

Abstract
Background
Throughout a long period of adaptation and selection, sheep have thrived in a diverse range of ecological environments. Mongolian sheep is the common ancestor of the Chinese short fat-tailed sheep. Migration to different ecoregions leads to changes in selection pressures and results in microevolution. Mongolian sheep and its subspecies differ in a number of important traits, especially reproductive traits. Genome-wide intraspecific variation is required to dissect the genetic basis of these traits.
Results
This research resequenced 3 short fat-tailed sheep breeds with a 43.2-fold coverage of the sheep genome. We report more than 17 million single nucleotide polymorphisms and 2.9 million indels and identify 143 genomic regions with reduced pooled heterozygosity or increased genetic distance to each other breed that represent likely targets for selection during the migration. These regions harbor genes related to developmental processes, cellular processes, multicellular organismal processes, biological regulation, metabolic processes, reproduction, localization, growth and various components of the stress responses. Furthermore, we examined the haplotype diversity of 3 genomic regions involved in reproduction and found significant differences in TSHR and PRL gene regions among 8 sheep breeds.
Conclusions
Our results provide useful genomic information for identifying genes or causal mutations associated with important economic traits in sheep and for understanding the genetic basis of adaptation to different ecological environments.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-016-3212-2) contains supplementary material, which is available to authorized users.
Keywords: Sheep, Microevolution, Whole-genome resequencing, Selection signal, Reproduction
Background
Sheep were initially reared mainly for meat and were subsequently specialized for other products approximately 4000–5000 years ago. With the development of animal husbandry and the application of directed mating technology, phenotypic radiation under selection has resulted in the spectrum of modern sheep breeds, adapted to a diverse range of environments and specialized for the production of meat, milk, and wool [1, 2]. China has a long history of sheep domestication and rich resources of sheep breeds [3, 4]. Based on tail type, the Chinese domesticated sheep can be divided into five types: short fat-tailed sheep, long fat-tailed sheep, short thin-tailed sheep, long thin-tailed sheep and buttock-tailed sheep. According to archaeological and genetic research, Mongolian sheep is the common ancestor of Chinese short fat-tailed sheep breeds. Mongolian sheep evolved from the wild Argali sheep in the mountain regions of Central Asia. More than 2000 years ago, with the development of free trade, inter-ethnic war and the southward migration of steppe tribes, a large number of populations had moved south of the Great Wall. As a result, Mongolian sheep also migrated to North China villages and were introduced into Gansu, Xinjiang, Qinghai, Shandong and other provinces. Thus, most modern Chinese sheep breeds have a relationship to Mongolian sheep. Small-tailed Han sheep, Duolang sheep, Hu sheep, Tan sheep and others are all Mongolian sheep subspecies [5] (Fig. 1). However, from the Mongolian plateau to various ecoregions around almost the entire country, Mongolian sheep have experienced changes in climate, environment and feeding conditions (from pastoral areas to rural areas) and been subjected to artificial selection in different directions [6]. All of these factors have the potential to drive changes in selection and thereby cause microevolution [7]. The subspecies of Mongolian sheep show significant differences in a number of traits, especially related to reproduction, but how species differ genetically in relation to these traits is not well understood [5]. Chinese short fat-tailed sheep are an excellent model for genetic studies on phenotypic evolution and adaptations to various ecoregions.
Fig. 1.
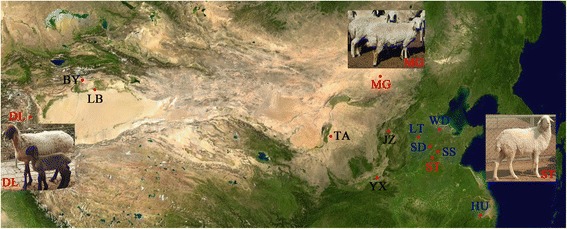
Geographic distribution of Chinese short fat-tailed sheep. MG, DL, LB, BY, JZ, TA, YX, ST, LT, WD, SD, SS and HU are abbreviations for Mongolian sheep, Duolang sheep, Luobu sheep, Buyinbuluk sheep, Jinzhong sheep, Tan sheep, Yuxi fat-tailed sheep, Small-tailed Han sheep, Large-tailed Han sheep, Wadi sheep, Luzhong Shandi sheep, Sishui Fur sheep and Hu sheep, respectively. The 3 breeds used for whole-genome resequencing are labeled in red, and the additional 5 breeds used for genotyping are labeled in blue. This figure has been modified from China 100.78713E 35.63718 N.jpg (https://commons.wikimedia.org/wiki/File:China_100.78713E_35.63718N.jpg). This image is in the public domain because it is a screenshot from NASA’s globe software World Wind using a public domain layer, such as Blue Marble, MODIS, Landsat, SRTM, USGS or GLOBE
In recent years, many studies have analyzed the genetic changes between wild and domestic animals, between species adapted to extreme environments and common domestic species and among different local breeds with phenotypic diversity. These studies have identified genes with key roles in domestication [8–12], adaptation to extreme environments [13–16] or prominent economic traits [17–24]. Studies on the domestication of livestock have demonstrated that genes affecting brain and neuronal development have often been targeted [8]. In addition, a striking selective sweep at the locus for thyroid-stimulating hormone receptor (TSHR), which likely affects seasonal reproduction, was identified in domestic chicken [9]. A coat color locus, MC1R, was identified in wild and domestic pigs [10]. A set of genes with key roles in starch digestion separating wolves from dogs were identified [11]. Research on domestic horse breeds has provided evidence of the importance of MSTN in racing breeds and selection for gait and size [12]. In the Tibetan Plateau, many species have become popular research topics, such as the Tibetan wild boar [13], Tibetan Mastiffs [14, 15] and ground tit [16]. A number of genes associated with adaptation to high altitudes and hypoxia have been revealed. To understand the molecular basis underlying phenotypic variation in economically important traits in livestock, many studies have also focused on genome-wide genetic variations among different local breeds, such as chicken [17], pig [18, 19] and cattle [20–24]. However, in Chinese short fat-tailed sheep, there have been few similar studies.
In this study we performed pooled whole-genome resequencing of 3 sheep breeds (Mongolian sheep, Small-tailed Han sheep and Duolang sheep) distributed over a wide range of geographical distance to examine the genetic variation among them. This effort identified a large number of single-nucleotide polymorphisms (SNPs) and short sequence insertions and deletions (indels) in sheep. Comparison of these variations defined potential genomic regions and metabolic pathways associated with important biological functions. This study revealed different signals of selection in the 3 sheep breeds and focused on the genetic relationships between Mongolian sheep and the two subspecies. Furthermore, to validate the presence of selection, we examined the haplotype diversity of 3 regions showed evidence of selective pressure related to reproduction among 8 sheep breeds. The substantial genomic resources provided here are useful for identifying genetic variations for phenotypic diversity and for revealing different signatures of selection associated with adaptation to various ecoregions.
Results
Data production
In total, more than 480 million 2 X 100-bp paired-end reads were generated and aligned to the sheep reference sequence (USUC oar_ref_Oar_v3.1) with a genome size of 2,587,507,083 bp. Thus, we achieved a 43.2-fold coverage of the reference genome, with a 14.4 X average sequencing depth for each breed. We identified 17,420,695 putative SNPs and 2,912,131 indels for the three breeds (Fig. 2, Additional file 1: Table S1; Additional file 2: Table S2; Additional file 3: Figure S1). We were able to experimentally validate 100 out of 102 tested SNPs (Additional file 4: Table S3).
Fig. 2.
