

Abstract
Accurate and complete analysis of genome variation in large populations will be required to understand the role of genome variation in complex disease. We present an analytical framework for characterizing genome deletion polymorphism in populations, using sequence data that are distributed across hundreds or thousands of genomes. Our approach uses population-level relationships to re-interpret the technical features of sequence data that often reflect structural variation. In the 1000 Genomes Project pilot, this approach identified deletion polymorphism across 168 genomes (sequenced at 4x average coverage) with sensitivity and specificity unmatched by other algorithms. We also describe a way to determine the allelic state or genotype of each deletion polymorphism in each genome; the 1000 Genomes Project used this approach to type 13,826 deletion polymorphisms (48 bp – 960 kbp) at high accuracy in populations. These methods offer a way to relate genome structural polymorphism to complex disease in populations.
Introduction
Describing genome variation in populations and identifying the alleles that influence complex phenotypes will require sequencing thousands of genomes. Genome sequencing will therefore increasingly be performed in clinical and reference cohorts of a substantial size 1. An important challenge will be to identify how genomes vary at large as well as fine scales.
Short sequence reads can reflect large variants in several ways: individual reads can span a variant’s breakpoints 2,3; molecularly paired sequences can flank a variant 4-6; and read depth is influenced by the underlying copy number of a genomic segment 7-9 (Fig. 1). However, identifying large variants from short sequence reads is error-prone: molecular libraries contain millions of chimeric molecules that masquerade as structural variants; read depth varies across the genome in ways that vary among sequencing libraries; and alignment algorithms are misled by the genome’s internal repeats. Illustrating this challenge, the 1000 Genomes Project found that even for deeply sequenced (> 30x) individual genomes, 14 published and novel methods for analyzing deletions generated false discovery rates (FDRs) of 9-89%, such that additional experiments (array CGH, PCR) were required to identify the real variants among the false discoveries 1,10.
Figure 1.
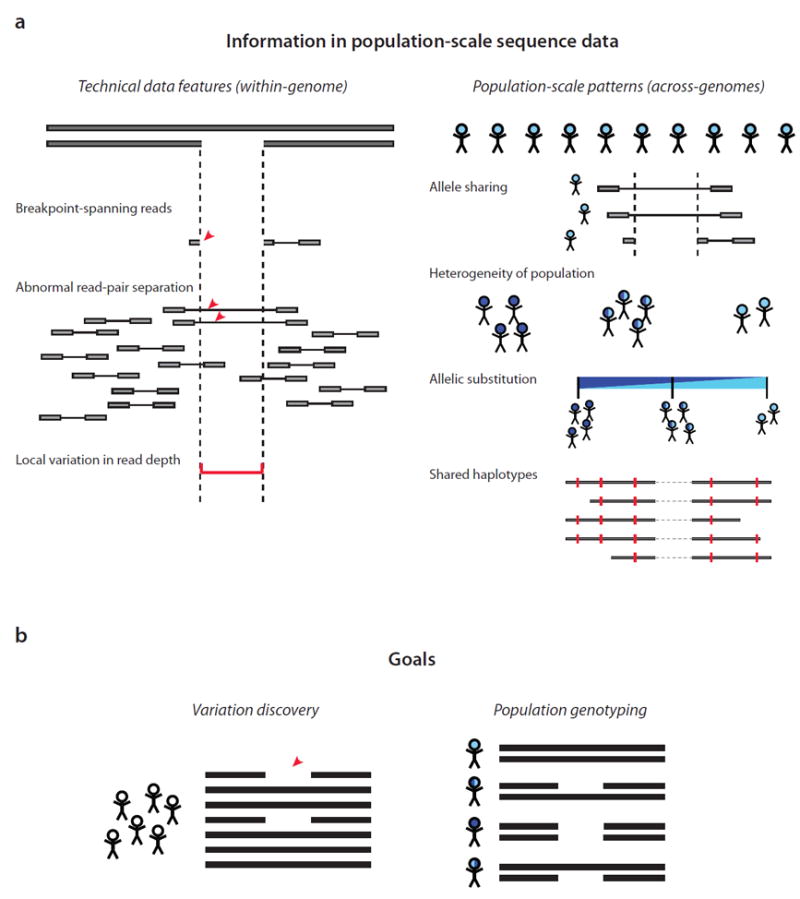
A population-aware analytical framework for analyzing Genome STRucture in Populations (Genome STRiP).
(a) Population-scale sequence data contain two classes of information: technical features of the sequence data within a genome, and population-scale patterns that span all the genomes analyzed. Technical features include breakpoint-spanning reads 2,3, paired-end sequences 4-6, and local variation in read depth of coverage 7-9. Genome STRiP combines these with population-scale patterns that span many genomes, including: the sharing of structural alleles by multiple genomes; the pattern of sequence heterogeneity within a population; the substitution of alternative structural alleles for each other; and the haplotype structure of human genome polymorphism.
(b) Goals of structural variation (SV) analysis in Genome STRiP. Variation discovery involves identifying the structural alleles that are segregating in a population. The power to observe a variant in any one genome is only partial, but the evidence defining a segregating site can be derived from many genomes at once. Population genotyping requires accurately determining the allelic state of each variant in every diploid genome in a population.
These difficulties are potentially more severe in sequence data that are generated on a population scale. As more genomes are sequenced, false discoveries accumulate more quickly than real variants do, since many real variants are simply rediscovered in more genomes. In addition, in population-based studies, investigators may use lower sequence coverage (across many more genomes) than is used for deeply sequenced personal genomes, as the resulting large sample size will allow studies to ascertain more low-frequency alleles and increase power for relating variation to phenotype. The high false discovery rate of structural variation algorithms in deeply sequenced individual genomes 1,10 has suggested that the problem of accurate inference at lower coverage will be challenging.
We hypothesized, however, that sequencing at a population scale will also enable new kinds of analytical approaches. True structural alleles might leave additional footprints in population-scale data (Fig. 1). Segregating alleles distinguish some genomes from others; they substitute for alternative structural alleles; they give rise to discrete allelic states in a diploid genome; they are often shared across genomes; and they segregate on haplotypes with other variants 11,12. Here we show that analysis of structural variation in populations is made far more accurate and powerful by apprehending patterns at a population level.
We present the results of an analysis applying these principles to map deletion polymorphism in the genomes of 168 individuals sequenced at low coverage (2-8x paired-end sequencing on the Illumina platform) in the 1000 Genomes Project pilot. We focus on deletion polymorphism, the most numerous and validated class of structural variation, though the population-level analytic concepts we describe can also be used to analyze other forms of genome variation. We show that population-aware analysis enables structural inference with far greater accuracy and allows the construction of an unprecedented resource on human genome deletion polymorphism – with few false discoveries, ascertainment of variants down to sub-kilobase sizes and low allele frequencies, localization of breakpoints at high resolution, accurate determination of genotype (allelic state) at each locus in each genome, and a high-resolution map of linkage disequilibrium between single-nucleotide and structural alleles. The resulting data set has been validated by independent experiments. It comprises a substantial fraction of the deletion loci and all of the structural variation genotype data released by the 1000 Genomes Project pilot 1.
