

Abstract
We describe a method for tracking RNA molecules in Escherichia coli that is sensitive to single copies of mRNA, and, using the method, we find that individual molecules can be followed for many hours in living cells. We observe distinct characteristic dynamics of RNA molecules, all consistent with the known life history of RNA in prokaryotes: localized motion consistent with the Brownian motion of an RNA polymer tethered to its template DNA, free diffusion, and a few examples of polymer chain dynamics that appear to be a combination of chain fluctuation and chain elongation attributable to RNA transcription. We also quantify some of the dynamics, such as width of the displacement distribution, diffusion coefficient, chain elongation rate, and distribution of molecule numbers, and compare them with known biophysical parameters of the E. coli system.
The bacterium Escherichia coli may well be the most thoroughly characterized model biological system. Much of its metabolism serves as the basic paradigm for DNA replication, RNA transcription, protein synthesis, and gene regulation (1, 2).
Recently, techniques have become available that allow us to study central problems in genome organization and expression in individual living cells (3, 4), rather than rely on the averaged properties of large populations. These studies have added to our knowledge in two main areas: the heterogeneity among cells in a supposedly homogeneous population (5–7) and the spatiotemporal organization of macromolecules in the bacterial cell (8). The subcellular location of a variety of different proteins involved in cell division, such as the MinCDE family (9), are now known to be localized in special regions, as is the location of replicating chromosomes (10) and several different plasmids (11, 12). Complexes of signaling proteins in Bacillus subtilis and Caulobacter crescentus have likewise been localized to the poles of dividing and differentiating cells (8, 13).
To our knowledge, there are no published studies on the localization and timing of mRNA synthesis in living bacterial cells. Our knowledge of RNA transcription and dynamics comes from population studies, or in vitro studies with purified components (14–17). A rare glimpse into cell-to-cell variability in message number was provided by the studies of Tolker-Nielsen et al. (18) with Salmonella using whole fixed mounts. However, there is no information at the single-cell level in living cells on message localization: whether mRNA is free to move throughout the cytoplasm, whether movement is active or passive, and, for RNA molecules present in low copy numbers, how the actual number of molecules is a function of cell state. This last information is of particular interest because it bears directly on the question of the regulation of proteins known to be present in low copy numbers (for example, many of the proteins that act to switch genes on and off) and how the cell successfully regulates copy number in the presence of considerable extrinsic and intrinsic noise (see ref. 7 for a review).
Here we describe a method for tracking RNA molecules in E. coli. We find that the method is sensitive to single copies of mRNA, and that individual molecules can be followed for many hours in living cells. This experimental system has unveiled new features about transcription in E. coli. We find that RNA motion is usually limited in space, perhaps because RNA remains tethered to the DNA from which it is transcribed. In other instances, transcripts appear to diffuse freely in the cytoplasm; occasionally, we can observe the polymer dynamics of single molecules and follow chain elongation during transcription. We quantify some of the parameters of RNA molecular dynamics, such as displacement of the tethered molecule, diffusion coefficient, and elongation rate, and find most of them in agreement with known biophysical parameters. We have also found that RNA movement is not inhibited by metabolic poisons, which leads us to suggest that the movement of RNA molecules in the cell is passive.
Methods
For a more detailed description of our methods, see Supporting Methods and Figs. 7 and 8, which are published as supporting information on the PNAS web site.
The Experimental System. Our approach is based on a method developed by Singer and coworkers (19, 20), which has been used to track RNA in various eukaryotic systems (21). The mRNA detection system is comprised of two elements, a fluorescence protein fused to the RNA bacteriophage MS2 coat protein (henceforth referred to as MS2) and a reporter RNA containing tandemly repeated MS2-binding sites. Our extensive modification of the Singer system has allowed us to image single RNA molecules in E. coli and to follow them as they move through the ≈2-μm cell.
A schematic description of the constructs used is shown in Fig. 1. MS2 interacts with a stem–loop structure in viral RNA to repress translation of the replicase and to encapsulate the viral genome. When an MS2–GFP fusion is coexpressed with a reporter RNA containing tandemly repeated MS2-binding sites, the fusion protein binds to the RNA, forming bright fluorescent particles. This method was first used in vivo to follow mRNA localization in yeast (19) and Drosophila (21) and was used to detect single mRNA molecules in mammalian cells (20). We found, not surprisingly, that several modifications of this system were required for use in E. coli. These are outlined below.
Fig. 1.

A schematic description of the constructs used in this study. The MS2 covalent dimer was fused to GFP variant mut3 (GFPmut3). The fusion protein was under the control of the P(LtetO-1) promoter in vector K133 with a ColE1 origin. A tandem array of 96 MS2-binding sites (96x MS2-bs), interspersed by random sequences, was placed under P(Lac) control in vector BAC2 with an F origin. The two plasmids were cotransformed into the E. coli strain DH5α-PRO, a constitutive producer of LacR and TetR repressors.
