

Abstract
Background
Pathogenic members of the Fusarium oxysporum species complex are responsible for vascular wilt disease on many important crops including legumes, where they can be one of the most destructive disease causing necrotrophic fungi. We previously developed a model legume-infecting pathosystem based on the reference legume Medicago truncatula and a pathogenic F. oxysporum forma specialis (f. sp.) medicaginis (Fom). To dissect the molecular pathogenicity arsenal used by this root-infecting pathogen, we sequenced its transcriptome during infection of a susceptible and resistant host accession.
Results
High coverage RNA-Seq of Fom infected root samples harvested from susceptible (DZA315) or resistant (A17) M. truncatula seedlings at early or later stages of infection (2 or 7 days post infection (dpi)) and from vegetative (in vitro) samples facilitated the identification of unique and overlapping sets of in planta differentially expressed genes. This included enrichment, particularly in DZA315 in planta up-regulated datasets, for proteins associated with sugar, protein and plant cell wall metabolism, membrane transport, nutrient uptake and oxidative processes. Genes encoding effector-like proteins were identified, including homologues of the F. oxysporum f. sp. lycopersici Secreted In Xylem (SIX) proteins, and several novel candidate effectors based on predicted secretion, small protein size and high in-planta induced expression. The majority of the effector candidates contain no known protein domains but do share high similarity to predicted proteins predominantly from other F. oxysporum ff. spp. as well as other Fusaria (F. solani, F. fujikori, F. verticilloides, F. graminearum and F. pseudograminearum), and from another wilt pathogen of the same class, a Verticillium species. Overall, this suggests these novel effector candidates may play important roles in Fusaria and wilt pathogen virulence.
Conclusion
Combining high coverage in planta RNA-Seq with knowledge of fungal pathogenicity protein features facilitated the identification of differentially expressed pathogenicity associated genes and novel effector candidates expressed during infection of a resistant or susceptible M. truncatula host. The knowledge from this first in depth in planta transcriptome sequencing of any F. oxysporum ff. spp. pathogenic on legumes will facilitate the dissection of Fusarium wilt pathogenicity mechanisms on many important legume crops.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-016-3192-2) contains supplementary material, which is available to authorized users.
Keywords: Fusarium wilt, Vascular wilt, RNA-Seq, DEG, root pathogen, Necrotroph, Hemibiotroph, Effector, Secreted In Xylem, Small secreted protein
Background
Fusarium oxysporum is a soil-borne fungal pathogen capable of causing widespread destructive losses on over 100 different plant species. Specialised pathogenic strains of this root-infecting fungus are classified into host-specific sub-species known as formae speciales (ff. spp.) (singular forma specialis, abbreviated: f. sp.) based on the host species they cause disease on, and are responsible for the disease known as Fusarium or vascular wilt [1–4]. The spores of this pathogen can survive in soil for decades, thus it is particularly difficult to eradicate following soil contamination [1]. Important agronomical crops which are affected by Fusarium wilt include cotton (Gossypium species), horticultural crops such as bananas (Musa species), cucurbits/melons (Cucurbitaceae species), strawberries (Fragaria × ananassa), lettuce (Lactuca sativa) and tomatoes (Solanum lycopersicum), and many grain and pasture legume species such as chickpea (Cicer arietinum), common bean (Phaseolus vulgaris), field pea (Pisum sativum), lentil (Lens culinaris) and lucerne/alfalfa (Medicago sativa) [1–6].
It was proposed for some isolates of a F. oxysporum f. sp. that their ability to cause disease on specific hosts arose through descent from a monophyletic origin. However, for others it was proposed their genetic heterogeneity was polyphyletic in origin and for several this has now been experimentally demonstrated [3, 7–11]. Comparative genomic studies across Fusaria identified ‘core’ chromosomes containing genes required for vegetative growth and metabolism, and ‘non-core’ chromosomes (or parts of) and gene content geared towards pathogenicity [9, 12]. The latter, also referred to as lineage-specific, accessory or conditionally dispensable chromosomes (CDCs), lack house-keeping genes and are poorly conserved across other Fusaria or other fungi, but may show higher levels of disparate conservation amongst specific F. oxysporum ff. spp. [5, 9, 13]. In a series of elegant experiments, it was demonstrated that the small CDC 14 (~1.6 Mb) and parts of other CDCs from some F. oxysporum f. sp. lycopersici (Fol) isolates (including the reference isolate Fol-4287) could be horizontally transferred to other isolates, enabling the transfer of pathogenicity [9, 10]. Conversely, a loss of pathogenicity or virulence can also result from the loss of all or parts of Fol Chr14 [10, 14–16]. The small Fol-4287 CDC 14 is referred to as the Fol ‘pathogenicity’ chromosome as it contains the majority of known Fol in planta expressed effectors, some of which have been shown to interfere with the host’s resistance response and/or are required for virulence [3, 9, 13, 14, 17, 18].
The CD chromosomes or scaffolds of F. oxysporum ff. spp. analysed in some detail to date (f. sp. lycopersici, melonis, medicaginis, ciceris, pisi, cubense) are enriched in repetitive elements with CDC encoded transposable elements (TEs) accounting for nearly 75 % of all TEs in the Fol-4287 genome [5, 9, 13, 19]. While only 20 % of Fol-4287 genes on these chromosomes can currently be functionally classified based on the presence of conserved domains, they are enriched for genes related to pathogenicity such as known and putative effectors, fungal transcription factors and genes with roles in signal transduction and secondary metabolism [9]. Similarly, over half of the predicted proteins from the legume-infecting ff. spp. medicaginis (Fom-5190a), ciceris (Foc-38-1) or pisi (Fop-37622) genome assemblies assigned to predicted dispensable scaffolds are unclassified proteins with no known function and those that could be assigned functional annotations grouped into similar categories as those enriched on Fol CDCs [5].
F. oxysporum is one of the major pathogens of legumes, particularly chickpea, the second most important global grain legume crop (FAO: www.fao.org). Typical annual yield losses due to pathogenic isolates of this host, F. oxysporum f. sp. ciceris, are upwards of 10 % but under favourable disease conditions yield loss can reach 100 % [20–23]. With the majority of the world’s chickpea production originating from one country (India) (FAO: www.fao.org), disease outbreaks and a lack of control mechanisms can have severe impact on global chickpea supplies. Various sources of host resistance in chickpeas and other legumes have been identified, but the underlying genetic or molecular mechanisms (e.g. Resistance or Pathogenesis-Related genes, signalling pathways) are yet to be fully elucidated [3, 20, 24, 25]. Parallel to this, genetic and molecular mechanisms responsible for individual F. oxysporum f. sp. pathogenicity on legumes is poorly understood. Towards the aim of transferring knowledge to complex legume species, pathosystems utilising the reference legume Medicago truncatula have been developed to dissect the interaction between F. oxysporum and legume hosts [5, 26–28]. Utilising the pathogenic F. oxysporum f. sp. medicaginis strain Fom-5190a (herein referred to as Fom) isolated from alfalfa (Lucerne, M. sativa) we developed a robust Medicago-F. oxysporum pathosystem and identified a resistant and highly susceptible accession [5, 29]. These are the reference M. truncatula accession A17 (resistant) and the accession DZA315 (susceptible).
Comparative genomics of a comprehensive draft Fom genome assembly against the genome assemblies of legume-infecting and other host pathogenic F. oxysporum ff. spp. as well as other Fusarium spp. and fungal plant pathogens, identified pathogenicity related gene content possibly geared towards legume host-specificity [5]. This information coupled with RNA-Seq data from an early time-point during infection of the susceptible M. truncatula accession (DZA315), facilitated the shortlisting of a set of 10 Fom key effector candidates. Herein, we set out to expand on this analysis and identify potential differences in the expressed pathogenicity profile of Fom during early infection of susceptible DZA315 versus resistant A17 M. truncatula hosts. One of the limiting factors in early detection of fungal transcripts in root colonised tissues is their poor relative abundance compared to host transcripts and to overcome this, high coverage sequencing or specific sequence capture of fungal transcripts is necessary. We overcame this constraint by conducting high coverage RNA-Seq and herein present one of the most comprehensive early in planta expressed F. oxysporum transcriptomes, and the first for a legume infecting formae speciales to our knowledge. Our RNA-Seq involved analysis of root samples collected at the early time-point of 48 h after infection and at a later time-point of 7 days when disease symptoms were starting to manifest in the susceptible plants. At the early time-point, although the degree of root colonisation between resistant and susceptible plants was alike, the number, level of induction, and composition of in planta expressed Fom genes was higher and more diverse in the susceptible interaction. By 7 days post inoculation a significant increase in colonisation of susceptible plants was evident coupled with increased expression of genes predicted as effectors or associated with protein, sugar and plant cell wall breakdown, membrane transport and nutrient uptake. We discuss these differences and the discovery of new candidate F. oxysporum effectors.
Results
Quantification of Fusarium growth in resistant and susceptible M. truncatula accessions
We previously identified the reference M. truncatula accession A17 to display moderate to strong resistance to Fom [29] whilst the DZA315 accession was susceptible [5]. To examine differences in disease progression between these two hosts we infected both alongside each other and quantified Fom growth in root and shoot tissues over an infection time-course (Fig. 1). Within 14 days post inoculation (dpi) 97 % of DZA315 plants had visible disease symptoms of wilting, and chlorotic or necrotic leaves, while only 30 % of A17 plants were diseased and of these, only 6 % of their leaves on average had visible disease symptoms (Fig. 1a, b). By 21 dpi all A17 plants survived, although the number of diseased plants had risen to 40 %, these again displayed limited symptoms (on average 5 % of leaves were diseased per plant, Fig. 1a). In contrast, all DZA315 plants were dead by 21 dpi (Fig. 1c). At 21 dpi the Fom inoculated A17 seedlings were visibly smaller than mock or control inoculated seedlings, showing a 30 % reduction in shoot fresh weight (Fig. 1d). The limited disease progression observed in A17 suggests Fom is able to colonise A17 seedlings but that the molecular resistance mechanisms employed by A17 plants to control pathogen spread results in reduced growth.
Fig. 1.
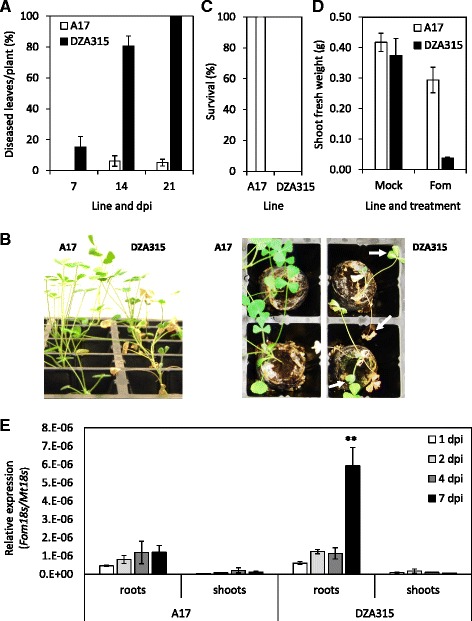
Disease symptoms and Fom colonisation of resistant and susceptible M. truncatula accessions. a-d Disease symptoms of Fom inoculated A17 and DZA315 M. truncatula seedlings. a Percentage of diseased seedlings at 7, 14 and 21 days post inoculation (dpi) with b) an image of representative seedlings at 14 dpi. White arrows highlight disease symptoms of wilting, chlorotic and necrotic leaves. c Average survival and d above ground fresh weight of mock (control) or Fom inoculated seedlings at 21 dpi. For a, c and d values are averages ± SE (n = 10). Similar results were obtained in independent experiments. e Relative Fom fungal abundance determined by qRT-PCR analysis of Fom_18S relative to M. truncatula_18S in samples harvested at 1, 2, 4 and 7 dpi. Samples are averages ± SE of 4 biological replicates consisting of pools of 10 seedlings. Asterisks indicate values that are significantly different (**P < 0.01 Student’s t-test) between A17 and DZA315 at the respective time point
To determine the extent of Fom colonisation in A17 and compare this to levels in susceptible accession DZA315, we quantified the relative amount of fungal biomass in root and shoot tissues of A17 and DZA315 seedlings by qRT-PCR over the early stages of infection between 1 and 7 dpi. Both A17 and DZA315 had similar levels of Fom biomass at 1, 2 and 4 dpi in roots, with some growth also detected in shoots (Fig. 1e). However, by 7 dpi the relative abundance of Fom had risen sharply and significantly in DZA315 root tissues to levels ~5-fold greater than levels in A17 which remained similar to those detected at its 4 dpi time-point.