Results
Coherence around shared alleles
Most of the variation in any human genome arises from alleles shared with other humans. In a sequencing study, allele sharing can arise in two ways. In studies of reference populations such as the 1000 Genomes Project, alleles that segregate in the general population at an appreciable frequency (>1% in the 1000 Genomes pilot) will generally be shared among multiple individuals sequenced. In medical sequencing studies that sequence many individuals with a particular phenotype, enrichment for high-risk alleles may cause such alleles to be present multiple times in a cohort of affected individuals, even when such alleles are rare in the general population.
We therefore sought to exploit shared variation wherever it exists, without filtering out rare, singleton variants that were private to individual genomes. We reasoned that making use of allele sharing would particularly increase power in low-coverage, population-scale sequencing, in which the evidence for a new allele in the data from any one genome may be insufficient to identify that variant with high confidence.
We applied this allele-sharing principle to increase power for ascertaining deletion alleles from discordant read pairs – paired-end reads that map to genomic locations that are unexpectedly far apart given a molecular library’s insert size distribution 4,6,13. Because the construction of molecular libraries produces millions of chimeric molecules (Fig. 2a), most such read pairs do not arise from real SVs. We identified sets of discordant read pairs, each set containing read pairs from 1-144 of the 168 genomes, that were coherent in the sense that all read pairs in a set could have arisen with high likelihood from the same deletion allele (Fig. 2b, Methods). Some 89% of the resulting sets included evidence from multiple genomes; the other 11% were supported by evidence from individual genomes.
Figure 2.
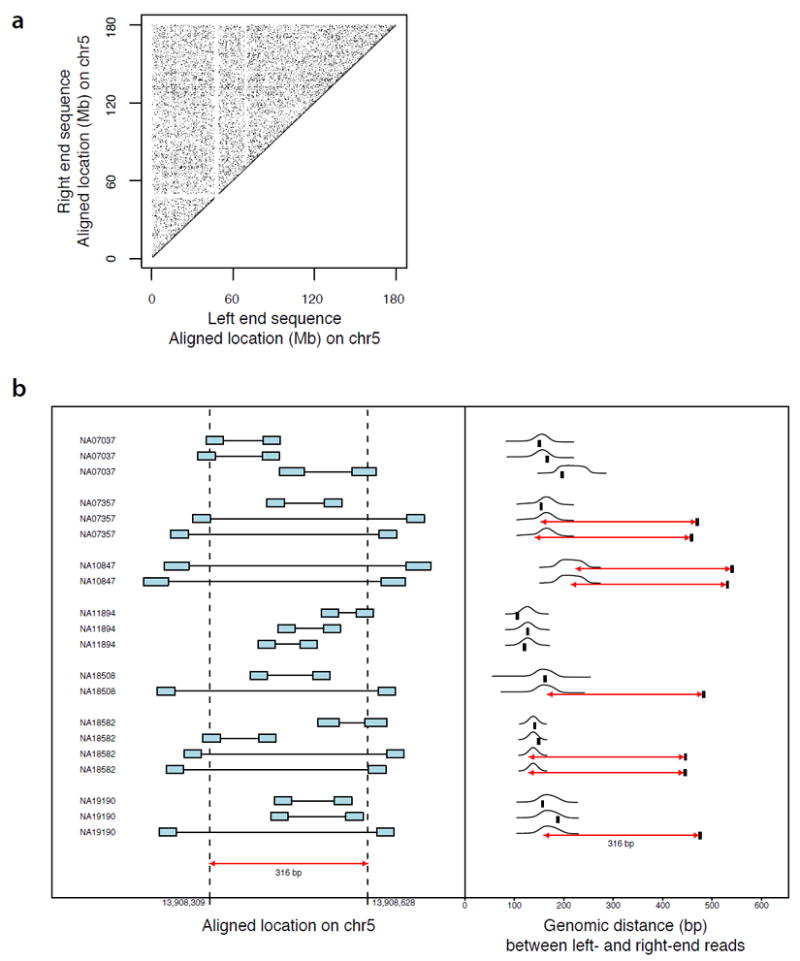
Identifying coherent sets of aberrantly mapping reads from a population of genomes.
(a) Millions of end-sequence pairs from sequencing libraries show aberrant alignment locations, appearing to span vast genomic distances. Almost all of these observations derive not from true structural variants but from chimeric inserts in molecular sequencing libraries. Data shown: paired-end alignments on chromosome 5, from 41 initial genome sequencing libraries from the 1000 Genomes Project.
(b) A set of “coherently aberrant” end-sequence pairs from many genomes. At this genomic locus, paired-end sequences (sequences of the two ends of the inserts in a molecular library) fall into two classes: (i) end-sequence pairs that show the genomic spacing expected given the insert size distribution of each sequencing library, such as the three read-pair alignments for genome NA07037; and (ii) end-sequence pairs that align to genomic locations unexpectedly far apart, but which relate to their expected insert size distributions by a shared correction factor (red arrows). A unifying model in which these eight read pairs from five genomes arise from a shared deletion allele (size of red arrows) converts all of these aberrant read pairs to likely observations. (In right panel, black tick marks indicate genomic distance between left and right end sequences; black curves indicate insert size distributions of the molecular library from which each sequence-pair is drawn.)
Utilizing coherence allowed our algorithm, Genome STRiP (Genome STRucture In Populations), to accumulate power across genomes without being misled by chimeric molecules or requiring multiple evidentiary read pairs in any one genome. However, it became clear that coherence was an insufficient criterion: the number of large putative deletions (coherent clusters of read pairs spanning > 10 kb) exceeded tenfold the number expected by extrapolation from the CNVs discovered by tiling-resolution array CGH in a recent study by Conrad et al. 12 of 40 of these same genomes (Supplementary Figure S1), though Conrad et al. had ample technical power to identify CNVs of this size. This indicated that additional criteria were necessary to distinguish real SVs from artifacts.
Heterogeneity in populations
A true polymorphism creates heterogeneity in a population in the sense that it is differentially present in the genomes of different individuals. We reasoned that, in population-scale sequencing, this principle could distinguish real variants from those molecular and alignment artifacts that can arise from any genome.
We analyzed how the evidentiary read-pairs supporting each putative deletion allele were distributed across the 168 genomes sequenced (Fig. 3). For each putative deletion, a chi-square test was used to evaluate the deviation of the observed distribution from a null model in which each genome was equally likely (per molecule sequenced) to yield deletion-suggestive reads from the locus (Fig. 3a,b). To evaluate the use of this statistic, we examined its distribution for a set of positive control deletions (from ref 12). The heterogeneity statistic yielded low p-values for almost all of these positive control regions (Fig. 3c); by contrast, the distribution of p-values for the “coherent” clusters of sequence reads included thousands that appeared to arise from a uniform distribution, consistent with the presence of thousands of artifacts that can arise with similar probability from any genome (Fig. 3d).
Figure 3.