The MS2–GFP Fusion Protein. Previous studies used an aggregation-deficient mutant (MS2 dlFG) of MS2 (22). After initial unsatisfactory results with this mutant, we fused a tandem dimer of the wild-type coat protein with an enhanced version of GFP. The fused dimer, MS2d, is known to be highly tolerant of structural perturbations and retains its in vivo functionality after insertion at various locations (23, 24). We fused MS2d to the N terminus of GFPmut3 (25) and placed the fusion under the control of the tetracycline promoter P(LtetO-1) (26) in the vector K133 (based on the PROTET.E vector; Clontech). This placed the MS2d–GFP fusion on a medium copy number plasmid with a ColE1 origin, under tight regulation by tetracycline. After induction with anhydrotetracycline (aTc), cells became bright green, and protein of the correct molecular size was made, as judged by Western blotting using anti-GFP antibodies (data not shown).
The RNA-binding capacity of the protein was examined in vitro by an RNA gel-shift assay of purified protein using RNA probes consisting of six tandem repeats of the MS2-binding site (see below). The MS2d–GFP bound at the predicted molarity (data not shown).
To test the activity of the protein in vivo, we used the blue/white β-galactosidase assay devised by Peabody (27). In this test, a functional coat protein represses the translation of an MS2 replicase–β-galactosidase fusion, thus yielding white (as opposed to blue) colonies. MS2d was active in vivo when fused to GFP. GFP fused to the mutant MS2 dlFG was inactive (data not shown).
The Target RNA. To detect single RNA molecules, a significant number of GFP molecules must be localized in space; hence, a tandem array of binding sites must be constructed. In the lacO/lacI–GFP system devised by Belmont and coworkers (28, 29), 256 copies of the lac operator sites were used. This number proved to be sufficient for tracking the location of chromosome and plasmid DNA sites in E. coli (11, 12). Singer and coworkers (20) describe the detection of single mRNA molecules in mammalian cells using 24 tandem MS2-binding sites. This allows the binding of 48 monomeric MS2–GFP proteins. In this case, however, probably only a small percentage of the target RNA molecules are detected.
Long tandem DNA repeats are known to be unstable (30, 31). This problem is worse in the MS2 system because each binding site contains a palindromic repeat. To increase the stability of the construct, we inserted random sequences between successive MS2-binding sites in the target RNA, thereby shortening the runs of perfect homology that serve as targets for recombinational instability (10). The construction of the 96 binding site (96 BS) tandem array is described in Supporting Methods. A 96-mer was then cloned in a bacterial artificial chromosome based on the F factor replication system (pTRUEBLUE-BAC2; Genomics One International, Buffalo, NY) (32, 33). This very-low-copy vector was chosen because it was expected to increase the stability of the inserts, it would enable us to induce a low number of RNA molecules, and its location is known in the cell (11). The 96-mer insert is placed downstream of a lac promoter and a ribosome-binding site, and upstream of a Rho-independent trpA terminator (34).
Bacterial Strains and Plasmids. All cloning and expression were performed in E. coli strain DH5α-PRO (identical to DH5α-Z1; Clontech) (26).
Construction of MS2d–GFP. GFPmut3 (25) was amplified from pKEN2 (provided by P. Wolanin, Princeton University, Princeton, NJ) and inserted into the NotI site of K133 (derived from pPROTET.E; Clontech). MS2d (24) was amplified from p2CTdl13 (provided by D. Peabody, University of New Mexico, Albuquerque) and inserted into the BamHI site of K133.
Construction of the 96 BS Arrays. An ensemble of oligonucleotides containing three MS2-binding sites, separated by random sequences, was synthesized. This oligonucleotide mixture was converted into double-stranded DNA and amplified by PCR. The product was then cut and inserted into pBS-SK (Stratagene), and sequential doublings of the array were carried out as shown in the Supporting Methods. A 96 BS array was then inserted between the NheI and HindIII sites of pTRUEBLUE-BAC2 (Genomics One International) to yield BAC2+bs96.
Bacterial Growth and Induction. Cells were grown in either LB or M63 minimal medium (35), supplemented by antibiotics according to the specific plasmids. For induction of protein and RNA, cells were grown overnight from a single colony, diluted into fresh medium, and grown at 37°C to mid-logarithmic phase (OD600, ≈0.3–0.5). The inducers isopropyl β-d-thiogalactopyranoside (IPTG; 1 mM) and aTc (10 ng/ml) were then added.