In vitro and in planta Fusarium RNA-sequencing
We hypothesized that increased Fom colonisation of susceptible DZA315 seedlings may be associated with changes in its pathogenicity gene expression profile compared to colonisation of a resistant A17 host. To capture Fom genes expressed in resistant and susceptible plants and to compare and contrast their expression profiles we generated high coverage RNA-Seq data from infected root and shoot tissues of A17 and DZA315 seedlings at 2 dpi where we detected no difference in host colonisation levels and at the later time-point of 7 dpi when disease symptoms start to manifest in susceptible plants (Fig. 1). We confirmed that disease progressed as expected in seedlings used for RNA-sequencing by scoring the percentage of diseased plants and survival at 7-21 dpi on plants from the same experiment that were not harvested for RNA extractions (Additional file 1). We also collected data from Fom mycelia grown vegetatively (in vitro) in order to compare levels of induction upon host detection and use this as a feature to facilitate identification of effectors or other pathogenicity-associated genes.
Based on our previous qRT-PCR results (Fig. 1e) we estimated the relative abundance of Fom transcripts to M. truncatula transcripts in harvested root tissues would range from 0.05-0.5 %. Using this as a guide we conducted stranded RNA-Seq on three biological replicates for each treatment/tissue on an Illumina HiSeq platform (2x100 bp) generating 10.76−12.82 Gb data for each sample. After read processing (quality trimming and adaptor removal), ~55 million paired end reads were obtained for each sample and mapped to our Fom reference genome assembly [5] using TopHat2 [30]. For the in vitro samples 93-94 % of reads could be mapped to the Fom genome assembly, while for infected root samples the percentage of reads mapped ranged from 0.02-0.04 % at 2 dpi to 0.02−0.19 % at 7 dpi (Table 1). Less than 0.01 % of reads could be mapped from the shoot samples, even at the later time point of 7 dpi (data not shown). Combined, the in vitro and in planta RNA-Seq supported expression of 16,473 of the 16,858 predicted Fom gene models with 12,312 expressed in planta based on mapping of one or more reads in any of the in planta samples. Correlating with our fungal biomass results (Fig. 1e), similar percentages of reads could be mapped to the Fom genome in samples harvested from A17 and DZA315 roots at 2 dpi but at 7 dpi the number and percentage of mapped Fom reads was up to 10 times greater in the DZA315 samples compared to A17.
Table 1.
Mapping results of RNA-Seq data from Fusarium infected A17 (resistant) or DZA315 (susceptible) root samples
| in vitro | Inf. A17 2 dpi | Inf. A17 7 dpi | Inf. DZA315 2 dpi | Inf. DZA315 7 dpi | |
|---|---|---|---|---|---|
| Pre-processed reads for each replicate (million pairs) | 58.7 ± 1.5 | 56.2 ± 1.4 | 55.5 ± 1.6 | 53.4 ± 0.9 | 55.4 ± 0.6 |
| Range of read pair numbers mapped | 54,789,566 ± 1,026,861.7 | 12,626.0 ± 951.5 | 30,343.2 ± 6,309.8 | 14,713.8 ± 1,745.2 | 91,061.5 ± 5,934.4 |
| % range of reads mapped | 92.6-94.2 | 0.016-0.028 | 0.021-0.080 | 0.021-0.038 | 0.139-0.194 |
| Pairs with multiple alignments (%) | 0.2 | 0.4-0.5 | 0.4-0.5 | 0.2-0.3 | 0.4-0.5 |
Inf Fom infected, dpi days post inoculation; ±: SE
Identification of differentially expressed Fusarium genes
To identify genes with a high potential for involvement in pathogenicity, we set out to identify Fom genes differentially expressed between vegetative (in vitro) and in planta growth conditions with the premise that genes involved in fungal pathogenicity would be switched on or more highly and rapidly expressed upon detection of a suitable host [6, 18, 19, 31–33]. Aligned read counts (generated from the maploci and genDEseq subprocesses within the BioKanga toolkit [http://sourceforge.net/projects/biokanga/files/]) were used with a normalisation step to identify differentially expressed genes (DEGs) between each dataset (growth condition, dpi, plant accession) with EdgeR [34]. DEGs were selected based on a ≥ 2-fold change and a False Discovery Rate (FDR) ≤ 0.05. Due to low proportion of Fom reads in the RNA-Seq datasets, to decrease the number of false positives based on mapping of only a few reads or mapping of multiple reads to only one location, we added an additional criteria of at least 25 % read coverage of the predicted gene model in each of the 3 in planta replicates. In the early 2 dpi dataset replicates this equated to on average 84−92 % of transcripts with a minimum of 5 mapped reads, with individual gene models covered on average by 85 reads. Similar additional DEG criteria have been used in other lowly in planta expressed pathogen or endophyte studies [35, 36]. Full details of DEGs and characteristics of their encoded proteins are listed in Additional files 2 and 3.
At both 2 and 7 dpi the number of DEGs induced/repressed in planta in DZA315 was greater than those detected in A17 suggesting a larger degree of transcriptional reprogramming in the susceptible plant interaction (Fig. 2, Table 2). This was particularly evident at 2 dpi even when Fom fungal biomass was comparable between susceptible and resistant plant roots (Fig. 1e) with 11 % more DEGs in the DZA315 up-regulated dataset compared to A17, and 30 % more genes with ≥ 95 % read coverage (Table 2). By 7 dpi the difference in number of DEGs between DZA315 and A17 increased by over 3.5-fold, likely associated with the greater fungal colonisation of DZA315 roots (Fig. 1e, Table 2). In DZA315 80 % of DEGs up-regulated at 2 dpi were also up-regulated at 7 dpi, while this was only observed for 57 % of DEGs in A17 (Figs. 2b, c). Considering the larger number of mapped reads and DEGs identified in DZA315 at 7 dpi, less overlap between the 2 and 7 dpi DZA315 datasets might be expected. This result suggests a fast, prolonged, and concerted expression of components of the Fom pathogenicity arsenal during infection of susceptible plants. With the exception of Fom_00898 (expression down at 2 dpi DZA315 but up at 7 dpi DZA315) there were no genes detected in both up- and down-regulated datasets of either genotype. Interestingly Fom_00898 encodes a protein with characteristics of a small secreted protein (SSP; protein length ≤ 300 amino acids, predicted to be secreted (SignalP) and containing ≤ one transmembrane domain in the N-terminal region [5]), contains a CFEM domain possibly associated with fungal pathogenicity [37], and is predicted as a putative effector by the fungal effector prediction software EffectorP [38].
Fig. 2.
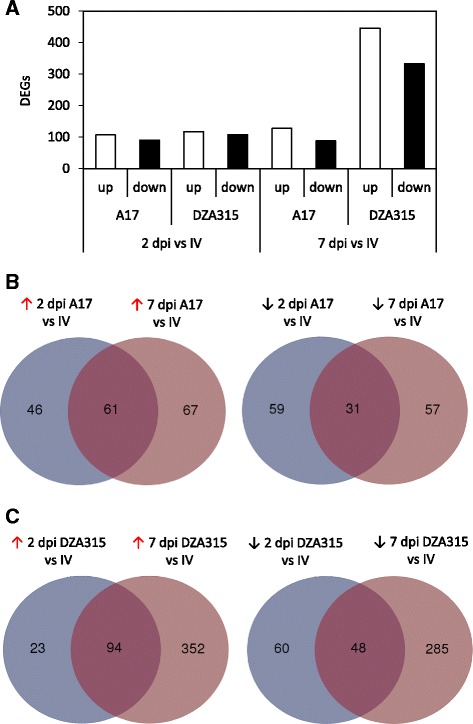
Number of Fom genes differentially expressed between in vitro and in planta samples. a DEGs detected between Fom grown in vitro (IV) and during infection of A17 or DZA315 roots at 2 or 7 days post inoculation (dpi). b-c Venn diagrams of DEGs in overlapping datasets from b) A17 and c) DZA315. Red and black arrows indicate up- or down-regulated in planta respectively
Table 2.
Differentially expressed Fusarium genes detected during infection of A17 or DZA315 roots
| Proportion of gene model coverage | A17 2 dpi vs IV | A17 7 dpi vs IV | DZA315 2 dpi vs IV | DZA315 7 dpi vs IV | ||||
|---|---|---|---|---|---|---|---|---|
| up | down | up | down | up | down | up | down | |
| ≥25 % | 107 | 90 | 128 | 88 | 117 | 108 | 446 | 333 |
| ≥50 % | 64 | 21 | 63 | 34 | 66 | 35 | 281 | 141 |
| ≥95 % | 13 | 1 | 25 | 5 | 17 | 4 | 105 | 28 |
IV in vitro. Note, DEGs in ≥ 95 % are also captured within the ≥ 50 % and ≥ 25 % datasets. Likewise, DEGs in ≥ 50 % are captured within the ≥ 25 % dataset
We also examined RNA-Seq data from the same experiment, at the same level of sample read coverage obtained from the shoots of the Fom inoculated DZA315 or A17 plants but due to the relative low abundance of fungal transcripts in shoot tissues at our early sampling time-points only a handful of DEGs could be identified (data not presented).
Protein characteristics of differentially expressed genes with a focus on in planta up-regulated datasets
To assess differences in pathogenicity profiles of Fom during interactions with susceptible or resistant Medicago hosts, we focussed the remainder of our studies on DEGs up-regulated in planta versus in vitro. To interpret putative functions, we interrogated their predicted protein characteristics including gene ontology (GO) terms, Pfam domains and pathogenicity associated characteristics such as encoding SSPs or similarity to known fungal effectors. Although fungal effectors generally lack similarity to other proteins, they may share common protein motifs or characteristics such as a secretion signal, small size (generally less than 300 aas), increased number of paired cysteines, proximity to repetitive DNA and these were therefore included in our analyses. Fom protein characteristics were previously determined by [5] and supported by annotations listed in publicly available Fusarium spp. genome projects, PHI-base [39, 40] and GenBank. We also incorporated an assessment of putative chromosomal location based on our previous study [5]. Fom scaffolds representing putative CD chromosomes were predicted based on the criteria of having no match to designated core chromosomes of F. oxysporum f. sp. lycopersici or F. solani, whose genome assemblies contain well characterised core and CD/accessory chromosome sequences.
One fifth of the predicted proteins in up-regulated DEG datasets could not be assigned functional annotations and were annotated as uncharacterised. This included 24 (22 % of total in the dataset) and 27 (21 %) in A17 at 2 and 7 dpi respectively. In DZA315 infected plants there were a similar number of uncharacterised proteins at 2 dpi (22 DEGs; 19 % of dataset) but within the 7 dpi dataset the total number of uncharacterised proteins more than tripled to 78 (representing 17 % of the dataset). Nearly half of the latter were predicted as secreted and as putative effectors by EffectorP.