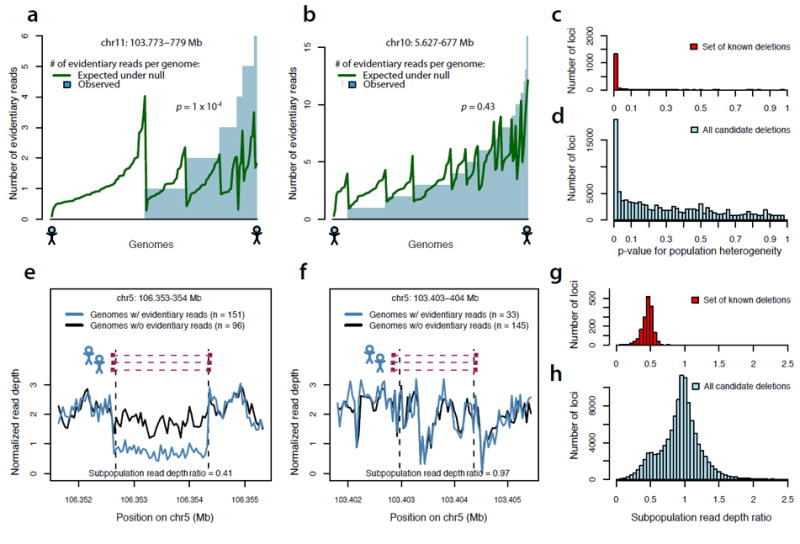
Evaluating the population-heterogeneity and allele-substitution properties of population-scale sequence data.
(a) At a candidate deletion locus, the distribution across genomes of “evidentiary reads” (read-pairs suggesting the presence of a deletion allele at a locus) (blue bars) is compared to a null model under which genomes are equally likely, per molecule sequenced, to give rise to such evidentiary reads (green curve). For the locus shown, the distribution of evidentiary reads across genomes differs from the null distribution (p = 1 × 10-4), confirming that evidentiary sequence data appears differentially within the population at this locus.
(b) At another genomic locus, putative SV-supporting read pairs arise from many genomes but in a pattern that does not significantly differ from a null distribution based on equal probability per molecule sequenced. Subsequent assays confirmed that this is not a true deletion.
(c) Distribution of a population-heterogeneity statistic (from a,b) for read-pair data at 1,420 sites of known deletion polymorphism.
(d) Distribution of the same population-heterogeneity statistic from read-pair data at 45 thousand candidate deletion loci nominated by read-pair analysis.
(e,f) If a putative deletion is real, then genomes with molecular evidence for the deletion allele would be expected to have less evidence for the reference allele (“allelic substitution”). A simple test of allelic substitution is to compare average read depth (across a putative deletion segment) between two subpopulations – the genomes with read-pair evidence for the deletion (blue curve), and the genomes lacking such evidence (black trace). The locus in (e) was subsequently validated as containing a real deletion; the locus in (f) was not.
(g) Distribution of this “subpopulation depth ratio” statistic (e,f) for sequence data at 1,420 sites of known deletion polymorphism.
(h) Distribution of the same statistic for sequence data at 45 thousand candidate deletion loci.
We evaluated the properties of putative common deletions for which sequence data were coherent but did not establish heterogeneity within the population. Many of these loci were flanked by homologous sequences that caused alignment algorithms to locate reads in the incorrect copy. Subsequent analysis of array-based copy number data confirmed that few of these putative deletions were real, and that the real ones generally passed our heterogeneity test. At other loci that lacked evidence for heterogeneity, we found almost no sequence support for the reference genome sequence, suggesting that the reference sequence is incorrect or represents a rare allele.
One consequence of the heterogeneity criterion is that, while Genome STRiP evaluates the evidence from all genomes at once, it is more convinced by putative variants for which supporting data arise multiple times from the same genome(s) than by putative variants for which support arises from many genomes in a thinly distributed way. The heterogeneity test therefore becomes far more powerful at intermediate and high levels of coverage.
Allelic substitution
Because genome variation does not generally change the number of copies of a chromosome, alternative structural alleles at the same locus are substitutes. If structural allele 1 (SA1) and structural allele 2 (SA2) are inconsistent with each other – for example, if SA1 contains genomic sequence that is deleted in SA2 – and both alleles are segregating in the same population, then there should be a negative correlation (across the genomes in a population) between evidence for SA1 and evidence for SA2. The nature of this molecular evidence need not be identical between SA1 and SA2, and this provides an opportunity to integrate multiple attributes of sequence data (such as read depth and read pairs) that have orthogonal error properties.
For the putative deletions in the 1000 Genomes pilot data, we evaluated the relationship between the presence of read-pair evidence for a deletion allele and the magnitude of sequence-depth evidence for the reference allele (Fig. 3e,f). To motivate use of this criterion, we evaluated its behavior for a set of positive control deletions (from ref 12). Genomes with read-pair evidence supporting these deletions invariably had diminished average read depth across the putatively deleted genomic segment (Fig. 3g). However, the candidate deletions from read-pair alignments (even those with coherent sequence data) appeared to arise from a mixture of real deletions and many more loci at which read-pair and read-depth data were uncorrelated in the population (Fig. 3h).
Most putative deletions for which supporting sequence data were coherent and established heterogeneity still failed this allelic substitution test, and turned out be false discoveries. At many such loci, cryptic sequence polymorphisms (often small indels) had caused sequence reads to misalign to nearby, paralogous sequences (Supplementary Figure S2). Another type of filtered site consisted of transposon insertion polymorphisms 14,15 that were not on the reference genome sequence; reads from such insertions often aligned to nearby transposon-derived sequences, causing sequence data to falsely suggest the presence of large deletions across the intervening genomic segment.
We combined the principles of coherence, heterogeneity, and substitution to infer the locations of deletion polymorphisms among the 168 genomes sequenced (Methods). The relative influences of coherence, heterogeneity, and substitution were optimized for this data set, which consisted mostly of 36-bp to 50-bp paired-end reads and sequencing coverage of 2-8x per genome (Methods). We identified 7,015 putative deletion polymorphisms, 100 bp to 471 kbp in size, with evidence for each deletion arising from 2–1,111 read pairs from 1–140 genomes (Fig. 4 a,b,c). Of these 7,015 deletions, 63% were novel relative to those discovered by tiling-resolution array CGH in Conrad et al.
Figure 4.
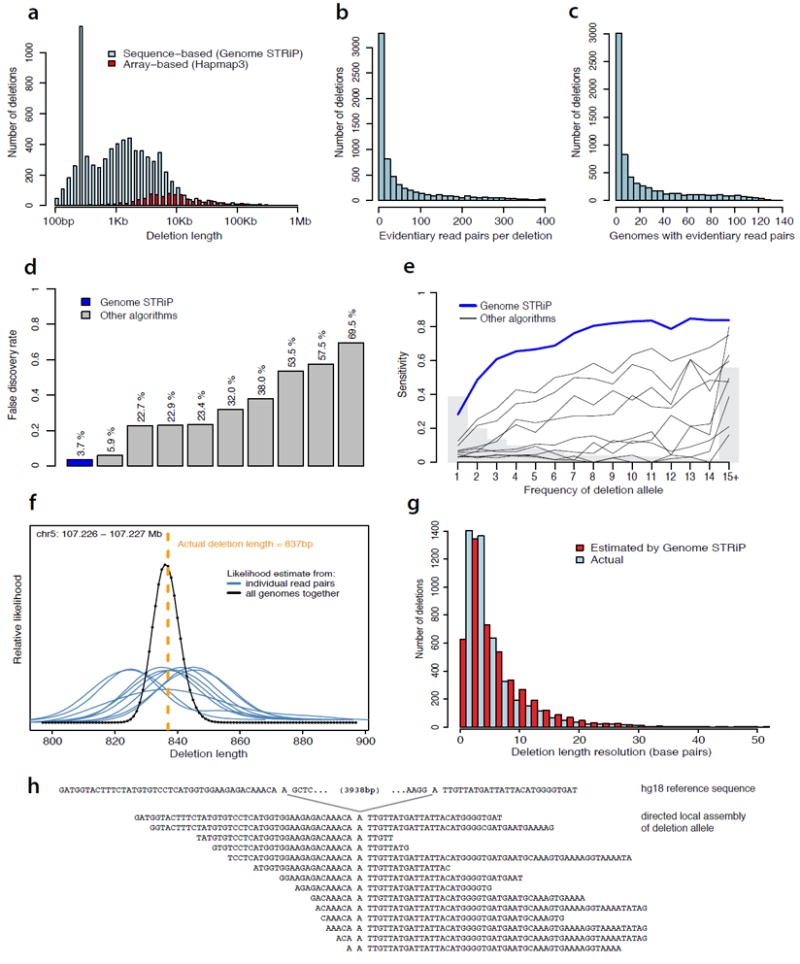
Deletion polymorphisms identified by Genome STRiP in low-coverage sequence data from 168 genomes.
