

Abstract
Deep hypothermic circulatory arrest is often required for the repair of complex congenital cardiac defects in infants. However, deep hypothermic circulatory arrest induces neuroapoptosis associated with later development of neurocognitive abnormalities. Selective cerebral perfusion theoretically provides superior neural protection possibly through modifications in cerebral substrate oxidation and closely integrated glutamate cycling. We tested the hypothesis that selective cerebral perfusion modulates glucose utilization, and ameliorates abnormalities in glutamate flux, which occur in association with neuroapoptosis during deep hypothermic circulatory arrest. Eighteen infant male Yorkshire piglets were assigned randomly to two groups of seven (deep hypothermic circulatory arrest or deep hypothermic circulatory arrest with selective cerebral perfusion for 60 minutes at 18℃) and four control pigs without cardiopulmonary bypass support. Carbon-13-labeled glucose as a metabolic tracer was infused, and gas chromatography–mass spectrometry and nuclear magnetic resonance were used for metabolic analysis in the frontal cortex. Following 2.5 h of cerebral reperfusion, we observed similar cerebral adenosine triphosphate levels, absolute levels of lactate and citric acid cycle intermediates, and carbon-13 enrichment among three groups. However, deep hypothermic circulatory arrest induced significant abnormalities in glutamate cycling resulting in reduced glutamate/glutamine and elevated γ-aminobutyric acid/glutamate along with neuroapoptosis, which were all prevented by selective cerebral perfusion. The data suggest that selective cerebral perfusion prevents these modifications in glutamate/glutamine/γ-aminobutyric acid cycling and protects the cerebral cortex from apoptosis.
Keywords: Energy metabolism, glucose, glutamate, γ-aminobutyric acid, neurotransmitters, apoptosis, magnetic resonance spectroscopy
Introduction
Deep hypothermic circulatory arrest (DHCA) is a standard operative procedure used during repair of neonatal aortic arch reconstruction and other specific complex congenital heart defects, particularly in neonates. This procedure requires cooling the patient to 18℃ and ceasing all flow to the entire body including brain. Unfortunately, DHCA is associated with high risk of brain injury and neurodevelopmental impairment.1 Selective cerebral perfusion (SCP) involves directing blood flow to the brain during DHCA and is postulated to provide superior neuroprotection by supplying oxygen and nutrients.2,3 This procedure, sometimes used with more moderate hypothermia, has emerged as the most common form of neuroprotection applied by surgeons during aortic arch reconstruction in infants.4 Yet, minimal evidence supports the superiority of SCP over DHCA alone with respect to neurological outcomes. Results in porcine models suggest some improved preservation of cerebral metabolic pathways and energy stores by SCP,5 although late neurological outcomes remained similar between the two therapies in one study.6 Results from small randomized clinical trials show no difference in late neurological outcomes between DHCA and SCP.7,8 However, the interpretations of results from clinical trials comparing these two strategies are hampered by many factors. The prevalence of preoperative morphological brain abnormalities detected by magnetic resonance imaging is extremely high in this population.9,10 Furthermore, relatively high incidence of genetic abnormalities associated with neurological impairments may occur in this population and confound data interpretation for these trials with small subject numbers.
Mechanistic or translational studies provide an alternative strategy for attacking this important clinical problem. Overall, the porcine and clinical studies have not identified the mechanisms of neurological injury during DHCA. Several investigations have shown that neurological impairment in newborns subjected to hypoxia relates to exaggerated neuronal death.11,12 At least, two modes of cell death, apoptosis and excitotoxicity, in the immature brain depend on interruption of normal synaptic excitation. Alterations in synaptic activity are caused by modifications in the glutamate and γ-aminobutyric acid (GABA) pathways.13 GABA serves as a major inhibitory neurotransmitter, while glutamate serves as an excitatory transmitter. Metabolism of glucose to pyruvate and then via the citric acid cycle (CAC) eventually produces glutamate via a dehydrogenase reaction in both neurons and glia cells. Glutamate cycles further to GABA via decarboxylation or to glutamine (via glutamine synthetase). Studies performed in adult canines suggest that increased glutamate excitotoxicity mediates neuronal apoptosis after hypothermic circulatory arrest.14,15 However, GABA was not explored in these models, and more importantly brain maturation modifies balance and influence for glutamate excitatory and GABA inhibitory inputs. These pathways are highly energy dependent and rely on substrate oxidation through the CAC (Figure 1). Alterations in flux to glutamate and GABA as well as glutamine can diminish the CAC intermediate pool, and limit adenosine triphosphate (ATP) production. Accordingly, we posed the hypothesis that an imbalance in flux from glutamate to GABA occurs during DHCA, which can be ameliorated with SCP. We tested the hypothesis in immature porcine brain in vivo by using the nonradioactive uniform carbon-13 (13C) isotope, [U-13C6]-glucose as a tracer to determine relative flux through the CAC, and glutamate cycling to GABA and glutamine. We also determined if alterations in glutamate cycling related to neuronal apoptosis.
Figure 1.
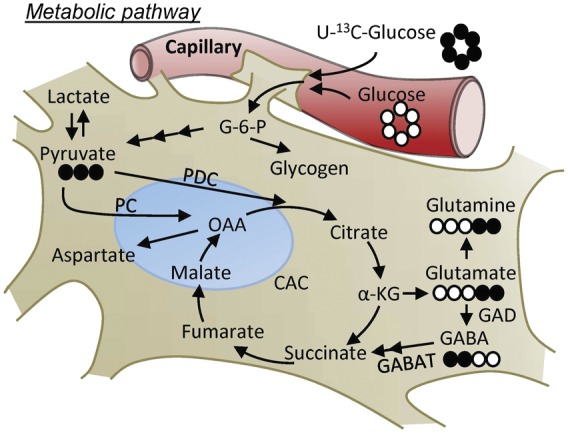
Schematic diagramming central role of glucose in glycolytic, CAC and accessory pathways. Full circle represent 13C and empty circle 12C. G-6-P: glucose 6-phosphate; CAC: citric acid cycle; PDC: pyruvate decarboxylase; PC: pyruvate carboxylase; α-KG: α-ketoglutarate; OAA: oxaloacetate; GABA: γ-aminobutyric acid; GAD: glutamate decarboxylase; GABAT: GABA aminotransferase.
