

Summary
HIV-1 integrase (IN) catalyzes viral DNA integration into the host genome and facilitates multifunctional steps including virus particle maturation. Competency of IN to form multimeric assemblies is functionally critical, presenting an approach for anti-HIV strategies. Multimerization of IN depends on interactions between the distinct subunit domains and amongst the flanking protomers. Here we elucidate an overlooked docking cleft of IN core domain that anchors the N-terminal helix-turn-helix (HTH)-motif in a highly preserved and functionally critical configuration. Crystallographic structure of IN core domain in complex with Fab specifically targeting this cleft reveals a steric overlap that would inhibit HTH-docking, C-terminal domain contacts, DNA binding and subsequent multimerization. While Fab inhibits in vitro IN integration activity, in vivo it abolishes virus particle production by specifically associating with preprocessed IN within Gag-Pol and interfering with early cytosolic Gag/Gag-Pol assemblies. The HTH-docking cleft may offer a fresh hotspot for future anti-HIV intervention strategies.
Graphical abstract
Introduction
The HIV-1 integrase (IN) protein, best recognized for the vital role it plays in integrating viral DNA into the host genome, also facilitates other important roles such as in reverse transcription of the viral RNA genome into DNA, nuclear import of the pre-integration nucleocomplex (PIC), recruiting vital host co-factors into the budding viral progeny and assisting viral particle assembly and maturation (reviewed in (Grandgenett et al., 2015)). IN functions as a multimer with discrete domains from each unit functionally complementing each other: the N-terminal domain (NTD) containing the helix-turn-helix (HTH) zinc-binding motif, the catalytic core domain (CCD) including the canonical catalytic triad D-D-E, and the C-terminal domain (CTD) with a SH3-like fold (Jaskolski et al., 2009; Li et al., 2011). All three domains have been shown to directly interact with the viral and host DNA, to mediate IN multimerization and to form extrinsic interactions which craft IN assemblies of virus and viral-host complexes (Ballandras-Colas et al., 2016; Grandgenett et al., 2015; Hare et al., 2009b; Kessl et al., 2009; Maertens et al., 2010; Taltynov et al., 2012; Yin et al., 2016). Reciprocal NTD and CTD domain swapping and contacts with CCDs of flanking dimers are indispensable for the stabilization of functional IN assemblies, which consists of multiple dimeric CCD units (2 in prototype foamy virus (PFV) (Maertens et al., 2010) and 4 in Rous sarcoma virus (RSV) (Yin et al., 2016) and mouse mammary tumor virus (MMTV) (Ballandras-Colas et al., 2016)). Amino acid compositions of interacting interfaces must therefore coevolve to complement each other by accumulating compensatory substitutions buffering deleterious effects of mutations and preserving functional interactions during multimeric complex assembly (Khwaja et al., 2016). Compensatory IN mutations have been shown to coevolve in response to variations in IN cellular partners such as with the IN-binding-domain (IBD) of the host cofactor lens epithelium–derived growth factor (LEDGF) (Wang et al., 2014). Interrupting the functional multimerization of IN is a rising trend in anti-IN intervention approaches (Desimmie et al., 2013; Feng et al., 2015; Gupta et al., 2014; Hayouka et al., 2007; Jurado et al., 2013). The production of non-infectious viral particles as a result of certain non-catalytic IN mutations or in response to allosteric IN inhibitors (ALLINIs) (Desimmie et al., 2013; Gupta et al., 2014; Jurado et al., 2013) emphasizes the multifunctional role facilitated by IN during HIV-1 replication and underscores the significance of the delicate tuning of IN multimerization.
The NTD/CCD interacting interfaces are especially interesting and functionally critical for IN multimerization and virus replication (Hare et al., 2009a; Hare et al., 2009b; Zheng et al., 1996). Intriguingly, available structures of IN functional intasomes and strand-transfer complexes (Ballandras-Colas et al., 2016; Maertens et al., 2010; Yin et al., 2016) now reveal the preservation of the NTD/CCD docking platform, which had previously been observed in structures of apo IN (Wang et al., 2001) and IN complexes with IBD (Hare et al., 2009a; Hare et al., 2009b). Here we feature the overlooked docking cleft of IN CCD, which anchors the NTD in a highly preserved and functionally critical configuration. We determined the crystallographic structure of HIV-1 IN CCD in complex with an antigen-binding fragment (Fab) specifically docked into this cleft. Fab binding blocks IN interfaces essential for DNA interactions and functional multimerization and, therefore, inhibits in vitro IN integration activity. In vivo targeting of this cleft using a single-chain variable fragment (ScFv), however, abolishes virus particle production. The ScFv specifically associates with the preprocessed IN within Gag-Pol, which potentially interferes with early cytosolic Gag/Gag-Pol assemblies and their trafficking. The highly preserved HTH-docking cleft of IN may provide a potential template for novel anti-HIV intervention strategies.
Results and Discussion
The HTH-docking cleft of retroviral IN CCD
Analysis of available structures of apo IN (PDB code 1K6Y) or in complexes with IBD protein (PDB codes 3F9K and 3HPH) or DNA of active intasomes (PDB codes 5EJK and 3JCA) and strand-transfer complexes (PDB code 3OS0) reveals a preserved binding configuration with the zinc- binding HTH-motif of NTD exclusively docked into a cleft within the CCD. This cleft is formed by the C-terminal end of α4-helix (residues 160-168, HIV-1 numbering) and the finger-loop connecting α5 and α6 (residues 186-195) (Figure 1A). The NTD docking in this configuration is stabilized through electrostatic interactions and hydrophobic stacking (Hare et al., 2009b; Lesbats et al., 2008). Intriguingly, at the canonical dimer interface, the HTH-motif, particularly through lentiviral K14, appears to bridge the two IN protomers extending the hydrophobic core centered around F185 (HIV-1 numbering) (Figure 1B). A single K14A substitution has been shown to destabilize IN tetramers and compromise IN catalytic activities (McKee et al., 2008). NTD bridging of the two protomers, within the canonical dimer, can potentially stabilize IN dimers during multimer assembly and reorganization upon DNA binding. We previously proposed that the hydrophobic core at the C-terminal tip of CCD α-5 (around F185 of HIV-1) is delicately tuned for optimal flexibility hinging the two protomers together during IN rearrangements, and a single Phe to Lys mutation at this hinge rendered IN of feline-lentivirus (FIV) monomeric (Galilee and Alian, 2014).
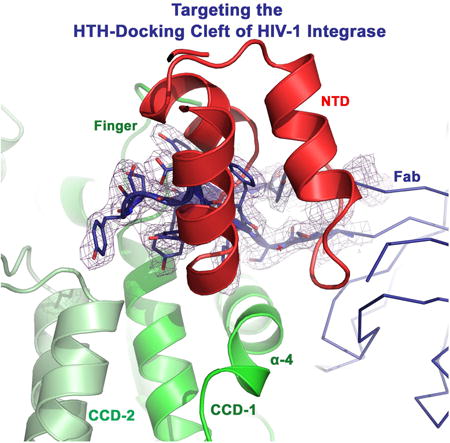
Figure 1. Crystal structure of IN CCD-Fab2 targeting the HTH-docking cleft.