Microscopy and Imaging. A few microliters of culture was placed between a coverslip and a thin slab of 0.8% agarose containing LB. Microscopy was performed in a room maintained at 22°C with a Nikon Eclipse (TE2000-U) inverted microscope equipped with a 100× (1.3 numerical aperture) objective and epifluorescence system. Images were taken with a Cascade:512B (Roper Scientific, Trenton, NJ) camera after an additional 6× magnification. Images and time-lapse Movies 1–6, which are published as supporting information on the PNAS web site, were acquired with metaview software (Universal Imaging, Downington, PA) and analyzed with matlab (Mathworks, Natick, MA).
Results and Discussion
MS2–RNA Complexes in Vitro. To examine the interaction between MS2d–GFP and RNA and to evaluate the possibility of detecting single RNA molecules, purified fusion protein and purified target RNA were mixed and examined under the microscope. The 96 BS RNA product and purified MS2d–GFP were mixed under conditions promoting protein–RNA binding (see the supporting information), with an ≈1,000 protein excess over RNA, and observed by epifluorescence. In addition, we imaged MS2d–GFP by itself, as well as MS2d–GFP mixed with both double- and single-stranded DNA coding for the 96 BS target sequence.
The combination of protein and RNA yields bright particles (total intensity 104–105 photons per sec; see the supporting information). These particles are long-lived (a lifespan of minutes or more). The MS2d–GFP protein by itself appears as a collection of weak point sources (≈103 photons per sec) that exhibit the typical “blinking” behavior of single GFP molecules over a timescale of seconds (36, 37). The mixture of MS2d–GFP and DNA (in either single- or double-stranded form) did not form complexes brighter than MS2d–GFP by itself; this result is consistent with our knowledge that MS2 does not interact with DNA but is a necessary control for the work reported below.
The intensity of these RNA–protein particles is ≈10-fold higher than that of single MS2d–GFP molecules. This is in contrast to the in vivo particles (see below), which apparently consist of almost fully occupied binding sites. A possible explanation for this result is that the 96-mer, because of its many direct and palindromic repeats, has many possible misfolded conformations, which may leave only a fraction of the binding sites available for MS2 binding in vitro. In contrast, as transcripts are synthesized in vivo, it is not unlikely that each binding site becomes occupied as soon as it is synthesized by the RNA polymerase and before inter- and intramolecular interactions can occur.
RNA Detection in Vivo. Cells containing both MS2d–GFP and 96 BS plasmids were grown to mid-logarithmic phase and induced with aTc and IPTG. After 45 min, many cells contained one or more fluorescent “particles” (Fig. 2). Some cells also contain “clusters” of a few particles (see also Fig. 5). Most are not fixed in space but move constantly (see Movies 1–6 and below). The observed particles are likely to be single RNA molecules for the following reasons.
Fig. 2.

GFP-tagged RNA molecules in E. coli cells. (A) Cells carrying BAC2 plus the 96 BS coding sequence and K133 plus MS2d–GFP were grown in LB at 37°C to an OD600 of ≈0.3–0.5. aTc (10 ng/ml) and IPTG (1 mM) were added. After 45–60 min, a few microliters of culture was placed between a coverslip and a thin slab of 0.8% agarose with LB. Cells were observed under epifluorescence, and images were taken at a 100-ms exposure time. As shown in Movies 1 and 2, most spots fluctuate around their mean location. The motion is not inhibited by the addition of 0.2% sodium azide (an inhibitor of ATPase) under conditions where cell growth is completely inhibited in 10 min, suggesting that the motion is passive rather than active. (Scale bar = 1 μm.) (Inset) Absence of spots in cells carrying K133 plus MS2d–GFP and BAC2 without the 96 BS coding sequence. Cells were grown and induced as in A. Cells exhibited low uniform fluorescence of varying levels. (B) Estimation of the number of GFP molecules in each fluorescent particle. Eighty spots were quantified: the histogram of N is shown, where N = (f_spot – f_cell)/(f_GFP). f denotes total photon flux per sec. Fluxes measured were from individual fluorescent spots in the cell (f_spot), background fluorescence in the same cell (f_cell), and individual MS2d–GFP molecules (30 measurements; see supporting information) (f_gfp). Exposure times were 250 msec for cell images and 2.5 sec for single molecules, under identical microscopy conditions. From the histogram we can estimate that each spot corresponds to 20–100 MS2d–GFP molecules (≈70 on average), consistent with our hypothesis that they correspond to single RNA molecules tagged by MS2d–GFP.
Fig. 5.