(a) Size distribution. Sensitivity for large deletions (>10 kb) is similar to that of the array-based approaches applied in large, population-scale studies (red); sensitivity for deletions smaller than 10 kb is much greater. A strong peak near 300 bp arises from ALU insertion polymorphisms; a smaller peak near 6 Kb arises from L1 insertion polymorphisms.
(b,c) Number of evidentiary sequence reads (b) and genomes (c) contributing to each deletion discovery in population-scale sequence data. 1,033 of these deletions (14.7%) were identified with evidentiary pairs from individual genomes.
(d) Specificity: false discovery rates of ten deletion discovery methods evaluated by the 1000 Genomes Project in the Project’s population-scale low-coverage sequence data.
(e) Sensitivity: power of the same ten discovery methods for identifying known deletions, as a function of the allele frequency of the deletion.
(f) Localization of the breakpoints of a common deletion allele using read-pair data from many genomes. The difference between (i) the genomic separation of each read-pair sequence and (ii) the insert-size distribution of the molecular library from which is it drawn (Fig. 2b) allows a likelihood-based estimate of deletion length from each read pair (blue curves). Combining this likelihood information across many genomes (black curve) allows fine-scale localization of the breakpoint.
(g) Resolution of breakpoint estimates from Genome STRiP, as estimated using Genome STRiP confidence intervals (red) and comparison to molecularly established breakpoint sequences (blue).
(h) Fine-scale localization of an SV breakpoint facilitates directed local assembly of the deletion allele from sequence data derived from many genomes.
Sensitivity and specificity
The fundamental challenge in population-scale sequencing is to efficiently discover genome variation while making as few false discoveries as possible. While diverse methods for identifying SVs have been described 2-9, to date their sensitivity and specificity have not been measured on empirical, population-scale sequence data sets.
To evaluate the putative deletions discovered by ten algorithms (Supplementary Table 1) from population-scale sequence data, investigators from the 1000 Genomes Project assayed thousands of these putative deletions using array comparative genome hybridization (CGH), hybrid SNP/CNV arrays, and PCR 1,10. In array-based analyses of several thousand deletion calls, deletions identified by Genome STRiP showed an estimated false discovery rate (FDR) of 2.9% (ref 1); this rate was confirmed (to within statistical sampling error) by independent PCR experiments on a randomly selected set of 100 deletion calls 1. A composite FDR estimate for Genome STRiP of 3.7% (obtained by applying the PCR-based FDR to all variants for which array-based data were uninformative) compared with rates of 5.9% for Spanner (DA Stewart, GT Marth) and 23-70% for the eight other approaches evaluated on low-coverage data (Fig. 4d and ref 1). A total of 5,833 (83%) of the deletions predicted by Genome STRiP were explicitly validated using PCR, array data or breakpoint assembly 10.
In addition to producing the most accurate predictions, Genome STRiP was also the most sensitive of the ten algorithms evaluated by the 1000 Genomes Project, on both of the following criteria: (i) discovery of the deletions identified in the highest-resolution array-based study, Conrad et al.12 (Fig. 4e); and (ii) the number of deletions explicitly validated in array- and PCR-based experiments (Supplementary Figure S3 and ref 1). An alternative way of estimating sensitivity is to use a set of CNVs ascertained in just one individual, though this approach heavily weights common variants because the probability of a variant being present in any one genome is proportional to the variant’s allele frequency. Against three such individual-genome reference data sets, Genome STRiP was the most sensitive against two (Mills et al. 16 and Tuzun et al.13) and the second most sensitive against the third one (Conrad et al.12, when downsampled to one person) 10.
A particularly vexing and important challenge in low-coverage sequencing is to efficiently ascertain low-frequency alleles 1. To evaluate sensitivity for low-frequency alleles, we used a gold-standard set of deletions that had been genotyped in these same 168 genomes to establish allele frequency (ref 12) and evaluated the power of SV discovery as a function of allele frequency (Fig. 4e). Genome STRiP was again the most sensitive of the ten algorithms – and more so at the lowest allele frequencies – though it only partially ameliorated the weakness of low-coverage sequencing for detecting rare alleles (Fig. 4e). To assess how Genome STRiP might perform for rare alleles at higher levels of sequence coverage, we utilized the fact that the “low-coverage” genomes in the 1000 Genomes pilot are in fact a mixture of genomes with different levels of coverage, ranging from 2x to 8x span coverage (Methods). Genome STRiP’s power to detect rare singletons (deletion alleles present only once among the surveyed genomes, according to genotype data from Conrad et al.) rose quickly with increasing sequence depth, from less than 10% in genomes with less than 2x coverage to more than 80% in genomes with more than 8x coverage (Supplementary Fig. S4). In the 8x genomes, Genome STRiP achieved a sensitivity comparable to the most sensitive high-coverage algorithm in deeply sequenced (>30x) individual genomes, though Genome STRiP’s FDR was far lower (Supplementary Fig. S4).
Genome STRiP still showed incomplete sensitivity in absolute terms. When all ten discovery methods (including Genome STRiP) were used together, many more deletions were identified 1. The most complementary method (Pindel, ref 2), is local-assembly-based and identifies more small (< 300 bp) deletions; for Genome STRiP, sensitivity fell below 50% for deletions smaller than 300 bp (Supplementary Figure S5). Given the above observations, a key direction for the evolution of Genome STRiP will be to increase sensitivity for rare and small SVs. Ongoing technical advances will facilitate this: longer sequence reads (100+ bp) and gapped alignments 17 will allow Genome STRiP to take advantage of breakpoint-spanning reads in the ab initio SV discovery step, increasing ascertainment of small and rare alleles.
Breakpoint localization
We estimated the breakpoint locations of common SVs, generally at 1–20 bp resolution, by combining data across all the individuals determined to share an SV allele in common (Fig. 4d-f). Breakpoint locations were estimated at each locus by evaluating the likelihood of the sequence data (the aberrant but coherent read pairs from all genomes) given each potential breakpoint model, all observed read pairs, and the insert size distributions of each library sequenced (Fig 2b, 4f). The resulting confidence intervals were generally tight, particularly for common deletions, since each informative read-pair contributed information (Fig. 4f,g). Comparison to validated breakpoints1 confirmed the accuracy of these predictions (Fig. 4g).