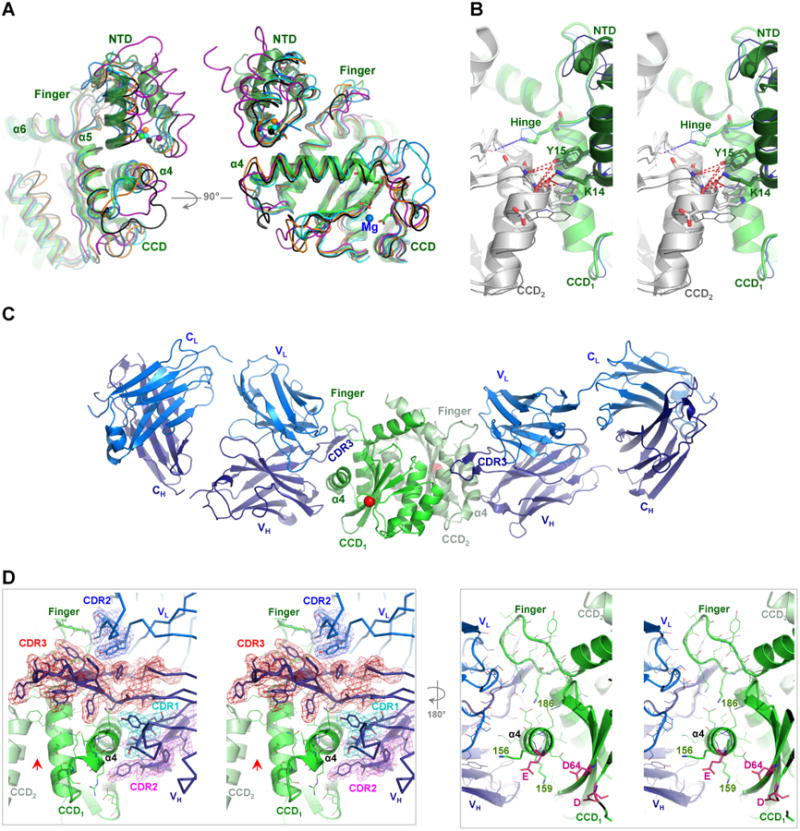
A) NTD bound in the HTH-cleft between α4 and finger-loop. Superposition of IN CCD and NTD structures from HIV-1 (1K6Y, green), HIV-2 (3F9K, blue), maedi visna virus (MVV, 3HPH, cyan), RSV intasome (5EJK, black), PFV strand-transfer complex (3OS0, magenta) and MMTV intasome (3JCA, orange). Spheres indicate Zn2+ of HTH-motif. Catalytic site residues (D64-D116-E152 of HIV-1) are shown with green sticks and Mg2+ (3F9K) is shown with blue sphere. B) Walleye stereo view showing the HTH-motif bridging the two protomers (CCD1 and CCD2) of IN dimer. Hinge (e.g. F185K of HIV-1), polar and hydrophobic interacting residues are shown in sticks. HIV-1 (green cartoon) and HIV-2 (blue ribbon) are shown. NTDs are shown with darker hues. Red dashed-lines indicate interactions within 4 Å distances. C) Structure of crystallographic asymmetric unit containing two IN CCD protomers (green and pale green, Cα of catalytic D64 in red sphere) each bound to one Fab2 molecule. Fab2 light and heavy chains containing constant [C] and variable [V] regions are labeled CL and VL for light chain (blue) and CH and VH for heavy chain (deep blue). D) Walleye stereo view around α4 of one IN protomer (CCD1). Colored meshes represent electron density maps (2FO-FC, 1.0 σ) rendered around heavy chain CDR3 (red), CDR2 (magenta) and CDR1 (cyan), as well as around CDR2 of light chain (blue). Red arrow indicates the interface of the canonical dimer. Catalytic residues D64, D116 and E152 are shown in magenta sticks.
The fundamental role of CCD helix α-4 in viral DNA recognition, binding and catalysis has been intensively investigated and is well established (reviewed in (Azzi et al., 2010; Hare et al., 2010)). Likewise, the flexible finger-loop has also been implicated in viral infectivity and IN functional tetramerization, nuclear localization, enzymatic activity and conformational rearrangement of IN upon DNA binding (Berthoux et al., 2007; Cellier et al., 2013; Hare et al., 2009a; Zhao et al., 2008). For example, K186 of the finger-loop has been shown to stabilize the tetrameric form of IN via a salt-bridge formed with the NTD E11 from the adjacent dimer (Berthoux et al., 2007; Hare et al., 2009a), and I191 has been proposed to support a crucial hydrophobic core at the dimer-dimer interface during tetramer formation. Mutations to either of these finger-loop residues inhibit IN enzymatic activity (Hare et al., 2009a). Likewise, the proper folding of NTD, which depends on Zn2+ binding to the HTH-motif, promotes IN multimerization and enhances the catalytic activity (Pandey et al., 2011; Zheng et al., 1996) underscoring the biologic relevance of the observed NTD/CCD docking configuration in HIV biology (Hare et al., 2009b). Therefore, disturbing the HTH-docking cleft can interfere with IN multifaceted functions during virus replication and may provide a template for novel anti-HIV targeting.
Structure of IN CCD in complex with Fab targeting the HTH-docking cleft
We previously used phage display to identify Fabs against HIV-1 IN (full-length) and, while two selected Fabs (Fab2 and Fab5) behaved similarly, a ∼10 Å Cryo-EM 3D model was reconstructed for Fab5 in complex with IN. Docking of the atomic structures of a Fab and IN CCD into the Cryo-EM density maps showed that two Fab molecules symmetrically bind a dimer of IN near the exposed catalytic site of CCD (Wu et al., 2012). Given the highly antigenic nature and global accessibility of α4 (Azzi et al., 2010), which runs across the entire surface of CCD, we anticipated these Fabs would target α4 and perhaps the flexible loops of CCD. To precisely describe the CCD targeted interfaces, details undetectable at the obtained low EM resolution, a complex of IN CCD with Fab2 was crystallized and diffraction data were collected to 2.6 Å (Table-1). The global structure of CCD–Fab2 complex is similar to the EM structure (Wu et al., 2012) with two Fab2 molecules binding to opposite sides of each CCD monomer within the preserved canonical IN dimer (Figure 1C). Fab2 does not induce apparent conformational changes to the overall structure of IN (RMSD 0.43 Å to apo, PDB code: 1BIS, (Goldgur et al., 1998)). As postulated, the variable regions of Fab2 mainly targeted α4 helix (residues 153-168) and most of the finger-loop (residues 186-191), burying a total of ∼ 960 Å2 of the CCD. These exposed surfaces are mostly required for DNA binding (∼60% of buried residues are polar) and, excitingly, for HTH-motif docking (Figure 1D). The complementarity-defining region-3 (CDR3) of the variable heavy fragment of Fab2, besides interacting with α4 helix, acquires a conformation highly similar to that of the IN finger-loop and this mimicry allows CD3 to dock into the HTH-cleft and to pair with the finger loop (Figure 1C-D and 2A). CDR3 can therefore compete with and prevent the binding of HTH-motif and finger-loop of flanking dimers during IN multimerization.
Table 1. Data collection and refinement statistics.
| HIV-1 IN-Fab2 | |
|---|---|
| Data collection | |
| Space group | P 1 21 1 |
| Mol/ASU | 2 |
| Cell dimensions | |
| a, b, c (Å) | 82.49 87.65 96.34 |
| α, β, γ (°) | 90 95.83 90 |
| Resolution (Å) | 95.84-2.64 (2.729-2.635) * |
| Rsym or Rmerge | 0.064 (0.156) |
| I / σI | 11.54 (5.92) |
| Completeness (%) | 98.70 (92.99) |
| Redundancy | 7.53 (3.1) |
| Refinement | |
| Resolution (Å) | 2.64 |
| No. unique reflections | 38035 (2578) |
| Rwork / Rfree (5% test set) | 0.181 (0.235) / 0.233 (0.332) |
| No. atoms | |
| Protein | 8883 |
| Water | 235 |
| B-factors | |
| Protein | 42.7 |
| Water | 35.2 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.015 |
| Bond angles (°) | 1.62 |
| Ramachandran (%) | |
| Favored | 97 |
| Outliers | 0 |
| PDB code | 5EU7 |
Number in parentheses is for highest resolution shell.