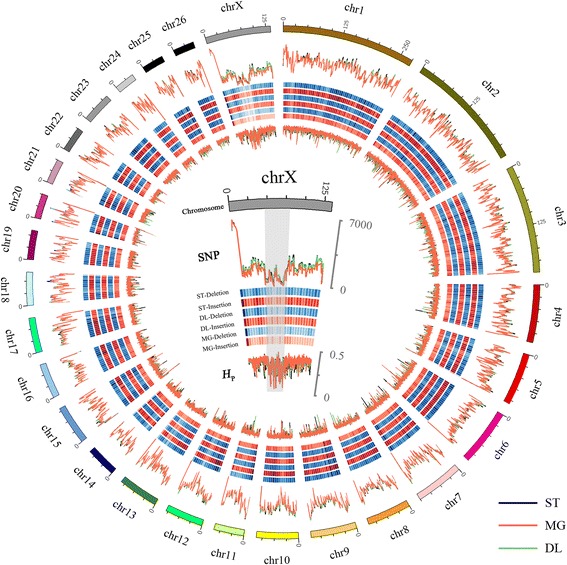
Summary of genome-wide genetic variation in the 3 Chinese short fat-tailed sheep breeds. Chromosomes are shown in different colors in the outermost circle, and the innermost circles show the distribution of SNPs (counted in 1-Mb windows), indels (counted in 5-Mb windows) and pooled heterozygosity (H P) in 200-kb windows of the Small-tailed Han sheep (ST), Mongolian sheep (MG) and Duolang sheep (DL) genomes relative to the sheep reference genome. For SNPs and H P scores, black represents Small-tailed Han sheep, red represents Mongolian sheep, and green represents Duolang sheep. At the center of the circular map, chr. X is shown separately. A remarkably homozygous region (in gray shadow) was observed from 43 to 78 Mb in Chinese short fat-tailed sheep. This figure was created using the Circos program [60]
SNP annotations
The numbers of SNPs and indels in the coding sequences (CDS) of the 3 breeds are shown in Additional file 5: Table S4. Small-tailed Han sheep and Duolang sheep both exhibit year-round estrous and prolificacy in breeding, whereas Mongolian sheep exhibits seasonal estrous and singleton breeding. Thus, we identified 3105 genes that contained missense SNPs or stop gained/loss variants in Mongolian sheep but not in Small-tailed Han sheep or Duolang sheep. And we also identified 803 genes that contained missense SNPs or stop gained/loss variants in both Small-tailed Han sheep and Duolang sheep, but not in Mongolian sheep. We speculated that these genes might contribute to the phenotypic differentiation. We searched for significantly overrepresented gene ontology (GO) terms in the above two groups of genes respectively and identified several categories (Additional file 6: Table S5; Additional file 7: Table S6; Additional file 8: Figure S2). In the former group one conspicuous cluster related to reproduction; 59 genes belonged to this category (13 genes were related to single fertilization and 24 genes were related to spermatogenesis). Another conspicuous cluster with 284 genes was related to response to stimulus (Table 1 and Additional file 6: Table S5); one of these genes TRPM8 has been shown to be a major determinant of cold perception in the mouse [25] and was reported to be related to adaptation to cold climate on sheep [26]; 12 of the 284 genes were related to response to light stimulus and we suggested these genes might had potential relationship with seasonal estrous. In the latter group one conspicuous cluster related to reproduction as well; 15 genes belonged to this category (12 genes were related to spermatogenesis). Ten Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways were also significantly enriched in the former group genes; four ones were enriched in the latter group genes among which “Progesterone-mediated oocyte maturation” contained 7 genes (RPS6KA3, MAD2L1, CCNB2, GNAI2, ADCY5, PIK3R5, CDC25B) that may be related to the reproductive traits of short fat-tailed sheep [27] (Additional file 9: Table S7; Additional file 10: Table S8; Additional file 8: Figure S2). We also identified 3916 genes that contained SNPs in promoter region (1 kb up- or downstream of genes) in Mongolian sheep but not in Small-tailed Han sheep or Duolang sheep and 136 genes that contained SNPs in promoter region (1 kb up- or downstream of genes) in both Small-tailed Han sheep and Duolang sheep, but not in Mongolian sheep. The GO term enrichment/KEGG analysis indicated that in the former 3916 genes, one conspicuous cluster containing 53 genes were related to reproduction (10 genes were related to fertilization) and another cluster containing 12 genes were related to rhythmic process (9 genes were related to circadian rhythm) (Additional file 11: Table S9; Additional file 12: Table S10; Additional file 13: Table S11; Additional file 14: Figure S3). These genes might be associated with the differences in reproductive performance of short fat-tailed sheep as well. In addition, the functional effects of missense SNPs were further investigated with SIFT and Provean. The result indicated 565, 490 and 320 SNPs were detected in protein coding genes from Mongolian sheep, Small-tailed Han sheep and Duolang sheep, respectively (Additional file 15: Table S12).
Table 1.
Enriched gene ontology terms related to response to stimulus among genes containing unique missense SNPs or stop gained/loss variants in Mongolian sheep
| Gene ontology term | Gene count | P value |
|---|---|---|
| Response to stress; | 140#1222 | 3.06488E-11 |
| Response to external stimulus; | 85#633 | 4.65E-11 |
| Response to wounding; | 61#423 | 1.46E-09 |
| Response to chemical stimulus; | 73#589 | 1.39E-07 |
| Response to hypoxia; | 13#48 | 0.000115 |
| Response to stimulus # regulation of response to stimulus; | 9#35 | 0.003204 |
| Response to biotic stimulus; | 33#289 | 0.007919 |
| Response to radiation; | 15#101 | 0.010492 |
| Response to abiotic stimulus; | 21#165 | 0.014159 |
| Response to drug; | 11#63 | 0.017014 |
| Response to lipopolysaccharide; | 3#5 | 0.018798 |
| Response to bacterium # response to molecule of bacterial origin; | 4#10 | 0.019823 |
| Response to fungus # response to molecule of fungal origin; | 2#2 | 0.027385 |
| Response to protein stimulus; | 10#59 | 0.027385 |
| Response to unfolded protein; | 10#59 | 0.027385 |
| Response to light stimulus; | 12#81 | 0.028201 |
Selective sweep analysis
To search genomic regions under selection during the migration of the short fat-tailed sheep, we measured the pooled heterozygosity (H P) in 200-kb windows, with half-step sliding along the genomes of the three breeds, and the fixation index (F ST) between any two breeds also in 200-kb windows with half-step sliding. The distributions of H P and F ST values, as well as the Z transformations of these values, Z(H P) and Z(F ST), for the 3 sheep breeds are plotted in Fig. 3, Additional file 16: Figure S4 and Additional file 17: Figure S5 (the Z transformation of H P in Mongolian sheep, Small-tailed Han sheep and Duolang sheep are Z(H P)M, Z(H P)S and Z(H P)D for short, respectively; the Z transformation of Z(F ST) between Mongolian sheep and Small-tailed Han sheep, between Mongolian sheep and Duolang sheep as well as between Small-tailed Han sheep and Duolang sheep are Z(F ST)M-S, Z(F ST)M-D and Z(F ST)S-D for short, respectively). The skewed distributions of the H P and F ST scores indicated the existence of selection on these sheep breeds. The windows with Z(H P) < -4 or (and) Z(F ST) > 4 were considered the target windows because these windows were at the extreme ends of the distribution (Fig. 3). Figure 3 shows intuitive evidence of different selection signals in each autosome among the 3 breeds. For example, on chromosome (chr.) 10, there were several windows with a low Z(H P)M, and high Z(F ST)M-S and Z(F ST)M-D, suggesting that a high selection signal was located on this chromosome. This finding is consistent with a previous study in which the genome-wide distribution of global F ST for 49,034 SNPs from 75 sheep breeds all over the world revealed the highest selection signal detected on chr. 10 [2]. The highest ranked SNP was located at Mb position 29.54 near Relaxin/insulin-like family peptide receptor 2 (RXFP2), which is linked to the absence of horns (poll) in sheep [2]. In our research, RXFP2 was also found in the selection region of Small-tailed Han sheep (F ST = 0.514, Z(F ST)M-S = 4.08).
Fig. 3.
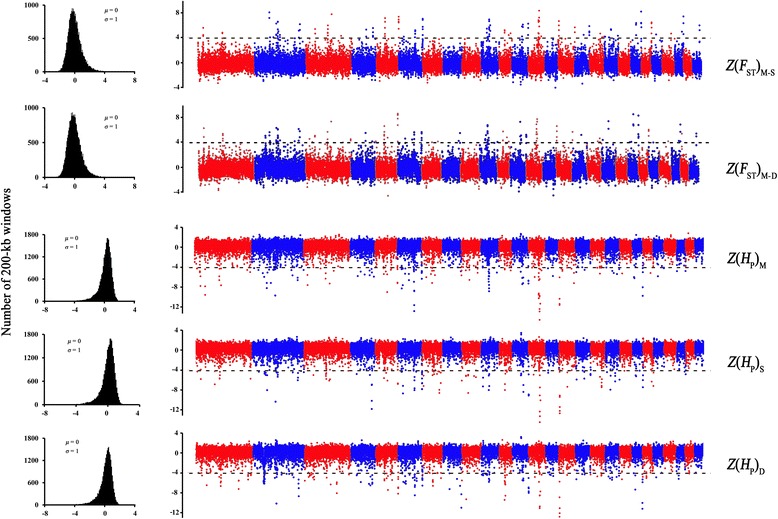
Selection analyses identified selection signals in 3 Chinese short fat-tailed sheep breeds. The distributions of Z-transformed average pooled heterozygosity (H P) as well as the average fixation index between any two breeds for autosomal 200-kb windows (σ, standard deviation; μ, average) are shown on the left. The Z-value distribution plotted along sheep autosomes 1–26 (chromosomes are separated by red and blue coloring) is shown on the right. The dashed horizontal line indicates the cut-off (Z > 4 or Z < -4) used for extracting outliers
We identified regions of selection in the genomes of the two subspecies Small-tailed Han sheep and Duolang sheep. As a result, in the autosomes of Small-tailed Han sheep, 108 regions with extremely high levels of homozygosity and 65 regions with increased genetic distance to Mongolian sheep were identified. In total, 134 regions containing 774 protein-coding genes were identified (Additional file 18: Table S13). Interestingly, the highest H P and the lowest F ST were located in the same window: 53.3–53.5 Mb on chr. 13 (F ST = 0.692, Z(F ST) = 8.158; H P = 0.0597, Z(H P) = -14.091). This window harbors 10 protein-coding genes, ZBTB46, ZGPAT, ARFRP1, RTEL1, STMN3, GMEB2, C20orf195, SRMS, PTK6 and EEF1A2, of which RTEL1 and EEF1A2 are related to the inhibition of apoptosis and programmed cell death. On chr. X, 9 regions containing 24 protein-coding genes were identified by applying a threshold of Z(H P) < -3 or (and) Z(F ST) > 3 (Additional file 18: Table S13). One of these genes, AR, which encodes the androgen receptor, is essential for prostate gland development, urogenital system development and reproductive development. In the whole genome, 143 regions with a total length of 45.7 Mb accounted for 1.77 % of the entire genome. Similarly, in the genome of Duolang sheep, 143 regions (134 autosomal regions and 9 regions on chr. X) with a total length of 51.7 Mb accounted for 2.0 % of the entire genome (Additional file 19: Table S14). There were 74 regions that overlapped each other between the 2 breeds, accounting for 25.1 Mb.