Dynamics of a single RNA molecule. (A) A series of consecutive epifluorescent images taken 1 sec apart. (IV) At the bottom left of the cell are multiple, barely distinguishable spots (1) that do not appear to move, as well as a single spot (2) exhibiting typical localized motion. At the top right is another cluster of spots (3), as well as a chain made from approximately three blobs (4). Based on measured photon fluxes, we estimate that the chain (4) and the single spot at the bottom left (2) consist of a single RNA molecule. The other clusters probably consist of two (3) and three to five (1) molecules each. The polymer chain can be seen to “wiggle” and change its conformation (see Movie 6). We believe that the apparent blob structure of the chain results from parts of the chain being outside the focal plane. (Scale bar = 1 μm.) (B) Estimated contour length of the polymer versus time. The contour was measured manually at different time points. Cells were grown and induced as in Fig. 2. A single cell was tracked by time-lapse photography for 10 min (one frame per sec, with an exposure time of 250 msec; see Movie 6).
First, cells containing the MS2d–GFP plasmid and an empty BAC2 plasmid (the parent of the plasmid carrying the 96 BS array) produce only uniform fluorescence. That fluorescence depends on aTc induction.
Second, most fluorescent spots are located either near the center or close to the quarter points of the cells, where F plasmids are known to reside (11, 38). As we discuss below, not only the spot location but also its motion are consistent with the known life history of RNA molecules: they are first tethered to the DNA from which they are transcribed and then released from the DNA template when an RNA polymerase termination site is reached.
Third, under conditions of promoter repression, when a small number of transcripts is expected, the numbers of particles in individual cells follow a Poisson distribution, in agreement with the assumption that these are discrete RNA molecules, made as independent, rare events.
Finally, quantification of the fluorescence intensity of each particle by photon counting allows us to estimate that the particles consist of ≈70 GFP molecules on average (see Fig. 2B). This occupancy ratio (bound/free sites) and the fluorescence histogram are very similar to those found by Singer and coworkers (20). We also note that this number may underestimate the number of bound MS2d–GFP molecules per particle because there may be partial quenching of adjacent GFP molecules by resonance energy transfer. Nonetheless, this rough agreement between the expected number of bound molecules and the number measured by photon counting suggests that most of the observed particles correspond to full-length messages, with occupied binding sites.
Single-Molecule RNA Dynamics in Living Cells. We have examined the motion of particles in many cells by time-lapse photography and photon counting. We can distinguish three distinct types of single-molecule behavior.
Localized Motion. As mentioned above, most fluorescent spots are located near the center or the quarter points of the cell. Measurements of 100 spot positions gave 0.51 ± 0.05 (15 spots) and 0.19 ± 0.07 (85 spots) cell length. These numbers correspond well with the location of F plasmids in the cell, as determined by Gordon et al. (11). Removing the ribosome-binding site upstream of the cloned 96-mer did not alter the patterns of particle location, ruling out the possibility that the observed position reflects RNA–ribosome interactions.
In most cells, the motion of fluorescent particles is limited to a small area of the cell. Fig. 3 (and see Movie 3) illustrates a typical example. Each spot appears to move randomly but never leaves its restricted area. The distribution of displacements on the long axis of the cell is bell-shaped, with a half-width of ≈50–200 nm (measurements in five other cells yielded similar results). The most natural explanation for these observations is that we are looking at an RNA molecule tethered to DNA during transcription and possibly afterward (39).
Fig. 3.

Localized motion of multiple RNA particles in a cell. (A) The measured locations of three individual spots, superimposed on an epifluorescence image of the cell. (Scale bar = 1 μm.) (B) Histograms of spot positions along the cell. Position along the cell was obtained by projecting the spot location (x,y) on the long axis of the cell (xl): xl = x cos θ + y sin θ, where θ is the angle between the long axis of the cell and x axis. Each spot is localized in a small region of the cell, with a bell-shaped distribution of positions. Cells were grown and induced as in Fig. 2. A single cell was tracked for 10 min (one frame per 2 sec, with an exposure time of 250 msec; see Movie 3).
It is interesting to compare the observed behavior with a simple biophysical model of the system, that of a particle tethered to a spot by a spring, with an effective spring constant ksp (40). At thermal equilibrium, it can be shown that the distribution for end-to-end distance is Gaussian, with a variance σ2 = 3 kb T/ksp (where kb is Boltzmann's constant and T is the temperature in K). For a polymer whose length L is much larger than the persistence length lp, the effective spring constant is ksp ≈ 3 kb T/2 Llp. We thus obtain 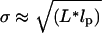 . In our system, the length of the RNA chain is L ≈ 4,000 bases or ≈ 2,500 nm. Single-stranded RNA is very flexible, with a persistence length lp of only a few bases (41, 42). In our case, the RNA molecule is tagged by MS2d–GFP, which probably stiffens it and increases lp to ≈5 nm [the estimated size of the protein plus the RNA stem–loop (43)]. We thus obtain an estimate of
. In our system, the length of the RNA chain is L ≈ 4,000 bases or ≈ 2,500 nm. Single-stranded RNA is very flexible, with a persistence length lp of only a few bases (41, 42). In our case, the RNA molecule is tagged by MS2d–GFP, which probably stiffens it and increases lp to ≈5 nm [the estimated size of the protein plus the RNA stem–loop (43)]. We thus obtain an estimate of 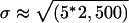 or ≈ 100 nm, within the range of the measured values.
or ≈ 100 nm, within the range of the measured values.