With this breakpoint localization and breakpoint-spanning reads from many genomes, it was often possible to assemble unmapped sequence reads into a precise breakpoint sequence (Fig. 4h). A comprehensive breakpoint assembly analysis was undertaken by the 1000 Genomes Project 1; in the genotyping analyses below we use this larger breakpoint library and the complete set of deletions discovered by all algorithms in high- and low-coverage sequence data.
Genotyping structural polymorphism in populations
To evaluate the relationship between structural variation and phenotypic variation, studies will need to go beyond variation discovery – making lists of alternative structural alleles that are observed in at least one genome – to population genotyping, precisely determining the allelic state (or genotype) of every SV in every genome (Fig. 1b).
Many sources of information in population-scale sequence data – paired-end alignments, read depth, and breakpoint-spanning reads – could in principle each supply partial information about the allelic state of each SV in each genome. We reasoned that combining such information might enable a powerful way to genotype SVs of all sizes, and developed a Bayesian framework for integrating all of this information into a calibrated measurement of genotype likelihood (Fig. 5).
Figure 5.
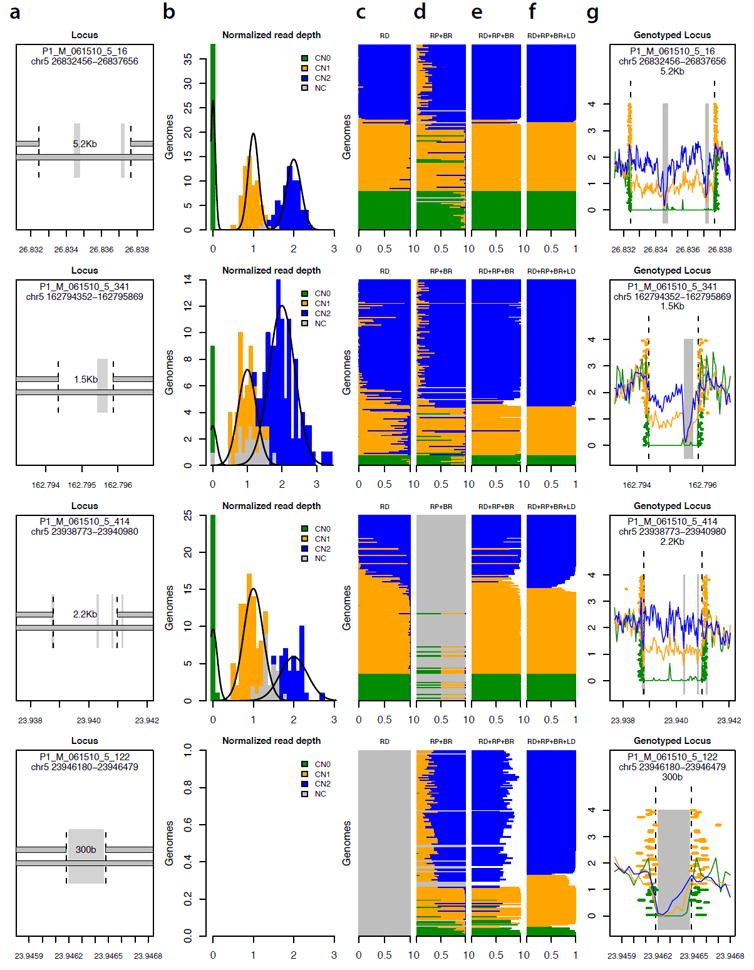
Determining the allelic state (genotype) of 13,826 deletions in 156 genomes.
(a) Four of the 13,826 deletion polymorphisms analyzed, representing diverse properties in terms of size and alignability of the affected sequence. Grey vertical rectangles indicate sequence that is repeat-masked or otherwise non-alignable. The locus in the bottom row is an ALU insertion polymorphism.
(b) Population-scale distribution of read depth across genomes, at each of the deletion loci in (a). For each locus, normalized measurements of read depth (across the deleted segment) from 156 genomes are fitted to a Gaussian mixture model. Colored squares represent genomes for which genotype could be called at 95% confidence based on read depth.
(c) Genotype likelihood from read depth. Each horizontal stripe (corresponding to one of the 156 genomes) is divided into three sections with length proportional to the estimated relative likelihood of the sequence data given each genotype model (blue: copy-number 2; green: copy-number 1; orange: copy-number 0).
(d) Genotype likelihood based on evidence from read pairs (RP) and breakpoint-spanning reads (BR). At the third locus from top, the absence of an established breakpoint sequence limits inference to read pairs.
(e) Genotype likelihood based on integrating evidence from read depth (RD), read pairs (RP) and breakpoint-spanning reads (BR).
(f) Genotype likelihood based on integrating evidence from (c-e) with flanking SNP data in a population haplotype model.
(g) Population-scale sequence data at each locus, as resolved into genotype classes. Traces indicate average read depth for genomes of each inferred genotype. Orange and green rectangles indicate evidentiary read pairs and breakpoint-spanning reads, colored by the genotype determination for the genome from which they arise.
To utilize read depth, we normalized measurements of locus-specific read depth for each of the 168 genomes, then clustered these measurements in a Bayesian mixture model (Fig. 5b, Methods). Importantly, genomes were clustered with and therefore calibrated to other genomes (Fig. 5b). The mixture model was used to estimate the relative likelihood of each potential underlying copy number (Fig. 5c).
To utilize breakpoint-spanning reads and read pairs, we aligned all unmapped reads to a breakpoint library that contained sequences of all alternative structural alleles identified by the 1000 Genomes Project pilot 1,10. At each locus in each genome, we determined the number of sequence reads corresponding to the reference and deletion alleles, and estimated the likelihood that this combination of read counts and read pairs could arise from each possible SV genotype (Fig. 5d).
Our Bayesian framework combined these three sources of information into an integrated measurement of the relative likelihood that the sequence data from each genome arose from each potential combination of structural allele at that locus (Fig. 5e, Methods). To assess the calibration of the resulting genotype likelihoods, we compared to CNV genotypes from the largest array-based study (ref 12). The calibrated confidence of each sequencing-based genotype call matched the concordance of such genotypes with array-based genotypes (Supplementary Table 2). For small (< 328bp) deletions, only a minority of genotypes could be inferred at high confidence; we therefore sought to extend genotype calling by drawing upon another population-based source of information, the haplotypes formed by SNPs and SVs together.
Most of the common SVs genotyped to date have been found to segregate on specific SNP haplotypes, reflecting the haplotype background on which each structural mutation occurred 11,12. We reasoned that a population-genetic haplotype model, such as that embedded in imputation algorithms 18-21, could help resolve the genotype uncertainty that remained for many genomes (Fig. 5e). We integrated the SV genotype likelihoods together with SNP genotypes in the same genomes 22 into SNP/SV haplotype models using the BEAGLE software 20, and used this to extend genotyping to more genomes (Fig. 5f). Intuitively, this used high-confidence genotypes to build models of the haplotypes segregating in a population, which were then used to resolve uncertainty about lower-confidence genotypes. This approach yielded genotypes that were consistent with all features of the sequence data (Fig. 5g) and also with the haplotype structure of the population. We generated genotypes for 13,826 of the deletion polymorphisms (48 to 959,782 bp in size) identified by the 1000 Genomes Project, with an average call rate of 94.1% (median 99.4%) (Methods). The genotyped loci included 1,123 mobile-element insertion polymorphisms, which have been refractory to genotyping by earlier, array-based methods. The remaining loci, for which we were unable to obtain high-quality genotypes, were mostly short deletions with less than 200 bp of uniquely mappable sequence and no assembled breakpoints.
