

Abstract
Understanding the genetic structure and transmission dynamics of Plasmodium falciparum parasites in malaria-endemic regions is crucial before the implementation of interventions. Located in a high-transmission region of western Kenya where P. falciparum is the predominant species, the Lake Victoria islands are ideal for feasibility of malaria elimination studies. We analyzed genetic variation in eight microsatellite loci to examine parasite population structure and gene flow patterns across five sites. High levels of genetic diversity were measured throughout the region (mean heterozygosity index = 0.84). The overall fixation index value between the sites was 0.044, indicating that approximately 5% of the overall allelic variation is due to differences between the populations. Based on these results, we concluded that parasite population structure in the studied islands is shaped by human migration patterns that maintain extensive parasite gene flow between the sites. Consequently, any malaria elimination and interventions strategies in the study area will have to be carried out broadly on all four islands and adjoining mainland region.
Introduction
Malaria arising from Plasmodium falciparum infection continues to be a major public health problem globally, causing approximately 438,000 deaths annually1 with sub-Saharan Africa experiencing the majority of morbidity and mortality.2–4 Efforts to control this devastating disease have been difficult due, in part, to high levels of P. falciparum genetic diversity, drug resistance, poor vector management, poor access to drug, and poverty.5–8 Global scale studies on P. falciparum genetic diversity show that high levels are generally predominant in African populations,9,10 low in South American populations,11 and intermediate in southeast Asian populations.12 In Africa, the diverse parasite populations are not strongly isolated from each other because of dynamic migratory patterns of the human host and different levels of geographic isolation.10,13,14 The high malaria transmission levels found in much of Africa lead to a large repertoire of genetically diverse parasites as a result of high genetic recombination rates.9,10,15 Exceptions are observed in remote islands, such as Comoros,16 which show fragmented parasite population structures due to restricted parasite gene flow. This diverse parasite population structure observed globally is directly implicated in the emergence of advantageous phenotypes such as drug resistance and antigenic variants,17,18 with persistent migration of vectors and humans playing an important role in the spread of these traits.19,20 These factors continue to curtail any progress made toward mitigating this disease.
Over the last decade, there has been increased commitment to eliminate malaria from selected areas where it is endemic.21,22 Islands are goods targets in determining feasibility of malaria elimination in different transmission settings due to their isolated nature. Vanuatu, an endemic archipelago in the southwest Pacific region has been studied to determine the feasibility of malaria elimination in low-transmission settings as well as how human movement within and between the islands affects intervention plans.20 The study in this region revealed that P. falciparum was largely isolated on individual islands, and human movement between the islands was responsible for malaria parasite gene flow. Human influx subsequently increased the rate of evolution and dissemination of parasites' novel traits.20,23 This implied that malaria interventions needed to be carried out on an island-by-island basis to curtail possible gene flow and subsequent parasite importation.24 Indeed, successful elimination of malaria has been registered in Caribbean, Cyprus, Mauritius, Maldives, Reunion, Taiwan, and Singapore islands where transmission is low and unstable.25
However, despite extensive malaria elimination feasibility studies on islands with low malaria transmission, not much has been done on islands in high-transmission regions. Past failures of malaria elimination experienced in malaria-endemic islands such as Zanzibar off the coast of Tanzania26 and Comoros archipelago off the eastern coast of Africa27 raised questions on the extent of human and vector migratory trends and parasite population structure in these intense transmission islands. Therefore, P. falciparum population genetic studies are crucial in evaluating the extent of parasite genetic diversity, which is an indicator of the parasite populations' resilience to control measures. Population genetic structure is pertinent, to help map spatial distribution of genetic diversity over geographical space and thus inferring parasite migratory patterns before implementation of malaria interventions.24
Although malaria transmission has decreased in some regions of Kenya, it still remains a major problem in low-elevation regions (0–1,300 m above sea level), around Lake Victoria, and along the coastal regions, with habitats that provide suitable breeding ground for Anopheles mosquitoes28–30 despite intense deployment of control measures. In the Lake Victoria basin, P. falciparum genetic variability and gene flows have been systematically compared only across the western highland regions (Kakamega) and malaria-endemic lowland sites (Kisumu), revealing high gene diversity in the highland populations despite their low-transmission intensity.31 This high genetic diversity in Kakamega was attributed to parasite-infected humans commuting between these two contrasting transmission regions. To date, few studies32–34 have examined the genetic divergence of malaria parasites in Lake Victoria islands where malaria transmission is stable. Such information is valuable to a country especially when developing a comprehensive elimination feasibility assessment and would provide a pertinent model for guiding future malaria elimination programs.
Thus, the aim of this study was to determine the genetic structure of P. falciparum within and between the Lake Victoria islands to establish whether it is feasible to eliminate malaria in highly endemic areas. Since the efficacy of any elimination program on islands may depend on the influx of new parasites, we performed population genetic analysis of P. falciparum isolates from four islands of Lake Victoria (Mfangano, Takawiri, Kibuogi, and Ngodhe), and one mainland region (Ungoye) using putatively neutral microsatellite markers35,36 to help identify routes of transmission and gene flow patterns. An early baseline survey by Idris and others37 revealed that the malaria prevalence in this study site is highly variable and is dependent on the season or geographic location. The highest malaria prevalence was observed in the mainland (Ungoye) with small islands reporting the lowest rates. We report results of P. falciparum genetic analysis and discuss ecological parameters that could explain the observed trends of parasite diversity and gene flow.
Materials and Methods
Ethics statement.
This study was derived from a cross-sectional epidemiological study of P. falciparum infection prevalence in resident populations of islands in Lake Victoria in Kenya. Ethical approval to conduct the study was granted by the joint Kenyatta National Hospital and University of Nairobi Ethical Review Board. Samples were obtained from participants after obtaining written informed consent.
Study sites and population characteristics.
The study was conducted on four inhabited islands of Lake Victoria: Mfangano, Ngodhe, Kibuogi, and Takawiri, as well as a mainland shoreline region (Ungoye) of Mbita District, western Kenya (Figure 1 ). These four islands are among 16 islands in Mbita District located along Lake Victoria. Mfangano Island is the largest (66 km2) and the most densely populated with a population of approximately 25,000 followed by Ungoye with a population of about 2,000.38 Ngodhe, Kibuogi, and Takawiri are small islands with estimated human populations of between 700 and 1,000 depending on the fishing season. These islands and the areas along the lakeshore are characterized by warm and wet climatic conditions, with long (March–May) and short (October–December) rain periods. Malaria prevalence peaks briefly in June, following the long rains and remain steady between September and February with more than 40% P. falciparum parasite rates reported in the overall population.29,30 Inhabitants of this lake basin belong to the Luo and Suba ethnic groups, although Luo is the most commonly spoken language. Members of the Luo tribe inhabit the shores of Mfangano and Ungoye, most of whom are fishermen. Ngodhe, Kibuogi, and Takawiri islands are entirely occupied by the Luo ethnic group who are migrant fishermen. Fishing activities in the region are at the peak during the dry spell (September and February). Subsistence farming is the main activity of the Suba tribe. Local transportation between the islands is mainly by small wooden dugout boats. Ferry service between mainland Ungoye and Mfangano Island is also available. Ungoye serves as a central trading hub for the surrounding islands.