Motion Spanning the Entire Cell. In some cases, motion of the particle spans the entire cell. This is the case shown in Fig. 4 (and Movies 4 and 5). Two cells were tracked, one for a long time (>1 h) at a low frame rate (30 sec per frame) and the other for a short time (≈2 min) with a faster frame rate (1 sec per frame). From the long-term tracking data (Fig. 4 A and B), we find that the RNA molecule traverses the entire cell. However, from the histogram of particle location (Fig. 4B), we learn that the molecule spends more time near the cell poles than at the center. This phenomenon might result from so-called hydrodynamic coupling between the RNA particle and the cell wall, because a particle close to a wall will sense increased drag and, therefore, will have a decreased “effective” diffusion coefficient (44). This will lead to the particle spending more time next to the cell poles. The short-term tracking results (Fig. 4C) reveal a similar trend. In addition, they enable us to estimate the diffusion coefficient of these particles. For this estimate, we use the Einstein–Smoluchowsky equation: 〈δ2〉= 2 dD τ, where δ = |r(t + τ) – r(t)| is the particle displacement between two time points, d is the dimension of the trajectory data, D is the diffusion coefficient, and τ is the time interval between the two measurements. As Fig. 4C shows, the squared displacement is linear in τ up to approximately τ = 5 sec, when the displacement becomes comparable with the cell width. From the slope of the graph, we obtain D ≈ 3*10–2 μm2/sec. Measurements in five other cells all yielded D values in the range of 1*10–3 to 3*10–2 μm 2/sec. Elowitz et al. (45) have measured D for a single GFP molecule and GFP fusion proteins in the E. coli cytoplasm. The values they obtained are in the range of ≈2–8 μm2/sec, i.e., 102–103 times higher than our value for the RNA–protein particles.
Fig. 4.

Two cells exhibiting motion of an RNA particle over the entire cell. Cells were grown and induced as in Fig. 2. (A and B) Cell 1 was tracked for >1h, at one frame per 30 sec (exposure time of 50 msec; see Movie 4). (Scale bars = 1 μm.) (A) A series of epifluorescent images of the cell. Images are 30 sec apart. During the 6 min covered, the RNA particle traveled the length of the cell twice. (B) Histogram of particle position along the cell. Position along the cell was obtained by projecting the spot location (x, y) on the long axis of the cell (see Fig. 3). The spot traversed the entire length of the cell, but the distribution of positions was not uniform. Rather, the particle spent more time away from the center of the cell, closer to the poles (see text). (Inset) Epifluorescence image of the cell, superimposed by measured locations of the RNA spot. (C) Cell 2 was tracked for 2 min, at one frame per sec, with an exposure time of 500 msec (see Movie 5). Mean displacement squared (see text), as a function-of-time interval between measurements. + indicates measurements; solid line indicates linear fit. The linear behavior up to τ ≈ 5 sec suggests that the motion is diffusive. (Inset) Epifluorescence image of the cell, superimposed by measured locations of the RNA spot.
Are these two estimates reconcilable? Our GFP-tagged RNA is much larger than a single GFP molecule (its mass, ≈6,000 kDa, is approximately twice the mass of an E. coli ribosome), but how should the diffusion coefficient scale with the size? Polymers are known to have various modes of motion, yielding very different relations between length and diffusion coefficient (46, 47). Under the simplest hypothesis, we can describe our tagged RNA molecule as a spherical particle whose diffusion coefficient, according to the Stokes–Einstein relation, scales as 1/R, where R is the particle radius. Assuming our molecule to be an ideal flexible polymer, its radius of gyration scales as N1/2, where N is the number of monomers. Thus D ≈ N–1/2. According to this picture, a 100-GFP particle should have a D coefficient only 10 times smaller than a single GFP molecule. However, our tagged RNA might not diffuse as a spherical particle but rather move by reptation, sliding through a tube whose contours are defined by the locus of entanglements with neighboring molecules. Under these conditions D ≈ N–2 (46), or a 10,000-fold factor between a single GFP and our tagged RNA chain. The fold difference between our measured D and the single-GFP value measured by Elowitz et al. (45) lies between these two estimates and suggests that the motion we observe may be closer to a partially extended reptating polymer than to a sphere whose radius of gyration is described by Flory “ideal chain” statistics.