To evaluate the accuracy of our genotype calls, we took several approaches. Across 1,970 common deletions for which high-quality genotype data existed (from ref 12), concordance of our genotypes with the array-based data was 99.1% (98.9% for homozygous deletions, 99.8% for homozygous reference allele, and 95.6% for heterozygous sites). Because this analysis included relatively few short deletions (< 1 kb) due to the resolution of the array-based genotyping used for comparison, we evaluated the linkage disequilibrium (LD) between our entire set of deletion genotypes and SNP genotypes from the same genomes. The LD properties of the full set of deletions (Supplementary Fig. S6, Supplementary Table 3) closely matched the known LD properties of SNPs 23 and multi-kilobase deletions 11, a relationship that was extremely unlikely to arise by chance or in data with a high genotyping error rate.
These data comprise the structural variation genotype data release of the 1000 Genomes Project pilot 1.
Discussion
We have described a new analytical framework for analyzing sequence data that arise from a large number of genomes. Our results show that re-interpreting the technical features of sequence data at a population level improves the quality and extends the power of inferences from sequence data. There are in principle many more ways in which these ideas could be combined with technical features of sequence data (Fig. 1) to ascertain and accurately type other forms of genome variation in populations.
We envision several ways in which our approach will be used. One will be to construct maps of the genome polymorphism that is segregating in populations. Such populations will include the human reference populations being analyzed in the 1000 Genomes Project, other human populations such as population isolates, and populations drawn from other species. Genome STRiP’s ability to discover polymorphisms efficiently and accurately, and to produce accurate genotypes – the substrate for haplotypes, measurements of allele frequency, and population genetic analysis – will increase the utility of genome variation data resources.
Another application of Genome STRiP will be in studies to uncover genome variation underlying complex phenotypes. Genome STRiP can be used in such studies in two ways. First, Genome STRiP can uncover structural alleles that are present among individuals with a particular phenotype but rare in the general population and therefore absent from resources such as 1000 Genomes. The low-coverage 1000 Genomes data analyzed here presented a more stringent test than will be presented by most disease studies, many of which will use intermediate or higher levels of sequence coverage that support greater sensitivity for rare variants. Second, Genome STRiP can be used to accurately determine the allelic state or genotype of each variant in each genome analyzed, allowing variation to be accurately correlated with phenotypes. The 13,826 deletion polymorphisms for which Genome STRiP produced genotypes here exceeds 10-fold the number of CNVs genotyped in our earlier efforts to develop hybrid SNP/CNV arrays for genome-wide association studies 11.
For common deletion alleles, many relationships to human phenotypic variation can be identified immediately by analyzing genome-wide association data together with the LD relationships identified here. Such relationships could be identified via tagSNPs (Supplementary Table 3) or imputation 18-20. To assess the potential yield of such approaches, we identified 70 reported phenotype-SNP associations (involving 56 unique phenotype-CNV pairs) that appear to be in LD (r2 > 0.8) with one or more of these variants (Supplementary Table 4), extending earlier efforts 12,24,25 that have identified 14 such relationships at this r2 threshold to date.
We have described ideas that could form the basis for new kinds of analytical approaches as sequencing-based studies are extended to large populations. Together with methods for analyzing single-nucleotide variation and small indels 26, these approaches will help realize the scientific potential of sequence data that are generated at a population scale.
Methods
Sequence data requirements
Genome STRiP requires paired-end sequence data that is generated from at least 10 genomes, at high, low, or intermediate levels of sequence coverage.
Sequence data used
Structural variation discovery was performed using the low-coverage Illumina sequence data for 168 individuals from the 1000 Genomes Project, including six higher-coverage genomes down-sampled to ~4x coverage for one CEU and one YRI trio. Two genomes were excluded due to data quality issues. The average genome-wide sequence coverage from the mapped Illumina data ranged from 0.8x to nearly 7x across different genomes, with 16 genomes sequenced at 2x or less average coverage. The “span coverage” – the amount of sequence physically flanked by paired-end reads (a better measurement of power for structural-variation methods that utilize paired ends, as Genome STRiP does) – ranged from 0.8x to nearly 9x. Of the 168 genomes, 24 had no paired-end data, which reduced the effective size of our discovery population to 144 genomes. Sequence reads were aligned by the 1000 Genomes Project to the hg18 reference human genome using the MAQ alignment algorithm 27. We reprocessed the data with Picard MarkDuplicates to achieve uniform removal of potential molecular duplicates.
Coherence and clustering
Candidate deletions were first identified as genomic clusters of at least two read pairs that were each “aberrant” in the sense that the left and right read aligned to the genome with unlikely (excessive) spacing, based on the empirical insert size distribution for each read group (corresponding to one sequencing lane). Each insert size distribution was characterized by the median value and a variance estimate (robust standard deviation, RSD) calculated as the width of the middle 68.2% of the distribution. Median insert sizes ranged from 108 to 469 (median 163) with RSD values ranging from 3% to 34% (median 11%) of the median insert size. Read pairs were used for clustering if they had correct orientation, a MAQ mapping quality of at least 10 on both ends and if the nominal insert size (measured by mapping to the reference genome) exceeded the median expected insert size by at least ten RSD. This threshold was motivated by the extremely large size of the data sets analyzed.
Clusters of aberrant read-pairs were formed using a connected components algorithm. Two read-pairs were considered connected if it was possible for them to share a breakpoint location such that the fragment length implied by the shared breakpoint was within the lower ~99% (median + 2.33 RSD) of both insert size distributions. Note that these criteria involve an implicit estimate of pairwise coherence.
After read-pair clustering, coherence was evaluated at the cluster level by first determining the most likely deletion length (dopt) that would explain the spacing and location of the read pair alignments, based on their insert size distributions (Supplementary Note).
Using dopt , we calculated an “incoherence” metric FC(dopt) where
This metric captures the degree to which a deletion event of length dopt would explain the observed cluster of read pairs. We tested by simulation the deviation of this metric from the null model
where u is uniformly distributed.
Assessment of population heterogeneity
To measure the heterogeneity in the distribution of evidence for each candidate deletion, we counted the number of evidentiary read pairs observed in each genome and then computed a chi-square test statistic
where Oi is the observed number of evidentiary read pairs for genome i and Ei is the expected number of evidentiary read pairs under a null model in which each genome is equally likely (per molecule sequenced) to produce an evidentiary read pair. Ei was calculated based on the genome-wide coverage depth and empirical insert size distribution of the reads for genome i. We estimated a p-value for this test statistic by Monte Carlo simulation.
Assessment of allelic substitution
An allelic substitution statistic was calculated for each candidate deletion by comparing average read depth (across the putatively deleted segment) for genomes containing evidentiary read pairs that supported this putative deletion (group A) to average read depth for genomes lacking such evidentiary read pairs (group B). A depth ratio DR was calculated as DA / DB. We additionally tested whether the numbers of reads (at the deletion versus elsewhere in the genome) differed between group A and group B genomes, using a 2x2 contingency table chi-square test on the number of reads in each category.