Figure 1.

Map of Lake Victoria region showing the study areas and the geographic distances between them. Black lines indicate the geographic distances between the sites.
Sample collection and DNA extraction.
Finger prick blood was collected from 393 asymptomatic study participants during a baseline survey conducted in January and February 2012. All 393 blood samples were screened in the field for P. falciparum by microscopy and rapid diagnostic test. Parasitemia levels of each blood sample were determined and recorded for archiving. Dried blood spots (DBS) were prepared by absorbing approximately 50 μL of the collected EDTA-blood onto Whatman filter paper (Whatman, Maidstone, United Kingdom) and air-drying. DBS were stored at room temperature in sealed zip-lock bags with desiccant.
Total genomic DNA was extracted from DBS in Osaka University, Japan, using the QIAamp DNA Mini Kit (Qiagen, Crawley, UK). Plasmodium falciparum identification was performed using ribosomal DNA-based polymerase chain reaction (PCR) method described by Singh and others.39 The DNA was kept under frozen conditions (−20°C) and shipped to CREATES, Nairobi, Kenya, for subsequent analysis.
Genotyping of P. falciparum microsatellite loci.
Malaria parasites were genotyped for eight polymorphic microsatellite marker loci (chromosome assignments are given in parentheses)35: TA81 (Chr5), TA87 (Chr6), TA109 (Chr6), ARA2 (Chr11), pfg377 (Chr12), TA1 (Chr6), TA40 (Chr10), and TA42 (Chr5). These loci consist of conserved locus-specific sequences flanking the tandem repeats region in P. falciparum haploid genome and have previously been used for both global and local population genetic studies.9,10,13,31 A two-round hemi-nested PCR protocol described by Anderson and others35 was adopted to amplify the eight loci, with slight modification for automated genotyping in 3730 DNA analyzer (Applied Biosystems, Foster City, CA). A protocol described by Li and others40 was adopted to allow these modifications. Details on the primer sequences and accession number are provided in Supplemental Table 1.
Both the first and second PCR reactions were carried out in a total volume of 20 μL per sample per locus and contained 1× Ex Taq buffer, 0.05 U of Ex Taq polymerase (Takara, Kyoto, Japan), 0.2 mM dNTPs mix, 0.2 μM of each locus-specific primer, and 3 μL of template DNA or 2 μL of primary PCR products. PCR was performed in a GeneAmp 9700 instrument (Applied Biosystems) under the following conditions: an initial denaturation phase at 94°C for 2 minutes, followed by 25 cycles of denaturation at 94°C for 20 seconds, annealing at 45°C for 30 seconds, and extension at 65°C for 40 seconds and a final extension at 65°C for 2 minutes. The second PCR reaction was carried out under the same conditions except that the amplification cycles were increased to 35. Negative controls (no DNA template) and P. falciparum positive controls consisting of K1, MAD20, FCR3, and THAI 838 laboratory clones were used in each run.
Capillary electrophoresis was done at the Macrogen Laboratory facility in South Korea using 3730 DNA Analyzer (Applied Biosystems). GeneScan™ 500LIZ (Applied Biosystems) was used as an internal size standard. Genemapper® (Applied Biosystems) software was used to quantify allele size on the basis of the height pattern of signal peaks with 100 relative fluorescent units set as the minimal peak threshold for scoring alleles.
Genetic data analyses.
Field isolates that successfully amplified at all the eight loci within the five populations were included in the respective analyses. For each individual isolate, only the predominant allele or the single allele at each locus was scored and counted for population genetic analyses. Since all eight markers are single-copy loci (haploid), presence of one or more additional alleles at a particular locus was interpreted as a multi-infection. Any additional allele(s) present due to multiple parasite infections was recorded if the peak was at least one-third the height of the predominant allele, but the allele(s) did not count toward the analyzed sample. Inclusion of these multiple alleles from multi-infected isolates would result in a biased estimation of allelic frequencies in the populations.35 In addition, it is also impossible to match the different alleles obtained at each locus to construct a valid genotype. The multiloci analyses mainly focused on population genetic diversity, genetic differentiation, multiple infection, and linkage disequilibrium (LD).
The overall genetic diversity in each of the geographic locations was assessed using Arlequin version 3.5.1.2 software (Bern, Switzerland)41 by determining the number of alleles per locus, allelic richness, and expected heterozygosity, calculated from allelic frequencies of the eight microsatellites. Number of alleles per locus, allelic frequencies, and expected heterozygosity were used as measures of level of polymorphism in the loci and to determine the diversity of the populations. The expected heterozygosity index (He), was calculated as follows:
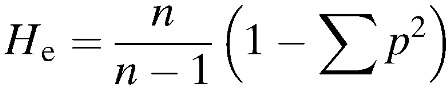 |
where p is the frequency of each different allele at each locus, n the number of alleles in the sample, and He the probability that two alleles sampled from a population are different. Thus heterozygosity has a potential range from 0 meaning that all alleles are identical (no polymorphism) to 1 that all alleles within the sampled population are unique (highly polymorphic). Since the number of alleles per locus is highly dependent on sample size and the fact that different numbers of isolates were analyzed in the five sites, it was important to normalize data on the basis of the smallest sample size (in this case, Ngodhe with 17 isolates). We therefore computed allelic richness based on a rarefaction method to allow the comparison of genetic diversity across different samples sizes using FSTAT version 2.9.3.2 software (Lausanne, Switzerland).42
The multiplicity of infection (MOI) was defined as the mean number of genetically distinct parasites genotypes coinfecting an individual. MOI was estimated for each isolate from locus with highest number of alleles. An infection was classed as a multiple genotype infection if more than one peak was detected by any of the markers and where the additional peak was at least one-third the height of the primary peak. A single infection was defined as one with only one peak detected at all the genotyped loci. Mean MOI was calculated based on the total number of parasites genotypes detected divided by the number of isolates analyzed.