Materials and methods
Study design
All experimental procedures were conducted according to the National Institute of Health’s Guide for the Care and Use of Laboratory Animals and approved by Seattle Children’s Institutional Animal Care and Use Committee. The manuscript is prepared in accordance with the ARRIVE guidelines. Fourteen male Yorkshire piglets (body weight 8.2–12.6 kg, age 34–44 days) were assigned randomly to two groups of seven animals. Pigs in one group had 60 min of DHCA, and they in another group had 60 min of DHCA with SCP as shown in Figure 2(a). Major time points were also expressed as follows, T0, baseline before starting cardiopulmonary bypass (CPB); T1, at just before starting DHCA; T2, at 60 min of DHCA; T3, completion of rewarming; T4, endpoint. In addition to these experimental groups, four pigs were used as a control group and they just received a median sternotomy without CPB.
Figure 2.

Diagram of perfusion protocol (a) and time course of cerebral and somatic regional oxygen saturation ((b), (c)). The two groups differ perfusion protocol after cooling for 45 min. Details are listed in the text. (b) The cerebral regional oxygen saturation (rSO2) after starting DHCA gradually dropped, while SCP maintained over 80% during DHCA (25 ± 1% in group DHCA vs. 92 ± 2% in group SCP, p < 0.001). (c) The somatic rSO2 values did not differ at any time points among two groups. CPB: cardiopulmonary bypass; DHCA: deep hypothermic circulatory arrest; SCP: selective cerebral perfusion; Glu: glucose; T0: baseline; T1: at just before starting DHCA; T2: at 60 min of DHCA; T3: completion of rewarming; T4: endpoint. *p < 0.001 vs. DHCA.
Perioperative management and operative technique
Pigs were initially sedated with an intramuscular injection of ketamine (33 mg/kg) and xylazine (2 mg/kg) and were mechanically ventilated with an oxygen (40%–50%) and isoflurane (1%–2%) mixture, as described previously.16–20 Near-infrared spectroscopy probes for measurement of cerebral and somatic regional oxygen saturation (rSO2) were placed on pig’s midline forehead and back at renal level (Samanetics INVOS 5100, Troy, MI). Esophageal and rectal temperatures were also continuously monitored. Angio catheters were inserted into the right axillary artery and the external jugular vein for continuous recording of the pressure, fluid infusion and blood sample. After median sternotomy, recordings of aortic flow (Transonic Systems Inc, Ithaca, NY) and left ventricular (LV) pressure (Millar Instruments, Houston, TX) were taken during experiment, and CPB with a roller peristaltic pump console (Sarns 8000) and a hollow fiber membrane oxygenator (CX-RX05RW, Terumo, Tokyo, Japan) was established by central cannulation via the ascending aorta (12-14 Fr) and right atrium (20-24 Fr) after heparinization (350 IU/kg). Arterial pump flow rate of 100–120 ml/kg/min was managed during CPB. A heat exchanger was used for core cooling. As blood gas management, pH-stat (temperature corrected) was utilized. CPB was initiated, and the animals were perfused for 10 min at normothermia. Animals were then cooled to esophageal temperature of 18℃ over 30 min. Before induction of circulatory arrest, the ascending aorta was clamped and cold cardioplegia was infused for myocardial arrest. DHCA was maintained for 60 min in group DHCA. In group SCP, the tip of aortic cannula was moved into the first branch of aortic arch (which then branched into common carotid and right subclavian arteries) and was snared at the proximal site of the first branch of aortic arch. CPB flow was maintained at 40 ml/kg/min during SCP. After removing the clamp on the ascending aorta and the repositioning of cannula into the aorta, CPB flow was increased gradually to 100–120 ml/kg/min until esophageal temperature of 36℃ was reached for over 45 min to rewarm. When necessary, cardiac defibrillation was performed. Blood arterial blood samples were collected at multiple time points. Baseline data were obtained after administration of heparin. Blood samples were immediately centrifuged and aliquots of plasma were stored at −80℃. Blood glucose was measured using a Bayer Contour point-of-care glucometer (Bayer HealthCare, Tarrytown, NY). Blood pH, pCO2, pO2, and hemoglobin were measured at regular intervals by a Radiometer ABL 800 (Radiometer America, Westlake, OH).
Infusion of labeled substrates
Once stable full-flow CPB was established after the completion of rewarming, 13C-labeled substrates were infused for metabolic analysis under continuous CPB support. [U-13C6]-glucose (90 mg/kg of body weight), purchased from Cambridge Isotope Laboratories (Andover, MA), was delivered as a metabolic tracer of glucose into the common carotid artery for the final 60 min of the protocol. Immediately upon completion of the 13C-labeled infusion for 60 min, the frontal cortex of the brain was harvested by a wide craniectomy and rapidly stored under liquid nitrogen for later extraction. After finishing the experimental protocol, the animals were euthanized with an overdose of inhaled isoflurane and intravenous potassium chloride.
Metabolic analysis
Our targeted metabolic pathways are in Figure 1. Glucose, the main fuel for cerebral energy synthesis, is incorporated into glycolytic and CAC intermediates, and is also the precursor of amino acid neurotransmitters glutamate, glutamine and GABA. We measured each key metabolite related to CAC metabolism by using gas chromatography–mass spectrometry (GC–MS) and nuclear magnetic resonance (NMR).