Figure 2. Modeling of Fab2 steric hindrance to IN multimerization and DNA binding.
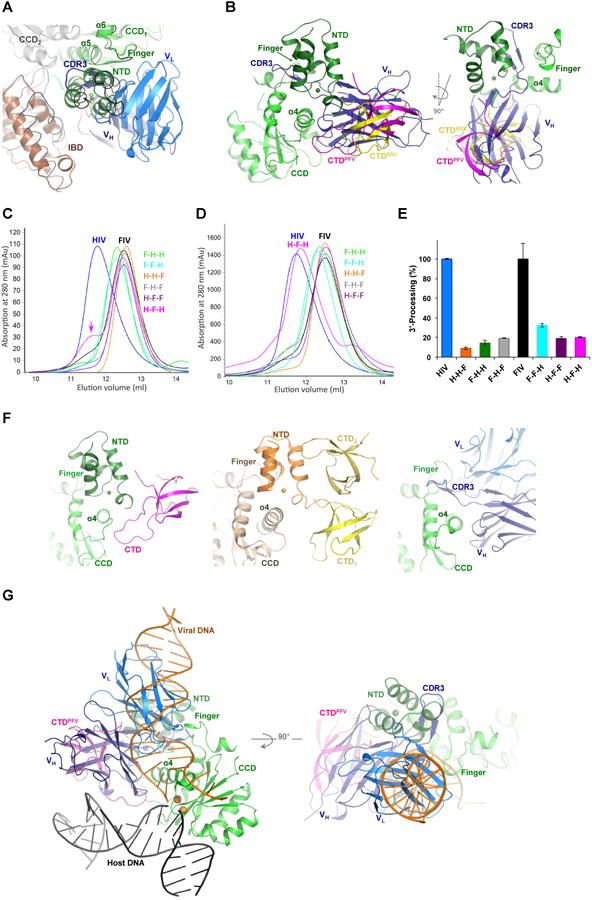
A) Superposition of HIV-1 IN-Fab2 complex to HIV-2 IN-IBD structure (3F9K). HIV-2 IN (gray and green cartoons) and IBD (brown cartoon) are shown together with Fab2 variable heavy (VH, deep blue) and light (VL, blue) chains. MVV IN-IBD complex structure (3HPH) is shown in black ribbon. B) Superposition of IN-Fab2 complex to PFV (3OS0) and RSV (5EJK) intasome structures showing PFV IN CCD (green), NTD (deep green), CTD (magenta) and the RSV CTD (yellow). C) Multimeric state of chimeric IN variants of HIV-1 “H” containing domains swapped from IN of FIV “F”, analyzed at low (0.25 mg/ml) and D) high (4.3 mg/ml) protein concentrations. E.g. H-H-F contains NTD and CCD of HIV-1 and CTD of FIV, while F-H-F contains NTD and CTD of FIV and CCD of HIV-1 and so forth. Color-code of IN variants in preserved through panel “E”. E) 3′-processing activity of IN variants. F) NTD and CTD wrapping of CCD a4 in intasome structures of PFV (3OS0, left) and RSV (5EJK, middle), which is mimicked by Fab2 binding (right panel). G) Superposition of IN-Fab2 complex to PFV strand-transfer complex structure (3OS0) showing PFV IN CCD (green), NTD (deep green) and CTD (magenta) bound to viral DNA (orange), host DNA (black) and two Mn2+ ions (gold spheres modeled from 3L2V).
Fab2 blocks IN interfaces essential for functional multimerization
We previously showed that Fab2 binding to full-length IN results in homogenous populations containing IN in the dimeric form (Wu et al., 2012). Although full-length IN exists in solution in various multimeric forms, including dimeric and predominantly tetrameric (Alian et al., 2009; Hayouka et al., 2007), Fab2 binding appears to have selected for the more stable and homogeneous dimeric form, either by dissociating existing tetramers or inhibiting their formation in solution. Superposition of IN-Fab2 structure with apo structures of tetrameric NTD-CCD two-domain HIV-1 IN (1K6Y, (Wang et al., 2001)) or IBD-bound HIV-2 IN (3F9K, (Hare et al., 2009b)) reveals a clear steric overlap that would prevent NTD docking but not the direct binding of IBD (Figure 2A). However, since the NTD – IBD interaction has previously been shown to critically enhance LEDGF functionality in vitro and during HIV replication (Hare et al., 2009b; Maertens et al., 2003), Fab2 interference with NTD binding may, therefore, indirectly attenuate IBD, and consequently LEDGF, binding to IN.
Likewise, the heavy-chain (VH) of Fab2 entirely overlaps with a flanking CTD, which would prevent CTD interactions with NTD and CCD (Figure 2B). The involvement of CTD in IN multimerization (Andrake and Skalka, 1995; Bojja et al., 2013; Jenkins et al., 1996) and stabilization of viral DNA binding (Ballandras-Colas et al., 2016; Maertens et al., 2010; Yin et al., 2016) is well recognized. Recent computational modeling has indeed predicted that CTD forms a dimer that binds between two CCD at the dimer-dimer interface, which should correctly space the two active CCD domains for the corresponding staggered integration of HIV-1 IN (Roberts, 2015), a prediction that has most recently been supported by the RSV and MMTV intasome structures (Ballandras-Colas et al., 2016; Yin et al., 2016). CTD has also been shown to play an important role in the aberrant IN multimerization and aggregation induced by allosteric IN inhibitors (ALLINIs), which bind to a pocket at the dimeric interface of IN CCD domains (Gupta et al., 2014; Shkriabai et al., 2014). Therefore, CTD interfaces vital for CCD and NTD contacts are functionally important and may offer potential targeting sites for anti-IN intervention.
We analyzed the functional complementarity among the three domains by assessing multimerization and catalytic activities of HIV-1 IN chimeras containing heterologous domains cloned from the FIV IN. We exploited the distinct multimerization features, tetrameric for HIV-1 IN and dimeric for the FIV IN (Galilee and Alian, 2014), to assess the effects of domain swapping on IN multimerization using size exclusion chromatography. Swapping CTD (H-H-F, CTD of FIV), NTD (F-H-H, NTD of FIV) or both domains (F-H-F, NTD and CTD of FIV) rendered HIV-1 IN chimeras dimeric, an intrinsic feature of the FIV IN (Galilee and Alian, 2014). Interestingly, while chimeric FIV IN containing either HIV-1 IN CTD (F-F-H) or NTD (H-F-F) remained dimeric, swapping both CTD and NTD (H-F-H) rendered FIV IN chimera tetrameric (Figure 2C, arrow) especially noticeable at higher protein concentrations (Figure 2D). Swapping any of the HIV-1 IN domains with heterologous FIV ones resulted in the complete inhibition of the strand-transfer activity (undetectable levels) and sever attenuation of the 3′-processing activity of all chimeric variants (Figure 2E). This is in agreement with previous HIV-1/FIV IN domain swapping studies reporting the inhibition of both 3′-processing and integration activities upon heterologous domain exchange (Shibagaki and Chow, 1997). Taken together, these results indicate that both NTD and CTD domains of HIV-1 IN are crucial for protein multimerization and that the compatibility between all three domains is delicately tuned for the assembly of functional multimers and integration activity. Analysis of available intasome structures shows that NTD and CTD pack against each other and delicately wrap around α4, a fine clench elegantly mimicked by the heavy chain of Fab2 (Figure 2F), which would overlap with the binding of these domains (Figure 2B). Detailed mutational analysis will be required to uncover the correlated interactions and the molecular basis underlying inter-domain complementarity highlighting fragile hotspots that may not tolerate mutational or therapeutic disruptions.
