

Abstract
Catalase, a conserved and abundant enzyme found in all domains of life, dissipates the oxidant hydrogen peroxide (H2O2). The gastric pathogen Helicobacter pylori undergoes host-mediated oxidant stress exposure, and its catalase contains oxidizable methionine (Met) residues. We hypothesized catalase may play a large stress-combating role independent of its classical catalytic one, namely quenching harmful oxidants through its recyclable Met residues, resulting in oxidant protection to the bacterium. Two Helicobacter mutant strains (katAH56A and katAY339A) containing catalase without enzyme activity but that retain all Met residues were created. These strains were much more resistant to oxidants than a catalase-deletion mutant strain. The quenching ability of the altered versions was shown, whereby oxidant-stressed (HOCl-exposed) Helicobacter retained viability even upon extracellular addition of the inactive versions of catalase, in contrast to cells receiving HOCl alone. The importance of the methionine-mediated quenching to the pathogen residing in the oxidant-rich gastric mucus was studied. In contrast to a catalase-null strain, both site-change mutants proficiently colonized the murine gastric mucosa, suggesting that the amino acid composition-dependent oxidant-quenching role of catalase is more important than the well described H2O2-dissipating catalytic role. Over 100 years after the discovery of catalase, these findings reveal a new non-enzymatic protective mechanism of action for the ubiquitous enzyme.
Keywords: antioxidant, bacterial metabolism, bacterial pathogenesis, infectious disease, microbiology, multifunctional enzyme, oxidative stress
Introduction
Catalase was described over 100 years ago (1), and the enzyme's role clearly is to detoxify H2O2 by converting it into H2O and O2. It is one of he most abundant proteins in cells, and it is present in most organisms, including in both plant and animal cells (2). It is oftentimes a highly expressed protein. For example, in the gastric pathogen Helicobacter pylori, catalase (KatA) levels are estimated to be 4–5% of the total protein content (3), and the katA gene is one of the most highly expressed genes in H. pylori cells recovered from the human stomach (4). HOCl-mediated oxidation of six identified Met residues in H. pylori KatA leads to methionine sulfoxide formation (Met-O),2 protein oligomerization, and loss of catalase activity (3). Most organisms, including Helicobacter, possess a peptide repair enzyme, methionine sulfoxide reductase (Msr), that reduces Met-O back to Met in certain oxidation-susceptible Msr-targeted proteins (5, 6). This Msr-mediated repair (along with added GroEL/ES) returns most of the catalase activity (3). Although the identified protein targets of repair are few among the total of all organisms, some of these repair targets are themselves stress-combating enzymes.
Purified KatA and Msr enzymes were shown to physically interact (6), and this interaction resulted in Msr-mediated repair of five out of the six oxidized KatA Met residues (3). In addition, H. pylori KatA is ubiquitous, present in both the cytoplasm and in the periplasm and on the cell surface as well as being readily detected (like Msr) extracellularly (7–9). Taken together, this information (reactivity toward HOCl; presence of multiple Msr-repairable Met residues, enzyme abundance) caused us to investigate the possibility that the catalase primary sequence or composition represents a Met-recycling sink providing oxidant protection per se to this bacterium. This Met-mediated quenching role would be separate from the enzyme's catalytic role, and no such whole cell protective mechanism has yet been assigned to any protein. However, the possibility that a Met-rich protein could exist to serve a protective oxidant-quenching role (via cyclic turnover of Met residues) was raised 20 years ago (10, 11), but results to support this have been unavailable. While our study identifies a new oxidant protective mechanism for catalase, the model organism we use, H. pylori, is an important pathogen. Although this bacterium is known to be the agent of human gastritis, which can develop into peptic ulcer disease (12), factors that allow it to persist in the host need to be identified, as such persistence is responsible for the most severe outcomes of the infection, namely gastric and duodenal cancers (13). In response to H. pylori infection, the host produces a battery of harmful reactive oxygen species (ROS), such as superoxide anion (O2˙̄), hydrogen peroxide (H2O2), hydroxyl radical (•HO), and hypochlorous acid (HOCl). Indeed, exposure of gastric cells (14) or phagocytes (15) to H. pylori increases host cell ROS production, and H. pylori-infected patients show elevated levels of ROS (16).