Population genetic structure was investigated using Wright's pairwise fixation index (FST) computed with Arlequin software.43 This index estimates genetic differentiation by calculating weighted F statistics also known as theta (θ) for each locus, and overall based on distinct number of alleles among the isolates.44 A random permutation test (N = 10,000) was performed to test whether the FST values observed significantly differed from zero. To validate the population genetic structure established by F statistics, Structure version 2.3.4 software (Oxford, UK) was used to assign each genotype from the five populations into genetically related clusters. The software assigns individual genotype to a predetermined number of clusters (K) based on allelic frequencies at each locus.45,46 We chose K values between 2 and 8 within these populations based on the calculation model by Evanno and others,47 where suitable K value ranges from K = 2 up to the true number of study populations plus 3. To choose the best K value, 20 replicate runs were performed after a burn-in period of 10,000 steps followed by 10,000 iterations under admixture model and assumed correlated allele frequencies. Structure Harvester version 0.6.94 software (Santa Cruz, CA) was applied to evaluate ΔK values from Structure output files.48 The most likely K value was computed by the higher number in the change for K (ΔK) according to the method described by Evanno and others.47 CLUMPP program v1.1.2 (Michigan, USA) was used to aid the interpretation of cluster results,49 and DISTRUCT program v1 (Michigan, USA) was used to facilitate graphical display of population clusters.50
To test for evidence of multilocus LD, the standardized index of association (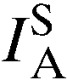 ) among alleles from all eight loci in each population was computed using LIAN version 3.5 web interface (Jena, Germany) developed for multilocus haploid data.51 LD is the nonrandom association of alleles across loci and can occur as a result of a range of processes including population substructure and selection. LD analyses were performed to determine the independent assortment of alleles in this study population. LD analyses were performed in two ways; first, a curtailed dataset that excluded all mixed-clone infections so that only single clone isolates with predominant allele at each locus was constructed and analyzed. Second, a dataset that contained multilocus genotypes found once in each population set (unique genotypes) was reconstructed and analyzed, and the results from the two analyses were compared. This association index was calculated, assuming a null hypothesis of complete random association among loci (
) among alleles from all eight loci in each population was computed using LIAN version 3.5 web interface (Jena, Germany) developed for multilocus haploid data.51 LD is the nonrandom association of alleles across loci and can occur as a result of a range of processes including population substructure and selection. LD analyses were performed to determine the independent assortment of alleles in this study population. LD analyses were performed in two ways; first, a curtailed dataset that excluded all mixed-clone infections so that only single clone isolates with predominant allele at each locus was constructed and analyzed. Second, a dataset that contained multilocus genotypes found once in each population set (unique genotypes) was reconstructed and analyzed, and the results from the two analyses were compared. This association index was calculated, assuming a null hypothesis of complete random association among loci (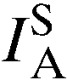 = 0) by Monte Carlo simulation at 10,000 per mutations, as follows:
= 0) by Monte Carlo simulation at 10,000 per mutations, as follows:
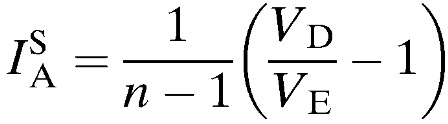 |
where VE is the expected mismatch variance under LD, VD the observed variance of the numbers of shared alleles in the population, and n the number of examined loci. Significant LD is detected if VD value is greater than 95% confidence interval of the values derived from the reshuffled datasets.
Statistical data analyses.
To test for evidence of statistically significant differences in the levels of genetic diversity as well as MOI between the five populations, pairwise comparisons were made using the Kruskal–Wallis test.
Results
Genetic diversity.
In total, 198 isolates from the five study sites were genotyped at eight microsatellite marker loci. Among these, 10 isolates (5%) had missing data at two loci (TA1 and TA42) and were excluded from subsequent analyses. The remaining 188 isolates (95%): Kibuogi (N = 35), Mfangano (N = 50), Ngodhe (N = 17), Takawiri (N = 36), and Ungoye (N = 50) were successfully genotyped at all the eight loci (TA81, TA87, TA109, ARA2, pfg377, TA1, TA42, and TA40), and a full genotype profile was generated (Supplemental Table 2). The eight markers examined were observed to be highly polymorphic, with overall number of distinct alleles per locus ranging from 11 (for locus ARA2) to 23 (for locus TA1; Table 1). There were no significant differences (P = 0.16) in the mean number of alleles per locus between the five parasite populations calculated using Kruskal–Wallis test. Allelic richness normalized as described in the methods did not differ significantly between populations. This implied that the chosen marker loci were equally informative for both island and mainland parasite populations.
Table 1.
Number of alleles (A), allelic richness (Rs), and allelic diversity (He) of eight loci from five study sites
| Locus | Kibuogi |
Mfangano |
Ngodhe |
Takawiri |
Ungoye |
Total |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | Rs | He | A | Rs | He | A | Rs | He | A | Rs | He | A | Rs | He | A | Rs | He | |
| ARA2 | 6 | 5.58 | 0.52 | 7 | 6.19 | 0.79 | 7 | 7.00 | 0.71 | 7 | 6.31 | 0.69 | 11 | 9.23 | 0.85 | 11 | 8.08 | 0.80 |
| PFG377 | 10 | 8.88 | 0.86 | 11 | 8.39 | 0.84 | 11 | 11.00 | 0.95 | 13 | 11.88 | 0.93 | 9 | 8.14 | 0.77 | 17 | 11.64 | 0.90 |
| TA1 | 11 | 9.80 | 0.86 | 11 | 9.69 | 0.89 | 7 | 7.00 | 0.82 | 11 | 9.55 | 0.88 | 15 | 11.51 | 0.88 | 23 | 12.02 | 0.89 |
| TA40 | 12 | 11.27 | 0.93 | 14 | 11.31 | 0.90 | 7 | 7.00 | 0.85 | 13 | 11.04 | 0.83 | 15 | 12.49 | 0.92 | 17 | 12.73 | 0.91 |
| TA42 | 7 | 6.40 | 0.72 | 10 | 7.68 | 0.63 | 8 | 8.00 | 0.90 | 6 | 5.42 | 0.69 | 7 | 5.77 | 0.48 | 14 | 7.52 | 0.66 |
| TA81 | 12 | 10.99 | 0.91 | 10 | 8.08 | 0.85 | 8 | 8.00 | 0.91 | 13 | 11.63 | 0.92 | 11 | 8.94 | 0.82 | 21 | 11.36 | 0.88 |
| TA87 | 8 | 7.47 | 0.76 | 9 | 7.58 | 0.81 | 8 | 8.00 | 0.87 | 10 | 8.86 | 0.87 | 8 | 6.88 | 0.76 | 12 | 7.93 | 0.81 |
| TA109 | 9 | 7.61 | 0.63 | 11 | 8.96 | 0.85 | 7 | 7.00 | 0.84 | 11 | 9.59 | 0.78 | 9 | 8.25 | 0.86 | 16 | 9.43 | 0.83 |
| Mean | 9.38 | 8.50 | 0.78 | 10.38 | 8.48 | 0.82 | 7.88 | 7.88 | 0.86 | 10.50 | 9.29 | 0.82 | 10.63 | 8.90 | 0.79 | 16.38 | 10.09 | 0.84 |
Allelic richness was normalized based on smallest sample size (N = 17).