Fab2 blocks DNA binding interfaces and inhibits integration activity of IN
Superposition of IN-Fab2 structure with DNA-bound structures (e.g. PFV (3OS0) or RSV (5EJK)) clearly shows that Fab2 would also clash with the viral DNA (Figure 2G). Fab2 binding, however, does not appear to block CCD catalytic residues or binding of DNA in an orientation similar to that of the host DNA in the strand-transfer complex (Figure 2G and 1C-D). The potentially different configurations of IN assemblies required during the distinct catalytic activities of 3′-processing and strand-transfer are well documented (Alian et al., 2009; Barsov et al., 1996; Hayouka et al., 2007; Lesbats et al., 2008). For example, a Fab specific to the CTD of IN inhibited 3′-processing and strand-transfer in vitro without affecting DNA binding (Barsov et al., 1996).
Probing for a potential inhibitory effect on IN catalytic activity revealed that while Fab2 did not interfere with the 3′-processing activity (118 ± 1.9%, n=3, at 1:1 molar ratio), it completely abolished the tetramerization-dependent single-site integration activity in a dose-dependent manner (Figure 3A). Retaining 3′-processing activity validates our structural observation that Fab2 does not cause severe inactivating conformational changes to IN. Fab2 inhibition of strand-transfer but not 3′-processing activity is in agreement with the modeling results above showing that while Fab2 can block the binding of viral DNA when in a strand-transfer orientation (Figure 2G, orange DNA), Fab2 does not appear to block CCD catalytic site residues. Therefore, for an active 3′-processing, the viral DNA may bind in an orientation different from that of strand-transfer, perhaps, an orientation similar to that of the host-DNA in the strand-transfer complex (Figure 2G, black DNA), an assumption that requires further experimental validation.
Figure 3. Fab2 inhibits IN activity and virus particle production.
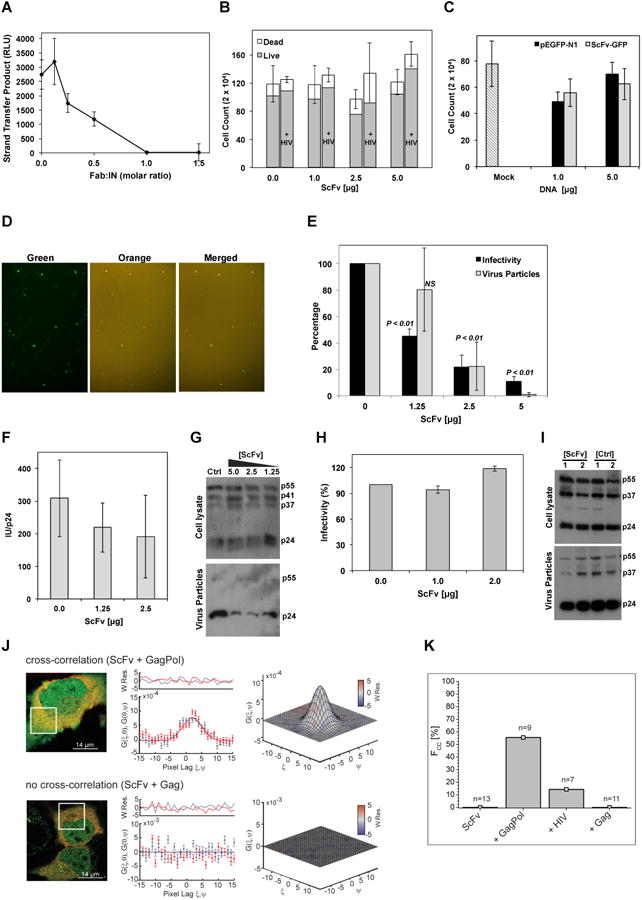
A) In vitro strand-transfer activity of IN-Fab2 complex at various Fab:IN molar ratios. RLU: relative luminescence units. Error bars represent standard errors of mean from three repeats. B) Viability of cells transfected with ScFv-GFP at various concentrations in the absence or presence of HIV-1 plasmid (+ HIV). Error bars represent standard error from mean (total cell count) of three biological repeats. C) HIV-1 infectivity of MAGI-cells expressing various concentrations of pEGFP-N1 or ScFv-GFP plasmids. Mock cells were not transfected with DNA. Error bars represent standard deviations from mean of four repeats. Cell counts indicate blue infected MAGI cells. D) Fluorescent microscopy analysis of 293T cells expressing ScFv-GFP and infected with Kusobira-Orange lentivirus. Superposition of green and orange panels (merged) shows that ScFv-GFP expressing cells (green) are also infected with the lentivirus (orange). E) HIV-1 infectivity assay of viruses produced from cells containing various ScFv-GFP concentrations (black bars) and the analysis of p24 released into the producer-cell medium (gray bars). Error bars represent standard deviation from mean of three biological repeats. P-values (one-way ANOVA) calculated in comparison to control samples. NS: insignificant. F) Relative infectivity (infectious units (IU) divided by the amount of p24 (ng/ml)). G) Western blot analysis (using α-p24 antibodies) of lysates from 293T HIV-1 producer cells (cell lysate) co-transfected with ScFv-GFP and HIV-1 plasmid as well as of released virus (viral particles) as affected by various DNA amounts (μg) of transfected ScFv-GFP or pEGFP-N1 as control (ctrl). Gag polyprotein (p55), p41 and p37 intermediates and mature capsid (p24) are labeled. H) Virus infectivity of HIV-1 produced in cells expressing Δ-IN virus, Vpr-IN, and ScFv-GFP or GFP as control. Error bars represent standard deviation from mean of three biological repeats. I) Western blot analysis of cell lysates and virus particles released to culture media of samples from panel-E. Plasmid DNA amounts used for transfection are indicated (μg). J) ccRICS analysis of ScFv-GFP cytosolic interactions with Gag/Gag-Pol polyproteins. Left panel: representative dual-color fluorescence images of cells expressing ScFv-GFP (green) and Gag-Pol.mCherry (red, top panel) or Gag.mCherry (red, bottom panel). White box indicates the region analyzed using ccRICS. Middle panel: cross-sections of experimental SCCF (data points) and fit (lines) for both fast (ξ) and slow (ψ) scanning axes. Right panel: 3D representations of SCCF. Color-coding represents values of weighted residuals of the fit (W.Res.). K) Percentage of cells (Fcc) showing positive cross-correlation amplitudes (n=number of cells measured).