Of proteins in the up-regulated datasets that could be assigned GO terms, 14−18 % were assigned to biological processes, 16−18 % molecular functions, and 3−4 % cellular components. Thirty-six and 33 GO terms could be assigned respectively to A17 and DZA315 2 dpi datasets, and 45 and 69 respectively in the 7 dpi datasets. Of these, metabolic and catalytic activity represented the majority of classified proteins in all datasets, followed by catabolism, binding, hydrolase activity and biosynthesis in different percentages (Additional file 4). The number of encoded proteins represented by these GO terms were 3-6 times more in the DZA315 7 dpi dataset compared to A17 at the same time-point. For example, DZA315:A17 carbohydrate metabolism 22:7, lipid metabolism 13:2, hydrolase activity 39:18, catabolism 37:10, signal transduction 5:0 (Additional file 5).
For Fom genes up-regulated in planta, a significant over-representation (Fisher’s exact test p ≤ 0.05) of domains associated with degradation of proteins and sugars/carbohydrates (e.g. glycoside hydrolase, pectate lyase, protease), membrane transport (e.g. sugar, amino acid and Major Facilitator Superfamily (MFS) transporters, nucleobase cation symporter-1, permease) and oxidative processes (e.g. 3-beta hydroxysteroid dehydrogenase, oxidoreductase, cytochrome p450s) was observed, particularly in DZA315 (Fig. 3, Additional file 6). Encapsulated, these results suggest host cell wall and membrane degradation along with nutrient transport are initiated earlier upon infection of susceptible plants, evident as early as 2 dpi compared to the resistant host interaction. By 7 dpi these processes were even more apparent during Fom infection of susceptible plants, correlating with an increase in fungal biomass at this time point (Fig. 1e).
Fig. 3.

Pfam domains more abundant in the in planta up-regulated datasets. Pfam domains enriched in the in planta up-regulated datasets from resistant (A17) and susceptible (DZA315) accessions relative to in vitro growth conditions are listed. Schematic figures illustrate the tissue sampled (represented as root tissues below the dashed line infected with Fom (purple spores), highlighting the chlorotic leaves visible at the later stage of the susceptible interaction). Enriched Pfam domains were identified based on comparisons against the total Fom protein set using Fisher’s exact test with a significance threshold of p ≤ 0.05. Values are ranked by representation of Pfam domains with colour coding signifying increasing abundance within each dataset. Cs: counts of Pfam domain containing proteins in DEG dataset. % D: % representation of Pfam domain containing proteins in DEG dataset; % G: % representation of Pfam domain containing proteins in whole genome. Further details are provided in Additional File 6. Aldeh: Aldehyde dehydrogenase; AIM24: Mitochondrial biogenesis; CIA30: mitochondrial Complex I intermediate-associated protein; Dabb: Stress responsive A/B Barrel Domain; EutQ: Ethanolamine utilisation protein; GH: glycoside hydrolase; Grp1_Fun34_YaaH: acetate transporter; GFA: Glutathione-dependent formaldehyde-activating enzyme; GST: Glutathione S-transferase; HAD: Haloacid dehalogenase-like hydrolase; LigB: LigB subunit of aromatic ring-opening dioxygenase; Meth_synthase: methionine synthases; NDT80_PhoG: DNA binding-family; PLAC8: Placenta-specific gene 8 protein; Pyr_redox: Pyridine nucleotide-disulphide oxidoreductase (includes oxidoreductases, NADH oxidases and peroxidases); SBP: Bacterial extracellular solute-binding protein; Thiamine4: thiamine biosynthetic enzyme
Searching for proteins involved in regulation of Fom pathogenicity-associated gene expression, we identified four proteins annotated as fungal transcription factors with one (Fom_15733) common to three of the datasets (2 dpi A17, 7 dpi A17 and DZA315). Fom_15733 is predicted CDC encoded and shares greatest similarity with proteins from F. oxysporum f. sp. raphani (Brassica pathogen, 90 %) and f. sp. pisi (pea pathogen, 87 %) but low similarity with other F. oxysporum ff. spp. (e.g. 26−36 % with ciceris, melonis, lycopersici, Fo5176, see [5]). This protein contains a fungal specific transcription factor domain (Pfam:PF04082) and a fungal Zn(2)-Cys(6) binuclear cluster domain (Pfam:PF00172). The other transcription factors identified in the up-regulated datasets were only found in the DZA315 7 dpi dataset (Fom_14279, 14162, 07202), are predicted to reside on core chromosomes and share most similarity to proteins from other F. oxysporum ff. spp.. The Fom homologue (Fom_08318) of Fol transcription factor SIX Gene Expression 1 (SGE1) whose expression is up-regulated during infection of tomato roots and is required for expression of most secreted Fol effectors [41, 42], was detected as expressed in vitro but not detected as significantly up-regulated in our in planta DEG datasets.
Next we applied several criteria to identify candidate effector and host-specific pathogenicity genes. These included characteristics such as predicted secretion, in planta up-regulated gene expression and chromosomal location, as F. oxysporum effectors have previously been identified on non-core or conditionally dispensable chromosomes (CD chromosomes). The majority of DEGs in all datasets were located on scaffolds predicted to form part of core Fom chromosomes [5] (Fig. 4a). Most DEGs predicted to lie on putative CD chromosomes were only identified within the up-regulated datasets (Figs. 4a, b). DEGs encoding SSPs were also predominantly identified within the in planta up-regulated datasets (Fig. 4c, Additional files 2 and 3). Interestingly the number of SSPs in A17 datasets didn’t differ much between the two sampled time-points but in DZA315 the number almost tripled between the 2 and 7 dpi up-regulated datasets (Fig. 4c). Further, DEGs within the susceptible DZA315 datasets also contained a larger proportion of genes expressed more highly in planta and these were enriched for SSPs (Additional file 7).
Fig. 4.

DEGs in up-regulated datasets contain an enrichment of genes located on putative non-core chromosomes and encoding predicted small secreted proteins. a-b Predicted putative chromosomal location of DEGs with a) total numbers in each dataset and b) percentage of DEGs predicted to reside on a predicted conditionally dispensable chromosome (CDC). c DEGs predicted to encode small secreted proteins within each dataset
Overall, the enrichment during the early stages of infection of genes encoding SSPs, uncharacterised proteins and proteins with roles in protein/sugar degradation and their transport, oxidative stress and other pathogenicity associated processes indicates substantial changes in pathogen gene expression upon colonisation of a susceptible host.
Highly up-regulated genes unique to in planta up-regulated datasets from a susceptible or resistant host and links to pathogenicity
As we were most interested in identifying pathogenicity factors we focused firstly on the differences between genes expressed during infection of resistant or susceptible host accessions (indicating changes that Fom may undergo when it detects a susceptible or resistant host) and secondly on those that were expressed during both infection of a susceptible and a resistant host (indicating key roles in pathogen attack).
Firstly we compared DEGs unique to infection of each host accession. Under this analysis, 11 and 6 DEGs were uniquely significantly up-regulated in A17 at 2 and 7 dpi respectively, while 16 and 280 were unique to DZA315 at the same time-points (Fig. 5). Of those expressed during infection of A17 at 2 dpi, 4 of the most highly induced (32-13,000-fold) were co-localised at the genomic scale (Table 3) and another, Fom_08477, is a predicted SSP showing similarity to an uncharacterized protein from the rice pathogen Fusarium fujikori. At 7 dpi in A17 all six unique DEGs were core scaffold located with two induced >100-fold and encoding SSPs (Fom_05133, Fom_09362, encoding a hypothetical protein and a glycoside hydrolase respectively).
Fig. 5.

Unique and overlapping Fom DEGs between A17 and DZA315 in planta up-regulated datasets. Venn diagram of DEGs in overlapping A17 and DZA315 in planta up-regulated datasets. Red arrows indicate up-regulated in planta
Table 3.
Examples of genomic co-localisation of in planta up-regulated differentially expressed Fom genes unique to DZA315 or A17 datasets. Full DEG lists are detailed in Additional files 2 and 3
| Dataset | Gene IDs | Chrom./ scaffolda | Pfam domain / best BlastP match / other characteristics |
|---|---|---|---|
| A17 2 dpi | Fom_11200, 11207, 11209, 11211 | core (scaffold_15; 1.38 Mb) | GST, MFS transporter, FAD-binding, uncharacterised protein |
| DZA315 7 dpi | Fom_01388, 01394, 01397 | core (scaffold_2; 3.35 Mb) | glycoside hydrolase, fasciclin, MFS transporter |
| Fom_01935, 01936, 01938 | core (scaffold_2; 3.35 Mb) | arca-like protein, MFS transporter, glycoside hydrolase | |
| Fom_03730, 03733, 03734 | core (scaffold_4; 2.62 Mb) | Hypothetical, MFS transporter, synthase | |
| Fom_15853, 15855, 15856, 15860 | core (scaffold_131; 0.03 Mb) | FAD-binding protein, ADH, FAD-binding protein, p450 | |
| Fom_14230, 14231, 14232, 14241 | CDC (scaffold_31; 0.21 Mb) | Oxidoreductase, MFS transporter, FAD-binding protein, oxidase |
apredicted core or CD chromosome (CDC) location and scaffold size based on [5]
The 16 unique DEGs from the DZA315 2 dpi dataset comprised of both core and non-core located genes with inductions ranging from 3.5 to >65,000-fold over in vitro conditions and encoding several FAD-binding proteins, hydrolases, peptidase, MFS transporter and p450 amongst others. Of the 280 significant DEGs in the DZA315 7 dpi unique dataset the majority were core scaffold located (243) and 43 were up-regulated in planta over 100-fold. Several were also co-localised (Table 3). Two Fom SSPs (Fom_08816, Fom_11981) with similarity to the phytotoxin cerato-platanin were also only identified in the DZA315 in planta 7 dpi up-regulated dataset. Some cerato-platanin members in other phytopathogens are implicated in disruption of host cell walls through expansin-like activity, chitin oligomer sequestration and the ability to induce plant cell necrosis [43, 44]. Other proteins only upregulated in DZA315 at 7 dpi included a xylosidase arabinosidase (Fom_12400) and an l-arabinitol 4-dehydrogenase (Fom_00399) with implied roles respectively in the hydrolysis of major hemicellulose component xylan to xylose and other sugars, and the catabolism of L-arabinose, an important constituent of plant cell wall polysaccharides.
Overlapping highly in planta up-regulated genes in susceptible and resistant host accessions and links to pathogenicity
Analysis of DEGs common to all four in planta up-regulated datasets identified 51 significantly induced DEGs (Fig. 5 and Table 4). These included 17 SSPs, 11 of which were predicted as effectors by EffectorP (8 CD and 3 core scaffold encoded). Thirty genes were predicted to be encoded on core scaffolds with 1/3 of these (21) not predicted as effectors by EffectorP and predominantly composed of proteins functioning in protein degradation and the breakdown of plant cell walls (hydrolases, peptidase, pectinesterase), transport or electron transfer (MFS transporter, p450s) or oxidative processes (oxidoreductases).
Table 4.