Allelic diversity per microsatellite loci was estimated by expected heterozygosity (He) based on frequency of alleles generated (Supplemental Table 3) at each of the eight loci. On average, He value was 0.84, reflecting high genetic diversity among the isolates. The highest mean He values were observed in Ngodhe (He = 0.86) and the lowest in Kibuogi (He = 0.78). This genetic variability though different by loci and by studied sites did not meet the threshold for statistical significance. The mean number of alleles per locus (A) was highest in Ungoye (A = 10.63) and lowest in Ngodhe (A = 7.88).
Multiplicity of infection.
The MOI was assessed based on the proportion of individual isolates with multiple alleles for each site. Since Plasmodium remains haploid in the human host, detection of multiple alleles at any of the genotyped loci in a sample indicates the presence of multiple genotypes. Of the 188 samples analyzed, 151 isolates had at least two or more alleles detected by one of the eight loci. The proportion of isolates with multiple genotypes was highest in Ungoye (0.88) and lowest in Mfangano (0.72; Table 2). Further, the highest number of parasite genotypes coinfecting an individual was six and tended to be higher in the islands particularly Mfangano and Takawiri. The highest and lowest mean MOIs were recorded in Ngodhe and Kibuogi, respectively, although the differences across the regions were not significant by the Kruskal–Wallis test (P = 0.81).
Table 2.
MOIs of Plasmodium falciparum infections in the five study areas
| Site | No. of isolates genotyped | Isolates* with particular number of genotypes |
Mean MOI† | Proportion of isolates with > 1 genotype | |||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | ||||
| Kibuogi | 35 | 7 | 10 | 12 | 5 | 1 | 0 | 2.51 | 0.80 |
| Mfangano | 50 | 14 | 9 | 13 | 12 | 1 | 1 | 2.60 | 0.72 |
| Ngodhe | 17 | 3 | 2 | 3 | 5 | 4 | 0 | 3.29 | 0.82 |
| Takawiri | 36 | 7 | 10 | 14 | 4 | 0 | 1 | 2.53 | 0.81 |
| Ungoye | 50 | 6 | 6 | 13 | 22 | 3 | 0 | 3.20 | 0.88 |
MOI = multiplicity of infection.
The isolates per study site with the minimum (1) and maximum (6) detected number of genotypes.
Mean MOI calculated as total number of parasite genotypes detected per number of isolates analyzed in each study site.
Genetic differentiation among the islands and population structure.
Individual pairwise differentiation (FST) values for all the eight marker loci across the five parasite populations ranged from 0.004 (for locus TA87) to 0.117 (for locus ARA2). Averaged across all marker loci, gene divergence among the different populations was 0.044, indicating that approximately 5% of the overall allelic variation is due to differences observed between the five populations. Figure 2 shows the FST values between the five parasite populations. Overall, the FST values were low ranging from 0.014 to 0.081, and were significantly different from zero for all population comparisons (P < 0.05). The low FST values observed virtually indicates the absence of population substructuring among the studied populations.
Figure 2.

Levels of genetic differentiation [(Wright's pairwise fixation index (FST)] between the five Plasmodium falciparum populations. Dotted lines indicate possible routes of parasite transmission as determined by low FST value between the sites.
We next performed cluster analysis using structure software to determine the most accurate number of parasites with similar microsatellite genotypes circulating in these populations. Structure analysis identified three putative clusters (ΔK = 32.45) with much admixture in the five regions (Figure 3 ). The analysis assigned malaria parasites to particular genetic cluster based on membership coefficients for each of the geographic regions representing parasite populations. As shown in Figure 3, each cluster is represented by color codes: dominant cluster (orange), common cluster (blue), and rare cluster (yellow). The common cluster of genotypes included most of the parasites from Ngodhe (60.68%). The dominant cluster was assigned 63.81% of isolates from Kibuogi, and the rare cluster grouped 48.94% of isolates from Ungoye.
Figure 3.

Structure analysis and assignment test for 188 Plasmodium falciparum microsatellite genotypes from Lake Victoria. Each bar represents the proportion of each genotype in the defined clusters, each cluster being indicated by a different color; dominant cluster (orange), common cluster (blue), and rare cluster (yellow).
Multilocus LD.
A measure of nonrandom association among loci (multilocus LD) was calculated on mixed-clone infections and separately for single-clone infections using index of association (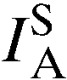 ). For this analysis, mixed-clone infections included the whole dataset comprising 151 isolates. All these isolates were single representatives of each genotype in each population dataset (unique genotypes). Single-clone infections comprised a curtailed dataset of 37 isolates with the predominant allele at each locus. The latter analysis was used to confirm LD in the absence of genotypes detected from multiple infections, which can result in higher estimates of recombination and thus bias against the detection of LD. The degree of LD was highly variable in the five parasite populations. The overall
). For this analysis, mixed-clone infections included the whole dataset comprising 151 isolates. All these isolates were single representatives of each genotype in each population dataset (unique genotypes). Single-clone infections comprised a curtailed dataset of 37 isolates with the predominant allele at each locus. The latter analysis was used to confirm LD in the absence of genotypes detected from multiple infections, which can result in higher estimates of recombination and thus bias against the detection of LD. The degree of LD was highly variable in the five parasite populations. The overall 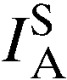 values ranged from 0.026 to 0.286 when single-clone infections were analyzed and −0.005 to 0.073 when mixed-clone infections were examined (Table 3). No evidence for multilocus LD (P = 0.18) was found in all the five populations when single-clone infections were analyzed. Mixed-clone infections only showed significant associations in Kibuogi and Takawiri populations.
values ranged from 0.026 to 0.286 when single-clone infections were analyzed and −0.005 to 0.073 when mixed-clone infections were examined (Table 3). No evidence for multilocus LD (P = 0.18) was found in all the five populations when single-clone infections were analyzed. Mixed-clone infections only showed significant associations in Kibuogi and Takawiri populations.
Table 3.