Chain Elongation. In a few cells, we observed a fluorescent “chain” behaving like a typical polymer in solution, stretching and writhing along the axis of the cell (see Fig. 5 and Movie 6). These chains are likely to be single-RNA molecules, both because their contour length matches the transcript length and because the total integrated photon counts from the chain area are very close to those of the fluorescent particles observed in other cells and the particle labeled 2 in Fig. 5A. The contour length of the chain increases with time, as is documented in Fig. 5B. The most straightforward explanation for the observed elongation is that we are watching RNA transcription. The maximal length measured is ≈2.6 μm, very close to the predicted transcript length. The observed elongation rate is not uniform, with a peak rate of ≈40 nm/sec (during ≈55–65 sec in Fig. 5B) as well as a period of apparent transcriptional halting that lasts ≈10 sec (during ≈25–35 sec in Fig. 5B). This behavior is consistent with the dynamics observed during transcription in vitro (16, 48). The average rate of ≈15 nm/sec (or ≈25 nucleotides per sec) is in good agreement with the known rate of transcription in E. coli at 22°C (49, 50). This phenomenon was observed in two additional cells.
Induction. To better control the timing and level of target RNA production, we replaced the original Plac promoter in BAC2 with Plac/ara-1 (26), designed to enable tighter regulation of transcription. The results of a typical induction experiment are shown in Fig. 6. Under conditions of promoter repression, most of the cells are devoid of RNA particles, and the number of particles per cell follows a Poisson distribution with a mean λ = 0.18. Three additional experiments (>1,000 cells counted) all gave a similar result: Poisson distribution of spot number, with λ values <1. The straightforward interpretation of this result is that we are observing transcripts that are rarely made under repression [Lutz and Bujard (26) have estimated that, under these conditions, a transcript is made less than once per cell per generation]. The Poisson distribution observed possibly reflects the rate-limiting step in the process of RNA polymerase binding and transcription (51). We note that the RNA particles are longer lived than E. coli RNA molecules. We suspect that the MS2d–GFP proteins bound to the RNA molecule might “immortalize” it, i.e., prevent or at least considerably slow down its degradation. The protein/RNA off-rate for the binding site sequence in use is on the order of many hours (52–54), which suggests that MS2d–GFP might not be displaced by the various RNA-degrading enzymes in the cell.
Fig. 6.

Induction of RNA transcription. Cells carrying BAC2 plus the 96 BS coding sequence (under control of Plac/ara-1) and K133 plus MS2d–GFP were grown in LB at 37°C to an OD600 ≈ 0.3–0.5. MS2d–GFP production was induced by adding aTc (100 ng/ml). After 1 h, cells were resuspended in the same medium without aTc. RNA transcription was then induced in half of the culture by adding IPTG (1 mM) and L(+)-arabinose (0.1%). Cells were then grown for an additional 1 h and observed as in Fig. 2; 150 cells were examined. (A) Distribution of the number of spots in cells not induced (no IPTG, no arabinose). Each bar represents cells actually counted; + indicates a fit to a Poisson distribution, with λ = 0.18, determined from the number of cells with no spots. Cells with three spots or more were grouped together, because clustered particles are sometimes hard to distinguish. (B) Distribution of the number of spots in induced cells (with IPTG and arabinose). Symbols are as in A. The distribution is bimodal, far from a Poisson distribution (λ= 1.18).
When cells are induced with IPTG and arabinose, most cells have multiple RNA particles, and the particle number distribution shifts to the right, becoming bimodal. (The increase in spot number was verified in three additional experiments.) The bimodal shape is reminiscent of the results obtained by Siegele and Hu (5), who examined gene expression under control of the araBAD promoter, from which the Plac/ara-1 promoter was derived.
Even under induction, there does not seem to be a steady accumulation of RNA particles. The RNA–protein particles might stay attached to the DNA template longer than normal RNA molecules, thus inhibiting further transcription from the same promoter. Neither can we rule out the possibility that, under induction, multiple transcripts are produced, which interact with each other through their bound proteins.
In sum, we report on the behavior of individual RNA molecules in live E. coli cells. We have observed three characteristic dynamics, all consistent with the known life history of RNA in prokaryotes: localized motion consistent with the Brownian motion of an RNA polymer tethered to its template DNA, free diffusion, and a few examples of polymer chain dynamics that appear to be a combination of polymer fluctuation and chain elongation attributable to RNA transcription. We have also quantified the dynamics of these molecules, including the width of the displacement distribution, their diffusion coefficient, and the chain elongation rate. These experimental results have been compared with the known biophysical parameters of our system and have been found to be in reasonable agreement with the literature.