Integration of coherence, heterogeneity and substitution criteria
For identifying deletions in the 1000 Genomes pilot data, we evaluated different combinations of thresholds based on the metrics described above; our evaluation criteria included the ability to ascertain known, positive-control SVs (from ref 12) and homozygosity for array-based SNP genotypes (from ref 22). We produced our call set for the 1000 Genomes Project pilot by applying the following thresholds: incoherence metric > 0.01; substitution p-value < 0.01; depth ratio <= 0.63, or depth ratio <= 0.8 and heterogeneity p-value < 0.01; and median normalized read depth of samples with observed evidentiary pairs < 1.0 (this last filter was used to remove calls in regions of unusually high sequence coverage across many samples). In general, the optimal choices of parameters and thresholds for this step in Genome STRiP analysis will be a function of the sample size, sequence coverage, read length, and insert size used in a study, and should therefore be optimized for each study using a gold standard SV data set (we used the data from ref 12; future studies may wish to use the much larger data set from the 1000 Genomes Project pilot 1) and the expectation of a realistic number of novel discoveries.
Evaluation of accuracy (specificity)
Experimental evaluation of SV discovery data sets was performed by a group of investigators for the 1000 Genomes Project and is described in detail in Supplementary Note and ref 1,15.
Evaluation of sensitivity
We evaluated sensitivity using multiple reference data sets to better understand the relationship of sensitivity to allele frequency and deletion size. (i) In Fig. 4e, which shows how sensitivity relates to allele frequency of the underlying SV, we used the largest array-based copy number data set for which genotype (and therefore allele-frequency) information are available (Conrad et al.). (ii) In Supplementary Fig. S4, which shows how sensitivity relates to the size of the underlying deletion, we used a reference data set (from ref 15) that was enriched for small deletions (<1 kb) and called most sub-kilobase variants at basepair resolution; these data consisted of deletions identified from a single genome (NA12156) by earlier analyses of fosmids, capillary sequence traces and tiling-resolution array CGH. Consistent with analyses of SNP-discovery sensitivity in ref 1, we considered the genomes as a set and did not require that the 1KG discovery arise specifically from NA12156. In these analyses, we considered a reference variant “discovered” if a callset variant overlapped with it. (iii) In Supplementary Fig. S3, which shows how FDR relates to the total number of experimentally validated discoveries from each method, we utilized data on the number of variants (predicted by each sequencing-based method) that validated at high stringency by array-based analysis, PCR or assembly of a breakpoint sequence; the raw data are reported in Supplementary Table 8 of ref 1.
Genotyping
Genotyping of deletion polymorphisms was attempted for 22,025 loci discovered by the 1000 Genomes Project. Genotyping was performed on loci discovered in both high-coverage and low-coverage sequencing and was not limited to SVs discovered by Genome STRiP. Genotyping was performed using the low-coverage Illumina sequencing data from the 1000 Genomes project for 168 individuals, including downsampled data at ~4x coverage for the CEU and YRI trios. Data curation and pre-processing steps were the same as for the sequencing data used for discovery, as described above.
Input to genotyping in Genome STRiP consisted of (i) a list of putative deletion loci with optional confidence intervals on the breakpoint locations, and (ii) a breakpoint sequence for each alternate structural allele, where available. Genotypes for each individual were determined by first calculating genotype likelihoods from three different sources of evidence from sequencing data (read depth, discordant read pairs and breakpoint-spanning reads, each described in subsequent sections) and then combining these likelihoods into a joint initial likelihood for each individual. Genotyping was performed independently for each putative deletion in the 1000 Genomes call set, even when there were other physically overlapping deletion calls. BEAGLE was used to combine these initial likelihoods with genotypes from nearby SNPs to arrive at final genotypes that benefit from taking into account LD between the deletions and nearby SNPs.
Utilization of read depth in genotyping
For each deletion locus, the number of sequenced fragments falling within the deleted region was counted for each sample, requiring a minimum mapping quality of 10, and correcting for the effective length of the deletion locus. The effective length excludes all base positions where less than half of the overlapping 35-mers were not unique (as defined by an alignability mask generated by the 1000 Genomes Project 1). The expected number of fragments for each sample was estimated based on the genome-wide sequencing coverage, the alignability mask and the effective length of the deleted region.
The read depth within the deletion locus was used to estimate the copy number at the locus using a constrained Gaussian mixture model applied to the observed and expected read counts for each sample (Supplementary Note). Based on the estimated model parameters and the observed read depths, we calculated the relative likelihood of each copy number class for each genome and converted this to genotype likelihoods (e.g. copy number zero corresponds to a homozygous deletion).
Utilization of discordant read pairs in genotyping
Discordant read pairs that spanned each deletion were utilized when the strand orientation was normal and the fragment length implied by the deletion was within the median expected insert size + 3 RSD. Discordant read pairs were not used as evidence of the alternate allele if they had a plausible alternative mapping (<= 0.25 mismatches/base after performing a quality-aware Smith-Waterman realignment) to the reference for either end of the read that would correct the nominal insert size to within the median expected insert size +/- 5 RSD. We used these stringent filters to select only read pairs that were (a) highly likely under the model of an intervening deletion and (b) unlikely to arise from mis-alignment, one important source of artifactual discordant read pairs that we were able to mitigate. Likelihoods from discordant read pairs were generated based on the mapping quality and the likelihood of the nominal insert size given the original mapping, in a model that implicitly made copy number 0 or 1 equiprobably much more likely than copy number 2 given a discordant pair observation.
Utilization of breakpoint-spanning reads in genotyping
Genotyping made use of a library of assembled breakpoints for 10,455 loci generated as part of the 1000 Genomes Project 1,15 by the algorithms TIGRA (L. Chen, unpublished) and Pindel 2. A non-redundant set of breakpoint sequences was extracted from this library and preprocessed to remove any alleles with inconsistent annotations and any mismatches to the reference sequence in the flanking regions of the alternate alleles. We also performed an automated procedure to detect inconsistencies in the mapping of the alternate allele assemblies to the breakpoints by testing whether small shifts in the alignment to the reference sequence reduced the number of mismatches. Assemblies with inconsistent annotations were not used. Reads from the unmapped BAM files from the 1000 Genomes Project were aligned to these alternate alleles using BWA 17 version 5.5 with default parameters.
For genotyping, we utilized any read that aligned across a breakpoint junction and would discriminate between the alternative alleles. Breakpoint-spanning reads were ascertained from three sources: (a) alignments to the breakpoint locations in the original BAM files, which were realigned against the alternate allele for comparison; (b) unmapped mates of paired reads that aligned near the breakpoints, which were aligned against the alternate allele for comparison; and (c) reads from the unmapped BAM files that aligned to the library of alternate alleles using BWA. The likelihoods for the three genotype classes (homozygous reference, heterozygote, homozygous alternate) for each read were determined based on the sum of base qualities of the mismatches to the reference and alternate alleles, the estimated mapping quality to both alleles, and the insert size distribution for paired reads. We corrected for the reference having two deletion breakpoint junctions, while the alternate allele has only one, taking into account both sequence homology at the junction and read length.
Genotype likelihood estimation
The likelihoods from these three sources of evidence (read depth, read pairs and breakpoint-spanning reads) were combined into joint initial likelihoods for the genotype of each sample. Likelihoods from breakpoint-spanning reads were computed only when an alternate allele sequence was available. Likelihoods from read depth were included only when the uniquely alignable (“effective”) length of the deletion exceeded 200 bp.