Multilocus linkage disequilibrium among Plasmodium falciparum populations
| Population | Mixed-clone infections | Single-clone infections | ||||
|---|---|---|---|---|---|---|
| No. | 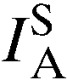 |
P value | No. | 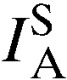 |
P value | |
| Kibuogi | 28 | 0.043* | 0.01 | 7 | 0.095 | 0.08 |
| Mfangano | 36 | 0.009 | 0.10 | 14 | 0.026 | 0.16 |
| Ngodhe | 14 | 0.036 | 0.09 | 3 | 0.286 | 0.31 |
| Takawiri | 29 | 0.073* | 0.01 | 7 | 0.027 | 0.31 |
| Ungoye | 44 | −0.005 | 0.63 | 6 | 0.068 | 0.11 |
| Total | 151 | 0.019* | 0.01 | 37 | 0.013 | 0.18 |
No. = number of isolates for each measure.
Significant levels for a test of departure from 0 for 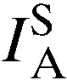 values (P < 0.05).
values (P < 0.05).
Discussion
This work presents the most extensive findings to date of the genetic structure of P. falciparum isolates from Lake Victoria islands using microsatellite markers analysis. Idris and others37 observed that the prevalence of malaria in this lake region is geographically and seasonally variable. We aimed to examine the levels of parasite genetic variability and gene flow, as this will allow tracking of parasite migratory routes in the study sites before the implementation of interventions. Our research reveals significantly high levels of genetic diversity and MOIs and low levels of genetic differentiation across the island study site populations. Although 10 isolates could not provide informative allelic data due to low parasitemia densities and were excluded, this exclusion is not uncommon in most epidemiologic studies12,43 and is unlikely to change the interpretation of the data as the exclusions are few in number.
This study revealed regional He values that were substantially higher than those observed in low-transmission areas of South America: Brazil (He = 0.14–0.62)52 and southeast Asia: Thailand (He = 0.65).17 However, these He values were comparatively similar to those previously described in other countries with high malaria transmission intensities such as Vietnam (He = 0.52–0.91), Nigeria (He = 0.79), Congo (He = 0.80), and Uganda (He = 0.76).9,53,54 Similarly, we demonstrated a high mean number of alleles for the five parasite populations. Generally, the number of alleles detected per locus is likely to be high in regions of high endemicity and low in regions of low endemicity.9 Based on this, the number of alleles recorded in this lake region were comparable to levels observed in other high-endemic regions of sub-Saharan Africa with allelic mean number range from 5.3 to 13.5.13,31
A large proportion of isolates reported here were multi-infected, with mean MOIs levels varying among the parasite populations. The high levels of multi-infections arguably resulted into high genetic diversity observed in this lake region. Overall, a mean MOI of 2.83 was recorded across populations and was comparatively similar to that described in other African areas.13 Assuming each genotype observed is transmissible to the vector (mosquito) during a blood meal, the rate of cross-over fertilization and genetic recombination is likely to be high. We argue that the high rates reported in this study could be attributed in most part to high-transmission rates in this malaria-endemic part of Kenya.
The genetic differentiation index (FST) revealed evidence of low substructuring among the studied populations. Most of the FST values were comparable to those revealed between P. falciparum populations from endemic regions in Africa9,10,15 but substantially lower than those from less endemic regions such as Philippines,12 Papua New Guinea,7 and Brazil.11 The level of genetic differentiation was high (FST = 0.081) between the mainland region (Ungoye) and Kibuogi Island despite their geographic proximity (9.6 km apart), whereas the next closest site from the mainland region (Mfangano, 15.8 km) recorded a relatively low level of genetic differentiation (FST = 0.023). We argue based on these observations, that there might be existence of gene flow barriers between these parasite populations. The probable mechanisms that could be responsible for parasite gene flow between these regions is through humans (carriers)7,20 and mosquito (vector) movements.55 Costantini and others55 demonstrated that the normal flight range of mosquito (Anopheles) is usually less than a kilometer. The geographic distances between the study sites range from 2.3 to 30.3 km, which is beyond the normal flight of the Anopheles vector. Thus, vector migration in this region is unlikely to be a contributor to parasite gene flow. Therefore, human movements could be the major factor responsible for parasite migration between these endemic sites. With no air and road networks, human movement across these geographically isolated islands is mainly through small fishing boats or ferry services.
Several epidemiological studies have shown that human movement and levels of genetic differentiation have inverse correlation.7,20,23 For example, Lum and others20 demonstrated that frequent human movements between geographically isolated islands were responsible for importation of new parasite genotypes into existing parasite population subsequently resulting in low FST indices. Based on this, our results strongly suggest frequent human traffic between the mainland (Ungoye) and Mfangano island than any other island as shown by FST value (FST = 0.023). Across the islands, human movement was high between Kibuogi and Takawiri (FST = 0.014). Lum and others20 and Pumpaibool and others17 have highlighted political and territorial conflicts, linguistic and cultural diversity as major barriers impeding human interaction, which is essential for malarial gene flow. However, with no such political and sociocultural barriers within residents of this lake basin, routine socio-activities or seasonal migrations of the residents could be the major factor influencing parasite migratory routes. Despite possible direct migratory route between Ungoye (mainland) and Kibuogi Island, human movements are minimal as compared with Ungoye and Mfangano Island, which are geographically far apart. This frequent human movement between Ungoye and Mfangano is attributed to a ferry service available on this seaway. Further, high human traffic trends inferred from FST value between Kibuogi and Takawiri (FST = 0.014) could be due to immense seasonal fishing activities by migrant fishermen. Although Ngodhe is the furthest site from Ungoye (mainland), the extent of human movement into the region is difficult to confirm due to the small sample size used in this study.
In this study, cluster analyses found a dominant cluster (orange) of genotypes circulating between the Kibuogi and Takawiri islands, which are consistent with the observed low levels of genetic differentiation (FST = 0.014) between the two sites. Furthermore, these dominant clusters appeared to be circulating between the Kibuogi, Takawiri, and Mfangano islands. We argue that this regular and informal human travel could accelerate the spread of these clusters of parasite genotypes and could have substantial influence on malaria epidemiology in the study areas. An earlier study in Papua New Guinea7 reported data that suggest the importance of human movement in mapping malaria parasite migration routes on islands. The study showed that the “mapped” human migratory routes lead to the spread of malaria parasite and was responsible for seasonal malaria cases in the catchment areas. Our study thus shows importance of mapping possible parasite transmission routes before implementation of malaria interventions.