Glycogen and ATP analysis
Glycogen, produced by glycogenesis within the brain, was measured using commercial kits (Cayman, Ann Arbor, MI). Intracellular ATP level in the frontal cortex tissue was measured using luminescence ATP detection assay (ATPlite PerkinElmer, Waltham, MA). Moreover, energy phosphate metabolites, [Phosphocreatine; PCr]/[ATP] and [ATP]/[ADP], were measured by using 1H-NMR.
Metabolic analyses by GC–MS
The 13C-enrichment and absolute concentrations of lactate, pyruvate, amino acids and CAC intermediates; citrate, α-ketoglutarate (α-KG), succinate, fumarate and malate in the extracted cerebral tissue were measured by GC–MS using Agilent 6890 N gas chromatograph equipped with a HP-5 column coupled to a 5975 N mass spectrometer (Agilent Technologies, Santa Clara, CA) as described elsewhere.19,21–23 13C-enrichment data were expressed as molar percent enrichment (MPE). For the 13C-enrichment of oxaloacetate (OAA), we assessed OAA moiety of citrate obtained after cleavage of citrate with citrate lyase. We also reported the flux rates of pyruvate decarboxylation (PDC; the conversion of pyruvate to acetyl CoA) and pyruvate carboxylation (PC; the conversion of pyruvate to OAA) relative to citrate synthesis (CS) using the following equations, previously described.24
Mass isotopomers of metabolites containing 1 to n 13C-atoms were identified as Mi with i = 1, 2, … n.
Metabolic analyses by NMR
13C-NMR was performed on the brain cortex for determination of specific carbon glutamate, glutamine and GABA labeling from [U-13C6]-glucose. 13C-NMR spectra were acquired on a Varian Direct Drive 600 MHz spectrometer (Agilent Technologies) equipped with a Dell Precision T3500 Linux workstation running VNMRJ 3.2 as previously described.22 The number of scans was 8000 on average for ∼9.5 h per spectrum. The NMR spectra were analyzed in spectra processed with 0.5 Hz line broadening, based on individual brain. The spectrometer system was outfitted with a Varian triple resonance salt-tolerant cold probe with a cold carbon preamplifier. Protons were decoupled with a Waltz decoupling scheme. Final spectra were obtained using a 45° excitation pulse (7.05 µs at 58 dB), with an acquisition time of 1.3 s, a recycle delay of 3 s, and a spectral width of 224.1 ppm. Fourier-transformed spectra were fitted with commercial software (NUTS; Acorn NMR, Livermore, CA). Isotopomers arising from [U-13C6]-glucose can lead to the formation of [4,5-13C]-glutamate, [4,5-13C]-glutamine or [1,2-13C]-GABA via PDC and CAC in the first turn. We calculated the relative pool size of these amino acid neurotransmitters derived from exogenous labeled glucose.
Energy metabolites and amino acids were measured by 1H-NMR spectra from the brain cortex tissues prepared by using methanol. Data were acquired on a Varian Direct Drive 600 MHz spectrometer equipped with a Dell Precision T3500 Linux workstation running VNMRJ 4.0. The spectrometer system was outfitted with a Varian triple resonance salt-tolerant cold probe with a cold carbon preamplifier. A Varian standard one-dimensional proton noesy with presaturation (tnnoesy) was collected on each sample, using a nonselective 90° excitation pulse (approximately 7 µs at 57 dB), a 100-ms mixing time, acquisition time of 4 s, a presaturation delay of 1.5 s, spectral width of 12 ppm, and temperature control set to 25℃. Collected spectra were analyzed using Chenomx 8.1 software (Edmonton, Alberta, Canada), with quantifications based on spectral intensities relative to 0.5 mmol/L 2,2-dimethyl-2-silapentane-5-sulfonate, which was added as a spike to each sample.
TUNEL staining and combined immunofluorescence staining
The four fresh frozen tissues covering the frontal cortexes of the brain followed a systematic, uniform, random sampling in each animal and were sectioned using a cryostat into 5-µm-thick slices, mounted onto slides, and then fixed in acetone. Apoptosis via DNA fragmentation was detected by using the terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) technique with a commercially available kit (In Situ Cell Death Kit; Roche, Indianapolis, IN), according to the manufacturer’s recommendations. Fixed frozen sections were treated with 3% H2O2 for 10 min to inactivate endogenous peroxidases at room temperature, and permeabilized with 0.1 % Triton X-100 in 0.1% sodium citrate for 2 min on ice. The sections were exposed to terminal deoxynucleotidyl transferase solution for 60 min at 37℃. Positive controls (with DNase I recombinant) and negative controls (without terminal deoxynucleotidyl transferase) were performed with each assay. Anti-fluorescein antibody conjugated with peroxidase was added for light microscopy analysis, and the peroxidase was visualized with diaminobenzidine. We also performed immunohistochemical staining of another apoptotic cell death marker, the cleaved caspase-3 (diluted 1:400; BD Biosciences, San Jose, CA) by using avidin-biotin-complex system kit (Santa Cruz Biotechnology, Santa Cruz, CA) and counterstaining in Gill’s hematoxylin. Tonsils from some pigs were used as positive control for cleaved-caspase-3, and negative control was treated identically with the primary antibody omitted. In addition, TUNEL combined immunofluorescence staining was performed with neuron-specific primary antibody against anti-NeuN (diluted 1:100, Millipore, MAB377). To verify the neuronal identity of TUNEL-positive cells, TUNEL staining was first carried out without anti-fluorescein peroxidase antibody and DAB, and then followed by exposure with anti-NeuN. This combination staining was followed by DAPI counterstaining to visualize nuclei. Images were obtained using fluorescence microscope and were analyzed by image software, Fiji. Cells were considered double-stained when co-staining was morphologically seen throughout the extent of the nucleus for nuclear markers, or when a cytoplasmic marker clearly surrounded a nuclear marker. Quantitative analysis represented counting of 10 fields from four independent samples per animal. Quantification was performed at 200 × magnification, and cell counts were averaged for the 40 200 × fields in each animal. A reproducibility assessment was carried out by randomly selecting and recounting one to four sections from each animal.