Differentially expressed Fom genes significantly up-regulated in planta during infection of DZA315 or A17 at 2 and 7 dpi. DEGs are ranked by DZA315 fold change (FC) over in vitro at 2 dpi. Bold FC indicates DZA315 fold changes greater than A17
| Protein ID | Chrom./scaffolda | Sec. pred.b | SSPc | Eff.P pred.d | AAe | Potential rolef | DZA315 FCg | A17 FCg | IV h | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 2dpi | 7dpi | 2dpi | 7dpi | ||||||||
| Fom_SIX13 | CDC | + | SSP | - | 292 | SIX effector | 397336 | 1290948 | 280959 | 1123836 | - |
| Fom_16257 | CDC | + | SSP | + | 91 | uncharacterised | 172951 | 741455 | 150562 | 301124 | - |
| Fom_16301 | CDC | + | SSP | + | 144 | LysM | 161369 | 912838 | 114105 | 425854 | - |
| Fom_16235 | CDC | + | SSP | + | 264 | uncharacterised | 37641 | 172951 | 35120 | 131072 | + |
| Fom_15517 | CDC | - | 567 | abhydrolase/peptidase | 12417 | 7132 | 7643 | 8780 | + | ||
| Fom_16729 | core | - | 54 | Protease (partial) | 11585 | 212927 | 23170 | 114105 | - | ||
| Fom_16326 | CDC | + | SSP | + | 111 | uncharacterised | 11585 | 106464 | 7132 | 57052 | + |
| Fom_SIX1 | CDC | + | SSP | - | 279 | SIX effector | 11585 | 61147 | 10809 | 40342 | + |
| Fom_SIX8 | CDC | + | SSP | + | 141 | SIX effector | 8780 | 106464 | 8192 | 70240 | + |
| Fom_16154 | CDC | + | - | 610 | GMC_oxidoreductase | 4390 | 20171 | 3566 | 12417 | + | |
| Fom_16208 | core | - | 185 | peptidase | 2896 | 37641 | 2048 | 15287 | + | ||
| Fom_12303 | core | + | SSP | + | 169 | uncharacterised | 2195 | 61147 | 2353 | 23170 | - |
| Fom_16263 | CDC | - | 235 | uncharacterised | 2195 | 14263 | 1911 | 6208 | + | ||
| Fom_15948 | CDC | + | SSP | + | 215 | peroxidase | 1911 | 16384 | 1097 | 6654 | + |
| Fom_10610 | core | + | - | 478 | oxidoreductase | 1552 | 5793 | 1448 | 2896 | + | |
| Fom_16592 | CDC | - | 87 | uncharacterised | 1261 | 5043 | 630 | 3566 | + | ||
| Fom_15730 | CDC | - | 199 | GST | 776 | 891 | 315 | 588 | + | ||
| Fom_15949 | CDC | - | 373 | peroxidase | 776 | 8192 | 776 | 4096 | + | ||
| Fom_09820 | core | - | 545 | p450 | 512 | 239 | 388 | 239 | + | ||
| Fom_09550 | core | - | 87 | uncharacterised | 446 | 3566 | 1097 | 3822 | + | ||
| Fom_SIX9 | CDC | + | SSP | + | 122 | SIX effector | 239 | 1552 | 208 | 1024 | + |
| Fom_15614 | CDC | + | SSP | - | 221 | glyco_hydrolase | 223 | 478 | 181 | 274 | + |
| Fom_06101 | core | - | 420 | uncharacterised | 194 | 239 | 181 | 128 | + | ||
| Fom_14503 | core | - | 138 | SnoaL | 169 | 119 | 223 | 45 | + | ||
| Fom_07856 | core | - | 533 | MFS | 147 | 169 | 97 | 84 | + | ||
| Fom_16718 | core | - | 79 | protease | 111 | 2702 | 208 | 1176 | + | ||
| Fom_07866 | core | - | 123 | uncharacterised | 79 | 69 | 60 | 34 | + | ||
| Fom_16085 | CDC | - | 403 | 2OG-FeII_Oxy | 60 | 446 | 52 | 315 | + | ||
| Fom_10284 | core | - | 112 | ligase | 60 | 239 | 49 | 338 | + | ||
| Fom_02574 | core | + | SSP | + | 87 | uncharacterised | 49 | 32 | 32 | 24 | + |
| Fom_04867 | core | - | 300 | NAD_binding | 39 | 23 | 32 | 9 | + | ||
| Fom_07859 | core | - | 292 | LigB, dioxyenase | 37 | 17 | 11 | 11 | + | ||
| Fom_01704 | core | - | 398 | methionine synth. | 24 | 26 | 32 | 23 | + | ||
| Fom_13701 | core | - | 391 | oxidoreductase | 24 | 24 | 20 | 11 | + | ||
| Fom_12076 | core | + | - | 324 | uncharacterised | 21 | 45 | 26 | 20 | + | |
| Fom_02353 | core | + | SSP | - | 295 | endoglucanase | 21 | 39 | 23 | 23 | + |
| Fom_15788 | CDC | + | SSP | + | 199 | uncharacterised | 18 | 69 | 11 | 49 | + |
| Fom_04087 | core | + | SSP | + | 158 | uncharacterised | 16 | 91 | 13 | 42 | + |
| Fom_10931 | core | + | - | 330 | pectinesterase | 11 | 60 | 20 | 26 | + | |
| Fom_08395 | core | + | SSP | - | 291 | abhydrolase | 9 | 17 | 6 | 5 | + |
| Fom_16385 | CDC | - | 195 | glyco_hydrolase | 9 | 39 | 10 | 30 | + | ||
| Fom_07662 | core | - | 438 | aminotransferase | 9 | 34 | 4 | 16 | + | ||
| Fom_08559 | core | + | - | 433 | glyco_hydrolase | 8 | 64 | 9 | 37 | + | |
| Fom_04883 | core | - | 514 | p450 | 7 | 9 | 7 | 5 | + | ||
| Fom_15521 | CDC | - | 660 | AMP-binding | 6 | 5 | 5 | 5 | + | ||
| Fom_05481 | core | + | SSP | - | 292 | uncharacterised | 6 | 5 | 5 | 3 | + |
| Fom_01040 | core | - | 124 | uncharacterised | 6 | 7 | 12 | 8 | + | ||
| Fom_16221 | CDC | - | 83 | glyco_hydrolase | 5 | 37 | 6 | 23 | + | ||
| Fom_16350 | core | - | 174 | abhydrolase | 5 | 2 | 5 | 4 | + | ||
| Fom_10260 | core | - | 255 | flavodoxin | 5 | 10 | 3 | 5 | + | ||
| Fom_09704 | core | + | - | 418 | glyco_hydrolase | 4 | 32 | 4 | 17 | + | |
apredicted core or CD chromosome (CDC) location
bpredicted secreted based on SignalP
cpredicted small secreted protein (SSP) based on ≤ 300 amino acids, SignalP, and ≤1 transmembrane domains
dEffectorP prediction (performed if predicted to be secreted): + effector, - non-effector
eBold font: protein length ≤ 300 amino acids
fPotential role based on Pfam domain, best BlastP match or other characteristics
gFold change in planta gene expression over in vitro conditions
hexpression in vitro, –: 0-10 % RNA-Seq read coverage over all replicates, +: >10 % RNA-Seq read coverage over all replicates
We first assessed the overlapping DEGs for the presence of those encoding the well documented F. oxysporum Secreted In Xylem (SIX) proteins, with known roles in virulence and/or avirulence described for some in F. oxysporum f. sp. lycopersici, F. oxysporum f. sp. melonis or Fo5176 (Brassica infecting) [6, 13, 14, 17, 18, 45–49]. SIX proteins can broadly be defined as small, generally cysteine rich, and possessing a secretion signal [13, 49]. Of the 14 F. oxysporum f. sp. lycopersici SIX proteins described so far, all four that we previously identified in the Fom genome (Fom SIX1, SIX8, SIX9 and SIX13) [5] were significantly upregulated in all in planta datasets where they were amongst the top in planta induced DEGs. Fom_SIX13 was the most highly induced at levels 1 million times greater than in vitro (Table 4). qRT-PCR verification of Fom SIX gene expression over a 1 to 7 day infection time-course revealed all but Fom_SIX13 could be reliably detected under in vitro conditions suggesting expression of this SIX gene in Fom responds specifically to detection of a possible host (Fig. 6, Additional file 8). Correlating with our RNA-Seq data (Table 4), qRT-PCR examination of all four Fom SIX genes confirmed these genes were overall more highly expressed in DZA315 than A17. Interestingly Fom_SIX13 and Fom_SIX1 didn’t meet the statistical cut-off of the EffectorP [38] prediction algorithm (as also observed for putative Fol effectors SIX7 and SIX13).
Fig. 6.
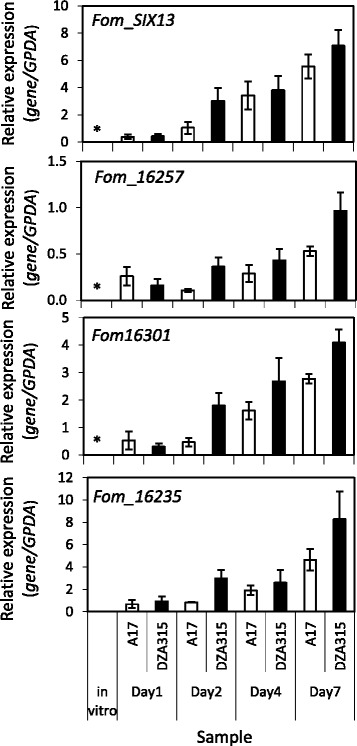
qRT-PCR validation of candidate effector genes in vitro versus in planta over an infection time-course. Expression of candidate effector genes as determined by qRT-PCR in in vitro samples and M. truncatula DZA315 and A17 root samples harvested at 1, 2, 4 and 7 days post inoculation (dpi) with Fom. in vitro samples are averages ± SE of 3 biological replicates. In planta samples are averages ± SE of 4 biological replicates each consisting of pools of 10 seedlings. Gene expression levels are relative to the fungal GPDA gene (Fom_05751). Note: * No detectable expression in any in vitro replicates
After Fom_SIX13, the next three most highly in-planta induced DEGs are strong effector candidates. These encode Fom_16257 a Fom-unique protein with unknown function, Fom_16301 a SSP encoding a LysM-domain (~40 residue lysin motif implicated in chitin binding [50–52]), and Fom_16235 an uncharacterised protein with orthologs in other F. oxysporum ff. spp.. qRT-PCR verification of Fom_16257 expression confirmed increased expression in DZA315 over A17 and absence of expression under in vitro conditions (Fig. 6). Similar results were observed for Fom_16301. Interestingly, Fom_16235 was lowly expressed under in vitro conditions. Along with Fom_16301, another LysM-domain containing SSP gene (Fom_04092) was also significantly upregulated, but only at 7 dpi. The Fom_16301 encoded protein contains one LysM domain, sharing best similarity to a protein from F. oxysporum f. sp. pisi (95 %), other F. oxysporum ff. spp. and Colletotrichum species. Fom_04092 contains two LysM domains and is most similar to proteins from other F. oxysporum ff.spp. and the rice pathogen F. fujikuroi.
Other highly in planta up-regulated genes included three proteases/peptidases and genes involved in oxidative stress responses (Table 4). Of the latter type, Fom_15948 and Fom_15949 both encode peroxidases predicted to reside alongside each other. Fom_15948 is a predicted effector and SSP sharing 95 % identity to other F. oxysporum peroxidases/catalases or hypothetical proteins, as well as to proteins from F. solani, F. fujikori, and Colletotrichum and Verticillium species.
Several uncharacterised proteins with properties of effectors were also identified as highly up-regulated in-planta during infection of both resistant and susceptible M. truncatula accessions (Table 4). Of those predicted as SSPs, only two were not predicted as effectors by EffectorP and these encode a putative endoglucanase (Fom_02353) and a cupredoxin domain-containing protein (Fom_05481) sharing 94-96 % similarity with several other serine rich proteins from F. oxysporum ff. spp. The serine rich nature of this protein may imply possible involvement in post-translational regulatory processes or protein-protein interactions. The remaining novel uncharacterised effector-like proteins identified contain no other known functional protein domains.