HOCl is produced in large amounts by neutrophil myeloperoxidases; its concentration can reach up to 5 mm at inflammatory sites (17), and it is 100 times more toxic than H2O2 (18). HOCl primarily targets sulfur-containing amino acids, cysteine (Cys) and methionine (Met), and indeed these residues are the preferred amino acids for oxidation under physiological conditions (19). Met oxidation by HOCl and similar small molecule oxidants (superoxide and hydroxyl radicals) leads to formation of Met-O in many proteins, and further oxidation results in methionine sulfone formation within the peptides, and either oxidized form at Met residues can lead to protein dysfunction (20). Oxidation of Met residues within a protein can “protect” other susceptible residues in that same protein from oxidation (5), but documentation of Met-containing residues within a protein to act as antioxidants in whole cell physiology is unavailable. Here, we address the roles of a catalase protein devoid of catalytic activity to provide oxidative stress protection.
Results and Discussion
To investigate whether catalase can quench oxidants independently of its H2O2 removal activity, we engineered two H. pylori mutants to synthesize only apo-catalase, e.g. devoid of catalase activity. Based on the published crystal structure of H. pylori catalase, two residues are essential for catalysis as follows: the proximal Tyr-339, coordinated to the heme iron, and the distal His-56, essential for the formation of the main reaction intermediate, compound I (21, 22). Therefore, two markerless chromosomal katA mutant versions, katAH56A and katAY339A, were constructed in two H. pylori wild-type (WT) strains, strain 43504 and the mouse colonizing strain X47 (23). Complete deletion of ΔkatA mutants were also constructed in both parental strains. When grown on plates, colonies of ΔkatA mutants, as well as katAH56A and katAY339A site-directed mutants, displayed strikingly different phenotypes compared with their individual parental strain (43504 or X47). While wild-type colonies were dark brown, the mutant strains were light brown (katAH56A) or yellow (katAY339A or ΔkatA) in color, suggesting that the heme b cofactor binding might be either disturbed (katAH56A) or even lacking (katAY339A or ΔkatA). Catalase assays revealed both 43504 ΔkatA and X47 ΔkatA mutants, as well as isogenic katAH56A and katAY339A site-directed mutants, had no detectable catalase activity (Table 1). However, catalase was still synthesized in both katAH56A and katAY339A strains, albeit to lower levels than WT (Fig. 1, A and B). No catalase protein was detected in ΔkatA gene deletion strains (Fig. 1, A and B).
TABLE 1.
Catalase activity of purified KatA proteins and wild-type and mutant H. pylori strains
| Proteina | |
| KatAWT | 45.15 ± 3.85 |
| KatAH56A | 0.06 ± 0.01 |
| KatAY339A | <0.01 (ND)b |
| H. pylori strainc | |
| 43504 (wild type) | 484 ± 38 |
| 43504 ΔkatA | <0.01 (ND) |
| 43504 katAH56A | <0.01 (ND) |
| 43504 katAY339A | <0.01 (ND) |
| X47 (wild type) | 306 ± 28 |
| X47 ΔkatA | <0.01 (ND) |
| X47 katAH56A | <0.01 (ND) |
| X47 katAY339A | <0.01 (ND) |
a Catalase activity was measured as μmol of H2O2/min/μg of protein. Results shown are average ± S.D. from two independent batches of purified proteins, and assays were done in triplicate (n = 6 total).
b ND means not detectable.
c Catalase activity was measured as μmol of H2O2/min/109 cells. Results shown are average ± S.D. from three biological replicates, and assays were done in triplicate (n = 9 total).
FIGURE 1.