Statistical analyses
Reported values are means ± standard error (SE) in text, figures and tables. Statistical analysis of the differences between DHCA and SCP on blood gas data (Table 1) and hemodynamics (Table 2 and Figure 3) was carried out with Mann–Whitney U test. Metabolic data among three groups were evaluated by one-way analysis of variance with Tukey’s post hoc test. Criterion for significance was p < 0.05 for all comparisons.
Table 1.
Hemodynamic data.
| Baseline |
DHCA (60 min) |
End |
||||
|---|---|---|---|---|---|---|
| DHCA | SCP | DHCA | SCP | DHCA | SCP | |
| HR (bpm) | 104 ± 4 | 99 ± 4 | 0 | 0 | 94 ± 9 | 107 ± 9 |
| SBP (mmHg) | 78 ± 9 | 74 ± 9 | 0 | 54 ± 3* | 65 ± 4 | 67 ± 4 |
| MBP (mmHg) | 57 ± 3 | 55 ± 2 | 0 | 42 ± 2* | 56 ± 3 | 57 ± 4 |
| CVP (mmHg) | 4.0 ± 0.4 | 4.0 ± 0.5 | 1.1 ± 0.3 | 1.7 ± 0.2 | 3.3 ± 0.6 | 3.6 ± 0.5 |
| CO (L/kg/min) | 1.07 ± 0.06 | 1.03 ± 0.08 | 0 | 0.40 ± 0.02* | 1.19 ± 0.05 | 1.15 ± 0.06 |
HR: heart rate; SBP: systolic systemic blood pressure; MBP: mean systemic blood pressure; CVP: central venous blood pressure; CO: cardiac output; DHCA: deep hypothermic circulatory arrest; SCP: selective cerebral perfusion.
p < 0.01 vs. DHCA.
Table 2.
Metabolite levels in extracts from cerebral cortex by 1H-NMR.
| Metabolite | Ctrl (n = 4) | DHCA (n = 7) | SCP (n = 7) | p |
|---|---|---|---|---|
| Alanine | 0.076 ± 0.007 | 0.082 ± 0.005 | 0.081 ± 0.006 | 0.86 |
| Lactate | 0.432 ± 0.048 | 0.444 ± 0.034 | 0.530 ± 0.050 | 0.17 |
| Pyruvate | 0.003 ± 0.001 | 0.004 ± 0.001 | 0.003 ± 0.001 | 0.26 |
| Glutamate | 1.029 ± 0.034 | 0.949 ± 0.026 | 1.043 ± 0.031 | 0.04 |
| Glutamine | 0.794 ± 0.043 | 0.832 ± 0.055 | 0.756 ± 0.034 | 0.25 |
| GABA | 0.078 ± 0.007 | 0.092 ± 0.003 | 0.077 ± 0.003 | 0.004 |
| NAA | 1.241 ± 0.036 | 1.141 ± 0.027 | 1.186 ± 0.019 | 0.19 |
| Aspartate | 0.267 ± 0.023 | 0.207 ± 0.015 | 0.210 ± 0.011 | 0.88 |
| Glycine | 0.175 ± 0.013 | 0.211 ± 0.040 | 0.211 ± 0.006 | 0.99 |
| [PCr]/[Cr] | 0.16 ± 0.02 | 0.19 ± 0.02 | 0.18 ± 0.03 | 0.71 |
| [PCr]/[ATP] | 1.13 ± 0.06 | 1.27 ± 0.10 | 1.18 ± 0.28 | 0.76 |
| [ATP]/[ADP] | 2.76 ± 0.41 | 2.62 ± 0.17 | 2.76 ± 0.29 | 0.68 |
| [NADH]/[NAD+] | 0.20 ± 0.01 | 0.20 ± 0.03 | 0.25 ± 0.02 | 0.28 |
| [NADPH]/[NADP+] | 0.82 ± 0.07 | 0.77 ± 0.06 | 0.59 ± 0.08 | 0.11 |
Values were normalized relative to total creatine. GABA: γ-aminobutyric acid; NAA: N-Acetylaspartate; Phosphocreatine, PCr. p-value (DHCA vs. SCP).
Figure 3.

Apoptosis cells and neurons identified by TUNEL staining in representative cerebral cortex sections. ((a), (b)) Detection of apoptosis and quantitation of apoptotic cells (brown/orange stains) from TUNEL staining. TUNEL assay showed no apoptosis in control pigs (Ctrl). Pigs undergoing DHCA had visibly more apoptosis compared with the SCP group, which also exhibited few apoptosis cells. Cleaved caspase-3 immunostaining (brown/orange stains) also showed same results as TUNEL staining. ((c), (d)) Double immunofluorescence staining for TUNEL (green) and the neuronal marker NeuN (red). Nuclei were counterstained with DAPI (blue). TUNEL-positive cells in DHCA group were detected in both neurons and other cells at the similar rate. TUNEL- and NeuN-double positive cells (arrowhead). TUNEL-positive and NeuN-negative cells (arrow). TUNEL, the terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling. Scale bar = 100 µm. *p < 0.001.
Results
A comparison of preoperative animal age (39 ± 1 days in group DHCA vs. 40 ± 1 days in group SCP) and weights (10.2 ± 0.5 kg in group DHCA vs. 10.1 ± 0.5 kg in group SCP) showed no differences between the groups. There were no operative or technical complications in any groups.