Global studies have reported LD to be inversely associated with high levels of malaria transmission.9,56 In areas with high transmission rates, LD is rapidly broken down due to increased proportion of mixed genotypes, leading to cross-breeding and meiotic recombination. On the other hand, low transmission rates decrease the frequency of mixed genotypes and so inbreeding, which increases LD. Within each population a large proportion of isolates were multi-infected and as expected, no significant deviation from random allelic association was observed on single-clone infections. The overall association index was 0.013 (P = 0.18) when single infections were analyzed, which was relatively weaker than that reported in low-transmission areas.17,43 This concurs with other epidemiological studies that have reported lack of LD in high-transmission regions when single-clone parasite infections are analyzed.10,53
As Kibuogi and Takawiri parasite populations had relatively low levels of MOI (2.51 and 2.53, respectively) compared with the other study sites, it was not surprising to find significant LD when mixed-clone infections were analyzed. Such significant LD could suggest high rates of ongoing inbreeding arising from self-fertilization or as a consequence of high genetic relatedness among the genotypes from the isolates. Indeed, separate studies from Kenya,32 Senegal,57 and the Democratic Republic of Congo54 have revealed significant LD despite their high transmission rates. However, caution is needed when interpreting such kind of observations. It has elsewhere been shown that individuals residing in high-endemic regions are prone to harbor a mixture of genetically distinct parasite genotypes, which may be obtained from the single bite of a mosquito infected with more than one parasite genotype.58,59 Since these parasites obtained from mixed-clone infections per isolate may be as a result of single recombination event in the mosquito, they are therefore expected to be closely related skewing the result to a higher and statistically significant LD. This bias is the reason why LD data are usually obtained from single-clone infections.
In conclusion, our results demonstrate that the population structure of P. falciparum in this lake region is fairly diverse and falls within the expectation of a high malaria transmission zone. The low levels of genetic differentiation observed between the study sites is likely to be a consequence of immense human traffic into and out of the islands as part of routine socioeconomic activities. Since islands are being considered as models of study for the feasibility of malaria elimination in endemic regions, studies of this nature are highly pertinent. The findings obtained in this study suggest that elimination strategies should be implemented broadly on the entire islands rather than on an island-by-island basis.
Supplementary Material
ACKNOWLEDGMENTS
We thank Roland Brandl, Washington Ochieng, and Jonathan Kariuki for reviewing and improving the manuscript. We also thank Geofrey Masankwa for providing technical support to this work.
Footnotes
Financial support: This work was supported by a Consortium for National Health Research (CNHR), Kenya, grant awarded to Carol W. Hunja and by Swedish Research Council grants (523-2009-3233, 348-2012-6346, and 348-2013-6311), Japan Society for Promotion of Science (JSPS) Core-to-Core Program B, Asia-Africa Science Platforms (JSPS KAKENHI) grant (26257504), and a collaborative research grant of Nagasaki University Institute of Tropical Medicine awarded to Akira Kaneko.
Authors' addresses: Felix M. Mulenge and Esther Magiri, Department of Biochemistry, Jomo Kenyatta University of Agriculture and Technology, Nairobi, Kenya, E-mails: felixmulenge@yahoo.com and emagiri@jkuat.ac.ke. Carol W. Hunja, South Eastern Kenya University, Kitui, Kenya, E-mail: chunja@seku.ac.ke. Richard Culleton, Malaria Unit, Institute of Tropical Medicine, Nagasaki University, Nagasaki, Japan, E-mail: richard@nagasaki-u.ac.jp. Akira Kaneko, Island Malaria Group, Department of Microbiology, Tumor and Cell Biology, Karolinska Institutet, Stockholm, Sweden, E-mail: akirakaneko555@gmail.com. Rashid A. Aman, Center for Research in Therapeutic Science, Strathmore University, Nairobi, Kenya, and African Center for Clinical Trials, Nairobi, Kenya, E-mail: raman@africaonline.co.ke.
References
- 1.World Health Organization . World Malaria Report 2014. Geneva, Switzerland: World Health Organization; 2014. [Google Scholar]
- 2.Snow RW. Global malaria eradication and the importance of Plasmodium falciparum epidemiology in Africa. BMC Med. 2015;13:14–16. doi: 10.1186/s12916-014-0254-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Murray CJL, Rosenfeld LC, Lim SS, Andrews KG, Foreman KJ, Haring D, Fullman N, Naghavi M, Lozano R, Lopez AD. Global malaria mortality between 1980 and 2010: a systematic analysis. Lancet. 2012;379:413–431. doi: 10.1016/S0140-6736(12)60034-8. [DOI] [PubMed] [Google Scholar]
- 4.World Health Organization . World Malaria Report 2010. Geneva, Switzerland: World Health Organization; 2010. [Google Scholar]
- 5.Kokwaro G. Ongoing challenges in the management of malaria. Malar J. 2009;8((Suppl 1)):S2. doi: 10.1186/1475-2875-8-S1-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaneko A. A community-directed strategy for sustainable malaria elimination on islands: short-term MDA integrated with ITNs and robust surveillance. Acta Trop. 2010;114:177–183. doi: 10.1016/j.actatropica.2010.01.012. [DOI] [PubMed] [Google Scholar]
- 7.Schultz L, Wapling J, Mueller I, Ntsuke PO, Senn N, Nale J, Kiniboro B, Buckee CO, Tavul L, Siba PM, Reeder JC, Barry AE. Multilocus haplotypes reveal variable levels of diversity and population structure of Plasmodium falciparum in Papua New Guinea, a region of intense perennial transmission. Malar J. 2010;9:336. doi: 10.1186/1475-2875-9-336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cohen JM, Smith DL, Cotter C, Ward A, Yamey G, Sabot OJ, Moonen B. Malaria resurgence: a systematic review and assessment of its causes. Malar J. 2012;11:122. doi: 10.1186/1475-2875-11-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anderson TJ, Haubold B, Williams JT, Estrada-Franco JG, Richardson L, Mollinedo R, Bockarie M, Mokili J, Mharakurwa S, French N, Whitworth J, Velez ID, Brockman AH, Nosten F, Ferreira MU, Day KP. Microsatellite markers reveal a spectrum of population structures in the malaria parasite Plasmodium falciparum. Mol Biol Evol. 2000;17:1467–1482. doi: 10.1093/oxfordjournals.molbev.a026247. [DOI] [PubMed] [Google Scholar]