Single-chain intrabody (ScFv) inhibits HIV-1 particle production by targeting Gag-Pol
To investigate a potential Fab2 inhibition of HIV-1 integration in vivo, a single-chain variable fragment (ScFv) was fused to the N-terminus of GFP (ScFv-GFP). Expression of ScFv-GFP did not disrupt overall cell viability as validated by Trypan blue exclusion assay and overall cell count (hemocytometer) (Figure 3B). ScFv-GFP expression (48 hr prior to infection) did not attenuate HIV-1 infectivity of MAGI-indicator cells (Figure 3C) or infectivity of 293T cells using single-cycle lentiviruses (Figure 3D). While this may implicate the inability of ScFv-GFP to interfere with IN within preassembled PICs during early stages of viral replication, detailed in vivo analysis of ScFv specific association with IN, as well as the assessment of integrated and unintegrated viral DNA, is required to rule out false signals caused by unintegrated circular viral DNA (Engelman et al., 1995).
Interestingly, co-transfection of ScFv-GFP with the HIV-1 expressing plasmid in virus producing cells inhibited the production of infectious virus particles as assessed by infectivity of MAGI-reporter cells (Figure 3E, black bars). To clarify if this inhibition is caused by abolished production of viruses or by the production of non-infectious defective viruses we measured the amount of viral particles, by analyzing p24, released into the culture medium of virus-producing cells coexpressing ScFv-GFP. The results show that virus particle production was inhibited (Figure 3E, gray bars). To rule out an effect of GFP, untagged lentiviral ScFv was similarly assessed and analogous to the GFP-fused ScFv, the Lenti-ScFv did not inhibit HIV-1 infectivity when expressed prior to HIV-1 infection but dramatically inhibited virus production when co-transfected with HIV-1 plasmids in the producer cells (95% inhibition, 1:5 ratio of ScFV to HIV-1 DNA). Normalizing infectivity to the amount of produced virus particles indicates that ScFv-GFP mainly inhibited virus production and release but did not result in defective virus particles (Figure 3F). ScFv-GFP expression does not appear to exclusively inhibit Gag polyprotein expression or proteolytic processing in the HIV-producing cells or in the limited number of released viral particles as evidenced by the presence of the Gag precursor (p55), intermediate proteolysis products (p41 and p37) and the mature p24 capsid protein (Figure 3G). However, detailed pulse-chase analysis of viral protein processing will be required to conclusively determine the precise effects of ScFv-GFP on Gag processing.
To assess whether ScFv-GFP targets the post-processed IN or IN within the context of immature Gag-Pol, we used HIV-1 virus lacking IN (Δ-IN) and functionally trans-complemented it with Vpr-IN fusion protein (Cano and Kalpana, 2011; Jurado et al., 2013). ScFv-GFP did not inhibit virus production or Gag processing of the Δ-IN HIV-1 complemented with Vpr-IN in trans, suggesting that ScFv-GFP exerts the inhibitory effect by targeting IN sequences within the Gag-Pol context (Figure 3H and I).
Direct interaction between ScFv-GFP and Gag-Pol was further validated in live cells using raster image cross-correlation spectroscopy (ccRICS). The positive cross-correlation amplitude (> 50% of all measured cells) obtained only with the Gag-Pol mutant (continuous frame-shifting construct and inactive protease) but not Gag alone, indicates that ScFv-GFP directly and specifically binds Gag-Pol in the cytosol (Figure 3J and K). Using HIV-1 viral construct, which produces 20:1 ratio of Gag:Gag-Pol, yielded similar results albeit with weaker signal (∼ 14% of total cells measured), presumably because of the 20 fold stronger Gag signal, which reduces the probability of detecting a cross-correlation signal (Figure 3K).
Unlike Gag, which itself can form viral like particles (VLP), Gag-Pol alone is incapable of plasma membrane targeting or particle production and can only be incorporated into viral particles by co-assembling with Gag (reviewed in (Haraguchi et al., 2012)). Although the mechanism by which the ScFv inhibits HIV-1 particle production is not established, the high-resolution structure of IN CCD in complex with Fab2 clearly indicates that Fab interferes with IN interfaces essential for CTD and HTH-docking and functional multimerization. While Fab binding did not hinder the canonical IN dimerization interface, blocking of α4 helix, the finger-loop and the HTH-docking cleft may have 1) interfered with the delicate assembly and reorganization of Gag/Gag-Pol oligomers that may utilize multimerization platforms similar to those used by processed IN, or 2) hindered interactions with cellular co-factors necessary for trafficking, and perhaps particle assembly and budding. However, binding of ScFv-GFP to Gag-Pol in the cytosol (Figure 3J and K), prior to any membrane binding, indicates that ScFv-GFP most probably acts on early cytosolic Gag/Gag-Pol complexes and their trafficking but not necessarily on budding sites at the membrane.
Alternatively to the specific inhibitory effect of ScFv-GFP, which implicates a specific functional role of IN during Gag/Gag-Pol assembly and trafficking, the additional size of Pol-bound ScFv-GFP may have impaired the Gag-Pol polyprotein. Increasing the size of Gag by C-terminal fusion of large protein extensions mimicking the size of Pol domain has indeed been shown to impair membrane affinity of Gag (Haraguchi et al., 2012). Nevertheless, specific targeting of IN has previously been shown to inhibit virus particle maturation or production. Allosteric IN inhibitors (ALLINIs) and class-II IN non-catalytic mutations, unlike Fab2 inhibition of particle production, result in the production of non-infectious viral particles (Gupta et al., 2014; Jurado et al., 2013). Similar to Fab2, however, is the expression of a dominant negative mutant of IN-interacting (INI1) host co-factor, harboring the minimal IN-binding domain (S6), which completely inhibits virus assembly prior to particle formation and budding (Cano and Kalpana, 2011).
Future studies, including high-resolution microscopy assessing virus assembly kinetics, will be required to detail the global functional role of Gag-Pol in virus assembly, the mechanisms by which ScFv inhibits virus production and whether the HTH-docking cleft can be therapeutically targeted during the late stages of virus replication. Here we brought to light the structural and functional importance of the HTH-docking cleft of IN core domain for crucial intrinsic and extrinsic interactions vital for IN activity and virus replication.
Experimental Procedures
Protein expression, purification and crystallization
HIV-1 IN CCD (residues 52-210), derived from the previously described IN (SF1; C56S, W131D, F185K, and C280S; (Alian et al., 2009)), and IN-specific Fab2 were prepared using protocols previously described for IN (Alian et al., 2009; Wu et al., 2012) and Fab (Fellouse et al., 2007). IN-Fab complex (mixed at 1:1) was purified using size exclusion chromatography (150 mM NaCl, 20 mM HEPES pH 7.0) and crystallized (using 9 mg/ml IN-Fab) in 20% PEG-3350, 0.1 M Bis-Tris Propane pH 7.0 (at 20°C). Chimeric IN constructs were generated by fusing the various IN domains, from HIV-1 and FIV (Petaluma), using primers overlapping the corresponding regions (NTD: 1-51/5-53, CCD: 52-209/54-211 and CTD: 210-288/212-281, HIV-1/FIV numbering). Chimeric variants were cloned into pET28b (Novagen) with a cleavable His-Tag, which was cleaved prior to analysis. Wild type and chimeric proteins were treated similarly during expression and purification according to previously described protocols (Alian et al., 2009). Solutions for size exclusion chromatography of IN and chimeric variants contained 20 mM HEPES pH 7.4, 1 mM dithiothreitol, 1 M NaCl and 1 mM CHAPS. Apparent molecular masses of full-length IN and chimeric variants (0.075 and 1.3 mg) were analyzed on a Superose-12 column (GE Healthcare Life Sciences).