Blood parameters and hemodynamics
Arterial and venous blood gas data including hemoglobin and glucose did not differ at baseline before starting CPB and endpoint of experiments between DHCA and SCP groups. Arterial pH, CO2 and O2 were normally maintained within a range of 7.35–7.48, 35–50 mmHg and > 150 mmHg, respectively. Table 1 demonstrated hemodynamic data at baseline (T0), 60 min of DHCA (T2) and endpoint of experiments (T4). Perfusion parameters including cardiac output directly measured by aortic flow were not significantly different at baseline and endpoint of experiments between two groups. During DHCA, SCP with 40 ml/kg/min produced around 40 mmHg of mean perfusion pressure for cerebral region and right upper limb. Furthermore, cerebral rSO2 in group DHCA was significantly dropped to around 25% during DHCA, whereas that in group SCP was maintained over 80% (Figure 2(b)). Somatic rSO2 was similar between two groups during experiments (Figure 2(c)).
Cerebral injury
TUNEL staining
TUNEL-positive cells and neurons occurred in both DHCA and SCP groups but were absent in control piglets. Piglets undergoing SCP showed substantially less TUNEL staining at 2.5 h compared to DHCA (Figure 3(a) and (b)). We also confirmed that the presence of cleaved caspase-3, indicative of apoptosis, was detected in DHCA group whereas very minimal staining was in other two groups. Combined immunofluorescence staining for TUNEL and the neuronal marker NeuN showed that DHCA increased in the number of TUNEL- and NeuN-double positive cells with approximately 35% of NeuN-positive cells and 27% of NeuN-negative cells positive for TUNEL staining (Figure 3(c) and (d)). DHCA-induced apoptosis were confirmed in NeuN-negative cells such as astrocytes as well as neurons.
Cerebral metabolism
Energy status
Intracellular ATP level, measured using luminescence ATP detection assay, in the extracted cerebral tissue was not statistically different among three groups (Figure 4(a)). Energy phosphate metabolism, [PCr]/[ATP] and [ATP]/[ADP], measured by 1H-NMR were similar among three groups (Table 2). Glycogen stores can be used for anaerobic glycolysis during an energy crisis induced by ischemia and were significantly low in DHCA group compared with control and SCP groups (Figure 4(b)).
Figure 4.

Cerebral energy metabolism. (a) Intracellular ATP level and (b) glycogen level were measured by detection assay kits. ATP level was not different among three groups, whereas glycogen level was lower in DHCA group than control and SCP groups. CAC intermediates and entering CAC in the cerebral tissue were measured by GC–MS (C–G). (c) 13C-MPE of pyruvate and lactate, derived from exogenous [U-13C6]-glucose. (d) Absolute quantity of pyruvate and lactate, derived from all carbohydrates including endogenous glucose and exogenous [U-13C6]-glucose. (e) Pyruvate decarboxylation (PDC) and pyruvate carboxylation (PC) relative to citrate synthase (CS) flux ratio. (f) Total 13C-MPE and (g) absolute quantity of CAC intermediates. These data were not statistically different among three groups. CAC: citric acid cycle; MPE: molar percent enrichment; α-KG: α-ketoglutarate; OAA: oxaloacetate. *p < 0.05.
Glycolysis
Glycolysis produces two molecules of pyruvate from one glucose molecule. Under anaerobic conditions, pyruvate is converted to lactate by the enzyme lactate dehydrogenase. We measured the concentration of pyruvate and lactate in the cerebral tissue by GC–MS. The 13C-enrichment and absolute concentrations of pyruvate and lactate were not significantly different between DHCA and SCP (Figure 4(c) and (d)).
CAC intermediates
The CAC is considered to be the central metabolic pathway for cerebral energetics. Pyruvate converted from glucose can enter into CAC through two pathways, PDC and PC. Both PDC/CS and PC/CS calculated by GC–MS did not differ between the two groups, and PC and PDC occurred in an ∼2:5 ratio in both groups (Figure 4(e)). The 13C-enrichment and absolute concentrations of each CAC intermediate were similar between two groups (Figure 4(f) and (g)).
Amino acids as neurotransmitters
We also measured glutamate, glutamine and aspartate, neurotransmitters derived from intermediates of the CAC. Absolute concentrations for glutamine and aspartate were similar between the two groups (Figure 5(a)), but glutamate concentration was significantly higher in SCP group (DHCA: 8.0 ± 0.2 nmol/mg, SCP: 9.0 ± 0.4, p = 0.03). [Glutamate]/[Glutamine] calculated from these data was also higher in SCP group but did not reach significant difference (DHCA: 1.07 ± 0.07, SCP: 1.29 ± 0.08, p = 0.08) (Figure 5(b)). Glutamate also metabolized to GABA in GABAergic neurons. Thus, we examined concentrations of GABA by NMR. The analysis of 13C-NMR spectra allowed the neurotransmitter pools of glutamine, glutamate and GABA derived from 13C-labeled glucose (Figure 5(c)). Analyzed data, calculated from C4 of glutamine and glutamate and C2 of GABA, showed low [Glutamate]/[Glutamine] and high [GABA]/[Glutamate] in DHCA group compared with other two groups (Figure 5(d)). 1H-NMR spectroscopy was used to determine the total concentration of cerebral metabolites (Table 2). 1H-NMR demonstrated that DHCA induced low glutamate and high GABA levels consistent with 13C-NMR data.
Figure 5.

Concentrations of amino acid neurotransmitters in the cerebral tissue. Absolute concentrations and 13C-labeled metabolite enrichment derived from [U-13C6]-glucose were measured by GC–MS ((a) and (b)) and 13C-NMR (typical spectrum in (c) and graphs in (d)), respectively. These data showed that glutamate in DHCA group was lower than in control and SCP groups, while GABA in DHCA group was higher than others. Chemical shifts were as follows: C4 of glutamine, 31.4 ppm; C4 of glutamate, 34.0 ppm; C2 of GABA, 34.9 ppm. GABA: γ-aminobutyric acid; ppm: parts per million. *p < 0.05.
