X. HMGA2 is associated with epithelial-mesenchymal transition and can predict poor prognosis in nasopharyngeal carcinoma. OncoTargets and therapy. 2015;8:169–176. doi: 10.2147/OTT.S74397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Motoyama K, Inoue H, Nakamura Y, Uetake H, Sugihara K, Mori M. Clinical significance of high mobility group A2 in human gastric cancer and its relationship to let-7 microRNA family. Clinical cancer research : an official journal of the American Association for Cancer Research. 2008;14:2334–2340. doi: 10.1158/1078-0432.CCR-07-4667. [DOI] [PubMed] [Google Scholar]
- 32.Lee CT, Wu TT, Lohse CM, Zhang L. High-mobility group AT-hook 2: an independent marker of poor prognosis in intrahepatic cholangiocarcinoma. Human pathology. 2014;45:2334–2340. doi: 10.1016/j.humpath.2014.04.026. [DOI] [PubMed] [Google Scholar]
- 33.Fedele M, Battista S, Kenyon L, Baldassarre G, Fidanza V, Klein-Szanto AJ, Parlow AF, Visone R, Pierantoni GM, Outwater E, Santoro M, Croce CM, Fusco A. Overexpression of the HMGA2 gene in transgenic mice leads to the onset of pituitary adenomas. Oncogene. 2002;21:3190–3198. doi: 10.1038/sj.onc.1205428. [DOI] [PubMed] [Google Scholar]
- 34.Thuault S, Valcourt U, Petersen M, Manfioletti G, Heldin CH, Moustakas A. Transforming growth factor-beta employs HMGA2 to elicit epithelial-mesenchymal transition. The Journal of cell biology. 2006;174:175–183. doi: 10.1083/jcb.200512110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen Z, Li Q, Wang S, Zhang J. miR4855p inhibits bladder cancer metastasis by targeting HMGA2. International journal of molecular medicine. 2015 doi: 10.3892/ijmm.2015.2302. [DOI] [PubMed] [Google Scholar]
- 36.Watanabe S, Ueda Y, Akaboshi S, Hino Y, Sekita Y, Nakao M. HMGA2 maintains oncogenic RAS-induced epithelial-mesenchymal transition in human pancreatic cancer cells. The American journal of pathology. 2009;174:854–868. doi: 10.2353/ajpath.2009.080523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lam K, Muselman A, Du R, Harada Y, Scholl AG, Yan M, Matsuura S, Weng S, Harada H, Zhang DE. Hmga2 is a direct target gene of RUNX1 and regulates expansion of myeloid progenitors in mice. Blood. 2014;124:2203–2212. doi: 10.1182/blood-2014-02-554543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mayr C, Hemann MT, Bartel DP. Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science. 2007;315:1576–1579. doi: 10.1126/science.1137999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rice SJ, Lai SC, Wood LW, Helsley KR, Runkle EA, Winslow MM, Mu D. MicroRNA-33a mediates the regulation of high mobility group AT-hook 2 gene (HMGA2) by thyroid transcription factor 1 (TTF-1/NKX2-1) The Journal of biological chemistry. 2013;288:16348–16360. doi: 10.1074/jbc.M113.474643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heo I, Joo C, Cho J, Ha M, Han J, Kim VN. Lin28 mediates the terminal uridylation of let-7 precursor MicroRNA. Molecular cell. 2008;32:276–284. doi: 10.1016/j.molcel.2008.09.014. [DOI] [PubMed] [Google Scholar]
- 41.Newman MA, Thomson JM, Hammond SM. Lin-28 interaction with the Let-7 precursor loop mediates regulated microRNA processing. Rna. 2008;14:1539–1549. doi: 10.1261/rna.1155108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rybak A, Fuchs H, Smirnova L, Brandt C, Pohl EE, Nitsch R, Wulczyn FG. A feedback loop comprising lin-28 and let-7 controls pre-let-7 maturation during neural stem-cell commitment. Nature cell biology. 2008;10:987–993. doi: 10.1038/ncb1759. [DOI] [PubMed] [Google Scholar]
- 43.Viswanathan SR, Daley GQ, Gregory RI. Selective blockade of microRNA processing by Lin28. Science. 2008;320:97–100. doi: 10.1126/science.1154040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee YS, Dutta A. The tumor suppressor microRNA let-7 represses the HMGA2 oncogene. Genes & development. 2007;21:1025–1030. doi: 10.1101/gad.1540407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bar EE, Lin A, Mahairaki V, Matsui W, Eberhart CG. Hypoxia increases the expression of stem-cell markers and promotes clonogenicity in glioblastoma neurospheres. The American journal of pathology. 2010;177:1491–1502. doi: 10.2353/ajpath.2010.091021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chu Q, Orr BA, Semenkow S, Bar EE, Eberhart CG. Prolonged inhibition of glioblastoma xenograft initiation and clonogenic growth following in vivo Notch blockade. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19:3224–3233. doi: 10.1158/1078-0432.CCR-12-2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kumar MS, Erkeland SJ, Pester RE, Chen CY, Ebert MS, Sharp PA, Jacks T. Suppression of non-small cell lung tumor development by the let-7 microRNA family. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:3903–3908. doi: 10.1073/pnas.0712321105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kaur H, Phillips-Mason PJ, Burden-Gulley SM, Kerstetter-Fogle AE, Basilion JP, Sloan AE, Brady-Kalnay SM. Cadherin-11, a marker of the mesenchymal phenotype, regulates glioblastoma cell migration and survival in vivo. Molecular cancer research : MCR. 2012;10:293–304. doi: 10.1158/1541-7786.MCR-11-0457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nature methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lal S, Lacroix M, Tofilon P, Fuller GN, Sawaya R, Lang FF. An implantable guide-screw system for brain tumor studies in small animals. Journal of neurosurgery. 2000;92:326–333. doi: 10.3171/jns.2000.92.2.0326. [DOI] [PubMed] [Google Scholar]
- 51.Weingart MF, Roth JJ, Hutt-Cabezas M, Busse TM, Kaur H, Price A, Maynard R, Rubens J, Taylor I, Mao XG, Xu J, Kuwahara Y, Allen SJ, Erdreich-Epstein A, Weissman BE, Orr BA, Eberhart CG, Biegel JA, Raabe EH. Disrupting LIN28 in atypical teratoid rhabdoid tumors reveals the importance of the mitogen activated protein kinase pathway as a therapeutic target. Oncotarget. 2015;6:3165–3177. doi: 10.18632/oncotarget.3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bar EE, Chaudhry A, Lin A, Fan X, Schreck K, Matsui W, Piccirillo S, Vescovi AL, DiMeco F, Olivi A, Eberhart CG. Cyclopamine-mediated hedgehog pathway inhibition depletes stem-like cancer cells in glioblastoma. Stem cells. 2007;25:2524–2533. doi: 10.1634/stemcells.2007-0166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, De Vitis S, Fiocco R, Foroni C, Dimeco F, Vescovi A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer research. 2004;64:7011–7021. doi: 10.1158/0008-5472.CAN-04-1364. [DOI] [PubMed] [Google Scholar]
- 54.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 55.Liu B, Pang B, Hou X, Fan H, Liang N, Zheng S, Feng B, Liu W, Guo H, Xu S, Pang Q. Expression of high-mobility group AT-hook protein 2 and its prognostic significance in malignant gliomas. Human pathology. 2014;45:1752–1758. doi: 10.1016/j.humpath.2014.02.028. [DOI] [PubMed] [Google Scholar]
- 56.Li Y, Zhang X, Chen D, Ma C. Let-7a suppresses glioma cell proliferation and invasion through TGF-beta/Smad3 signaling pathway by targeting HMGA2. Tumour biology : the journal of the International Society for Oncodevelopmental Biology and Medicine. 2015 doi: 10.1007/s13277-015-4674-6. [DOI] [PubMed] [Google Scholar]
- 57.Akai T, Ueda Y, Sasagawa Y, Hamada T, Date T, Katsuda S, Iizuka H, Okada Y, Chada K. High mobility group I-C protein in astrocytoma and glioblastoma. Pathology, research and practice. 2004;200:619–624. doi: 10.1016/j.prp.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 58.Lee J, Kotliarova S, Kotliarov Y, Li A, Su Q, Donin NM, Pastorino S, Purow BW, Christopher N, Zhang W, Park JK, Fine HA. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer cell. 2006;9:391–403. doi: 10.1016/j.ccr.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 59.De Witt Hamer PC, Van Tilborg AA, Eijk PP, Sminia P, Troost D, Van Noorden CJ, Ylstra B, Leenstra S. The genomic profile of human malignant glioma is altered early in primary cell culture and preserved in spheroids. Oncogene. 2008;27:2091–2096. doi: 10.1038/sj.onc.1210850. [DOI] [PubMed] [Google Scholar]