We divided all of the protein-coding genes residing in the genomic regions with a Z(H P) < -4 into 3 groups: I, Z(H P)M < -4 (605 genes); II, Z(H P)S < -4 (567 genes); and III, Z(H P)D < -4 (605 genes) (Fig. 4a), and then analyzed significantly enriched GO terms among these groups. In the 208 genes belonging to all the 3 groups, nervous system development, reproductive processes and other biological functions were enriched (Fig. 4b). These findings were in agreement with the notion that genes affecting brain and neuronal development have often been targeted during animal domestication [8]. In addition, 5 genes involved in ear development were pinpointed in the 3 sheep breeds: OTX1 and SOD1, in both group II and III (Fig. 4c), and LHFPL5, HOXA2 and GJB6, in group I (Fig. 4d). This result indicated that Chinese indigenous sheep breeds might have been subjected to different selection for ear development than Texel sheep, which were sequenced for the sheep reference genome. In relation to adaptation to different ecoregions, the three Chinese short fat-tailed sheep breeds show different selection signals. Among the subspecies, Small-tailed Han sheep and Duolang sheep both show signals of selection for immune system development and regulation, reproduction, muscle contraction and stress responses (Fig. 4c). The stress responses could be divided to two kinds: first, superoxide metabolic processes including genes ALOX12 and SOD1 in both group II and III (Fig. 4c); second, DNA repair including genes POLH and UBE2N only in group II (Fig. 4e) and the response to chemical stimuli including genes TGM7 and OXSR1 only in group III (Fig. 4f).
Fig. 4.
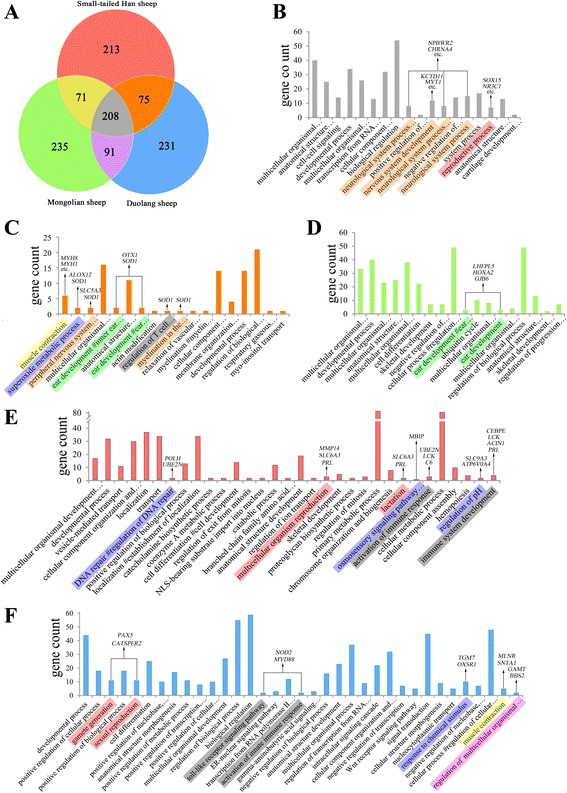
Biological processes enrichment of genes located in regions with a Z(H P) < -4. a All of the genes in regions with a Z(H P) < -4 were divided into 3 groups: I, Z(H P)M < -4 (605 genes); II, Z(H P)S < -4 (567 genes); and III, Z(H P)D < -4 (605 genes). b, c, d, e and f indicate the biological process enrichment of genes belonging to all 3 groups; belonging to both group II and III; belonging to only group I; belonging to only group II and belonging to only group III. Important enriched terms are color coded to reflect the relatedness of biological processes. Orange, nervous system development; red, reproductive process; yellow, muscle contraction; blue, various processes related to stress responses; green, ear development; grey, immune system development and regulation; pink, regulation of multicellular organism growth
The protein-coding genes in the regions of selection with a Z(F ST)M-S > 4 or (and) Z(F ST)M-D > 4 were also compared and used to detect significantly enriched GO terms. A total of 361 genes located in regions with a Z(F ST)M-S > 4 and a Z(F ST)M-D > 4 were related to nervous system development, ear development, fatty acid biosynthetic and metabolic processes, regulation of growth, sexual reproduction and other biological processes (Additional file 20: Table S15). Additionally, 91 genes located in regions with a Z(F ST)M-S > 4 were related to vesicle-mediated transport, protein digestion, regulation of protein polymerization, chemical homeostasis, reproductive processes and other biological processes (Additional file 21: Table S16), while 43 genes located in regions with a Z(F ST)M-D > 4 were related to spermatogenesis, single fertilization, post-translational protein modification, regulation of protein kinase activity, regulation of fatty acid beta-oxidation, blastoderm segmentation, cell proliferation and other biological processes (Additional file 22: Table S17).
Interestingly, gene HOXA10 met both Z(F ST)M-S > 4 and Z(H P)M < -4 (Z(F ST)M-S = 4.168, Z(H P)M = -5.39), and also contained missense SNPs in both Small-tailed Han sheep and Duolang sheep, but not in Mongolian sheep. In previous reports HOXA10 is a well-known transcriptional factor gene, shown to be one of the most promising candidate genes that play major roles in endometrial differentiation and development, establishing the conditions required for implantation and normal pregnancy maintenance [28] and have been widely studied in human, mouse and other species [29–33]. In this research, HOXA10 showed strong selection signatures in Chinese short fat-tailed sheep breeds. We suggested this gene is an important factor causing the differences in reproductive performance among Chinese short fat-tailed sheep breeds.
On chr. X, we found that the average H P score was lower (H P-chrX = 0.371 < H P-chrA = 0.386) and the average F ST score was higher (F ST-chrX = 0.427 > F ST-chrA = 0.337) than the autosomes (Additional file 16: Figure S4 and Additional file 17: Figure S5). Furthermore, we noted that the distributions of the standard deviations of H P (σX = 0.049 > σA = 0.023) and F ST (σX = 0.089 > σA = 0.043) were larger on chr. X. As shown in Additional file 17: Figure S5, a larger proportion of the windows resided in the tails of the distributions on chr. X compared to the autosomes.
Three putative sweeps related to reproductive traits
Reproductive traits showed the most significant phenotypic differences among the Chinese short fat-tailed sheep breeds. Therefore, we chose 3 genomic regions related to reproduction to validate the different signals of selection among 8 sheep breeds. TSHR showed evidence of selective pressure in both Small-tailed Han sheep and Duolang sheep (Z(H P)S = -6.55; Z(H P)D = -4.9; Z(H P)M = 0.6) and has been reported to have pivotal roles in metabolic regulation and photoperiod control of reproduction in vertebrates [9, 34] (Fig. 5a; Additional file 23: Figure S6). We studied haplotype diversity across the partial gene region (89.35–89.48 Mb on chr. 7) to validate its selection and genotyped 19 randomly selected SNPs in 95 individuals from 8 sheep breeds. These 95 tested individuals carried 123 copies of a 119-kb haplotype (Fig. 5b). The haplotype frequency of Mongolian sheep was significantly lower than those of the other 7 breeds (p < 0.01; Fig. 5c). This remarkable genetic diversity indicated that TSHR showed strong evidence of selective pressure. In selection signal analysis of sheep breeds from all over the world, positive selection was also detected surrounding TSHR [2, 26].
Fig. 5.