Discussion
The success of a pathogen’s attack requires the co-ordinated early expression of its pathogenicity arsenal. While an increasing list of putative arsenal components from various F. oxysporum ff. spp. have been predicted via whole genome sequencing or more classical PCR-based approaches, the relatively low abundance of fungal transcripts in host root tissues during early stages of infection has meant that very few whole transcriptome based identification studies have been accomplished. To overcome the constraint of low F. oxysporum RNA abundance in M. truncatula root tissues early in infection, we coupled our draft Fom genome assembly [5] and the pathogen’s vegetative (in vitro) transcriptome with high coverage in planta RNA-Seq of inoculated host roots to identify genes specifically up-regulated upon host infection. Comparison of a susceptible versus a resistant M. truncatula accession at 48 h post infection and at the later time-point of 7 dpi when disease symptoms on susceptible M. truncatula plants start to manifest, facilitated the identification of DEGs either unique or common to resistant or susceptible F. oxysporum-legume host interactions. These studies revealed a greater degree of transcriptional reprogramming during the susceptible plant interaction including expression of genes encoding SSPs and genes associated with plant cell wall and membrane degradation, nutrient uptake and oxidative processes. In addition we identified novel candidate effectors and pathogenicity-associated genes highly induced during infection of both susceptible and resistant plants.
To date, several transcriptome studies (e.g. RNA-Seq, ESTs/cDNAs) examining the F. oxysporum-host interaction have been reported, including those from infected roots of Arabidopsis, cabbage, banana, melon, cotton, chickpea, soybean or Medicago. The majority of these studies only included comparative analyses amongst plant transcripts due to insufficient sequencing depth for fungal transcript detection [19, 31, 48, 50–59]. Transcriptome analysis of this pathogen is also hindered by inherent variations in F. oxysporum root-colonisation dynamics at early stages of host colonisation and subsequent variations in disease progression amongst individual plants. Those studies with probes unique to the pathogen or enough coverage to detect pathogen transcripts are limited and have typically involved analysis of later time-points in the pathogen-host interaction process. For example, Schmidt and colleagues [48] analysed the expression of F. oxysporum f. sp. melonis transcripts at 10 dpi on melon roots. Here we were able to overcome these constraints by pooling root tissues from Fom inoculated M. truncatula seedlings and combining high coverage RNA-Seq with our stringent DEG selection criteria.
In addition to in planta F. oxysporum transcriptome analysis, in planta proteomics has been used to identify F. oxysporum pathogenicity components. The combined studies of Rep, Houterman, Schmidt and their colleagues [13, 49] which identified several avirulence and virulence products of Fol in the xylem sap proteome of infected tomato plants, were the first to identify the key Secreted in Xylem (SIX) effectors. Two studies have been conducted on the root proteomes of F. oxysporum infected legumes (chickpea-F. oxysporum f. sp. ciceris [24] and pea-F. oxysporum f. sp. pisi [60]) however, neither of these studies assessed the presence of Fusarium proteins.
Via RNA-Seq Guo and colleagues [19] were successful in comparing the vegetative transcriptomes of two races (virulent and avirulent) of F. oxysporum f. sp. cubense against their transcriptomes at 2 days post-infection of Cavendish banana. As with our in planta up-regulated Fom analysis, Guo and colleagues [19] found the largest groups of expressed genes were significantly enriched for catalytic and metabolic activities. However, the next most enriched GO term assignments differed between our studies, with the F. oxysporum f. sp. cubense datasets enriched for primary/cellular/nitrogen compound metabolic and biosynthetic processes, while Fom in planta datasets were most enriched for binding/catabolism/biosynthesis/and hydrolase activity, followed by cell/carbohydrate metabolism/and nucleic acid metabolism (Additional file 4). This suggests that at the early time-point of 2 dpi of Medicago roots there is a substantial bias towards host cell wall and membrane degradation and the metabolism of released nutrient sources such as sugars. Furthermore, these processes were more pronounced during the susceptible M. truncatula DZA315-Fom interaction (Fig. 3) even though at 2 dpi the degree of Fom colonisation was not significantly different between the susceptible DZA315 and resistant A17 M. truncatula hosts (Fig. 1e). It is interesting to speculate that differences observed between Fom and F. oxysporum f. sp. cubense may potentially be due to differences in root tissue or cell structure between a dicot/legume or monocot host.
Functional analysis revealed enrichment during infection of susceptible DZA315 plants of Fom DEGs encoding protein domains (Pfam) associated with degradation of proteins and carbohydrates/sugars as well as membrane transport and oxidative processes (Fig. 3). Lower numbers of these protein features were detected in the A17 up-regulated DEG datasets, with more pronounced differences observed at 7 dpi when a significant increase in Fom biomass was recorded in susceptible plants (Fig. 1e). Like other pathogenic plant-fungal pathogens, F. oxysporum genomes are enriched in plant cell wall degrading enzymes (CWDEs) [5, 9, 61–65] and are known to secrete these extracellular degradative enzymes during host colonization [66, 67]. These include enzymes such as polygalacturonases, pectate lyases, xylanases and proteases to break down cell walls and membranes to release nutrient sources such as sugars [20, 68] with expression during infection and roles in virulence demonstrated for some via double deletion mutants (e.g. double Fol polygalacturonase and endopolygalacturonase Δpg1Δpgx6 deletions) [19, 31, 64, 69, 70]. Fom genes highly expressed during infection of susceptible plants included several proteases/peptidases (including metallo- and serine-proteases) and pectate lyases with proposed roles in degradation of pectin and other cell wall components. Recently it was demonstrated that a secreted Fol metalloprotease and serine protease could cleave tomato chitinases with double deletion mutants of these two genes impaired in virulence on tomato [71]. Other Fom highly in planta expressed genes included several glycoside hydrolases that may act on the substrates sucrose, pectin, hemicellulose and chitin (Fig. 3). Fungal chitinases belong exclusively to the glycosyl hydrolase (GH) 18 family, but apart from a fundamental role in chitin hydrolysis little is known about their role during pathogen-plant interactions [72, 73]. It is suggested that during host infection they may have roles in fungal cell wall remodelling including involvement in processes such as spore germination, hyphal tip growth and branching [72, 73]. Transcripts encoding proteins with roles in membrane transport such as permeases, and sugar, amino acid and Major Facilitator Superfamily (MFS) transporters were also highly up-regulated and enriched in the DZA315 DEG datasets implying enhanced uptake of nutrient sources such as sugars released from plant roots by the aforementioned enzymes.
Cytochrome p450s were overrepresented in the in planta up-regulated Fom DEG datasets suggesting important roles in pathogenicity, with two identified in the overlapping datasets (Table 4). Located adjacent to one of these (Fom_09820) is a putative effector (Fom_09819) whose expression was up-regulated in both A17 and DZA315 7 dpi and shares 69 % protein identity to the Verticillium dahliae isochorismatase effector VdIsc1 which is required for full pathogenesis and disrupts plant defense responses by interfering with salicylate production [74]. Three oxidoreductases and two peroxidases (including EffectorP predicted Fom_15948) were identified in the overlapping in planta up-regulated DEG dataset (Table 4). A secreted Fol catalase-peroxidase (FOXG_17460) and an oxidoreductase (Orx1) were detected in the xylem sap proteome of Fol-infected tomato plants suggesting they are important for infection [13, 49]. Fom_15948 shares 84 % amino acid identity with FOXG_17460 and similar levels of identify with other F. oxysporum ff. spp. catalase/peroxidases, but differs by only one amino acid from a predicted catalase/peroxidase from the pea-infecting F. oxysporum f. sp. pisi HDV247 (FOVG_19731). Other genes expressed within the DZA315 7 dpi unique dataset included hydrolases, oxidases, oxidoreductases, phytotoxic cerato-platanins and several cytochrome p450s with similarity to pisatin demethylases (PDAs) (Fom_15860, Fom_09645, Fom_13458) that may degrade phytoalexin pterocarpans produced by Medicago species. While some of these genes were detected during infection of A17 they did not meet the minimum read coverage criteria.
A significant proportion of the in planta up-regulated Fom DEGs were enriched for proteins with no functional annotation including the tripling of abundance of uncharacterised genes in DZA315 infection between 2 and 7 dpi. We applied several criteria to narrow in on proteins within these datasets that may have roles in host-specific pathogenicity. One of these, up-regulated in planta expression during host infection, is a stringent criteria that has been successfully applied to identify and/or verify avirulence (Avr) proteins and putative effectors in other ff. spp. of F. oxysporum [5, 6, 13, 19, 32, 33, 65, 75]. For example, expression of Fol Avr3 (also referred to as SIX1) and Fol SIX6 whose gene products are respectively recognised by the tomato F. oxysporum Resistance gene I3 or suppress I2-mediated cell death, are strongly induced in the presence of living plant cells [17, 32, 41]. It should be noted that not all fungal effectors are exclusively expressed in planta (reviewed in [33, 65]) and we therefore did not ignore any potential effector candidates that were expressed in our in vitro dataset. We also placed emphasis on genes likely to be SSPs and CDC encoded as in the genus Fusarium CDCs are often enriched in rapidly evolving genes, can be horizontally transferred and encode novel effector candidates detected in the xylem sap of Fol infected tomato plants [9, 10, 13, 76]. The majority of candidate effectors we identified from our overlapping expression analysis (Table 4) were either unique to Fom and thus may define its host specificity, or were conserved to some degree with proteins of other F. oxysporum ff. spp. or other Fusaria and thus may have conserved roles in plant pathogenicity. We identified two proteins (Fom_16301, Fom_15948) that share a high degree of similarity to proteins from anthracnose disease causing Colletotrichum species. This similarity has been reported previously between effectors from Fol or F. oxysporum f. sp. melonis (e.g. SIX6) or candidate effectors from other Fusaria [17, 76]. Fom_16301 contains a lysin motif (LysM) and shares some similarity to extracellular protein7 (Ecp7) of unknown function from the tomato fungal pathogen Cladosporium fulvum and a LysM containing effector candidate from F. oxysporum f. sp. melonis [48, 77]. It is suggested that other C. fulvum effectors containing the LysM-domain may suppress chitin triggered immunity by protecting fungal hyphae against host chitinases or sequestering fungal cell wall derived chitin fragments from host detection [77–80]. The Fom genome encodes 11 proteins with LysM-domains and in addition to up-regulated in planta expression of the predicted CDC encoded Fom_16301, we also identified a core chromosome encoded LysM-domain containing protein (Fom_04092) sharing this expression profile. The vascular wilt pathogen Verticillium dahliae also contains an expanded LysM effector family but the expression of only one of these is induced in planta [81]. This effector, VDAG_05180, is CDC encoded and contributes to virulence on tomato, while the remaining LysM-domain proteins are core chromosome located, not expressed during infection and single deletion mutants have no effect on virulence [18, 81]. While the roles of the latter putative V. dahliae LysM effectors remains ambiguous, the in planta up-regulated expression of Fom_16301 and Fom_04092 suggests these two SSPs may contribute to pathogenicity.
The most highly in planta up-regulated gene detected in all Fom datasets (Table 4) was Fom_SIX13 whose expression could not be detected under vegetative conditions by either RNA-Seq or qRT-PCR (Fig. 6). The functional role of SIX13 homologues across F. oxysporum ff. spp. is unknown. Previously we found Fom_SIX13 shares most similarity with a SIX13 homologue from a melon-infecting F. oxysporum isolate and less similarity with its legume-infecting counterparts in f. sp. pisi or f. sp. ciceris, although it was the only SIX gene common to the legume-infecting ff. spp. analysed [5]. Recently, additional SIX13 sequences have been identified in other F. oxysporum f. sp. pisi isolates (races 2 and 5) [75] and BLASTN analysis [82] revealed the Fom_SIX13 gene sequence shares 98 % identity with these two f. sp. pisi SIX13 sequences suggesting potentially conserved roles on legume hosts. Within the overlapping in planta up-regulated DEGs we also identified seven genes unique to Fom and sharing no orthologues with other fungal species. This includes three uncharacterised proteins, a glycoside hydrolase, two proteases and a peptidase (Table 4 and Additional files 2 and 3). Of these, the CDC encoded SSP Fom_16257 contains a motif resembling a zinc finger domain suggesting this protein may bind host DNA sequences [5]. These features combined with its expression only during infection, earmarks Fom_16257 as a host-specific effector for future functional studies and identification of its possible host target(s).