Discussion
We showed that reperfusion after DHCA for 1 h does not induce significant alterations in glucose flux to the CAC. However, DHCA does disrupt cycling along the glutamate/glutamine/GABA pathways, which closely integrate with CAC metabolism. Abnormalities in glutamate cycling occur in association with neuronal injury and death indexed by TUNEL assay. Furthermore, SCP prevents both these modifications in glutamate cycling and neuronal apoptosis, suggesting that these adverse processes are linked.
Glutamate and GABA are respectively the major excitatory and inhibitory neurotransmitters in the mature cerebral cortex. Cycling through these transmitters accounts for ∼70% of total brain energy consumption at baseline cortical stimulation levels.25,26 Together these transmitters account for ∼90% of total synapses with excitatory outnumbering inhibitory in the adult brain in a ratio ∼5:1.27–29 Therefore, excitation holds the energetically dominant role in the mature cortex. Importantly, these relationships change with development, and disruptions in balance between glutamatergic and GABAergic activity are associated with neuronal apoptosis in rodent models of hypoxic-ischemic injury or post-traumatic stress disorder.30,31 Increased glutamate excitotoxicity is implicated in neuronal and glial injury in an age selective pattern after hypoxia-ischemia.12,32 However, glutamate synaptic inhibition by N-methyl-D-Aspartate antagonists promotes neuronal apoptosis in the developing cerebral cortex exposed to hypoxia-ischemia.33,34
In the current study, we used a model in immature swine, which emulates clinical circulatory arrest and SCP for infants and children undergoing specific types of cardiac surgeries. Cerebral rSO2 data show that DHCA alone dramatically decreases oxygen supply to the brain. ATP levels did not differ significantly among the groups. However, the DHCA group data demonstrate enhanced glycogen depletion suggesting over reliance on anaerobic ATP generation during ischemia with inability to restore glycogen after reperfusion. Following reperfusion and reoxygenation, we showed that SCP preserves relative flux rates from glucose towards glutamate and GABA. Preservation of these fluxes is associated with a reduction in apoptosis after DHCA. While synaptic glutamate and GABA is generated predominantly from CAC intermediates in neurons, glutamine is primarily synthesized from glutamate in astrocytes and other glial cells.35 Thus, the overall absolute and 13C-isotopomer data suggest that DHCA induces impairments in neuronal glutamate cycling, which also promotes GABA production. The data also show that glutamate cycling differences occur despite similarity in 13C cycling from glucose through the CAC in all three experimental groups.
The differences in glutamate–glutamine cycling are statistically significant among the experimental groups but of relatively small magnitude. Nevertheless, these differences should be considered in context of the deep anesthetic requirements within these protocols and very low level of brain stimulation. Shulman and coworkers25,36 have generated data supporting a linear relationship between rates of glucose oxidation in excitatory glutamatergic neurons and total neurotransmitter cycling within glutamate and GABA systems, measured over a wide range of activity in the anesthetized rat brain extending from higher levels of activity down to isoelectric conditions. The fraction of energy consumed by the brain that is devoted to neurotransmitter cycling decreases with increasing depth of anesthesia until, at the isoelectric condition, all the energy supports “housekeeping needs.” Thus, we detected differences in glutamate–glutamine cycling at extremely low levels of brain stimulation. Conceivably, the impairments in cycling could be more dramatic if we had imposed higher cortical stimulation rates during the reperfusion period.
Limitations
Several limitations of this study should be noted. As the techniques used in this study require rapid tissue extraction and freeze clamping for metabolic analysis, we were able to sample from only frontal cortex location, not the depths of the cerebrum such as hippocampus and basal ganglia by a craniectomy. Therefore, we cannot be certain that these findings occur throughout the brain. Brain injury after DHCA was detected in various regions. Some previous studies in large animals showed that the highest degree of brain damage was detected in the hippocampus.37,38 These reports also detected neuronal cell damage in the frontal cortex. The hippocampus is highly vulnerable to hypoxic/ischemic as well as cortex, and its damage is strongly related to long-term cognitive impairments. The assessment of hippocampus and long-term cognitive function could not be studied further with the current experimental design. Tiwari et al.39 reported that neurotransmitter cycling fluxes were different in various regions of mouse brain. Their data showed higher glutamatergic and lower GABAergic neurotransmissions in the cerebral cortex compared with other regions suggesting variable vulnerability to DHCA throughout the brain. Second, we did not specifically evaluate potential affects of cerebral events such as embolism or edema caused by hyperperfusion on cerebral metabolism. Furthermore, the aortic arch branch anatomy of piglets differs slightly from human aortic arch branching. SCP through first branch of aorta bilateral covers the whole brain and right upper limb in our pig model. We thought that SCP flow rate (40 ml/kg/min) in our study was reasonable and equal to general clinical strategy used in human infants, because it led to suitable mean BP (around 40 mmHg) at the right upper limb.40 In this study, we monitored cerebral oxygenation continuously by near-infrared spectroscopy but did not acquire cerebral venous blood gas from the sagittal sinus. Therefore, cerebral oxygen consumption data could not be calculated. Lastly, we used 60 min of DHCA, which is generally longer than the duration used by most surgeons clinically. However, the Boston circulatory arrest trial suggested that neurodevelopmental outcomes were not adversely affected unless the duration of circulatory arrest exceeded a threshold of 41 min.41 Thus, our extended time period was necessary to test our hypothesis.