- 10.Mobegi VA, Loua KM, Ahouidi AD, Satoguina J, Nwakanma DC, Amambua-Ngwa A, Conway DJ. Population genetic structure of Plasmodium falciparum across a region of diverse endemicity in west Africa. Malar J. 2012;11:223. doi: 10.1186/1475-2875-11-223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Machado RLD, Povoa MM, Calvosa VSP, Ferreira MU, Rossit ARB, dos Santos EJM, Conway DJ. Genetic structure of Plasmodium falciparum populations in the Brazilian Amazon region. J Infect Dis. 2004;190:1547–1555. doi: 10.1086/424601. [DOI] [PubMed] [Google Scholar]
- 12.Iwagami M, Rivera PT, Villacorte EA, Escueta AD, Hatabu T, Kawazu S, Hayakawa T, Tanabe K, Kano S. Genetic diversity and population structure of Plasmodium falciparum in the Philippines. Malar J. 2009;8:96. doi: 10.1186/1475-2875-8-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bogreau H, Renaud F, Bouchiba H, Durand P, Assi S-B, Henry M-C, Garnotel E, Pradines B, Fusai T, Wade B, Adehossi E, Parola P, Kamil MA, Puijalon O, Rogier C. Genetic diversity and structure of African Plasmodium falciparum populations in urban and rural areas. Am J Trop Med Hyg. 2006;74:953–959. [PubMed] [Google Scholar]
- 14.Lynch C, Roper C. The transit phase of migration: circulation of malaria and its multidrug-resistant forms in Africa. PLoS Med. 2011;8:e1001040. doi: 10.1371/journal.pmed.1001040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oyebola MK, Idowu ET, Olukosi YA, Iwalokun BA, Agomo CO, Ajibaye OO, Tola M, Otubanjo AO. Genetic diversity and complexity of Plasmodium falciparum infections in Lagos, Nigeria. Asian Pac J Trop Biomed. 2014;4:87–91. doi: 10.12980/APJTB.4.2014C1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rebaudet S, Bogreau H, Silaï R, Lepere JF, Bertaux L, Pradines B, Delmont J, Gautret P, Parola P, Rogier C. Genetic structure of Plasmodium falciparum and elimination of malaria, Comoros archipelago. Emerg Infect Dis. 2010;16:1686–1694. doi: 10.3201/eid1611.100694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pumpaibool T, Arnathau C, Durand P, Kanchanakhan N, Siripoon N, Suegorn A, Sitthi-Amorn C, Renaud F, Harnyuttanakorn P. Genetic diversity and population structure of Plasmodium falciparum in Thailand, a low transmission country. Malar J. 2009;8:155. doi: 10.1186/1475-2875-8-155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Talisuna AO, Okello PE, Erhart A, Coosemans M, D'Alessandro U. Intensity of malaria transmission and the spread of Plasmodium falciparum-resistant malaria: a review of epidemiologic field evidence. Am J Trop Med Hyg. 2007;77:170–180. [PubMed] [Google Scholar]
- 19.Roper C, Pearce R, Nair S, Sharp B, Nosten F, Anderson T. Intercontinental spread of pyrimethamine-resistant malaria. Science. 2004;305:1124. doi: 10.1126/science.1098876. [DOI] [PubMed] [Google Scholar]
- 20.Lum JK, Kaneko A, Tanabe K, Takahashi N, Björkman A, Kobayakawa T. Malaria dispersal among islands: human mediated Plasmodium falciparum gene flow in Vanuatu, Melanesia. Acta Trop. 2004;90:181–185. doi: 10.1016/j.actatropica.2003.09.022. [DOI] [PubMed] [Google Scholar]
- 21.Alonso PL, Tanner M. Public health challenges and prospects for malaria control and elimination. Nat Med. 2013;19:150–155. doi: 10.1038/nm.3077. [DOI] [PubMed] [Google Scholar]
- 22.Feachem RGA, Phillips AA, Hwang J, Cotter C, Wielgosz B, Greenwood BM, Sabot O, Rodriguez MH, Abeyasinghe RR, Ghebreyesus TA, Snow RW. Shrinking the malaria map: progress and prospects. Lancet. 2010;376:1566–1578. doi: 10.1016/S0140-6736(10)61270-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lum JK, Kaneko A, Taleo G, Amos M, Reiff DM. Genetic diversity and gene flow of humans, Plasmodium falciparum, and Anopheles farauti s.s. of Vanuatu: inferred malaria dispersal and implications for malaria control. Acta Trop. 2007;103:102–107. doi: 10.1016/j.actatropica.2007.05.012. [DOI] [PubMed] [Google Scholar]
- 24.Reid H, Vallely A, Taleo G, Tatem AJ, Kelly G, Riley I, Harris I, Henri I, Iamaher S, Clements ACA. Baseline spatial distribution of malaria prior to an elimination programme in Vanuatu. Malar J. 2010;9:150. doi: 10.1186/1475-2875-9-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.World Health Organization World Malaria Report 2009. World Heal Organ. 2009;12:66. [Google Scholar]
- 26.Le Menach A, Tatem AJ, Cohen JM, Hay SI, Randell H, Patil AP, Smith DL. Travel risk, malaria importation and malaria transmission in Zanzibar. Sci Rep. 2011;1:93. doi: 10.1038/srep00093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.World Health Organization . World Health Statistics 2008. Geneva, Switzerland: World Health Organization; 2008. [Google Scholar]
- 28.Okara RM, Sinka ME, Minakawa N, Mbogo CM, Hay SI, Snow RW. Distribution of the main malaria vectors in Kenya. Malar J. 2010;9:69. doi: 10.1186/1475-2875-9-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Minakawa N, Dida GO, Sonye GO, Futami K, Njenga SM. Malaria vectors in Lake Victoria and adjacent habitats in western Kenya. PLoS One. 2012;7:32725. doi: 10.1371/journal.pone.0032725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Noor AM, Gething PW, Alegana VA, Patil AP, Hay SI, Muchiri E, Juma E, Snow RW. The risks of malaria infection in Kenya in 2009. BMC Infect Dis. 2009;9:180. doi: 10.1186/1471-2334-9-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhong D, Afrane Y, Githeko A, Yang Z, Cui L, Menge DM, Temu EA, Yan G. Plasmodium falciparum genetic diversity in western Kenya highlands. Am J Trop Med Hyg. 2007;77:1043–1050. [PubMed] [Google Scholar]