Conclusions
In summary, our findings greatly improve the current understanding of Fusarium wilt pathogen molecular responses during early stages of legume colonisation and infection. Our analysis of in planta up-regulated genes represents a significant resource that can be leveraged to hone in on candidate effectors and pathogenicity associated genes from other F. oxysporum ff. spp.. Prioritisation for functional follow up studies to elucidate the role(s) of candidate Fom effectors listed within Table 4 will focus on those that are Fom specific and thus have implied roles in host specificity or those that are highly conserved amongst other legume infecting F. oxysporum ff. spp. and will expedite the dissection of their pathogenicity mechanisms on important global legume crops.
Methods
Isolate sources and growth conditions
F. oxysporum f. sp. medicaginis (Weimer) W.C. Snyder & H.N. Hansen, (Fom-5190a, BRIP 5190a/IMI 172838, collection number 19911) was obtained from the Queensland Plant Pathology Herbarium (BRIP) and has been described previously [5]. Fom was maintained on sterile filter paper and grown on ½ strength potato dextrose agar at 22 °C in the dark. M. truncatula accessions A17 and DZA315 were at least second generation inbred lines derived from seed obtained from the South Australian Research and Development Institute (SARDI). M. truncatula seeds were germinated on damp filter paper, transplanted into 30 mm Jiffy-7 peat pellets and grown under a short day-light regime (8-h light/16-h dark) at 21 °C.
F. oxysporum disease assays
Fom disease assays of M. truncatula were conducted as described previously [5, 29]. Briefly, a 1 × 106 spores mL–1 spore suspension was used to inoculate the roots of two week old seedlings which had roots protruding from the peat pellets trimmed prior to inoculation. Seedlings in peat pellets were placed in a petri dish of spore suspension for 5 min, followed by addition of a further 1 mL of spore suspension to the base of the hypocotyl. Inoculated pellets were transferred to growth trays lined with a plastic sheet and a thin layer of damp vermiculite, covered with a clear plastic dome to maintain humidity, and incubated under a long-daylight regime (16-h light/8-h dark) at 28 °C. Plants were scored every 7 days over 4 weeks to assess disease progression.
RNA isolation
RNA for qRT-PCR and RNA-Seq experiments on Fom infected M. truncatula root tissue was conducted as described previously [5]. Briefly, root tissue was collected from 10 plants and pooled per replicate at 1, 2, 4 and 7 days post inoculation. For Fom in vitro samples, mycelia were grown in a petri dish containing one-half-strength potato dextrose broth for 7 days at 22 °C and mycelia harvested by filtering through Miracloth. RNA extraction was performed independently for each of 3 replicates using a Trizol extraction (Sigma-Aldrich, St. Louis, MO) followed by DNase treatment using TURBO DNase (Ambion). RNA samples were cleaned via RNeasy mini spin columns (Qiagen).
qRT-PCR
Following RNA isolation and DNase treatment, complementary DNA synthesis was performed with 1ug of input RNA followed by qRT-PCR performed using SsoFast EvaGreen Supermix (Bio-Rad) on a CFX384 (Bio-Rad) system as described previously [5]. Absolute gene expression levels relative to F. oxysporum housekeeping gene GPDA (Fom_05751) were used for each complementary DNA sample using the equation: relative ratio gene of interest/GPDA = (Egene-Ct gene)/(EGPDA-Ct GPDA) where Ct is the cycle threshold value. Medicago root samples were verified for even abundance of plant input material using the M. truncatula B-tubulin reference gene [5, 83] which was found to be within ±1 Ct across all samples. Quantification of Fom biomass in M. truncatula tissues was conducted as described previously by determining the abundance of Fom_18S relative to M. truncatula_18S [5]. Primer sequences have been previously published [5] and/or are also listed in Additional file 9.
RNA-Seq library construction, Illumina sequencing and read-mapping
RNA integrity was confirmed using the Agilent 2100 Bioanalyser Plant Nano system (Agilent Biotechnologies). Stranded Illumina TruSeq libraries were generated from 1 μg of total RNA and sequenced (100 bp paired end reads) on an Illumina HiSeq platform by the Australian Genome Research Facility (AGRF). As per [5], RNA-Seq paired-end reads were trimmed for low-quality base-calls (≥ q30) and Illumina adapter sequences using Cutadapt v1.1 [84] (parameters: --quality-cutoff 30 --overlap 10 --times 3 –minimum-length 25) with reads trimmed to less than 25 bp discarded and remaining reads sorted into pairs and singleton reads. RNA-Seq reads were then mapped to the Fom-5190a genome assembly via Tophat2 (TopHat2 v2.0.9) (parameters: --b2-very-sensitive -r 80 --mate-std-dev 40 -i 20 -I 4000 -g 20 --report-secondary-alignments --report-discordant-pair-alignments -m 0 --min-coverage-intron 20 --microexon-search --library-type fr-firststrand) [30]. BAM files generated from Tophat2 output were sorted using SAMtools version 0.1.19 [85] and visualised using IGV (version 2.3) [86]. Trimmed sequencing data is available from the NCBI/GenBank database under BioProject number PRJNA294248 (http://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA294248).
Differential gene expression analysis
BAM files generated by TopHat2 [30] were used by BioKanga maploci [http://sourceforge.net/projects/biokanga/files/] to assign aligned reads to known loci. This step was followed by the genDEseq subprocess of BioKanga to generate counts files for differential expression. Read counts from different biological replicates and samples were combined for each gene and the resulting count matrix was normalized and analysed for differential expression using the EdgeR [34] package in R version 3.2.2 http://www.r-project.org/ using a false discovery rate cutoff of 0.05 and a minimum fold change of log2 ≥ 1. Identification of unique or overlapping genes within the DEG datasets and the generation of Venn diagrams was determined using Draw Venn Diagram http://bioinformatics.psb.ugent.be/webtools/Venn/ (accessed 12-01-16). Gene coverage was calculated from TopHat2 mapped RNA-Seq reads using BEDTools (v2.21.0) coverageBed [87].
Protein characteristics
Protein characteristics were previously reported by [5] with GO term counts determined via CateGOrizer [88] (accessed 15-01-16). Statistical examination of over-represented protein functional attributes based on the number of proteins with specific Pfam domains were compared between DEG datasets and those from the whole annotated Fom genome using Fisher’s exact test with a significance threshold of p ≤ 0.05.
Acknowledgments
This research was undertaken with the assistance of resources from the Australian Genome Research Facility (AGRF) and the National Computational Infrastructure Specialised Facility for Bioinformatics (NCI-SF Bioinformatics) which are both supported by the Australian Government, and through the resources located at iVEC within the Pawsey Supercomputing Centre. We also thank Elaine Smith, Lindsey Spiller and Nicole Arielli for assistance with tissue sampling and preparation for RNA extractions, and Drs Jana Sperschneider and Andrew Spriggs for critical reading of the manuscript and useful suggestions.
Funding
This research was undertaken with funding provided by Grains Research and Development Corporation (GRDC) (Grant ID UWA00145) and Commonwealth Scientific and Industrial Research Organisation (CSIRO).
Availability of data and materials
The authors declare all data and material is available with all datasets deposited in publicly available NCBI repositories. Trimmed RNA-sequencing data is available from the NCBI/GenBank BioProject database under BioProject number PRJNA294248 and can be accessed via the following link http://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA294248.
Authors’ contributions
KS and LT conceived and designed the study. LT prepared RNA and drafted the manuscript. LT and AW conducted Fom experiments. AW and GG processed and mapped the RNA-Seq data. GG performed DEG analysis. AW, GG and LT analysed the RNA-Seq and DEG data. LT and SB conducted and analysed the qRT-PCR transcript data. All authors contributed to the draft and read and approved the final version.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
All authors of the manuscript have read and agreed to its content and are accountable for all aspects of the accuracy and integrity of the manuscript.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Abbreviations
- CDC(s)
Conditionally dispensable chromosome(s)
- DEG(s)
Differentially expressed gene(s)
- dpi
Days post inoculation
- f. sp
forma specialis
- ff. spp
formae speciales
- Fol
F. oxysporum f. sp. lycopersici
- Fom
F. oxysporum f. sp. medicaginis
- RNA-Seq
RNA sequencing
- SIX
Secreted in xylem
Additional files
Confirmation of disease progression in experiment from which plants were sampled for RNA-Seq. (PPTX 42 kb)
Fom genes up or down-regulated in DZA315 plants versus in vitro. (XLSX 464 kb)
Fom genes up or down-regulated in A17 plants versus in vitro. (XLSX 167 kb)
Proportion of Fom proteins in A17 and DZA315 DEGs in planta up-regulated datasets with Gene Ontology (GO) assignments. (PPTX 165 kb)
Gene Ontology (GO) assignments associated with in planta upregulated DEGs. (XLSX 13 kb)
Pfam domains more abundant in the in planta up-regulated DEG datasets. (XLSX 17 kb)
Plots of DEG significance versus in planta up-regulated fold induction. (PPTX 77 kb)
qRT-PCR validation of candidate pathogenicity-associated genes in vitro versus in planta over an infection time-course. (PPTX 556 kb)
qRTPCR_primers. (XLSX 9 kb)
Contributor Information
Louise F. Thatcher, Email: Louise.Thatcher@csiro.au
Angela H. Williams, Email: Angela.Williams2@csiro.au
Gagan Garg, Email: Gagan.Garg@csiro.au.