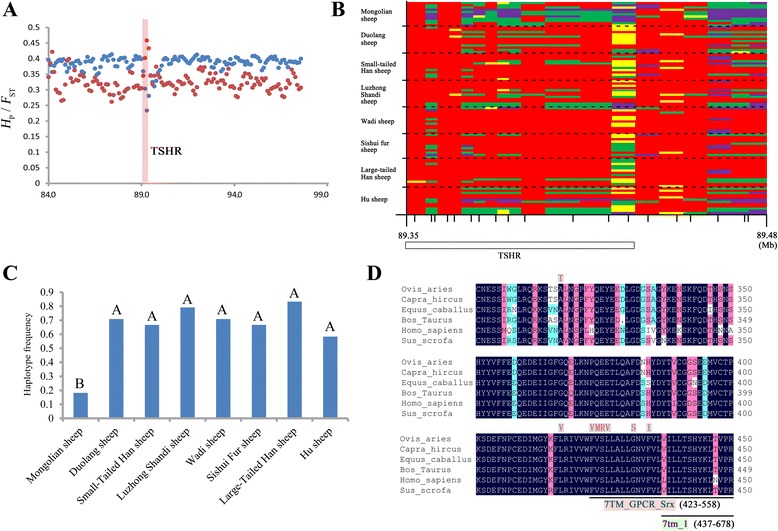
The haplotype diversity and candidate mutations of TSHR. a Pooled heterozygosity, H P (in blue), and average fixation index between Mongolian sheep and Small-tailed Han sheep, F ST (in red), plotted for 200-kb windows spanning the region harboring TSHR (in pink shadow) on chr. 7. b Genetic variation in the region 89.35–89.48 Mb on chr. 7 across partial TSHR. Individual sheep (95 from 8 breeds) were genotyped using WaferGen genotyping. Dashed horizontal lines separate the 8 breeds. At the bottom of the figure, short tick marks represent individual SNPs. Long tick marks indicate the position in Mb. Red color: homozygous A-allele; green color: heterozygous; purple color: homozygous a-allele; yellow color: missing genotype call. c The haplotype frequencies of the 8 sheep breeds. The haplotype frequency of Mongolian sheep was significantly less than those of the other 7 breeds (p < 0.01). d A total of 8 missense variants in TSHR were identified by an amino acid sequence comparison (300–450) from 6 species: Ovis aries, Capra hircus, Equus caballus, Bos Taurus, Homo sapiens and Sus scrofa. Asterisks, double dots, and single dots denote fully, strongly, and weakly conserved residues, respectively. Conserved Protein Domain: 7TM_GPCR_Srx and 7tm_1 are both underlined
In addition to haplotype diversity, we identified 8 missense variants in the TSHR genic region, 7 of which were unique to Mongolian sheep (Duolang sheep also has p. 315Ala > Thr). As shown in Fig. 5d, p.423Phe > Val, p.424Val > Met, p.425Ser > Arg, p.426Leu > Val, p.431Gly > Ser and p.434Phe > Ile were located in the Conserved Protein Domain 7TM_GPCR_Srx (Serpentine type seven-transmembrane G-protein-coupled receptor class chemoreceptor Srx) and close to 7tm_1 (7-transmembrane receptor (rhodopsin family)). The seven-transmembrane structure has a decisive effect on the biological function of TSHR. Thus, these 6 missense variants were considered candidate causal mutations for the TSHR sweep. It is possible that selection for these variants could be associated with photoperiod control of reproduction in Chinese short fat-tailed sheep.
PRL (Z(H P)S = -4.16; Z(H P)D = -0.91; Z(H P)M = -0.93) and HMGCR (Z(H P)S = -5.14; Z(H P)D = -2.42; Z(H P)M = -1.57) showed evidence of selective pressure in Small-tailed Han sheep only (Additional file 24: Figure S7A and S7B). PRL encodes prolactin and is essential for female pregnancy and lactation. In sheep, the luteinizing hormone-secretory response to gonadotropin-releasing hormone is completely suppressed by the combined actions of PRL and dopamine in the nonbreeding season [35]. To characterize haplotype diversity among different sheep breeds, we genotyped 17 randomly chosen SNPs in this selection region and identified a 38.2-kb region (34.22–34.26 Mb on chr. 20) spanning the PRL section and the entire LOC443319 encoding placental lactogen, which was highly divergent among the 8 tested sheep breeds (Additional file 24: Figure S7C). As shown in Additional file 24: Figure S7E, the haplotype frequencies of Small-tailed Han sheep, Luzhong Shandi sheep and Hu sheep were significantly higher than those of Mongolian sheep and Duolang sheep (p < 0.05). This result is in agreement with the H P scores of the 3 resequenced breeds in this region and may demonstrate the selection of PRL in Small-tailed Han sheep. HMGCR encodes 3-hydroxy-3-methylglutaryl-CoA reductase and is a key enzyme in cholesterol biosynthesis. This gene is related to germ cell migration and gonad development. We screened 16 SNPs across the HMGCR region (6.53–6.57 Mb on chr. 7) using 95 individuals from 8 breeds. However, no significant difference was found among the haplotype frequencies of the 8 tested breeds (Additional file 24: Figure S7F). We speculated that the selection on HMGCR might be due to genetic variation in other regulatory regions and sequences.
Discussion
In this study, we performed whole-genome resequencing of 45 sheep from 3 Chinese short fat-tailed breeds. We catalogued millions of SNPs from each breed to advance evolutionary and genetic research. This is the first whole-genome study to characterize genetic polymorphisms in Chinese short fat-tailed sheep breeds. Our selection analyses revealed that Mongolian sheep, Small-tailed Han sheep and Duolang sheep contain many common selection signatures unique to the Chinese short fat-tailed sheep and also many different selection signatures among their genomes. The 3 Chinese indigenous sheep breeds were suggested to have experienced differential selection for nervous system development, ear development and reproductive processes compared with Texel sheep, which were sequenced for the sheep reference genome. The regions of selection identified in Small-tailed Han sheep and Duolang sheep harbor different protein-coding genes with important biological functions, including roles in development, reproduction, growth and stress responses. Furthermore, we used WaferGen Genotyping to genotype 53 randomly chosen SNPs in 95 additional individuals from 8 sheep breeds across 3 genomic regions related to reproduction to validate the selection of these regions.
The most significant phenotypic difference between Mongolian sheep and the 2 subspecies was related to reproduction. In the recent large scale selection signature studies on sheep, reproduction has been identified as one of the selected targets and candidate genes associated with reproduction are detected in selection regions [2, 26, 36]. Kijas (2012) reported 31 genome regions with extreme differentiation among 74 world-wide sheep breeds by computing the globe F ST at all SNP in the genome, which included candidate genes related to coat pigmentation, skeletal morphology, body size, growth, and reproduction [2]. Fariello (2014) used a new approach FLK/hapFLK to re-scan the sheep genome for selection based on the Sheep HapMap dataset among 7 broad geographical groups and also identified 71 new selection signatures, with candidate genes related to coloration, morphology or production traits [26]. The candidate gene related to reproduction TSHR were both detected in the above two studies and identified as ancestral signatures of selection in the latter one. TSHR and other genes such as HOXA10, PRL and HMGCR were also located in the selection regions identified in this research. Besides, we identified genes containing SNPs causing missense mutations or stop gained/loss variants and genes with SNPs located in promoter regions in Mongolian sheep but not in Small-tailed Han sheep or Duolang sheep and vice-versa. The GO term enrichments and KEGG analysis both showed evidences for selection in reproduction and provided important additional candidate genes and pathways associated with reproduction such as genes related to single fertilization, spermatogenesis, response to light stimulus and circadian rhythm and the pathway “Progesterone-mediated oocyte maturation”. For the past several decades, there has been increasing interest in the identification and utilization of major genes for prolificacy in sheep. Genes with mutations that increase the ovulation rate (BMPR-1B, BMP15 and GDF9) and related to seasonal reproduction (MTNR1A) have been discovered in sheep [37–44]. However, in this study, we found that only GDF9 showed evidence of selective pressure in Duolang sheep (Z(H P)D = -4.103). The other genes appeared to not be under selection in Small-tailed Han sheep, Mongolian sheep or Duolang sheep. We checked the H P and F ST within genes BMPR1B and MTNR1A (the number of SNPs within BMP15 and GDF9 is less than 10) and the results showed the same conclusion (Additional file 25: Table S18). In contrast, according to recent research, the BMPR1B gene showed strong evidence of selection in highly prolific breeds, Hu sheep and Large-tailed Han sheep [3].
Besides reproductive performance, we also paid attention to the adaptation of sheep to different environments. The subspecies of Mongolian sheep have adapted to various ecoregions during the domestication process. Correspondingly, we detected several genes located in the selection regions or containing unique function-altering mutations with potential relationship with adaption such as the genes in Table 1. In the precious research, the selection near the TRPM8 gene was reported to be related to adaptation to cold climate [26] and this gene also showed selection evidence in this study. The adaption process is complex and the potential role of many selection signatures in the adaptation of sheep breeds remains unclear. In the meantime selection response for adaptation and welfare traits may be expected to continue [2]. The findings in this research confirmed the existence of selection during the adaption of Chinese short fat-tailed sheep and provided a large number of variants to be further investigated.