IN in vitro fluorescence activity
IN was preincubated with Fab2, at the indicated ratios, and the DNA was subsequently added to start the reactions. For 3′-processing, the DNA substrate was prepared by annealing two fragments: 5′-TACAAAATTCCATAGCAGT-6FAM and 5′-ACTGCTATGGAATTTTGTA. For integration assay, the donor DNA was prepared by annealing 5′-TACAAAATTCCATAGCA and 5′- ACTGCTATGGAATTTTGTA-6FAM, and the acceptor DNA was annealed from 5′-Biotin-TATCCGCGATAAGCTTTAATGCGGTAG and 5′-Biotin-CTACCGCATTAAAGCTTATCGCGGATA. Single-site integration and 3′-processing activities of IN were assessed (1 hr in 37°C) using the previously described in vitro fluorescence assay (Merkel et al., 2009). We previously reported the inefficient in vitro strand-transfer activity of recombinant FIV IN (Galilee and Alian, 2014).
Structure determination
Diffraction data were collected at the Advance Light Source (Lawrence Berkeley Laboratory, Berkeley, CA) and processed using HKL-3000 (Minor et al., 2006) (Table 1). The structure was solved with Phaser (McCoy et al., 2007) molecular replacement using IN CCD coordinates extracted from 1EX4 (Chen et al., 2000) and Fab coordinates from 1FVD (Eigenbrot et al., 1993) as search models. Electron densities were fitted using COOT (Emsley et al., 2010) and refined, in the CCP4 suite (Winn et al., 2011), using REFMAC5 (Murshudov et al., 2011). Table-1 summarizes data collection and refinement statistics. Figures presenting structures and structural superposition were prepared using PyMOL Molecular Graphics System (Schrödinger, LLC).
Quantification of virus infectivity
Single-chain variable fragment (ScFv) was constructed using a flexible linker ((Gly4-Ser)3) fusing the variable regions of heavy and light chains as previously described (Huston et al., 1988). Expression of ScFv-GFP fusion construct was verified by Western blot using anti-GFP antibodies. 293T and HIV-indicator TZM-bl cells (NIH AIDS reagent program) were transfected (1× 106 cells) with untagged lenti-ScFv (in pCMV-DsRed-Express, Clontech) or ScFv-GFP fusion (in pEGFP-N1, Clontech) at various concentrations with pSVC21-HIV-HXB2 (5 μg) (Ratner et al., 1985) or pSG3-ΔIN/Vpr-RT-IN (Liu et al., 1997) plasmids using a standard calcium phosphate protocol. 48 hr post-transfection, cells and cell-culture media were harvested, and following ultra-centrifugation of culture supernatants at 100,000 g, the samples were analyzed by Western blot (using anti-p24/capsid antibodies) or subjected to virus infectivity assay (MAGI assay) (Kimpton and Emerman, 1992). Briefly, TZM-bl indicator cells were exposed to concentrated supernatants harvested from HIV-1 producing cells, cultured for 48 hr, after which cells were fixed with formaldehyde and stained with X-gal substrate (5-bromo-4-chloro-3-indolyl β-d -galactopyranoside). Blue cells were counted under microscope examination with each blue cell corresponding to one infectious unit. For the p24 quantification, virus-containing cell-culture media were examined with the ELISA-based p24 Antigen Capture assay kit ((Leidos, Frederic National Laboratory for Cancer Research – AIDS and Cancer Virus Program)). For lentivirus infectivity assay, 239T cells expressing ScFv-GFP, or pEGFP-N1 control, plasmids (5 μg) were infected (48 hr post-transfection) with lentiviruses containing humanized Kusobira-Orange fluorescent protein (CSII-EF-MCS-IRES2-hKO1, RIKEN, Tsukuba, Japan) and 24 hr post-infection were analyzed using fluorescent microscopy with green or orange light-filters.
Cross Correlation Raster Image Correlation Spectroscopy (ccRICS)
HeLa cells (3.6×104) (NIH AIDS reagent program) were transfected with ScFv-GFP DNA (40 ng/well; equivalent to 1.1 μg in the virus infectivity assay) and 24 hr post-transfection cells were further transfected with the various HIV-1 constructs: pKHIV(GagPolonly).mCherry, pKHIV(Gagonly).mCherry or pKHIV.mCherry (full viral construct), as previously described (Muller et al., 2004). 12 hr later, live cells were washed and imaged (at 37° C) by a home-built PIE fluctuation imaging (PIE-FI) microscope (Hendrix et al., 2013) using a Nikon 100× Apo TIRF 1.49 oil objective and a perfect focus system for maximal z-stability. For ccRICS, a z-plane in the center of each cell was chosen to avoid any influence of the cell membrane on the analysis and subsequently 250 frames of a selected region of interest (14 × 14 μm with 300 × 300 pixels) were recorded (frame time: 1s, line time: 3.33 ms, pixel dwell time: 11.11 μs, pixel size: 46.667 nm). Each region of interest was selected outside the nucleus and by avoiding cell areas with a high number of vesicles or organelles to reduce potential artifacts, which might be caused by correlations from diffusing vesicles. Slow fluctuations and inhomogeneity were additionally removed by using a moving average of ΔF=3. The spatial autocorrelation function (SACF) and spatial cross-correlation function (SCCF) were calculated using a two-dimensional discrete Fourier transform algorithm and fitted by one-component correlation function with a slow-diffusing (immobile) fraction added as an additional correlation term according to our previously described protocol (Hendrix et al., 2015).
Supplementary Material
Highlights.
Crystal structure of HIV-1 integrase core domain in complex with Fab
The configuration of HTH-motif docking is conserved and functionally imperative
Helix α4 mutually binds viral DNA and crucially mediates integrase mutlimerization
Intrabody targeting of HTH-cleft within Gag-Pol inhibits HIV particle production
Acknowledgments
This research was partly supported by the Rubin Scientific and Medical Research Fund (to A.A.), the National Institutes of Health Grant P50 GM082250 via the HARC Center (to R.M.S.), and U01 GM094588 (to A.A.K). D.C.L. and V.B. acknowledge financial support of the Deutsche Forschungsgemeinschaft (DFG) through the Excellence Initiatives Nanosystems Initiative Munich, Center for Integrated Protein Science Munich, and of the Ludwig-Maximilians-Universität München through the Center for Nano Science and the LMUinnovative BioImaging Network. They also thank B. Müller from the Department of Infectious Diseases and Virology, University Hospital Heidelberg for providing fluorescently tagged HIV, Gag and Gag-Pol constructs.
Footnotes
Accession Numbers: Atomic coordinates and structure factors were deposited in the Protein Data Bank with the accession number 5EU7.