Sally-Anne G. Buck, Email: sallyanne.buck@gmail.com
Karam B. Singh, Email: Karam.Singh@csiro.au
References
- 1.Agrios GN. chapter eleven - PLANT DISEASES CAUSED BY FUNGI. In: Agrios GN, editor. Plant Pathology. Fifth. San Diego: Academic; 2005. pp. 385–614. [Google Scholar]
- 2.Takken F, Rep M. The arms race between tomato and Fusarium oxysporum. Mol Plant Pathol. 2010;11(2):309–314. doi: 10.1111/j.1364-3703.2009.00605.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Michielse CB, Rep M. Pathogen profile update: Fusarium oxysporum. Mol Plant Pathol. 2009;10(3):311–324. doi: 10.1111/j.1364-3703.2009.00538.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dombrecht B, Kazan K, Manners JM. Improved resistance to Fusarium wilt through genetic engineering of defense signaling pathways. In: da Silva JA T, editor. Floriculture, ornamental and plant biotechnology. London: Global Science Books; 2006. pp. 388–398. [Google Scholar]
- 5.Williams AH, Sharma M, Thatcher LF, Azam S, Hane JK, Sperschneider J, Kidd BN, Anderson JP, Ghosh R, Garg G, et al. Comparative genomics and prediction of conditionally dispensable sequences in legume-infecting Fusarium oxysporum formae speciales facilitates identification of candidate effectors. BMC Genomics. 2016;17(1):191. doi: 10.1186/s12864-016-2486-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thatcher LF, Gardiner DM, Kazan K, Manners JM. A highly conserved effector in Fusarium oxysporum is required for full virulence on Arabidopsis. Mol Plant Microbe Interact. 2012;25(2):180–190. doi: 10.1094/MPMI-08-11-0212. [DOI] [PubMed] [Google Scholar]
- 7.Gordon TR, Martyn RD. The evolutionary biology of Fusarium oxysporum. Annu Rev Phytopathol. 1997;35(1):111–128. doi: 10.1146/annurev.phyto.35.1.111. [DOI] [PubMed] [Google Scholar]
- 8.O'Donnell K, Kistler HC, Cigelnik E, Ploetz RC. Multiple evolutionary origins of the fungus causing Panama disease of banana: concordant evidence from nuclear and mitochondrial gene genealogies. Proc Natl Acad Sci U S A. 1998;95(5):2044–2049. doi: 10.1073/pnas.95.5.2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ma LJ, van der Does HC, Borkovich KA, Coleman JJ, Daboussi MJ, Di Pietro A, Dufresne M, Freitag M, Grabherr M, Henrissat B, et al. Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature. 2010;464(7287):367–373. doi: 10.1038/nature08850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vlaardingerbroek I, Beerens B, Rose L, Fokkens L, Cornelissen BJC, Rep M. Exchange of core chromosomes and horizontal transfer of lineage-specific chromosomes in Fusarium oxysporum. Environ Microbiol. 2016:n/a-n/a. doi: 10.1111/1462-2920.13281 [DOI] [PubMed]
- 11.Laurence MH, Summerell BA, Liew ECY. Fusarium oxysporum f. sp. canariensis: evidence for horizontal gene transfer of putative pathogenicity genes. Plant Pathol. 2015;64(5):1068–1075. doi: 10.1111/ppa.12350. [DOI] [Google Scholar]
- 12.Ma L-J. Horizontal chromosome transfer and rational strategies to manage Fusarium vascular wilt diseases. Mol Plant Pathol. 2014;15(8):763–766. doi: 10.1111/mpp.12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schmidt SM, Houterman PM, Schreiver I, Ma L, Amyotte S, Chellappan B, Boeren S, Takken FL, Rep M. MITEs in the promoters of effector genes allow prediction of novel virulence genes in Fusarium oxysporum. BMC Genomics. 2013;14:119. doi: 10.1186/1471-2164-14-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rep M, van der Does HC, Meijer M, van Wijk R, Houterman PM, Dekker HL, de Koster CG, Cornelissen BJ. A small, cysteine-rich protein secreted by Fusarium oxysporum during colonization of xylem vessels is required for I-3-mediated resistance in tomato. Mol Microbiol. 2004;53(5):1373–1383. doi: 10.1111/j.1365-2958.2004.04177.x. [DOI] [PubMed] [Google Scholar]
- 15.Rep MS S, van Dam P, de Sain M, Vlaardingerbroek I, Shahi S, Widinugraheni S, Fokkens L, Tintor N, Beerens B, Houterman P, van der Does C. Effectors of Fusarium oxysporum: identification, function, evolution and regulation of gene expression. In: Sachs MS, editor. Fungal Genetics Reports. Pacific Grove, California: Fungal Genetics Stock Center; 2015. p. 26. [Google Scholar]
- 16.Vlaardingerbroek I, Beerens B, Schmidt SM, Cornelissen BJC, Rep M. Dispensable chromosomes in Fusarium oxysporum f.sp. lycopersici. Mol Plant Pathol. 2016:n/a-n/a. doi: 10.1111/mpp.12440 [DOI] [PMC free article] [PubMed]
- 17.Gawehns F, Houterman PM, Ichou FA, Michielse CB, Hijdra M, Cornelissen BJ, Rep M, Takken FL. The Fusarium oxysporum effector Six6 contributes to virulence and suppresses I-2-mediated cell death. Mol Plant Microbe Interact. 2014;27(4):336–348. doi: 10.1094/MPMI-11-13-0330-R. [DOI] [PubMed] [Google Scholar]
- 18.de Sain M, Rep M. The Role of Pathogen-Secreted Proteins in Fungal Vascular Wilt Diseases. Mol Plant Microbe Interact. 2015;16(10):23970. doi: 10.3390/ijms161023970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guo L, Han L, Yang L, Zeng H, Fan D, Zhu Y, Feng Y, Wang G, Peng C, Jiang X, et al. Genome and transcriptome analysis of the fungal pathogen Fusarium oxysporum f. sp. cubense causing banana vascular wilt disease. PLoS ONE. 2014;9(4):e95543. doi: 10.1371/journal.pone.0095543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gupta S, Bhar A, Das S. Understanding the molecular defence responses of host during chickpea–Fusarium interplay: where do we stand? Funct Plant Biol. 2013;40(12):1285–1297. doi: 10.1071/FP13063. [DOI] [PubMed] [Google Scholar]
- 21.Trapero-Casas AJ-DR. Fungal wilt and root rot diseases of chickpea in southern Spain. Phytopathology. 1985;75(1):1146–1151. doi: 10.1094/Phyto-75-1146. [DOI] [Google Scholar]
- 22.Abera M, Sakhuja PK, Fininsa C, Ahmed S. Status of chickpea fusarium wilt (Fusarium oxysporum f. sp. ciceris) in northwestern Ethiopia. Arch Phytopathol Plant Protect. 2011;44:1261-72.
- 23.Navas-Cortés JA, Hau B, Jiménez-Díaz RM. Yield Loss in Chickpeas in Relation to Development of Fusarium Wilt Epidemics. Phytopathology. 2000;90(11):1269–1278. doi: 10.1094/PHYTO.2000.90.11.1269. [DOI] [PubMed] [Google Scholar]
- 24.Chatterjee M, Gupta S, Bhar A, Chakraborti D, Basu D, Das S. Analysis of root proteome unravels differential molecular responses during compatible and incompatible interaction between chickpea (Cicer arietinum L.) and Fusarium oxysporum f. sp. ciceri Race1 (Foc1) BMC Genomics. 2014;15:949. doi: 10.1186/1471-2164-15-949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xue R, Wu J, Zhu Z, Wang L, Wang X, Wang S, Blair MW. Differentially Expressed Genes in Resistant and Susceptible Common Bean (Phaseolus vulgaris L.) Genotypes in Response to Fusarium oxysporum f. sp. phaseoli. PLoS ONE. 2015;10(6):e0127698. doi: 10.1371/journal.pone.0127698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rispail N, Bani M, Rubiales D. Resistance reaction of Medicago truncatula genotypes to Fusarium oxysporum: effect of plant age, substrate and inoculation method. Crop Pasture Sci. 2015;66(5):506–515. doi: 10.1071/CP14216. [DOI] [Google Scholar]
- 27.Rispail N, Rubiales D. Identification of Sources of Quantitative Resistance to Fusarium oxysporum f. sp. medicaginis in Medicago truncatula. Plant Dis. 2014;98(5):667–673. doi: 10.1094/PDIS-03-13-0217-RE. [DOI] [PubMed] [Google Scholar]
- 28.Ramírez-Suero M, Khanshour A, Martinez Y, Rickauer M. A study on the susceptibility of the model legume plant Medicago truncatula to the soil-borne pathogen Fusarium oxysporum. Eur J Plant Pathol. 2010;126(4):517–530. doi: 10.1007/s10658-009-9560-x. [DOI] [Google Scholar]
- 29.Thatcher LF, Gao L-L, Singh KB. Jasmonate Signalling and Defence Responses in the Model Legume Medicago truncatula—A Focus on Responses to Fusarium Wilt Disease. Plants. 2016;5(1):11. doi: 10.3390/plants5010011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14(4):R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McFadden HG, Wilson IW, Chapple RM, Dowd C. Fusarium wilt (Fusarium oxysporum f. sp. vasinfectum) genes expressed during infection of cotton (Gossypium hirsutum) Mol Plant Pathol. 2006;7(2):87–101. doi: 10.1111/j.1364-3703.2006.00327.x. [DOI] [PubMed] [Google Scholar]
- 32.van der Does HC, Duyvesteijn RG, Goltstein PM, van Schie CC, Manders EM, Cornelissen BJ, Rep M. Expression of effector gene SIX1 of Fusarium oxysporum requires living plant cells. Fungal Genet Biol. 2008;45(9):1257–1264. doi: 10.1016/j.fgb.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 33.Stergiopoulos I, de Wit PJ. Fungal effector proteins. Annu Rev Phytopathol. 2009;47:233–263. doi: 10.1146/annurev.phyto.112408.132637. [DOI] [PubMed] [Google Scholar]
- 34.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hacquard S, Kracher B, Hiruma K, Munch PC, Garrido-Oter R, Thon MR, Weimann A, Damm U, Dallery J-F, Hainaut M, et al. Survival trade-offs in plant roots during colonization by closely related beneficial and pathogenic fungi. Nat Commun. 2016;7:13072. doi: 10.1038/ncomms13072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.O'Connell RJ, Thon MR, Hacquard S, Amyotte SG, Kleemann J, Torres MF, Damm U, Buiate EA, Epstein L, Alkan N, et al. Lifestyle transitions in plant pathogenic Colletotrichum fungi deciphered by genome and transcriptome analyses. Nat Genet. 2012;44(9):1060–1065. doi: 10.1038/ng.2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Z-N, Wu Q-Y, Zhang G-Z, Zhu Y-Y, Murphy RW, Liu Z, Zou C-G. Systematic analyses reveal uniqueness and origin of the CFEM domain in fungi. Sci Rep. 2015;5:13032. doi: 10.1038/srep13032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sperschneider J, Gardiner DM, Dodds PN, Tini F, Covarelli L, Singh KB, Manners JM, Taylor JM. EffectorP: predicting fungal effector proteins from secretomes using machine learning. New Phytol. 2016;210(2):743–761. doi: 10.1111/nph.13794. [DOI] [PubMed] [Google Scholar]
- 39.Winnenburg R, Baldwin TK, Urban M, Rawlings C, Köhler J, Hammond-Kosack KE. PHI-base: a new database for pathogen host interactions. Nucleic Acids Res. 2006;34(suppl 1):D459–D464. doi: 10.1093/nar/gkj047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Winnenburg R, Urban M, Beacham A, Baldwin TK, Holland S, Lindeberg M, Hansen H, Rawlings C, Hammond-Kosack KE, Köhler J. PHI-base update: additions to the pathogen–host interaction database. Nucleic Acids Res. 2008;36(suppl 1):D572–D576. doi: 10.1093/nar/gkm858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Michielse CB, van Wijk R, Reijnen L, Manders EM, Boas S, Olivain C, Alabouvette C, Rep M. The nuclear protein Sge1 of Fusarium oxysporum is required for parasitic growth. PLoS Pathog. 2009;5(10):e1000637. doi: 10.1371/journal.ppat.1000637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Michielse CB, van Wijk R, Reijnen L, Cornelissen BJ, Rep M. Insight into the molecular requirements for pathogenicity of Fusarium oxysporum f. sp. lycopersici through large-scale insertional mutagenesis. Genome Biol. 2009;10(1):R4. doi: 10.1186/gb-2009-10-1-r4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pazzagli L, Seidl-Seiboth V, Barsottini M, Vargas WA, Scala A, Mukherjee PK. Cerato-platanins: Elicitors and effectors. Plant Sci. 2014;228:79–87. doi: 10.1016/j.plantsci.2014.02.009. [DOI] [PubMed] [Google Scholar]
- 44.Baccelli I. Cerato-platanin family proteins: one function for multiple biological roles? Front Plant Sci. 2015;5:769. doi: 10.3389/fpls.2014.00769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ma L, Houterman PM, Gawehns F, Cao L, Sillo F, Richter H, Clavijo-Ortiz MJ, Schmidt SM, Boeren S, Vervoort J, et al. The AVR2–SIX5 gene pair is required to activate I-2-mediated immunity in tomato. New Phytol. 2015;208(2):507–518. doi: 10.1111/nph.13455. [DOI] [PubMed] [Google Scholar]