F ST is widely used in the selection signature detection of the whole genome of the domestic animals [2, 11, 19, 36, 45]. However, this approach was reported to have potential limitations: first, when applied to hierarchically structured data sets, F ST analysis may lead to a large proportion of false positives and false negatives; second, the heterogeneity of effective population size among breeds implies that some breeds are more prone to contribute large locus-specific F ST values than others [26, 46, 47]. In view of these limitations, a new strategy to evaluate the haplotype differentiation between populations was proposed to increase the detection power of selective sweeps and also enable to detect soft or incomplete sweeps, FLK/hapFLK [26]. In this study, we performed pooled whole genome resequencing and used three approaches to detect the selection signature on the Chinese short fat-tailed sheep genome, unique function-altering mutations, the pooled heterozygosity (H P) and the average F ST between breeds. We expect the combination of these three approaches could breakthrough the limitation of the single method to a certain extent. The detection results of the three approaches all confirmed that reproduction is important selection target of Chinese short fat-tailed sheep.
Chr. X differs from autosomes in several aspects of population genetics, including reductions in effective population size and recombination rate. Due to genetic drift, these differences on chr. X are expected to cause a greater reduction in the level of genetic variation and increased genetic differentiation among different sheep breeds compared to the autosomes [11]. On chr. X of Chinese short fat-tailed sheep, a remarkably homozygous region (43–78 Mb) was observed in which the number of SNPs and indels was significantly less than other regions (Fig. 2).
Large-tailed Han sheep has a long history, and its origins might lie in the fat-tailed sheep in Ancient Central Asia and West Asia, which were imported to China via the Silk Road [5]. Due to the high reproductive performance of Large-tailed Han sheep, we compared it to 7 breeds of Chinese short fat-tailed sheep in this study. The results indicated that the haplotype frequencies of 3 tested gene regions related to reproduction were not significantly different between Large-tailed Han sheep and Small-tailed Han sheep. We speculate that Large-tailed Han sheep and Small-tailed Han sheep might have been affected by similar selection during the long period of microevolution.
With the advance of high-throughput sequencing technologies, the detection of variants in domesticated animals on a large scale offers great opportunities to study genome evolution in response to phenotypic selection. The present study provided genome scan for selection in Chinese native sheep breeds and is an attempt to investigate unique evolution direction and gene resources. Full use of these resources and searching the valuable genes for sheep genetic improvement will become the targets of future research. In the meantime, more extensive range of breeds, larger population size, more in-depth sequencing, and more advanced statistical methods are the directions of our improvements in this study.
Conclusions
In summary, this study detected large amounts of genetic variations and different genomic regions under selection in 3 Chinese short fat-tailed sheep breeds. Small-tailed Han sheep is a valued local Chinese variety, famous for its high reproduction performance and strong adaptability. Genome-wide comparison studies revealed genes with unique selective signals that are associated with reproduction and other traits of this sheep breed. Our results are a valuable resource for future studies of genotype-phenotype and for the improvement of sheep breeding.
Methods
Animals
We obtained genomic DNA samples from whole blood from 8 sheep populations, with 20 female individuals from each population. Three breeds were used for resequencing: Mongolian sheep from Siziwang Banner in Inner Mongolia province, Small-tailed Han sheep from Jiaxiang in Shandong province, and Duolang sheep from Maigaiti in Xinjiang province. The environment, body size and reproductive performance of these three breeds were shown in Table 2. These three breeds came from different ecoregions distributed over a wide range of geographical distance with different climate and feeding conditions. WaferGen genotyping was performed on all 8 breeds. Except the three breeds used for resequencing, the other five breeds were: Luzhong Shandi sheep from Pingyin in Shandong province, Wadi sheep from Zhanhua in Shandong province, Sishui Fur sheep from Sishui in Shandong province, Large-tailed Han sheep from Linqing in Shandong province and Hu sheep from Huzhou in Zhejiang province.
Table 2.
Comparison of the environment, body size and productive performance of the breeds used for resequencing
| Mongolian sheep | Small-tailed Han sheep | Duolang sheep | |
|---|---|---|---|
| Ecoregion | Mongolian-Manchurian grassland | Huang He Plain mixed forests | Tarim Basin deciduous forests and steppe |
| Coordinates | 41°10′–43°22′N 110°20′–113°E | 35°11′–35°38′N 116°06′–116°27′E | 38°25′–39°22′N 77°28′–79°05′E |
| Climate | Plateau sub temperate continental monsoon | Warm temperate monsoon | Temperate continental dry |
| Feeding conditions | Grazing | Drylot | Drylot |
| Male adult weight (kg) | 51.3–71.1 | 78.2–129.6 | 80.6–112 |
| Female adult weight (kg) | 44.4–55.2 | 56–72.8 | 61.6–86.6 |
| Estrous characteristics | Seasonal | Annual | Annual |
| Lambing rate (%) | 103 | 267.1 | 250 |
The data shown were collected from Animal genetic resources in China: Sheep and Goats
DNA extraction and sequencing
DNA was extracted from EDTA-preserved blood using a QIAamp DNA Blood Mini Kit (Qiagen). We performed pooled whole-genome resequencing of three sheep breeds. We pooled DNA from 15 individuals of each breed into one pool before library construction (six paired-end sequencing libraries with an insert size of 200–500 bp, two for each breed) and sequencing of 100-bp paired-end reads with a HiSeq2000 instrument (Illumina, USA). The raw sequence reads were filtered by removing the index sequences and low-quality paired reads. Specifically, we filtered sequences in which the single-ended N content exceeded 10 % of the length of the entire read or the single-ended number of bases with less than 5X depth exceeded 50 % of the entire read. Clean reads were mapped to the Ovis aries (sheep) genome (USUC oar_ref_Oar_v3.1; http://www.livestockgenomics.csiro.au/sheep) using the Burrows-Wheeler Alignment tool [48]. Duplicate reads were removed.
SNP identification and annotation
SNPs are small differences but have a great impact on variation in genomes and biological traits [49]. Therefore, we investigated SNP annotations in detail and paid special attention to those in genic regions. To detect genomic regions under selection in Chinese short fat-tailed sheep, we identified SNPs from the 3 pools of resequenced breeds relative to the sheep reference genome, respectively, using SAMTools. Firstly SAMTools collects summary information from the input BAM files (Binary Alignment/Map) and computes the likelihood of data given each possible genotype and stores the likelihoods in the BCF (Binary Variant Call Format). Secondly Bcftools applies the prior and does the SNP calling and converts BCF to VCF (Variant Call Format) which can be used in the following analysis [50]. Additional filters were applied as follows: minimum read depth = 5, minimum read depth for SNP identification = 2, and VarQuality ≥ 30. After SNP identification, we annotated and predicted the effects of the SNPs using SnpEff toolbox [51] and furtherly predicted whether the amino acid substitutions affect protein function using SIFT and Provean [52].
Selection analysis
Two approaches were used to search the sheep genome for regions that may have been affected by selection during the migration of Chinese short fat-tailed sheep. First, we calculated the average pooled heterozygosity (H P) in 200-kb windows sliding 100 kb at a time, following the formula described in Rubin et al. [9, 11]. We Z-transformed the distribution of H P and extracted putatively selected windows at the extreme ends of the distribution by applying a Z(H P) < −4 cut-off. We divided the protein-coding genes in the putative regions of selection that met this cut-off of Small-tailed Han sheep into 3 groups: Z(H P)S < -4, Z(H P)M < -4 and Z(H P)D < -4. Genes belonging to one (2 or 3) of the 3 groups were considered under selection in the corresponding breed(s). Second, we calculated F ST values between any two breeds for individual SNPs [53, 54]. We averaged F ST values across 200-kb windows, sliding 100 kb at a time, and Z-transformed the distribution. Putative selection targets were extracted from the extreme ends of the distribution by applying a Z(F ST) > 4 cut-off.
Due to the single-window pass cut-off Z(H P) < −4 and the no-windows pass Z(F ST) > 4 on chr. X, we performed extraction by applying Z(H P) < −3 or Z(F ST) > 3 for this chromosome.
Gene ontology functional enrichment and KEGG analysis
Ensembl gene annotations were used to identify protein-coding genes located in target regions [55] (extending 100 kb up- and downstream). All of these genes were classified into the categories of molecular function in the GO database using the GOstat program (P-Value Cutoff: 0.1, GO-Cluster Cutoff: -1 and Correct-Method: Benjamini) [56] and were mapped to the KEGG database using DAVID Bioinformatics Resources [27, 57].