- 32.Razakandrainibe FG, Durand P, Koella JC, De Meeüs T, Rousset F, Ayala FJ, Renaud F. “Clonal” population structure of the malaria agent Plasmodium falciparum in high-infection regions. Proc Natl Acad Sci USA. 2005;102:17388–17393. doi: 10.1073/pnas.0508871102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Annan Z, Durand P, Ayala FJ, Arnathau C, Awono-Ambene P, Simard F, Razakandrainibe FG, Koella JC, Fontenille D, Renaud F. Population genetic structure of Plasmodium falciparum in the two main African vectors, Anopheles gambiae and Anopheles funestus. Proc Natl Acad Sci USA. 2007;104:7987–7992. doi: 10.1073/pnas.0702715104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Prugnolle F, Durand P, Jacob K, Razakandrainibe F, Arnathau C, Villarreal D, Rousset F, de Meeûs T, Renaud F. A comparison of Anopheles gambiae and Plasmodium falciparum genetic structure over space and time. Microbes Infect. 2008;10:269–275. doi: 10.1016/j.micinf.2007.12.021. [DOI] [PubMed] [Google Scholar]
- 35.Anderson TJ, Su XZ, Bockarie M, Lagog M, Day KP. Twelve microsatellite markers for characterization of Plasmodium falciparum from finger-prick blood samples. Parasitology. 1999;119:113–125. doi: 10.1017/s0031182099004552. [DOI] [PubMed] [Google Scholar]
- 36.Su XZ, Wellems TE. Toward a high-resolution Plasmodium falciparum linkage map: polymorphic markers from hundreds of simple sequence repeats. Genomics. 1996;33:430–444. doi: 10.1006/geno.1996.0218. [DOI] [PubMed] [Google Scholar]
- 37.Md Idris Z, Chim WC, Chang SD, Masatsugu K, Isao T, Ahmediin O, Willis A, James K, Akira K. Geographic and seasonal variation in malaria prevalence on islands in Lake Victoria (western Kenya): results from three cross sectional studies. Malar J. 2014;13:1. [Google Scholar]
- 38.Kenya National Bureau of Statistics . Population and Housing Census 2009. Nairobi, Kenya: Kenya National Bureau of Statistics; 2010. [Google Scholar]
- 39.Singh B, Bobogare A, Cox-Singh J, Snounou G, Abdullah MS, Rahman HA. A genus- and species-specific nested polymerase chain reaction malaria detection assay for epidemiologic studies. Am J Trop Med Hyg. 1999;60:687–692. doi: 10.4269/ajtmh.1999.60.687. [DOI] [PubMed] [Google Scholar]
- 40.Li J, Zhang Y, Sullivan M, Hong L, Huang L, Lu F, McCutchan TF, Su XZ. Typing Plasmodium yoelii microsatellites using a simple and affordable fluorescent labeling method. Mol Biochem Parasitol. 2007;155:94–102. doi: 10.1016/j.molbiopara.2007.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Excoffier L, Laval G, Schneider S. Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinforma. 2005;1:47–50. [PMC free article] [PubMed] [Google Scholar]
- 42.Goudet J. FSTAT (version 1.2): a computer program to calculate F-statistics. J Hered. 1995;86:485–486. [Google Scholar]
- 43.Larrañaga N, Mejía RE, Hormaza JI, Montoya A, Soto A, Fontecha GA. Genetic structure of Plasmodium falciparum populations across the Honduras-Nicaragua border. Malar J. 2013;12:354. doi: 10.1186/1475-2875-12-354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Michalakis Y, Excoffier L. A generic estimation of population subdivision using distances between alleles with special reference for microsatellite loci. Genetics. 1996;142:1061–1064. doi: 10.1093/genetics/142.3.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pritchard JK. Documentation for structure software : version 2.3. In Pract. 2010;6:321–326. [Google Scholar]
- 47.Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol. 2005;14:2611–2620. doi: 10.1111/j.1365-294X.2005.02553.x. [DOI] [PubMed] [Google Scholar]
- 48.Earl DA, vonHoldt BM. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour. 2011;4:359–361. [Google Scholar]
- 49.Jakobsson M, Rosenberg NA. CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics. 2007;23:1801–1806. doi: 10.1093/bioinformatics/btm233. [DOI] [PubMed] [Google Scholar]
- 50.Rosenberg NA. DISTRUCT: a program for the graphical display of population structure. Mol Ecol Notes. 2004;4:137–138. [Google Scholar]
- 51.Haubold B, Hudson RR. LIAN 3.0: detecting linkage disequilibrium in multilocus data. Linkage analysis. Bioinformatics. 2000;16:847–848. doi: 10.1093/bioinformatics/16.9.847. [DOI] [PubMed] [Google Scholar]
- 52.Hoffmann EHE, Ribolla PEM, Ferreira MU. Genetic relatedness of Plasmodium falciparum isolates and the origin of allelic diversity at the merozoite surface protein-1 (MSP-1) locus in Brazil and Vietnam. Malar J. 2003;2:24. doi: 10.1186/1475-2875-2-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Oyebola MK, Idowu ET, Nyang H, Olukosi YA, Otubanjo OA, Nwakanma DC, Awolola ST, Amambua-ngwa A. Microsatellite markers reveal low levels of population sub-structuring of Plasmodium falciparum in southwestern Nigeria. Malar J. 2014;13:493. doi: 10.1186/1475-2875-13-493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Durand P, Michalakis Y, Cestier S, Oury B, Leclerc MC, Tibayrenc M, Renaud F. Significant linkage disequilibrium and high genetic diversity in a population of Plasmodium falciparum from an area (Republic of the Congo) highly endemic for malaria. Am J Trop Med Hyg. 2003;68:345–349. [PubMed] [Google Scholar]
- 55.Costantini C, Li SG, Della Torre A, Sagnon N, Coluzzi M, Taylor CE. Density, survival and dispersal of Anopheles gambiae complex mosquitoes in a west African Sudan savanna village. Med Vet Entomol. 1996;10:203–219. doi: 10.1111/j.1365-2915.1996.tb00733.x. [DOI] [PubMed] [Google Scholar]
- 56.Anthony TG, Conway DJ, Cox-Singh J, Matusop A, Ratnam S, Shamsul S, Singh B. Fragmented population structure of Plasmodium falciparum in a region of declining endemicity. J Infect Dis. 2005;191:1558–1564. doi: 10.1086/429338. [DOI] [PubMed] [Google Scholar]
- 57.Leclerc MC, Durand P, De Meeûs T, Robert V, Renaud F. Genetic diversity and population structure of Plasmodium falciparum isolates from Dakar, Senegal, investigated from microsatellite and antigen determinant loci. Microbes Infect. 2002;4:685–692. doi: 10.1016/s1286-4579(02)01587-3. [DOI] [PubMed] [Google Scholar]
- 58.Fraser-Hurt N, Felger I, Edoh D, Steiger S, Mashaka M, Masanja H, Smith T, Mbena F, Beck HP. Effect of insecticide-treated bed nets on haemoglobin values, prevalence and multiplicity of infection with Plasmodium falciparum in a randomized controlled trial in Tanzania. Trans R Soc Trop Med Hyg. 1999;93:47–51. doi: 10.1016/s0035-9203(99)90327-9. [DOI] [PubMed] [Google Scholar]
- 59.Rosenberg R, Wirtz RA, Schneider I, Burge R. An estimation of the number of malaria sporozoites ejected by a feeding mosquito. Trans R Soc Trop Med Hyg. 1990;84:209–212. doi: 10.1016/0035-9203(90)90258-g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.