- 46.Houterman PM, Cornelissen BJ, Rep M. Suppression of plant resistance gene-based immunity by a fungal effector. PLoS Pathog. 2008;4(5):e1000061. doi: 10.1371/journal.ppat.1000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Houterman PM, Ma L, Ooijen G, Vroomen MJ, Cornelissen BJ, Takken FL, Rep M. The effector protein Avr2 of the xylem-colonizing fungus Fusarium oxysporum activates the tomato resistance protein I-2 intracellularly. Plant J. 2009;58(6):970–978. doi: 10.1111/j.1365-313X.2009.03838.x. [DOI] [PubMed] [Google Scholar]
- 48.Schmidt SM, Lukasiewicz J, Farrer R, van Dam P, Bertoldo C, Rep M. Comparative genomics of Fusarium oxysporum f. sp. melonis reveals the secreted protein recognized by the Fom-2 resistance gene in melon. New Phytol. 2016;209(1):307–318. doi: 10.1111/nph.13584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Houterman PM, Speijer D, Dekker HL CGDEK, Cornelissen BJ, Rep M. The mixed xylem sap proteome of Fusarium oxysporum-infected tomato plants. Mol Plant Pathol. 2007;8(2):215–221. doi: 10.1111/j.1364-3703.2007.00384.x. [DOI] [PubMed] [Google Scholar]
- 50.Lanubile A, Muppirala UK, Severin AJ, Marocco A, Munkvold GP. Transcriptome profiling of soybean (Glycine max) roots challenged with pathogenic and non-pathogenic isolates of Fusarium oxysporum. BMC Genomics. 2015;16(1):1–14. doi: 10.1186/s12864-015-2318-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bai TT, Xie WB, Zhou PP, Wu ZL, Xiao WC, Zhou L. Transcriptome and expression profile analysis of highly resistant and susceptible banana roots challenged with Fusarium oxysporum f. sp. cubense tropical race 4. Plos ONE. 2013;8(9):e73945. doi: 10.1371/journal.pone.0073945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li C, Shao J, Wang Y, Li W, Guo D, Yan B, Xia Y, Peng M. Analysis of banana transcriptome and global gene expression profiles in banana roots in response to infection by race 1 and tropical race 4 of Fusarium oxysporum f. sp. cubense. BMC Genomics. 2013;14:851. doi: 10.1186/1471-2164-14-851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li CY, Deng GM, Yang J, Viljoen A, Jin Y, Kuang RB. Transcriptome profiling of resistant and susceptible Cavendish banana roots following inoculation with Fusarium oxysporum f. sp. cubense tropical race 4. BMC Genom. 2012;13:374. doi: 10.1186/1471-2164-13-374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lyons R, Rusu A, Stiller J, Powell J, Manners JM, Kazan K. Investigating the Association between Flowering Time and Defense in the Arabidopsis thaliana-Fusarium oxysporum Interaction. PLoS ONE. 2015;10(6):e0127699. doi: 10.1371/journal.pone.0127699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lyons R, Stiller J, Powell J, Rusu A, Manners JM, Kazan K. Fusarium oxysporum Triggers Tissue-Specific Transcriptional Reprogramming in Arabidopsis thaliana. PLoS ONE. 2015;10(4):e0121902. doi: 10.1371/journal.pone.0121902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhu QH, Stephen S, Kazan K, Jin G, Fan L, Taylor J. Characterization of the defense transcriptome responsive to Fusarium oxysporum-infection in Arabidopsis using RNA-seq. Gene. 2013;512. [DOI] [PubMed]
- 57.Xing M, Lv H, Ma J, Xu D, Li H, Yang L, Kang J, Wang X, Fang Z. Transcriptome Profiling of Resistance to Fusarium oxysporum f. sp. conglutinans in Cabbage (Brassica oleracea) Roots. PLoS ONE. 2016;11(2):e0148048. doi: 10.1371/journal.pone.0148048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang Z, Zhang J, Jia C, Liu J, Li Y, Yin X, Xu B, Jin Z. De Novo characterization of the banana root transcriptome and analysis of gene expression under Fusarium oxysporum f. sp. cubense tropical race 4 infection. BMC Genomics. 2012;13(1):1–9. doi: 10.1186/1471-2164-13-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ashraf N, Ghai D, Barman P, Basu S, Gangisetty N, Mandal MK, Chakraborty N, Datta A, Chakraborty S. Comparative analyses of genotype dependent expressed sequence tags and stress-responsive transcriptome of chickpea wilt illustrate predicted and unexpected genes and novel regulators of plant immunity. BMC Genomics. 2009;10(1):1–21. doi: 10.1186/1471-2164-10-415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Castillejo MA, Bani M, Rubiales D. Understanding pea resistance mechanisms in response to Fusarium oxysporum through proteomic analysis. Phytochemistry. 2015;115:44–58. doi: 10.1016/j.phytochem.2015.01.009. [DOI] [PubMed] [Google Scholar]
- 61.Zhao Z, Liu H, Wang C, Xu JR. Comparative analysis of fungal genomes reveals different plant cell wall degrading capacity in fungi. BMC Genomics. 2013;14:274. doi: 10.1186/1471-2164-14-274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sestili S, Polverari A, Luongo L, Ferrarini A, Scotton M, Hussain J. Distinct colonization patterns and cDNA-AFLP transcriptome profiles in compatible and incompatible interactions between melon and different races of Fusarium oxysporum f. sp. melonis. BMC Genom. 2011;12:122. doi: 10.1186/1471-2164-12-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lowe RG, Cassin A, Grandaubert J, Clark BL, Van de Wouw AP, Rouxel T, Howlett BJ. Genomes and transcriptomes of partners in plant-fungal-interactions between canola (Brassica napus) and two Leptosphaeria species. PLoS ONE. 2014;9(7):e103098. doi: 10.1371/journal.pone.0103098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kubicek CP, Starr TL, Glass NL. Plant cell wall–degrading enzymes and their secretion in plant-pathogenic fungi. Annu Rev Phytopathol. 2014;52(1):427–451. doi: 10.1146/annurev-phyto-102313-045831. [DOI] [PubMed] [Google Scholar]
- 65.Lo Presti L, Lanver D, Schweizer G, Tanaka S, Liang L, Tollot M, Zuccaro A, Reissmann S, Kahmann R. Fungal effectors and plant susceptibility. Ann Rev Plant Biol. 2015;66(1):513–545. doi: 10.1146/annurev-arplant-043014-114623. [DOI] [PubMed] [Google Scholar]
- 66.Beckman CH. The nature of wilt diseases of plants / C.H. Beckman. St. Paul, Minn: APS Press; 1987. [Google Scholar]
- 67.Roncero MIG, Hera C, Ruiz-Rubio M, Garcıa Maceira FI, Madrid MP, Caracuel Z, Calero F, et al. Fusarium as a model for studying virulence in soilborne plant pathogens. Physiol Mol Plant Pathol. 2003;62(2):87–98. doi: 10.1016/S0885-5765(03)00043-2. [DOI] [Google Scholar]
- 68.Yadeta K, Thomma B. The xylem as battleground for plant hosts and vascular wilt pathogens. Front Plant Sci. 2013;4:97. doi: 10.3389/fpls.2013.00097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bravo Ruiz G, Di Pietro A, Roncero MIG. Combined action of the major secreted exo- and endopolygalacturonase is required for full virulence of Fusarium oxysporum. Mol Plant Pathol. 2016;17(3):339-53. [DOI] [PMC free article] [PubMed]
- 70.Recorbet G, Steinberg C, Olivain C, Edel V, Trouvelot S, Dumas-Gaudot E, Gianinazzi S, Alabouvette C. Wanted: pathogenesis-related marker molecules for Fusarium oxysporum. New Phytol. 2003;159(1):73–92. doi: 10.1046/j.1469-8137.2003.00795.x. [DOI] [PubMed] [Google Scholar]
- 71.Jashni MK, Dols IHM, Iida Y, Boeren S, Beenen HG, Mehrabi R, Collemare J, de Wit PJGM. Synergistic Action of a Metalloprotease and a Serine Protease from Fusarium oxysporum f. sp. lycopersici Cleaves Chitin-Binding Tomato Chitinases, Reduces Their Antifungal Activity, and Enhances Fungal Virulence. Mol Plant Microbe Interact. 2015;28(9):996–1008. doi: 10.1094/MPMI-04-15-0074-R. [DOI] [PubMed] [Google Scholar]
- 72.Langner T, Göhre V. Fungal chitinases: function, regulation, and potential roles in plant/pathogen interactions. Curr Genet. 2016;62(2):243–254. doi: 10.1007/s00294-015-0530-x. [DOI] [PubMed] [Google Scholar]
- 73.Huang Q-S, Xie X-L, Liang G, Gong F, Wang Y, Wei X-Q, Wang Q, Ji Z-L, Chen Q-X. The GH18 family of chitinases: Their domain architectures, functions and evolutions. Glycobiology. 2012;22(1):23–34. doi: 10.1093/glycob/cwr092. [DOI] [PubMed] [Google Scholar]
- 74.Liu T, Song T, Zhang X, Yuan H, Su L, Li W, Xu J, Liu S, Chen L, Chen T, et al. Unconventionally secreted effectors of two filamentous pathogens target plant salicylate biosynthesis. Nat Commun. 2014;5:4686. doi: 10.1038/ncomms5686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Taylor A, Vagany V, Jackson AC, Harrison RJ, Rainoni A, Clarkson JP. Identification of pathogenicity-related genes in Fusarium oxysporum f. sp. cepae. Mol Plant Pathol 2016;17(7):1032-47. [DOI] [PMC free article] [PubMed]
- 76.Sperschneider J, Gardiner DM, Thatcher LF, Lyons R, Singh KB, Manners JM, Taylor JM. Genome-wide analysis in three Fusarium pathogens identifies rapidly evolving chromosomes and genes associated with pathogenicity. Genome Biol Evol. 2015;7(6):1613–1627. doi: 10.1093/gbe/evv092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bolton MD, van Esse HP, Vossen JH, de Jonge R, Stergiopoulos I, Stulemeijer IJ, van den Berg GC, Borras-Hidalgo O, Dekker HL, de Koster CG, et al. The novel Cladosporium fulvum lysin motif effector Ecp6 is a virulence factor with orthologues in other fungal species. Mol Microbiol. 2008;69(1):119–136. doi: 10.1111/j.1365-2958.2008.06270.x. [DOI] [PubMed] [Google Scholar]
- 78.Kombrink A, Thomma BPHJ. LysM Effectors: Secreted Proteins Supporting Fungal Life. PLoS Pathog. 2013;9(12):e1003769. doi: 10.1371/journal.ppat.1003769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.de Jonge R, van Esse HP, Kombrink A, Shinya T, Desaki Y, Bours R, van der Krol S, Shibuya N, Joosten MH, Thomma BP. Conserved fungal LysM effector Ecp6 prevents chitin-triggered immunity in plants. Science. 2010;329(5994):953–955. doi: 10.1126/science.1190859. [DOI] [PubMed] [Google Scholar]
- 80.Sánchez-Vallet A, Mesters JR, Thomma BPHJ. The battle for chitin recognition in plant-microbe interactions. FEMS Microbiol Rev. 2015;39(2):171–183. doi: 10.1093/femsre/fuu003. [DOI] [PubMed] [Google Scholar]
- 81.de Jonge R, Bolton MD, Kombrink A, van den Berg GCM, Yadeta KA, Thomma BPHJ. Extensive chromosomal reshuffling drives evolution of virulence in an asexual pathogen. Genome Res. 2013;23(8):1271–1282. doi: 10.1101/gr.152660.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–10. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 83.Gao L-L, Anderson JP, Klingler JP, Nair RM, Edwards OR, Singh KB. Involvement of the Octadecanoid Pathway in Bluegreen Aphid Resistance in Medicago truncatula. Mol Plant-Microbe Interact. 2007;20(1):82–93. doi: 10.1094/MPMI-20-0082. [DOI] [PubMed] [Google Scholar]
- 84.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal. 2011;17:1. [Google Scholar]
- 85.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat Biotech. 2011;29(1):24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26(6):841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hu Z-L, Bao J, Reecy JM. CateGOrizer: a web-based program to batch analyze gene ontology classification categories. Online J Bioinformatics. 2008;9(2):108–112. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The authors declare all data and material is available with all datasets deposited in publicly available NCBI repositories. Trimmed RNA-sequencing data is available from the NCBI/GenBank BioProject database under BioProject number PRJNA294248 and can be accessed via the following link http://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA294248.