Genotyping validation
We used WaferGen genotyping, targeting 53 SNPs located in regions showing a high level of homozygosity or population differentiation. A total of 95 sheep, representing 8 different breeds, were genotyped using standard protocols provided by the manufacturer (WaferGen, USA). Haplotypes were phased using PHASE software [58].
Multiple sequence alignment and conserved domain analysis
DNAman software was used to perform multiple sequence alignments. Conserved domains in TSHR were detected using the NCBI bioinformatics tools [59].
Acknowledgments
This work was financially supported by the Shandong Provincial Modern Agriculture Industry Technology System Sheep Industry Innovation Team (No. SATS2012263) and the Innovation Research of Agriculture and Biology Resources (No. 2011186125). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Availability of data and materials
Raw sequence data of 3 sheep breeds in the bam format from this study were deposited in the NCBI Short Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra) under accession numbers SRR2420291, SRR2420281 and SRR2420287.
Authors’ contributions
JMW managed the project. JMW, ZHL, ZBJ and LH designed the analyses. ZHL and TLC carried out the bioinformatics analyses and other works of this study. ZHL, GZW, ZBJ and LH collected the samples. ZHL wrote the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
The methods were carried out in accordance with the approved guidelines of Guidelines for Experimental Animals of the Ministry of Science and Technology (Beijing, China). All experimental procedures and animal collections were conducted under a permit (No. 2004006) approved by the Institutional Animal Care and Use Ethics Committee of Shandong Agricultural University.
Abbreviations
- 7tm_1
7-transmembrane receptor (rhodopsin family)
- 7TM_GPCR_Srx
Serpentine type seven-transmembrane G-protein-coupled receptor class chemoreceptor Srx
- ADCY5
Adenylate cyclase 5
- ALOX12
Arachidonate 12-lipoxygenase
- AR
Androgen receptor
- ARFRP1
ADP-ribosylation factor related protein 1
- BAM
Binary Alignment/Map
- BCF
Binary Variant Call Format
- BMP15
Bone morphogenetic protein 15
- BMPR-1B
Bone morphogenetic protein receptor, type 1B
- bp
Base pairs
- C20orf195
Chromosome 20 open reading frame 195
- CCNB2
Cyclin B2
- CDC25B
Cell division cycle 25B
- CDS
Sequences encoding amino acids in proteins
- Chr.
Chromosome
- DNA
Deoxyribonucleic acid
- EDTA
Ethylene diamine tetraacetic acid
- EEF1A2
Eukaryotic translation elongation factor 1 alpha 2
- FST
Fixation index
- FST-chrA
The average F ST on autosomes
- FST-chrX
The average F ST on chromosome X
- GDF9
Growth differentiation factor 9
- GJB6
Gap junction protein beta 6
- GMEB2
Glucocorticoid modulatory element binding protein 2
- GNAI2
Guanine nucleotide binding protein (G protein), alpha inhibiting 2
- GO
Gene ontology
- HMGCR
3-hydroxy-3-methylglutaryl-CoA reductase
- HOXA10
Homeobox A10
- HOXA2
Homeobox A2
- HP
Heterozygosity
- HP-chrA
The average H P on autosomes
- HP-chrX
The average H P on chromosome X
- indel
Insertion/deletion
- KEGG
Kyoto encyclopedia of genes and genomes
- LHFPL5
Lipoma HMGIC fusion partner-like 5
- LOC443319
Placental lactogen
- MAD2L1
MAD2 mitotic arrest deficient-like 1
- MC1R
Melanocortin 1 receptor
- MSTN
Myostatin
- MTNR1A
Melatonin receptor 1A
- NCBI
The National Center for Biotechnology Information
- OTX1
Orthodenticle homolog 1
- OXSR1
Oxidative Stress Responsive 1
- PIK3R5
Phosphoinositide-3-kinase, regulatory subunit 5
- POLH
Polymerase (DNA) Eta
- PRL
Prolactin
- PTK6
Protein tyrosine kinase 6
- RPS6KA3
Ribosomal protein S6 kinase, 90 kDa, polypeptide 3
- RTEL1
Regulator of telomere elongation helicase 1
- RXFP2
Relaxin/insulin-like family peptide receptor 2
- SAM
Sequence Alignment/Map
- SNP
Single-nucleotide polymorphism
- SOD1
Superoxide dismutase 1, soluble
- SRMS
Src-related kinase lacking C-terminal regulatory tyrosine and N-terminal myristylation sites
- STMN3
Stathmin-like 3
- TGM7
Transglutaminase 7
- TRPM8
Transient Receptor Potential Cation Channel Subfamily M Member 8
- TSHR
Thyroid stimulating hormone receptor
- UBE2N
Ubiquitin Conjugating Enzyme E2 N
- VCF
Variant Call Format
- Z(FST)
The Z transformation of F ST
- Z(FST)M-S
The Z transformation of F ST between Mongolian sheep and Small-tailed Han sheep
- Z(HP)
The Z transformation of H P
- Z(HP)D
The Z transformation of H P in Duolang sheep
- Z(HP)M
The Z transformation of H P in Mongolian sheep
- Z(HP)S
The Z transformation of H P in Small-tailed Han sheep
- ZBTB46
Zinc finger and BTB domain containing 46
- ZGPAT
Zinc finger, CCCH-type with G patch domain
- σA
The standard deviation of H P or F ST on autosomes
- σX
The standard deviation of H P or F ST on chromosome X
Additional files
Numbers and distribution of SNPs in the resequenced sheep breeds. (DOC 34 kb)
Numbers and distribution of indels in the resequenced sheep breeds. (DOC 34 kb)
The number of SNPs detected from the three Chinese short fat-tailed sheep breeds. The numbers of common SNPs between Mongolian sheep and Small-tailed Han sheep, between Mongolian sheep and Duolang sheep, between Small-tailed Han sheep and Duolang sheep and across the three breeds are 6,292,070/ 6,205,458/ 6,255,570 and 4,381,068, respectively. The numbers of unique SNPs belong to the three breeds are 2,326,015/ 2,530,094/ 2,573,624. (DOC 115 kb)
Ninety-eight percent of SNPs validated using WaferGen technology. (DOC 32 kb)
Numbers and distribution of coding region SNPs and indels in the resequenced sheep breeds. (DOC 32 kb)
Enriched GO terms among genes containing missense SNPs or stop gained/loss variants in Mongolian sheep but not in Small-tailed Han sheep or Duolang sheep. (DOC 673 kb)
Enriched GO terms among genes containing missense SNPs or stop gained/loss variants in both Small-tailed Han sheep and Duolang sheep, but not in Mongolian sheep. (DOC 159 kb)
Biological processes and KEGG pathway enrichment in genes containing missense SNPs or stop gained/loss variants. A, Biological processes enrichment in gene containing missense SNPs or stop gained/loss variants in Mongolian sheep but not in Small-tailed Han sheep or Duolang sheep. B, Biological processes enrichment in gene containing missense SNPs or stop gained/loss variants in both Small-tailed Han sheep and Duolang sheep, but not in Mongolian sheep. C, KEGG pathway enrichment in genes containing missense SNPs or stop gained/loss variants in Mongolian sheep but not in Small-tailed Han sheep or Duolang sheep. D, KEGG pathway enrichment in genes containing missense SNPs or stop gained/loss variants in both Small-tailed Han sheep and Duolang sheep, but not in Mongolian sheep. Biological processes and KEGG pathway related to reproduction are labeled in red. (DOC 955 kb)
Enriched KEGG pathways among genes containing missense SNPs or stop gained/loss variants in Mongolian sheep but not in Small-tailed Han sheep or Duolang sheep. (DOC 44 kb)
Enriched KEGG pathways among genes containing missense SNPs or stop gained/loss variants in both Small-tailed Han sheep and Duolang sheep, but not in Mongolian sheep. (DOC 34 kb)
Enriched GO terms among genes containing missense SNPs in promoter regions in Mongolian sheep but not in Small-tailed Han sheep or Duolang sheep. (DOC 570 kb)
Enriched KEGG pathways among genes containing missense SNPs in promoter regions in Mongolian sheep but not in Small-tailed Han sheep or Duolang sheep. (DOC 50 kb)
Enriched GO terms among genes containing missense SNPs in promoter regions in both Small-tailed Han sheep and Duolang sheep, but not in Mongolian sheep. (DOC 71 kb)
Biological processes and KEGG pathway enrichment in genes containing SNPs in promoter regions. A, Biological processes enrichment in genes containing SNPs in promoter regions in Mongolian sheep but not in Small-tailed Han sheep or Duolang sheep. B, Biological processes enrichment in genes containing SNPs in promoter regions in both Small-tailed Han sheep and Duolang sheep, but not in Mongolian sheep. C, KEGG pathway enrichment in genes containing SNPs in promoter regions in Mongolian sheep but not in Small-tailed Han sheep or Duolang sheep. Biological processes related to reproduction are labeled in red. (DOC 1117 kb)
The protein coding SNPs detected by SIFT and Provean analysis. (DOC 29 kb)
The distributions of heterozygosity (H P) and the average fixation index (F ST) between any two of the 3 resequenced breeds for autosomal 200-kb windows. (DOC 107 kb)
The distributions of heterozygosity (H P) and the average fixation index (F ST) between any two of the 3 resequenced breeds and their Z transformations for 200-kb windows on chr. X. (DOC 124 kb)
Genomic regions under selection in Small-tailed Han sheep. (DOC 126 kb)
Genomic regions under selection in Duolang sheep. (DOC 127 kb)
Enriched GO terms among genes located in the selection regions with Z(F ST)M-S > 4 and Z(F ST)M-D > 4. (DOC 166 kb)
Enriched GO terms among genes located in the selection regions with Z(F ST)M-S > 4. (DOC 145 kb)
Enriched GO terms among genes located in the selection regions with Z(F ST)M-D > 4. (DOC 145 kb)
Pooled heterozygosity of Duolang sheep, H P (in blue), and average fixation index between Mongolian sheep and Duolang sheep, F ST (in red), plotted for 200-kb windows spanning the region harboring TSHR (in shadow) on chr. 7. (DOC 308 kb)
Haplotype diversities of genomic regions harboring PRL and HMGCR. A and B indicate the H P of the 3 resequenced breeds, plotted for 200-kb windows spanning the region harboring PRL and HMGCR. C and D indicate the genetic variation in the region 24.22–24.26 Mb on chr. 20 across PRL and LOC443319 and the region 6.53–6.57 Mb on chr. 7 across HMGCR. Individual sheep (95 from 8 breeds) were genotyped using WaferGen genotyping. Dashed horizontal lines separate the 8 breeds. At the bottom of the figure, short tick marks represent individual SNPs. Long tick marks indicate the position in Mb. Red color: homozygous A-allele; green color: heterozygous; purple color: homozygous a-allele, yellow color: missing genotype call. E and F indicate the haplotype frequencies of the 8 sheep breeds at the 2 genomic regions. (DOC 214 kb)
The H P and F ST within BMPR1B and MTNR1A. (DOC 32 kb)
Contributor Information
Zhaohua Liu, Email: 18606366066@163.com.