Supplementary Material
Acknowledgments
We thank D. Peabody, H. Diamant, J. Paulsson, R. Segev, Y. Zhang, R. Austin, P. Wolanin, and J. Puchalla for generous advice; D. Peabody (University of New Mexico, Albuquerque), K. Forrest (Princeton University, Princeton), and P. Wolanin (Princeton University, Princeton) for strains and plasmids; L. Guo for technical assistance; and members of the Cox and Gavis laboratories. This work was supported by National Institutes of Health Grant HG 001506. I.G. is a Louis Thomas Fellow.
Abbreviations: aTc, anhydrotetracycline; 96 BS, 96 binding site(s); IPTG, isopropylβ-d-thiogalactopyranoside.
References
- 1.Neidhardt, F. C., Ingraham, J. L. & Schaechter, M. (1990) Physiology of the Bacterial Cell: A Molecular Approach (Sinauer, Sunderland, MA).
- 2.Neidhardt, F. C. (1996) Escherichia coli and Salmonella: Cellular and Molecular Biology (Am. Soc. Microbiol., Washington, DC).
- 3.Errington, J. (2003) Nat. Cell Biol. 5, 175–178. [DOI] [PubMed] [Google Scholar]
- 4.Southward, C. M. & Surette, M. G. (2002) Mol. Microbiol. 45, 1191–1196. [DOI] [PubMed] [Google Scholar]
- 5.Siegele, D. A. & Hu, J. C. (1997) Proc. Natl. Acad. Sci. USA 94, 8168–8172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Elowitz, M. B., Levine, A. J., Siggia, E. D. & Swain, P. S. (2002) Science 297, 1183–1186. [DOI] [PubMed] [Google Scholar]
- 7.Paulsson, J. (2004) Nature 427, 415–418. [DOI] [PubMed] [Google Scholar]
- 8.Shapiro, L. & Losick, R. (2000) Cell 100, 89–98. [DOI] [PubMed] [Google Scholar]
- 9.Shih, Y. L., Le, T. & Rothfield, L. (2003) Proc. Natl. Acad. Sci. USA 100, 7865–7870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lau, I. F., Filipe, S. R., Soballe, B., Okstad, O. A., Barre, F. X. & Sherratt, D. J. (2003) Mol. Microbiol. 49, 731–743. [DOI] [PubMed] [Google Scholar]
- 11.Gordon, G. S., Sitnikov, D., Webb, C. D., Teleman, A., Straight, A., Losick, R., Murray, A. W. & Wright, A. (1997) Cell 90, 1113–1121. [DOI] [PubMed] [Google Scholar]
- 12.Gordon, S., Rech, J., Lane, D. & Wright, A. (2004) Mol. Microbiol. 51, 461–469. [DOI] [PubMed] [Google Scholar]
- 13.Lewis, P. J., Thaker, S. D. & Errington, J. (2000) EMBO J. 19, 710–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Forde, N. R., Izhaky, D., Woodcock, G. R., Wuite, G. J. & Bustamante, C. (2002) Proc. Natl. Acad. Sci. USA 99, 11682–11687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harada, Y., Funatsu, T., Murakami, K., Nonoyama, Y., Ishihama, A. & Yanagida, T. (1999) Biophys. J. 76, 709–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shaevitz, J. W., Abbondanzieri, E. A., Landick, R. & Block, S. M. (2003) Nature 426, 684–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Skinner, G. M., Baumann, C. G., Quinn, D. M., Molloy, J. E. & Hoggett, J. G. (2004) J. Biol. Chem. 279, 3239–3244. [DOI] [PubMed] [Google Scholar]
- 18.Tolker-Nielsen, T., Holmstrom, K., Boe, L. & Molin, S. (1998) Mol. Microbiol. 27, 1099–1105. [DOI] [PubMed] [Google Scholar]
- 19.Bertrand, E., Chartrand, P., Schaefer, M., Shenoy, S. M., Singer, R. H. & Long, R. M. (1998) Mol. Cell 2, 437–445. [DOI] [PubMed] [Google Scholar]
- 20.Fusco, D., Accornero, N., Lavoie, B., Shenoy, S. M., Blanchard, J. M., Singer, R. H. & Bertrand, E. (2003) Curr. Biol. 13, 161–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Forrest, K. M. & Gavis, E. R. (2003) Curr. Biol. 13, 1159–1168. [DOI] [PubMed] [Google Scholar]
- 22.Peabody, D. S. & Ely, K. R. (1992) Nucleic Acids Res. 20, 1649–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peabody, D. S. & Lim, F. (1996) Nucleic Acids Res. 24, 2352–2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peabody, D. S. (1997) Arch. Biochem. Biophys. 347, 85–92. [DOI] [PubMed] [Google Scholar]
- 25.Cormack, B. P., Valdivia, R. H. & Falkow, S. (1996) Gene 173, 33–38. [DOI] [PubMed] [Google Scholar]