To improve the genotype calls, LD between the deletion events and nearby SNPs was utilized. The initial genotype likelihoods were combined with SNP genotype data from the International Hapmap Project 22 (for 156 of the 168 samples for which SNP genotypes were available in Hapmap Phase III, release 2) using BEAGLE 20 3.1. The SNP genotypes were input to BEAGLE assuming a genotyping error rate of 0.1%. BEAGLE was run separately on each population (CEU, YRI and CHB+JPT). The trio parents and children were run separately. The BEAGLE output was converted back to normalized relative likelihoods of the three genotype classes for each genome.
Genotyped loci
A set of genotypable deletions for the 1000 Genomes Project was selected that met the following two criteria: (a) at least 50% of the genomes had a genotype call that was >95% confident; and (b) the genotype calls were in Hardy-Weinberg equilibrium in each of the three populations (p > 0.01, trio offspring excluded).
Overall, 10,742 of 15,893 deletion sites discovered in the low coverage samples met these criteria, as did 6,317 of 11,248 sites discovered in the high coverage trios. This yielded 13,826 sites out of 22,025 after merging discoveries that were determined by the 1000 Genomes analysis to be redundant between the low- and high-coverage discovery sets. Our genotyping analysis also suggests that some of the 1000 Genomes deletion calls are potentially redundant with other, physically overlapping 1000 Genomes deletion calls, as revealed by their yielding identical genotypes across the genomes analyzed; this reflects that these deletions were discovered by different algorithms with varying levels of resolution 15, and that efforts in the 1000 Genomes pilot to combine potentially redundant calls from different algorithms were not completely successful. We have reported all supplementary data and statistics here in a way that is synchronized with the 1000 Genomes pilot data sets and analyses as reported in ref 1,10.
Supplementary Material
Acknowledgments
The authors wish to thank the 1000 Genomes Structural Variation Analysis Group for helpful conversations throughout this work and for collaborative work to evaluate the sensitivity and specificity of SV discovery algorithms. We would particularly like to acknowledge Ken Chen for creation of a high-quality breakpoint library for the 1000 Genomes Project, on which Genome STRiP’s genotyping algorithm drew, and Ryan Mills for managing the 1000 Genomes deletion discovery sets and validation data. We also thank Chip Stewart, Klaudia Walter, Matt Hurles, and Nick Patterson for helpful conversations during the course of this work; David Altshuler, Mark DePristo, and Mark Daly for helpful comments on the manuscript and figures; and the anonymous reviewers of this manuscript, whose feedback improved it. This work was supported by the National Human Genome Research Institute (U01HG005208-01S1) and by startup funds from the Department of Genetics at Harvard Medical School.
Footnotes
Author Contributions
R.E.H., J.M.K., J.N., and S.A.M. conceived the analytical approaches. R.E.H. implemented the algorithms and performed the data analysis. R.E.H. and S.A.M. wrote the manuscript.
URLs
1000 Genomes Project: http://www.1000genomes.org
Genotypes generated by Genome STRiP: ftp://ftp-trace.ncbi.nih.gov/1000genomes/ftp/pilot_data/paper_data_sets
GenomeSTRiP web site: http://www.broadinstitute.org/gsa/wiki/index.php/Genome_STRiP
References
- 1.1000_Genomes_Project_Consortium. A map of human genome variation from population scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ye K, Schulz MH, Long Q, Apweiler R, Ning Z. Pindel a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009;25:2865–2871. doi: 10.1093/bioinformatics/btp394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lam HY, et al. Nucleotide-resolution analysis of structural variants using BreakSeq and a breakpoint library. Nat Biotechnol. 2010;28:47–55. doi: 10.1038/nbt.1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Korbel JO, et al. Paired-end mapping reveals extensive structural variation in the human genome. Science. 2007;318:420–426. doi: 10.1126/science.1149504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Korbel JO, et al. PEMer: a computational framework with simulation-based error models for inferring genomic structural variants from massive paired-end sequencing data. Genome Biol. 2009;10:R23. doi: 10.1186/gb-2009-10-2-r23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen K, et al. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat Methods. 2009;6:677–681. doi: 10.1038/nmeth.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chiang DY, et al. High-resolution mapping of copy-number alterations with massively parallel sequencing. Nat Methods. 2009;6:99–103. doi: 10.1038/nmeth.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yoon S, Xuan Z, Makarov V, Ye K, Sebat J. Sensitive and accurate detection of copy number variants using read depth of coverage. Genome Res. 2009;19:1586–1592. doi: 10.1101/gr.092981.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alkan C, et al. Personalized copy number and segmental duplication maps using next-generation sequencing. Nat Genet. 2009;41:1061–1067. doi: 10.1038/ng.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mills RE, et al. Mapping copy number variation by population scale sequencing. Nature. doi: 10.1038/nature09708. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McCarroll SA, et al. Integrated detection and population-genetic analysis of SNPs and copy number variation. Nat Genet. 2008;40:1166–1174. doi: 10.1038/ng.238. [DOI] [PubMed] [Google Scholar]
- 12.Conrad DF, et al. Origins and functional impact of copy number variation in the human genome. Nature. 2009 doi: 10.1038/nature08516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tuzun E, et al. Fine-scale structural variation of the human genome. Nat Genet. 2005;37:727–732. doi: 10.1038/ng1562. [DOI] [PubMed] [Google Scholar]
- 14.Iskow RC, et al. Natural mutagenesis of human genomes by endogenous retrotransposons. Cell. 2010;141:1253–1261. doi: 10.1016/j.cell.2010.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang CR, et al. Mobile interspersed repeats are major structural variants in the human genome. Cell. 2010;141:1171–1182. doi: 10.1016/j.cell.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mills RE, et al. An initial map of insertion and deletion (INDEL) variation in the human genome. Genome Res. 2006;16:1182–1190. doi: 10.1101/gr.4565806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marchini J, Howie B, Myers S, McVean G, Donnelly P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet. 2007;39:906–913. doi: 10.1038/ng2088. [DOI] [PubMed] [Google Scholar]
- 19.Li Y, Willer C, Sanna S, Abecasis G. Genotype imputation. Annu Rev Genomics Hum Genet. 2009;10:387–406. doi: 10.1146/annurev.genom.9.081307.164242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Browning BL, Browning SR. A unified approach to genotype imputation and haplotype-phase inference for large data sets of trios and unrelated individuals. Am J Hum Genet. 2009;84:210–223. doi: 10.1016/j.ajhg.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coin LJ, et al. cnvHap: an integrative population and haplotype-based multiplatform model of SNPs and CNVs. Nat Methods. 2010;7:541–546. doi: 10.1038/nmeth.1466. [DOI] [PubMed] [Google Scholar]
- 22.International_HapMap3_Consortium. Integrating common and rare genetic variation in diverse human populations. Nature. 2010;467:52–58. doi: 10.1038/nature09298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.International_HapMap_Consortium. A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McCarroll SA, et al. Deletion polymorphism upstream of IRGM associated with altered IRGM expression and Crohn’s disease. Nat Genet. 2008;40:1107–1112. doi: 10.1038/ng.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Willer CJ, et al. Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat Genet. 2009;41:25–34. doi: 10.1038/ng.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.DePristo M, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. submitted manuscript, under review at Nature Genetics. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li H, Ruan J, Durbin R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008;18:1851–1858. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.