Zhibin Ji, Email: 594005988@qq.com.
Guizhi Wang, Email: wgz@sdau.edu.cn.
Tianle Chao, Email: 2398901017@qq.com.
Lei Hou, Email: 250741420@qq.com.
Jianmin Wang, Phone: +86-538-8241448, Email: wangjm@sdau.edu.cn.
References
- 1.Chessa B, Pereira F, Arnaud F, Amorim A, Goyache F, Kao RR, et al. Revealing the history of sheep domestication using retrovirus integrations. Science. 2009;324:532–6. doi: 10.1126/science.1170587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kijas JW, Lenstra JA, Hayes B, Boitard S, Porto Neto LR, San Cristobal M, et al. Genome-wide analysis of the world’s sheep breeds reveals high levels of historic mixture and strong recent selection. PLoS Biol. 2012;10:e1001258. doi: 10.1371/journal.pbio.1001258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wei C, Wang H, Liu G, Wu M, Cao J, Liu Z, et al. Genome-wide analysis reveals population structure and selection in Chinese indigenous sheep breeds. BMC Genomics. 2015;16:194. doi: 10.1186/s12864-015-1384-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen SY, Duan ZY, Sha T, Xiangyu J, Wu SF, Zhang YP. Origin, genetic diversity, and population structure of Chinese domestic sheep. Gene. 2006;376:216–23. doi: 10.1016/j.gene.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 5.China national commission of animal genetic resources. Animal genetic resources in China: Sheep and Goats. 1st ed. Beijing: China Agriculture Press; 2011.
- 6.Olson DM, Dinerstein E, Wikramanayake ED, Burgess ND, Powell GVN, Underwood EC, et al. Terrestrial ecoregions of the world: a new map of life on earth. Bioscience. 2001;51:933–8. doi: 10.1641/0006-3568(2001)051[0933:TEOTWA]2.0.CO;2. [DOI] [Google Scholar]
- 7.Karell P, Ahola K, Karstinen T, Valkama J, Brommer JE. Climate change drives microevolution in a wild bird. Nat Commun. 2011;2:208. doi: 10.1038/ncomms1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carneiro M, Rubin CJ, Di Palma F, Albert FW, Alföldi J, Barrio AM, et al. Rabbit genome analysis reveals a polygenic basis for phenotypic change during domestication. Science. 2014;345:1074–9107. doi: 10.1126/science.1253714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rubin CJ, Zody MC, Eriksson J, Meadows JR, Sherwood E, Webster MT, et al. Whole-genome resequencing reveals loci under selection during chicken domestication. Nature. 2010;464:587–91. doi: 10.1038/nature08832. [DOI] [PubMed] [Google Scholar]
- 10.Fang M, Larson G, Ribeiro HS, Li N, Andersson L. Contrasting mode of evolution at a coat color locus in wild and domestic pigs. PLoS Genet. 2009;5:e1000341. doi: 10.1371/journal.pgen.1000341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Axelsson E, Ratnakumar A, Arendt ML, Maqbool K, Webster MT, Perloski M, et al. The genomic signature of dog domestication reveals adaptation to a starch-rich diet. Nature. 2013;495:360–4. doi: 10.1038/nature11837. [DOI] [PubMed] [Google Scholar]
- 12.Petersen JL, Mickelson JR, Rendahl AK, Valberg SJ, Andersson LS, Axelsson J, et al. Genome-wide analysis reveals selection for important traits in domestic horse breeds. PLoS Genet. 2013;9:e1003211. doi: 10.1371/journal.pgen.1003211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li M, Tian S, Jin L, Zhou G, Li Y, Zhang Y, et al. Genomic analyses identify distinct patterns of selection in domesticated pigs and Tibetan wild boars. Nat Genet. 2013;45:1431–8. doi: 10.1038/ng.2811. [DOI] [PubMed] [Google Scholar]
- 14.Gou X, Wang Z, Li N, Qiu F, Xu Z, Yan D, et al. Whole-genome sequencing of six dog breeds from continuous altitudes reveals adaptation to high-altitude hypoxia. Genome Res. 2014;24:1308–15. doi: 10.1101/gr.171876.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang GD, Fan RX, Zhai W, Liu F, Wang L, Zhong L, et al. Genetic convergence in the adaptation of dogs and humans to the high-altitude environment of the tibetan plateau. Genome Biol Evol. 2014;6:2122–8. doi: 10.1093/gbe/evu162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qu Y, Zhao H, Han N, Zhou G, Song G, Gao B, et al. Ground tit genome reveals avian adaptation to living at high altitudes in the Tibetan plateau. Nat Commun. 2013;4:2071. doi: 10.1038/ncomms3071. [DOI] [PubMed] [Google Scholar]
- 17.Fan WL, Ng CS, Chen CF, Lu MY, Chen YH, Liu CJ, et al. Genome-wide patterns of genetic variation in two domestic chickens. Genome Biol Evol. 2013;5:1376–92. doi: 10.1093/gbe/evt097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang S, Li X, Li K, Fan B, Tang Z. A genome-wide scan for signatures of selection in Chinese indigenous and commercial pig breeds. BMC Genet. 2014;15:7. doi: 10.1186/1471-2156-15-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang C, Wang H, Zhang Y, Tang Z, Li K, Liu B. Genome-wide analysis reveals artificial selection on coat colour and reproductive traits in Chinese domestic pigs. Mol Ecol Resour. 2015;15:414–24. doi: 10.1111/1755-0998.12311. [DOI] [PubMed] [Google Scholar]
- 20.Kawahara-Miki R, Tsuda K, Shiwa Y, Arai-Kichise Y, Matsumoto T, Kanesaki Y, et al. Whole-genome resequencing shows numerous genes with nonsynonymous SNPs in the Japanese native cattle Kuchinoshima-Ushi. BMC Genomics. 2011;12:103. doi: 10.1186/1471-2164-12-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stothard P, Choi JW, Basu U, Sumner-Thomson JM, Meng Y, Liao X, et al. Whole genome resequencing of black Angus and Holstein cattle for SNP and CNV discovery. BMC Genomics. 2011;12:559. doi: 10.1186/1471-2164-12-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee KT, Chung WH, Lee SY, Choi JW, Kim J, Lim D, et al. Whole-genome resequencing of Hanwoo (Korean cattle) and insight into regions of homozygosity. BMC Genomics. 2013;14:519. doi: 10.1186/1471-2164-14-519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liao X, Peng F, Forni S, McLaren D, Plastow G, Stothard P. Whole genome sequencing of Gir cattle for identifying polymorphisms and loci under selection. Genome. 2013;56:592–8. doi: 10.1139/gen-2013-0082. [DOI] [PubMed] [Google Scholar]
- 24.Choi JW, Liao X, Stothard P, Chung WH, Jeon HJ, Miller SP, et al. Whole-genome analyses of Korean native and Holstein cattle breeds by massively parallel sequencing. PLoS One. 2014;9:e101127. doi: 10.1371/journal.pone.0101127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bautista DM, Siemens J, Glazer JM, Tsuruda PR, Basbaum AI, Stucky CL, et al. The menthol receptor TRPM8 is the principal detector of environmental cold. Nature. 2007;448:204–8. doi: 10.1038/nature05910. [DOI] [PubMed] [Google Scholar]
- 26.Fariello MI, Servin B, Tosser-Klopp G, Rupp R, Moreno C, International Sheep Genomics Consortium et al. Selection signatures in worldwide sheep populations. PLoS One. 2014;9:e103813. doi: 10.1371/journal.pone.0103813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Modi D, Godbole G. HOXA1O signals on the highway through pregnancy. J Reprod Immunol. 2009;83:72–8. doi: 10.1016/j.jri.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 29.Murthi P, Hiden U, Rajaraman G, Liu H, Borg AJ, Coombes F, et al. Novel homeobox genes are differentially expressed in placental microvascular endothelial cells compared with macrovascular cells. Placenta. 2008;29:624–30. doi: 10.1016/j.placenta.2008.04.006. [DOI] [PubMed] [Google Scholar]