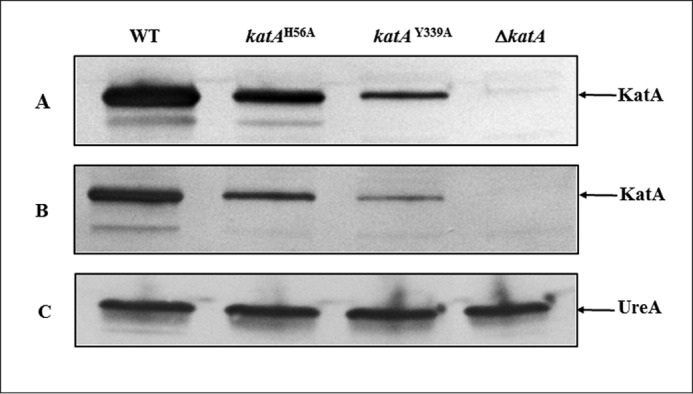
Catalase protein in crude extracts of wild-type and katA mutant strains. Approximately 107 H. pylori whole cells were loaded per lane. Proteins were separated on a SDS-12.5% polyacrylamide gel, along with prestained mass standards, and the proteins were then transferred onto a nitrocellulose membrane and subjected to immunoblotting. Strains are indicated on the top. Proteins detected on each immunoblot are indicated by the arrow on the right. A, anti-KatA immunoblot, strain 43504 (WT) and 43504 katA isogenic mutants. B, anti-KatA immunoblot, strain X47 (WT) and X47 katA isogenic mutants. C, anti-UreA immunoblot, strain 43504 (WT) and 43504 katA isogenic mutants (control to verify equal amounts of cell extracts were loaded in each lane).
To investigate the respective involvement of each residue in the heme-binding ability of the Helicobacter H2O2-dissipating enzyme (24), we likewise studied specific mutant versions. KatAH56A and KatAY339A as well as the native catalase KatAWT (control) were expressed as recombinant proteins in Escherichia coli and purified to near homogeneity. Purified proteins were phenotypically different. KatAWT was light brown; KatAH56A was dark brown, and KatAY339A was colorless (Fig. 2A). When protein samples were loaded on a gel in a non-reducing buffer without prior heating, both KatAWT and KatAH56A ran as apparent tetramers (apparent molecular mass above 200 kDa) (Fig. 2B), in agreement with previous studies (3, 24). By contrast, KatAY339A was unable to tetramerize. When subjected to reducing conditions and boiling, KatAWT, KatAH56A, and KatAY339A proteins migrated as monomers with an apparent molecular mass below 60 kDa, in agreement with their calculated mass (58.6 kDa). Catalase activity of the purified KatAH56A was less than 1% of the activity of purified KatAWT, although KatAY339A had no detectable activity (Table 1). The three proteins were analyzed by UV-visible scan spectrophotometry (Fig. 2C). A heme b-specific absorption Soret peak centered at λ = 410 nm was observed for KatAWT; this peak was shifted for KatAH56A (λ = 402 nm) and was essentially absent for KatAY339A, indicating that the heme b environment is modified in KatAH56A, and it is abolished in KatAY339A protein. These results were expected based on heme ligands predicted from the crystal structure (21, 22).
FIGURE 2.

Purification and characterization of native and variant catalase proteins. Native KatAWT and variant KatAH56A and KatAY339A catalase proteins were expressed in E. coli, FPLC-purified, and analyzed by visual inspection, SDS-PAGE, and UV-visible spectroscopy. A, purified catalase protein solutions at 34 μm. B, purified catalase proteins were either mixed with a non-reducing buffer and not heated (0.66 μg per lane) or mixed with a reducing buffer and boiled (0.33 μg per lane) before being subjected to SDS-12.5% PAGE and stained with Coomassie Blue. Molecular mass standard sizes are indicated on the right. C, UV-visible spectroscopy scan of purified native and variant catalase proteins. UV-visible scans were run from 250 to 600 nm for each protein (10 μm) in a final volume of 500 μl. This scan is representative of three experimental repeats (independent purifications).