Conclusions
We have demonstrated that SCP prevents abnormalities in glutamate/glutamine/GABA cycling, which are induced by DHCA. Consistent with prior studies, DHCA promotes neuronal cell death (apoptosis) in association with these impairments in neurotransmitter recycling. However, the apoptosis occurs without increased flux towards glutamate, which has been proposed as a mechanism for neurological injury associated with hypothermic circulatory arrest in adult cerebral cortex.14,15 The discrepancy in our study compared to some prior investigations may relate to developmental differences as antagonism of glutamate synaptic transmission causes neuronal apoptosis in immature cortex. Thus, neuroprotective strategies such as N-methyl-D-Aspartate antagonism may not be appropriate for the immature brain undergoing DHCA. Rather, strategies which preserve glutamate/glutamine/GABA cycling may prevent apoptosis and neuronal injury after DHCA. These data suggest a novel paradigm for potential neuroprotection in infants undergoing aortic arch procedures.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Research reported in this publication was supported by the National Heart Lung and Blood Institute of the National Institutes of Health under award number R01HL60666. A portion of the research was performed using EMSL, a DOE Office of Science User Facility sponsored by the Office of Biological and Environmental Research and located at Pacific Northwest National Laboratory.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
MK devised the study, performed the surgical and experimental components of the study, analyzed and interpreted the data, designed the figures, and drafted the manuscript. DL and AO performed the surgical and experimental components of the study. NI performed the NMR studies and analysis. IRF performed the GC–MS study and analysis. CDR performed the GC–MS study and analysis, analyzed and interpreted all data, and provided critical comments on the manuscript. MP devised the study, obtained the funding, analyzed and interpreted all data and wrote the manuscript. All authors have seen and approved the manuscript.
References
- 1.Bellinger DC, Wypij D, Rivkin MJ, et al. Adolescents with d-transposition of the great arteries corrected with the arterial switch procedure: neuropsychological assessment and structural brain imaging. Circulation 2011; 124: 1361–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Asou T, Kado H, Imoto Y, et al. Selective cerebral perfusion technique during aortic arch repair in neonates. Ann Thorac Surg 1996; 61: 1546–1548. [DOI] [PubMed] [Google Scholar]
- 3.Pigula FA, Siewers RD, Nemoto EM. Regional perfusion of the brain during neonatal aortic arch reconstruction. J Thorac Cardiovasc Surg 1999; 117: 1023–1024. [DOI] [PubMed] [Google Scholar]
- 4.Ohye RG, Goldberg CS, Donohue J, et al. The quest to optimize neurodevelopmental outcomes in neonatal arch reconstruction: the perfusion techniques we use and why we believe in them. J Thorac Cardiovasc Surg 2009; 137: 803–806. [DOI] [PubMed] [Google Scholar]
- 5.Salazar JD, Coleman RD, Griffith S, et al. Selective cerebral perfusion: real-time evidence of brain oxygen and energy metabolism preservation. Ann Thorac Surg 2009; 88: 162–169. [DOI] [PubMed] [Google Scholar]
- 6.Sasaki H, Guleserian KJ, Rose R, et al. Hypothermic extracorporeal circulation in immature swine: a comparison of continuous cardiopulmonary bypass, selective antegrade cerebral perfusion and circulatory arrest. Eur J Cardiothorac Surg 2009; 36: 992–997. [DOI] [PubMed] [Google Scholar]
- 7.Goldberg CS, Bove EL, Devaney EJ, et al. A randomized clinical trial of regional cerebral perfusion versus deep hypothermic circulatory arrest: outcomes for infants with functional single ventricle. J Thorac Cardiovasc Surg 2007; 133: 880–887. [DOI] [PubMed] [Google Scholar]
- 8.Algra SO, Jansen NJ, van der Tweel I, et al. Neurological injury after neonatal cardiac surgery: a randomized, controlled trial of 2 perfusion techniques. Circulation 2014; 129: 224–233. [DOI] [PubMed] [Google Scholar]
- 9.Miller SP, McQuillen PS, Hamrick S, et al. Abnormal brain development in newborns with congenital heart disease. N Engl J Med 2007; 357: 1928–1938. [DOI] [PubMed] [Google Scholar]
- 10.Mahle WT, Tavani F, Zimmerman RA, et al. An MRI study of neurological injury before and after congenital heart surgery. Circulation 2002; 106(12 Suppl. 1): I109–I114. [PubMed] [Google Scholar]
- 11.Rivkin MJ. Hypoxic-ischemic brain injury in the term newborn. Neuropathology, clinical aspects, and neuroimaging. Clin Perinatol 1997; 24: 607–625. [PubMed] [Google Scholar]
- 12.Johnston MV, Trescher WH, Ishida A, et al. Neurobiology of hypoxic-ischemic injury in the developing brain. Pediatr Res 2001; 49: 735–741. [DOI] [PubMed] [Google Scholar]
- 13.Walls AB, Waagepetersen HS, Bak LK, et al. The glutamine-glutamate/GABA cycle: function, regional differences in glutamate and GABA production and effects of interference with GABA metabolism. Neurochem Res 2015; 40: 402–409. [DOI] [PubMed] [Google Scholar]
- 14.Redmond JM, Gillinov AM, Zehr KJ, et al. Glutamate excitotoxicity: a mechanism of neurologic injury associated with hypothermic circulatory arrest. J Thorac Cardiovasc Surg 1994; 107: 776–786; discussion 786–787. [PubMed] [Google Scholar]
- 15.Tseng EE, Brock MV, Kwon CC, et al. Increased intracerebral excitatory amino acids and nitric oxide after hypothermic circulatory arrest. Ann Thorac Surg 1999; 67: 371–376. [DOI] [PubMed] [Google Scholar]