- 30.Taylor HS, Arici A, Olive D, Igarashi R. HOXA1O is expressed in response to sex steroids at the time of implantation in the human endometrium. J Clin Invest. 1998;101:1379–84. doi: 10.1172/JCI1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Taylor HS, Vanden Heuvel GB, Igarashi P. A conserved Hox axis in the mouse and human female reproductive system: late establishment and persistent adult expression of the Hoxa cluster genes. Biol Reprod. 1997;57:1338–45. doi: 10.1095/biolreprod57.6.1338. [DOI] [PubMed] [Google Scholar]
- 32.Bagot CN, Troy PJ, Taylor HS. Alteration of maternal Hoxa10 expression by in vivo gene transfection affects implantation. Gene Ther. 2000;7:1378–84. doi: 10.1038/sj.gt.3301245. [DOI] [PubMed] [Google Scholar]
- 33.Lim H, Ma L, Ma WG, Maas RL, Dey SK. Hoxa-10 regulates uterine stromal cell responsiveness to progesterone during implantation and decidualization in the mouse. Mol Endocrinol. 1999;13:1005–17. doi: 10.1210/mend.13.6.0284. [DOI] [PubMed] [Google Scholar]
- 34.Nakao N, Ono H, Yamamura T, Anraku T, Takagi T, Higashi K, et al. Thyrotrophin in the pars tuberalis triggers photoperiodic response. Nature. 2008;452:317–22. doi: 10.1038/nature06738. [DOI] [PubMed] [Google Scholar]
- 35.Hodson DJ, Henderson HL, Townsend J, Tortonese DJ. Photoperiodic modulation of the suppressive actions of prolactin and dopamine on the pituitary gonadotropin responses to gonadotropin-releasing hormone in sheep. Biol Reprod. 2012;86:122. doi: 10.1095/biolreprod.111.096909. [DOI] [PubMed] [Google Scholar]
- 36.Zhang L, Mousel MR, Wu X, Michal JJ, Zhou X, Ding B, et al. Genome-wide genetic diversity and differentially selected regions among Suffolk, Rambouillet, Columbia, Polypay, and Targhee sheep. PLoS One. 2013;8:e65942. doi: 10.1371/journal.pone.0065942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Davis GH. Fecundity genes in sheep. Anim Reprod Sci. 2004;82-83:247–53. doi: 10.1016/j.anireprosci.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 38.Davis GH. Major genes affecting ovulation rate in sheep. Genet Sel Evol. 2005;37(Suppl 1):S11–23. doi: 10.1186/1297-9686-37-S1-S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kumar D, Joshi A, Naqvi SM, Kumar S, Mishra AK, Maurya VP, et al. Sperm motion characteristics of GarolexMalpura sheep evolved in a semi-arid tropical environment through introgression of FecB gene. Anim Reprod Sci. 2007;100:51–60. doi: 10.1016/j.anireprosci.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 40.Abdoli R, Zamani P, Deljou A, Rezvan H. Association of BMPR-1B and GDF9 genes polymorphisms and secondary protein structure changes with reproduction traits in Mehraban ewes. Gene. 2013;524:296–303. doi: 10.1016/j.gene.2013.03.133. [DOI] [PubMed] [Google Scholar]
- 41.Eghbalsaied S, Ghaedi K, Shahmoradi S, Pirestani A, Amini H, Saiedi T, et al. Presence of SNPs in GDF9 mRNA of Iranian Afshari Sheep. Int J Fertil Steril. 2012;5:225–30. [PMC free article] [PubMed] [Google Scholar]
- 42.Martínez-Royo A, Lahoz B, Alabart JL, Folch J, Calvo JH. Characterisation of the Melatonin Receptor 1A (MTNR1A) gene in the Rasa Aragonesa sheep breed: association with reproductive seasonality. Anim Reprod Sci. 2012;133:169–75. doi: 10.1016/j.anireprosci.2012.06.018. [DOI] [PubMed] [Google Scholar]
- 43.Mateescu RG, Lunsford AK, Thonney ML. Association between melatonin receptor 1A gene polymorphism and reproductive performance in Dorset ewes. J Anim Sci. 2009;87:2485–8. doi: 10.2527/jas.2008-1688. [DOI] [PubMed] [Google Scholar]
- 44.Mura MC, Luridiana S, Vacca GM, Bini PP, Carcangiu V. Effect of genotype at the MTNR1A locus and melatonin treatment on first conception in Sarda ewe lambs. Theriogenology. 2010;74:1579–86. doi: 10.1016/j.theriogenology.2010.06.028. [DOI] [PubMed] [Google Scholar]
- 45.McRae KM, McEwan JC, Dodds KG, Gemmell NJ. Signatures of selection in sheep bred for resistance or susceptibility to gastrointestinal nematodes. BMC Genomics. 2014;15:637. doi: 10.1186/1471-2164-15-637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bonhomme M, Chevalet C, Servin B, Boitard S, Abdallah J, Blott S, et al. Detecting selection in population trees: the Lewontin and Krakauer test extended. Genetics. 2010;186:241–62. doi: 10.1534/genetics.110.117275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bierne N, Roze D, Welch JJ. Pervasive selection or is it…? Why are FST outliers sometimes so frequent? Mol Ecol. 2013;22:2061–4. doi: 10.1111/mec.12241. [DOI] [PubMed] [Google Scholar]
- 48.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zheng LY, Guo XS, He B, Sun LJ, Peng Y, Dong SS, et al. Genome-wide patterns of genetic variation in sweet and grain sorghum (Sorghum bicolor) Genome Biol. 2011;12:R114. doi: 10.1186/gb-2011-12-11-r114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cingolani P, Platts A, le Wang L, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, snpeff: Snps in the genome of drosophila melanogaster strain w(1118); iso-2; iso-3. Fly (Austin) 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–81. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 53.Cockerham CC, Weir BS. Covariances of relatives stemming from a population undergoing mixed self and random mating. Biometrics. 1984;40:157–64. doi: 10.2307/2530754. [DOI] [PubMed] [Google Scholar]
- 54.Weir BS, Hill WG. Estimating F-statistics. Annu Rev Genet. 2002;36:721–50. doi: 10.1146/annurev.genet.36.050802.093940. [DOI] [PubMed] [Google Scholar]
- 55.Smedley D, Haider S, Durinck S, Pandini L, Provero P, Allen J, et al. The BioMart community portal: an innovative alternative to large, centralized data repositories. Nucleic Acids Res. 2015;43:W589–98. doi: 10.1093/nar/gkv350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Beissbarth T, Speed TP. GOstat: find statistically overrepresented Gene Ontologies within a group of genes. Bioinformatics. 2004;20:1464–5. doi: 10.1093/bioinformatics/bth088. [DOI] [PubMed] [Google Scholar]
- 57.da Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 58.Scheet P, Stephens M. A fast and flexible statistical model for large-scale population genotype data: applications to inferring missing genotypes and haplotypic phase. Am J Hum Genet. 2006;78:629–44. doi: 10.1086/502802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Marchler-Bauer A, Derbyshire MK, Gonzales NR, Lu S, Chitsaz F, Geer LY, et al. CDD: NCBI’s conserved domain database. Nucleic Acids Res. 2015;43:D222–6. doi: 10.1093/nar/gku1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, et al. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639–45. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Raw sequence data of 3 sheep breeds in the bam format from this study were deposited in the NCBI Short Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra) under accession numbers SRR2420291, SRR2420281 and SRR2420287.