- 60.Rape A, Ananthanarayanan B, Kumar S. Engineering strategies to mimic the glioblastoma microenvironment. Advanced drug delivery reviews. 2014;79-80:172–183. doi: 10.1016/j.addr.2014.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Natarajan S, Hombach-Klonisch S, Droge P, Klonisch T. HMGA2 inhibits apoptosis through interaction with ATR-CHK1 signaling complex in human cancer cells. Neoplasia. 2013;15:263–280. doi: 10.1593/neo.121988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Singh I, Mehta A, Contreras A, Boettger T, Carraro G, Wheeler M, Cabrera-Fuentes HA, Bellusci S, Seeger W, Braun T, Barreto G. Hmga2 is required for canonical WNT signaling during lung development. BMC biology. 2014;12:21. doi: 10.1186/1741-7007-12-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hovestadt V, Jones DT, Picelli S, Wang W, Kool M, Northcott PA, Sultan M, Stachurski K, Ryzhova M, Warnatz HJ, Ralser M, Brun S, Bunt J, Jager N, Kleinheinz K, Erkek S, Weber UD, Bartholomae CC, von Kalle C, Lawerenz C, Eils J, Koster J, Versteeg R, Milde T, Witt O, Schmidt S, Wolf S, Pietsch T, Rutkowski S, Scheurlen W, Taylor MD, Brors B, Felsberg J, Reifenberger G, Borkhardt A, Lehrach H, Wechsler-Reya RJ, Eils R, Yaspo ML, Landgraf P, Korshunov A, Zapatka M, Radlwimmer B, Pfister SM, Lichter P. Decoding the regulatory landscape of medulloblastoma using DNA methylation sequencing. Nature. 2014;510:537–541. doi: 10.1038/nature13268. [DOI] [PubMed] [Google Scholar]
- 64.Picard D, Miller S, Hawkins CE, Bouffet E, Rogers HA, Chan TS, Kim SK, Ra YS, Fangusaro J, Korshunov A, Toledano H, Nakamura H, Hayden JT, Chan J, Lafay-Cousin L, Hu P, Fan X, Muraszko KM, Pomeroy SL, Lau CC, Ng HK, Jones C, Van Meter T, Clifford SC, Eberhart C, Gajjar A, Pfister SM, Grundy RG, Huang A. Markers of survival and metastatic potential in childhood CNS primitive neuro-ectodermal brain tumours: an integrative genomic analysis. The Lancet Oncology. 2012;13:838–848. doi: 10.1016/S1470-2045(12)70257-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Spence T, Perotti C, Sin-Chan P, Picard D, Wu W, Singh A, Anderson C, Blough MD, Cairncross JG, Lafay-Cousin L, Strother D, Hawkins C, Narendran A, Huang A, Chan JA. A novel C19MC amplified cell line links Lin28/let-7 to mTOR signaling in embryonal tumor with multilayered rosettes. Neuro-oncology. 2014;16:62–71. doi: 10.1093/neuonc/not162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liu H, Li P, Li B, Sun P, Zhang J, Wang B, Jia B. RKIP inhibits gastric cancer cell survival and invasion by regulating the expression of HMGA2 and OPN. Tumour biology : the journal of the International Society for Oncodevelopmental Biology and Medicine. 2014;35:11949–11958. doi: 10.1007/s13277-014-2486-8. [DOI] [PubMed] [Google Scholar]
- 67.Sun M, Gomes S, Chen P, Frankenberger CA, Sankarasharma D, Chung CH, Chada KK, Rosner MR. RKIP and HMGA2 regulate breast tumor survival and metastasis through lysyl oxidase and syndecan-2. Oncogene. 2014;33:3528–3537. doi: 10.1038/onc.2013.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lei X, Chang L, Ye W, Jiang C, Zhang Z. Raf kinase inhibitor protein (RKIP) inhibits the cell migration and invasion in human glioma cell lines in vitro. International journal of clinical and experimental pathology. 2015;8:14214–14220. [PMC free article] [PubMed] [Google Scholar]
- 69.Jiang W, Gu W, Qiu R, Shen C, YaohaoWu EY, Zhang J, Zhou J, Guo Y, Li Z, Deng J, Zeng L, Tang J, Zhi Q, Deng X. miRNA-101 suppresses epithelial-to-mesenchymal transition by targeting HMGA2 in pancreatic cancer cells. Anti-cancer agents in medicinal chemistry. 2015 doi: 10.2174/1871520615666150507122142. [DOI] [PubMed] [Google Scholar]
- 70.Lin Y, Liu AY, Fan C, Zheng H, Li Y, Zhang C, Wu S, Yu D, Huang Z, Liu F, Luo Q, Yang CJ, Ouyang G. MicroRNA-33b Inhibits Breast Cancer Metastasis by Targeting HMGA2. SALL4 and Twist1, Scientific reports. 2015;5:9995. doi: 10.1038/srep09995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang P, Bai H, Liu G, Wang H, Chen F, Zhang B, Zeng P, Wu C, Peng C, Huang C, Song Y, Song E. MicroRNA-33b, upregulated by EF24, a curcumin analog, suppresses the epithelial-to-mesenchymal transition (EMT) and migratory potential of melanoma cells by targeting HMGA2. Toxicology letters. 2015;234:151–161. doi: 10.1016/j.toxlet.2015.02.018. [DOI] [PubMed] [Google Scholar]
- 72.Kim TH, Song JY, Park H, Jeong JY, Kwon AY, Heo JH, Kang H, Kim G, An HJ. miR-145, targeting high-mobility group A2, is a powerful predictor of patient outcome in ovarian carcinoma. Cancer letters. 2015;356:937–945. doi: 10.1016/j.canlet.2014.11.011. [DOI] [PubMed] [Google Scholar]
- 73.Zhou H, Guo W, Zhao Y, Wang Y, Zha R, Ding, Liang L, Hu J, Shen H, Chen Z, Yin B, Ma B. MicroRNA-26a acts as a tumor suppressor inhibiting gallbladder cancer cell proliferation by directly targeting HMGA2. International journal of oncology. 2014;44:2050–2058. doi: 10.3892/ijo.2014.2360. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.