Author contributions: M.G., A.M. and A.A. designed and performed research, analyzed data and wrote the manuscript. R.M.S. contributed crystallography reagents/analytic tools, and S.L.G. performed initial crystallization experiments. M.K. contributed virus infectivity reagents/analytic tools, and E.BR. performed infectivity assays. A.A.K and S.U. contributed Fab2 protein. D.C.L. and V.B. contributed fluorescence correlation analysis.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alian A, Griner SL, Chiang V, Tsiang M, Jones G, Birkus G, Geleziunas R, Leavitt AD, Stroud RM. Catalytically-active complex of HIV-1 integrase with a viral DNA substrate binds anti-integrase drugs. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:8192–8197. doi: 10.1073/pnas.0811919106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrake MD, Skalka AM. Multimerization determinants reside in both the catalytic core and C terminus of avian sarcoma virus integrase. The Journal of biological chemistry. 1995;270:29299–29306. doi: 10.1074/jbc.270.49.29299. [DOI] [PubMed] [Google Scholar]
- Azzi S, Parissi V, Maroun RG, Eid P, Mauffret O, Fermandjian S. The HIV-1 integrase alpha4-helix involved in LTR-DNA recognition is also a highly antigenic peptide element. PloS one. 2010;5:e16001. doi: 10.1371/journal.pone.0016001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballandras-Colas A, Brown M, Cook NJ, Dewdney TG, Demeler B, Cherepanov P, Lyumkis D, Engelman AN. Cryo-EM reveals a novel octameric integrase structure for betaretroviral intasome function. Nature. 2016;530:358–361. doi: 10.1038/nature16955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barsov EV, Huber WE, Marcotrigiano J, Clark PK, Clark AD, Arnold E, Hughes SH. Inhibition of human immunodeficiency virus type 1 integrase by the Fab fragment of a specific monoclonal antibody suggests that different multimerization states are required for different enzymatic functions. Journal of virology. 1996;70:4484–4494. doi: 10.1128/jvi.70.7.4484-4494.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthoux L, Sebastian S, Muesing MA, Luban J. The role of lysine 186 in HIV-1 integrase multimerization. Virology. 2007;364:227–236. doi: 10.1016/j.virol.2007.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bojja RS, Andrake MD, Merkel G, Weigand S, Dunbrack RL, Jr, Skalka AM. Architecture and assembly of HIV integrase multimers in the absence of DNA substrates. The Journal of biological chemistry. 2013;288:7373–7386. doi: 10.1074/jbc.M112.434431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cano J, Kalpana GV. Inhibition of early stages of HIV-1 assembly by INI1/hSNF5 transdominant negative mutant S6. Journal of virology. 2011;85:2254–2265. doi: 10.1128/JVI.00006-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cellier C, Moreau K, Gallay K, Ballandras A, Gouet P, Ronfort C. In vitro functional analyses of the human immunodeficiency virus type 1 (HIV-1) integrase mutants give new insights into the intasome assembly. Virology. 2013;439:97–104. doi: 10.1016/j.virol.2013.02.001. [DOI] [PubMed] [Google Scholar]
- Chen JC, Krucinski J, Miercke LJ, Finer-Moore JS, Tang AH, Leavitt AD, Stroud RM. Crystal structure of the HIV-1 integrase catalytic core and C-terminal domains: a model for viral DNA binding. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:8233–8238. doi: 10.1073/pnas.150220297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desimmie BA, Schrijvers R, Demeulemeester J, Borrenberghs D, Weydert C, Thys W, Vets S, Van Remoortel B, Hofkens J, De Rijck J, et al. LEDGINs inhibit late stage HIV-1 replication by modulating integrase multimerization in the virions. Retrovirology. 2013;10:57. doi: 10.1186/1742-4690-10-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eigenbrot C, Randal M, Presta L, Carter P, Kossiakoff AA. X-ray structures of the antigen-binding domains from three variants of humanized anti-p185HER2 antibody 4D5 and comparison with molecular modeling. J Mol Biol. 1993;229:969–995. doi: 10.1006/jmbi.1993.1099. [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta crystallographica Section D, Biological crystallography. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman A, Englund G, Orenstein JM, Martin MA, Craigie R. Multiple effects of mutations in human immunodeficiency virus type 1 integrase on viral replication. Journal of virology. 1995;69:2729–2736. doi: 10.1128/jvi.69.5.2729-2736.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellouse FA, Esaki K, Birtalan S, Raptis D, Cancasci VJ, Koide A, Jhurani P, Vasser M, Wiesmann C, Kossiakoff AA, et al. High-throughput generation of synthetic antibodies from highly functional minimalist phage-displayed libraries. J Mol Biol. 2007;373:924–940. doi: 10.1016/j.jmb.2007.08.005. [DOI] [PubMed] [Google Scholar]
- Feng L, Larue RC, Slaughter A, Kessl JJ, Kvaratskhelia M. HIV-1 integrase multimerization as a therapeutic target. Curr Top Microbiol Immunol. 2015;389:93–119. doi: 10.1007/82_2015_439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galilee M, Alian A. Identification of Phe187 as a crucial dimerization determinant facilitates crystallization of a monomeric retroviral integrase core domain. Structure. 2014;22:1512–1519. doi: 10.1016/j.str.2014.08.001. [DOI] [PubMed] [Google Scholar]
- Goldgur Y, Dyda F, Hickman AB, Jenkins TM, Craigie R, Davies DR. Three new structures of the core domain of HIV-1 integrase: an active site that binds magnesium. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:9150–9154. doi: 10.1073/pnas.95.16.9150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandgenett DP, Pandey KK, Bera S, Aihara H. Multifunctional facets of retrovirus integrase. World J Biol Chem. 2015;6:83–94. doi: 10.4331/wjbc.v6.i3.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta K, Brady T, Dyer BM, Malani N, Hwang Y, Male F, Nolte RT, Wang L, Velthuisen E, Jeffrey J, et al. Allosteric inhibition of human immunodeficiency virus integrase: late block during viral replication and abnormal multimerization involving specific protein domains. The Journal of biological chemistry. 2014;289:20477–20488. doi: 10.1074/jbc.M114.551119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haraguchi H, Noda T, Kawaoka Y, Morikawa Y. A large extension to HIV-1 Gag, like Pol, has negative impacts on virion assembly. PloS one. 2012;7:e47828. doi: 10.1371/journal.pone.0047828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hare S, Di Nunzio F, Labeja A, Wang J, Engelman A, Cherepanov P. Structural basis for functional tetramerization of lentiviral integrase. PLoS pathogens. 2009a;5:e1000515. doi: 10.1371/journal.ppat.1000515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hare S, Gupta SS, Valkov E, Engelman A, Cherepanov P. Retroviral intasome assembly and inhibition of DNA strand transfer. Nature. 2010;464:232–236. doi: 10.1038/nature08784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hare S, Shun MC, Gupta SS, Valkov E, Engelman A, Cherepanov P. A novel co-crystal structure affords the design of gain-of-function lentiviral integrase mutants in the presence of modified PSIP1/LEDGF/p75. PLoS pathogens. 2009b;5:e1000259. doi: 10.1371/journal.ppat.1000259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayouka Z, Rosenbluh J, Levin A, Loya S, Lebendiker M, Veprintsev D, Kotler M, Hizi A, Loyter A, Friedler A. Inhibiting HIV-1 integrase by shifting its oligomerization equilibrium. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:8316–8321. doi: 10.1073/pnas.0700781104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrix J, Baumgartel V, Schrimpf W, Ivanchenko S, Digman MA, Gratton E, Krausslich HG, Muller B, Lamb DC. Live-cell observation of cytosolic HIV-1 assembly onset reveals RNA-interacting Gag oligomers. J Cell Biol. 2015;210:629–646. doi: 10.1083/jcb.201504006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrix J, Schrimpf W, Holler M, Lamb DC. Pulsed interleaved excitation fluctuation imaging. Biophys J. 2013;105:848–861. doi: 10.1016/j.bpj.2013.05.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huston JS, Levinson D, Mudgett-Hunter M, Tai MS, Novotny J, Margolies MN, Ridge RJ, Bruccoleri RE, Haber E, Crea R, et al. Protein engineering of antibody binding sites: recovery of specific activity in an anti-digoxin single-chain Fv analogue produced in Escherichia coli. Proceedings of the National Academy of Sciences of the United States of America. 1988;85:5879–5883. doi: 10.1073/pnas.85.16.5879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaskolski M, Alexandratos JN, Bujacz G, Wlodawer A. Piecing together the structure of retroviral integrase, an important target in AIDS therapy. The FEBS journal. 2009;276:2926–2946. doi: 10.1111/j.1742-4658.2009.07009.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins TM, Engelman A, Ghirlando R, Craigie R. A soluble active mutant of HIV-1 integrase: involvement of both the core and carboxyl-terminal domains in multimerization. The Journal of biological chemistry. 1996;271:7712–7718. doi: 10.1074/jbc.271.13.7712. [DOI] [PubMed] [Google Scholar]