- 26.Lutz, R. & Bujard, H. (1997) Nucleic Acids Res. 25, 1203–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peabody, D. S. (1990) J. Biol. Chem. 265, 5684–5689. [PubMed] [Google Scholar]
- 28.Belmont, A. S. & Straight, A. F. (1998) Trends Cell Biol. 8, 121–124. [DOI] [PubMed] [Google Scholar]
- 29.Robinett, C. C., Straight, A., Li, G., Willhelm, C., Sudlow, G., Murray, A. & Belmont, A. S. (1996) J. Cell Biol. 135, 1685–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Albertini, A. M., Hofer, M., Calos, M. P. & Miller, J. H. (1982) Cell 29, 319–328. [DOI] [PubMed] [Google Scholar]
- 31.Bzymek, M. & Lovett, S. T. (2001) Proc. Natl. Acad. Sci. USA 98, 8319–8325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Slilaty, S. N. & Lebel, S. (1998) Gene 213, 83–91. [DOI] [PubMed] [Google Scholar]
- 33.Shizuya, H., Birren, B., Kim, U. J., Mancino, V., Slepak, T., Tachiiri, Y. & Simon, M. (1992) Proc. Natl. Acad. Sci. USA 89, 8794–8797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Christie, G. E., Farnham, P. J. & Platt, T. (1981) Proc. Natl. Acad. Sci. USA 78, 4180–4184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miller, J. H. (1992) A Short Course in Bacterial Genetics: A Laboratory Manual and Handbook for Escherichia coli and Related Bacteria (Cold Spring Harbor Lab. Press, Plainview, NY).
- 36.Dickson, R. M., Cubitt, A. B., Tsien, R. Y. & Moerner, W. E. (1997) Nature 388, 355–358. [DOI] [PubMed] [Google Scholar]
- 37.Moerner, W. E., Peterman, E. J., Brasselet, S., Kummer, S. & Dickson, R. M. (1999) Cytometry 36, 232–238. [DOI] [PubMed] [Google Scholar]
- 38.Pogliano, J. (2002) Curr. Opin. Microbiol. 5, 586–590. [DOI] [PubMed] [Google Scholar]
- 39.Hamkalo, B. A. & Miller, O. L., Jr. (1973) Annu. Rev. Biochem. 42, 379–396. [DOI] [PubMed] [Google Scholar]
- 40.Qian, H. & Elson, E. L. (1999) Biophys. J. 76, 1598–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liphardt, J., Onoa, B., Smith, S. B., Tinoco, I. J. & Bustamante, C. (2001) Science 292, 733–737. [DOI] [PubMed] [Google Scholar]
- 42.Rippe, K. (2001) Trends Biochem. Sci. 26, 733–740. [DOI] [PubMed] [Google Scholar]
- 43.Valegard, K., Murray, J. B., Stockley, P. G., Stonehouse, N. J. & Liljas, L. (1994) Nature 371, 623–626. [DOI] [PubMed] [Google Scholar]
- 44.Happel, J. & Brenner, H. (1973) Low Reynolds Number Hydrodynamics: With Special Applications to Particulate Media (Noordhoff International, Leiden, The Netherlands).
- 45.Elowitz, M. B., Surette, M. G., Wolf, P. E., Stock, J. B. & Leibler, S. (1999) J. Bacteriol. 181, 197–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Doi, M. & Edwards, S. F. (1986) The Theory of Polymer Dynamics (Oxford Univ. Press, New York).
- 47.de Gennes, P. G. (1979) Scaling Concepts in Polymer Physics (Cornell Univ. Press, Ithaca, NY).
- 48.Davenport, R. J., Wuite, G. J., Landick, R. & Bustamante, C. (2000) Science 287, 2497–2500. [DOI] [PubMed] [Google Scholar]
- 49.Mathews, C. K., Van Holde, K. E. & Ahern, K. G. (2000) Biochemistry (Benjamin Cummings, San Francisco).
- 50.Ryals, J., Little, R. & Bremer, H. (1982) J. Bacteriol. 151, 879–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lutz, R., Lozinski, T., Ellinger, T. & Bujard, H. (2001) Nucleic Acids Res. 29, 3873–3881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Talbot, S. J., Goodman, S., Bates, S. R., Fishwick, C. W. & Stockley, P. G. (1990) Nucleic Acids Res. 18, 3521–3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Johansson, H. E., Dertinger, D., LeCuyer, K. A., Behlen, L. S., Greef, C. H. & Uhlenbeck, O. C. (1998) Proc. Natl. Acad. Sci. USA 95, 9244–9249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Romaniuk, P. J., Lowary, P., Wu, H. N., Stormo, G. & Uhlenbeck, O. C. (1987) Biochemistry 26, 1563–1568. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.