- 16.Kajimoto M, O’Kelly Priddy CM, Ledee DR, et al. Extracorporeal membrane oxygenation promotes long chain fatty acid oxidation in the immature swine heart in vivo. J Mol Cell Cardiol 2013; 62: 144–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kajimoto M, Ledee DR, Olson AK, et al. Differential effects of octanoate and heptanoate on myocardial metabolism during extracorporeal membrane oxygenation in an infant swine model. Am J Physiol Heart Circ Physiol 2015; 309: H1157–H1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kajimoto M, Ledee DR, Xu C, et al. Triiodothyronine activates lactate oxidation without impairing Fatty Acid oxidation and improves weaning from extracorporeal membrane oxygenation. Circ J 2014; 78: 2867–2875. [PMC free article] [PubMed] [Google Scholar]
- 19.Kajimoto M, O’Kelly Priddy CM, Ledee DR, et al. Myocardial reloading after extracorporeal membrane oxygenation alters substrate metabolism while promoting protein synthesis. J Am Heart Assoc 2013; 2: e000106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kajimoto M, O’Kelly Priddy CM, Ledee DR, et al. Effects of continuous triiodothyronine infusion on the tricarboxylic acid cycle in the normal immature swine heart under extracorporeal membrane oxygenation in vivo. Am J Physiol Heart Circ Physiol 2014; 306: H1164–H1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Des Rosiers C, Lloyd S, Comte B, et al. A critical perspective of the use of (13)C-isotopomer analysis by GCMS and NMR as applied to cardiac metabolism. Metab Eng 2004; 6: 44–58. [DOI] [PubMed] [Google Scholar]
- 22.Kajimoto M, Atkinson DB, Ledee DR, et al. Propofol compared with isoflurane inhibits mitochondrial metabolim in immature swine cerebral cortex. J Cereb Blood Flow Metab 2014; 34: 514–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O’Kelly Priddy CM, Kajimoto M, Ledee D, et al. Myocardial oxidative metabolism and protein synthesis during mechanical circulatory support by extracorporeal membrane oxygenation. Am J Physiol Heart Circ Physiol 2012; 304: H406–H414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Comte B, Vincent G, Bouchard B, et al. A 13C mass isotopomer study of anaplerotic pyruvate carboxylation in perfused rat hearts. J Biol Chem 1997; 272: 26125–26131. [DOI] [PubMed] [Google Scholar]
- 25.Sibson NR, Dhankhar A, Mason GF, et al. Stoichiometric coupling of brain glucose metabolism and glutamatergic neuronal activity. Proc Natl Acad Sci U S A 1998; 95: 316–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oz G, Berkich DA, Henry PG, et al. Neuroglial metabolism in the awake rat brain: CO2 fixation increases with brain activity. J Neurosci 2004; 24: 11273–11279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beaulieu C, Colonnier M. A laminar analysis of the number of round-asymmetrical and flat-symmetrical synapses on spines, dendritic trunks, and cell bodies in area 17 of the cat. J Comparat Neurol 1985; 231: 180–189. [DOI] [PubMed] [Google Scholar]
- 28.Nicholls DG. Release of glutamate, aspartate, and gamma-aminobutyric acid from isolated nerve terminals. J Neurochem 1989; 52: 331–341. [DOI] [PubMed] [Google Scholar]
- 29.Patel AB, de Graaf RA, Mason GF, et al. The contribution of GABA to glutamate/glutamine cycling and energy metabolism in the rat cortex in vivo. Proc Natl Acad Sci U S A 2005; 102: 5588–5593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Andine P, Sandberg M, Bagenholm R, et al. Intra- and extracellular changes of amino acids in the cerebral cortex of the neonatal rat during hypoxic-ischemia. Dev Brain Res 1991; 64: 115–120. [DOI] [PubMed] [Google Scholar]
- 31.Gao J, Wang H, Liu Y, et al. Glutamate and GABA imbalance promotes neuronal apoptosis in hippocampus after stress. Med Sci Monit 2014; 20: 499–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Macri MA, D’Alessandro N, Di Giulio C, et al. Region-specific effects on brain metabolites of hypoxia and hyperoxia overlaid on cerebral ischemia in young and old rats: a quantitative proton magnetic resonance spectroscopy study. J Biomed Sci 2010; 17: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Desfeux A, El Ghazi F, Jegou S, et al. Dual effect of glutamate on GABAergic interneuron survival during cerebral cortex development in mice neonates. Cereb Cortex 2010; 20: 1092–1108. [DOI] [PubMed] [Google Scholar]
- 34.Roux C, Aligny C, Lesueur C, et al. NMDA receptor blockade in the developing cortex induces autophagy-mediated death of immature cortical GABAergic interneurons: an ex vivo and in vivo study in Gad67-GFP mice. Exp Neurol 2015; 267: 177–193. [DOI] [PubMed] [Google Scholar]
- 35.Norenberg MD, Martinez-Hernandez A. Fine structural localization of glutamine synthetase in astrocytes of rat brain. Brain Res 1979; 161: 303–310. [DOI] [PubMed] [Google Scholar]
- 36.Shulman RG, Rothman DL, Behar KL, et al. Energetic basis of brain activity: implications for neuroimaging. Trends Neurosci 2004; 27: 489–495. [DOI] [PubMed] [Google Scholar]
- 37.Arnaoutakis GJ, George TJ, Wang KK, et al. Serum levels of neuron-specific ubiquitin carboxyl-terminal esterase-L1 predict brain injury in a canine model of hypothermic circulatory arrest. J Thorac Cardiovasc Surg 2011; 142: 902e1–910e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schubert S, Gerlach F, Stoltenburg-Didinger G, et al. Cerebral expression of neuroglobin and cytoglobin after deep hypothermic circulatory arrest in neonatal piglets. Brain Res 2010; 1356: 1–10. [DOI] [PubMed] [Google Scholar]
- 39.Tiwari V, Ambadipudi S, Patel AB. Glutamatergic and GABAergic TCA cycle and neurotransmitter cycling fluxes in different regions of mouse brain. J Cereb Blood Flow Metab 2013; 33: 1523–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fraser CD, Jr, Andropoulos DB. Principles of antegrade cerebral perfusion during arch reconstruction in newborns/infants. Semin Thorac Cardiovasc Surg Pediatr Card Surg Annu 2008; 2008: 61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wypij D, Newburger JW, Rappaport LA, et al. The effect of duration of deep hypothermic circulatory arrest in infant heart surgery on late neurodevelopment: the Boston Circulatory Arrest Trial. J Thorac Cardiovasc Surg 2003; 126: 1397–1403. [DOI] [PubMed] [Google Scholar]