- Jurado KA, Wang H, Slaughter A, Feng L, Kessl JJ, Koh Y, Wang W, Ballandras-Colas A, Patel PA, Fuchs JR, et al. Allosteric integrase inhibitor potency is determined through the inhibition of HIV-1 particle maturation. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:8690–8695. doi: 10.1073/pnas.1300703110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessl JJ, McKee CJ, Eidahl JO, Shkriabai N, Katz A, Kvaratskhelia M. HIV-1 Integrase-DNA Recognition Mechanisms. Viruses. 2009;1:713–736. doi: 10.3390/v1030713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khwaja A, Galilee M, Marx A, Alian A. Structure of FIV capsid C-terminal domain demonstrates lentiviral evasion of genetic fragility by coevolved substitutions. Scientific Reports. 2016;6:24957. doi: 10.1038/srep24957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimpton J, Emerman M. Detection of replication-competent and pseudotyped human immunodeficiency virus with a sensitive cell line on the basis of activation of an integrated beta-galactosidase gene. Journal of virology. 1992;66:2232–2239. doi: 10.1128/jvi.66.4.2232-2239.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesbats P, Metifiot M, Calmels C, Baranova S, Nevinsky G, Andreola ML, Parissi V. In vitro initial attachment of HIV-1 integrase to viral ends: control of the DNA specific interaction by the oligomerization state. Nucleic acids research. 2008;36:7043–7058. doi: 10.1093/nar/gkn796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Krishnan L, Cherepanov P, Engelman A. Structural biology of retroviral DNA integration. Virology. 2011;411:194–205. doi: 10.1016/j.virol.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Wu X, Xiao H, Conway JA, Kappes JC. Incorporation of functional human immunodeficiency virus type 1 integrase into virions independent of the Gag-Pol precursor protein. Journal of virology. 1997;71:7704–7710. doi: 10.1128/jvi.71.10.7704-7710.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maertens G, Cherepanov P, Pluymers W, Busschots K, De Clercq E, Debyser Z, Engelborghs Y. LEDGF/p75 is essential for nuclear and chromosomal targeting of HIV-1 integrase in human cells. The Journal of biological chemistry. 2003;278:33528–33539. doi: 10.1074/jbc.M303594200. [DOI] [PubMed] [Google Scholar]
- Maertens GN, Hare S, Cherepanov P. The mechanism of retroviral integration from X-ray structures of its key intermediates. Nature. 2010;468:326–329. doi: 10.1038/nature09517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee CJ, Kessl JJ, Shkriabai N, Dar MJ, Engelman A, Kvaratskhelia M. Dynamic modulation of HIV-1 integrase structure and function by cellular lens epithelium-derived growth factor (LEDGF) protein. The Journal of biological chemistry. 2008;283:31802–31812. doi: 10.1074/jbc.M805843200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkel G, Andrake MD, Ramcharan J, Skalka AM. Oligonucleotide-based assays for integrase activity. Methods. 2009;47:243–248. doi: 10.1016/j.ymeth.2008.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. HKL-3000: the integration of data reduction and structure solution--from diffraction images to an initial model in minutes. Acta crystallographica Section D, Biological crystallography. 2006;62:859–866. doi: 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- Muller B, Daecke J, Fackler OT, Dittmar MT, Zentgraf H, Krausslich HG. Construction and characterization of a fluorescently labeled infectious human immunodeficiency virus type 1 derivative. Journal of virology. 2004;78:10803–10813. doi: 10.1128/JVI.78.19.10803-10813.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA. REFMAC5 for the refinement of macromolecular crystal structures. Acta crystallographica Section D, Biological crystallography. 2011;67:355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey KK, Bera S, Grandgenett DP. The HIV-1 integrase monomer induces a specific interaction with LTR DNA for concerted integration. Biochemistry. 2011;50:9788–9796. doi: 10.1021/bi201247f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratner L, Haseltine W, Patarca R, Livak KJ, Starcich B, Josephs SF, Doran ER, Rafalski JA, Whitehorn EA, Baumeister K, et al. Complete nucleotide sequence of the AIDS virus, HTLV-III. Nature. 1985;313:277–284. doi: 10.1038/313277a0. [DOI] [PubMed] [Google Scholar]
- Roberts VA. C-Terminal Domain of Integrase Binds between the Two Active Sites. J Chem Theory Comput. 2015;11:4500–4511. doi: 10.1021/ct501125r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibagaki Y, Chow SA. Central core domain of retroviral integrase is responsible for target site selection. The Journal of biological chemistry. 1997;272:8361–8369. doi: 10.1074/jbc.272.13.8361. [DOI] [PubMed] [Google Scholar]
- Shkriabai N, Dharmarajan V, Slaughter A, Kessl JJ, Larue RC, Feng L, Fuchs JR, Griffin PR, Kvaratskhelia M. A critical role of the C-terminal segment for allosteric inhibitor-induced aberrant multimerization of HIV-1 integrase. The Journal of biological chemistry. 2014;289:26430–26440. doi: 10.1074/jbc.M114.589572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taltynov O, Desimmie BA, Demeulemeester J, Christ F, Debyser Z. Cellular cofactors of lentiviral integrase: from target validation to drug discovery. Mol Biol Int. 2012;2012:863405. doi: 10.1155/2012/863405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Shun MC, Li X, Di Nunzio F, Hare S, Cherepanov P, Engelman A. Efficient Transduction of LEDGF/p75 Mutant Cells by Gain-of-Function HIV-1 Integrase Mutant Viruses. Mol Ther Methods Clin Dev. 2014;1 doi: 10.1038/mtm.2013.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JY, Ling H, Yang W, Craigie R. Structure of a two-domain fragment of HIV-1 integrase: implications for domain organization in the intact protein. EMBO J. 2001;20:7333–7343. doi: 10.1093/emboj/20.24.7333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, et al. Overview of the CCP4 suite and current developments. Acta crystallographica Section D, Biological crystallography. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Avila-Sakar A, Kim J, Booth DS, Greenberg CH, Rossi A, Liao M, Li X, Alian A, Griner SL, et al. Fabs enable single particle cryoEM studies of small proteins. Structure. 2012;20:582–592. doi: 10.1016/j.str.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Z, Shi K, Banerjee S, Pandey KK, Bera S, Grandgenett DP, Aihara H. Crystal structure of the Rous sarcoma virus intasome. Nature. 2016;530:362–366. doi: 10.1038/nature16950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, McKee CJ, Kessl JJ, Santos WL, Daigle JE, Engelman A, Verdine G, Kvaratskhelia M. Subunit-specific protein footprinting reveals significant structural rearrangements and a role for N-terminal Lys-14 of HIV-1 Integrase during viral DNA binding. The Journal of biological chemistry. 2008;283:5632–5641. doi: 10.1074/jbc.M705241200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng R, Jenkins TM, Craigie R. Zinc folds the N-terminal domain of HIV-1 integrase, promotes multimerization, and enhances catalytic activity. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:13659–13664. doi: 10.1073/pnas.93.24.13659. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.