Previous work demonstrated that HOCl reacts with H. pylori catalase, leading to catalase oligomerization, methionine oxidation, and enzyme inactivation (3). To determine whether purified wild-type or variant catalase could quench HOCl, subsequently protecting H. pylori cells from oxidant-mediated death, non-growing H. pylori 43504 WT cells were incubated either with PBS buffer, or with HOCl, or with HOCl that had been pre-incubated for 15 min with either purified KatAWT, purified KatAH56A, or purified KatAY339A. In addition, two unrelated protein controls, HypC (high Met content, 6%) and UreE (low Met content, 1%), were included in the study. Exposure to HOCl only (no protein added) killed all the cells, e.g. we could not recover any CFU following this treatment (n = 3) (Fig. 3). In contrast, when HOCl was incubated with purified native or either variant catalase, the final CFU count was in the same range as the PBS only control, indicating that each purified catalase protein, whether active (KatAWT) or inactive (KatAH56A and KatAY339A), can protect H. pylori in vitro against the deadly effect of the oxidant (Fig. 3). When the same experiment was repeated with decreasing concentrations of catalase, cell survival rates decreased accordingly; however, there was no significant difference in protection between the native (WT) and mutant versions of KatA (data not shown). Furthermore, addition of HypC to the HOCl mixture conferred levels of protection similar to catalase, whereas addition of UreE was not protective, suggesting a correlation between Met content and HOCl quenching. This protective effect is apparently independent of Msr and Met recycling, as the Msr repair mixture was not added for this experiment. Nevertheless, it demonstrates the oxidant quenching capacity of catalase that is inherent in its sequence and independent of its catalytic activity. A previous biochemical study almost 30 years ago suggested that pure bovine catalase, at high concentration, and either active or azide-inactivated, could protect bovine α1-antiproteinase enzyme against inactivation by HOCl (25); but it was concluded the heme ring of catalase likely reacted with HOCl to somehow dissipate the oxidant.
FIGURE 3.
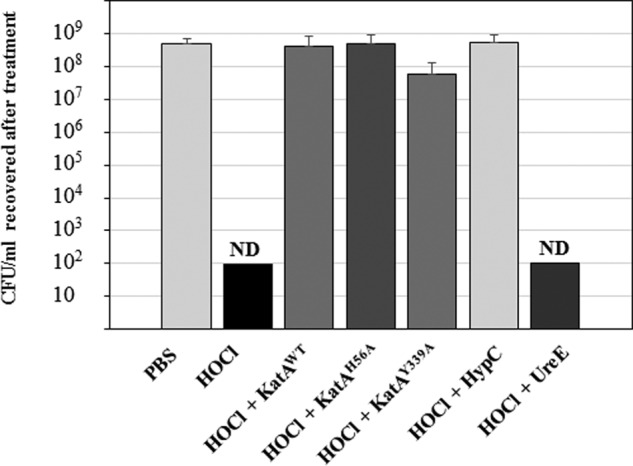
Purified apo- or holo-catalase prevents HOCl-mediated whole cell death. H. pylori wild-type cells (∼5 × 108 CFU/ml) were incubated for 1 h in PBS buffer, or PBS with HOCl, or PBS with HOCl that had been previously incubated for 15 min at 37 °C with either purified KatAWT, KatAH56A, KatAY339A, HypC (Met-rich protein), or UreE (Met-poor protein). Final protein concentration was 0.25 μm, and final NaOCl concentration was 200 μm in each reaction. Results (CFU/ml recovered after 1 h) represent the mean and standard deviation from three independent challenge experiments, with each serial dilution plated in triplicate. ND, no CFU could be detected (detection limit, 102 CFU/ml).
To determine whether holo- or apo-catalase can protect H. pylori cells against HOCl in vivo, disk inhibition assays with HOCl were conducted using wild-type strain 43504 as well as ΔkatA, katAH56A, and katAY339A mutant strains (Fig. 4). In addition, because Msr was shown to interact with and repair KatA in H. pylori (3, 6), Δmsr single mutants, as well as Δmsr ΔkatA, Δmsr katAH56A, and Δmsr katAY339A double mutants were included in this study. The sensitivity (diameter of inhibition) of the ΔkatA mutant was significantly greater than the parental strain (43504) or the mutant strains katAH56A and katAY339A (p < 0.01, ANOVA). There was no significant difference between WT, katAH56A, and katAY339A strains (Fig. 4). These results suggest that catalase, even when inactive and lacking the heme moiety, enables H. pylori cells to combat HOCl. In addition, Δmsr single mutants were significantly (p < 0.01) more sensitive to HOCl than the WT strain, in agreement with previously published results (26). Double mutants ΔkatA Δmsr, katAH56A Δmsr, and katAY339A Δmsr were also more sensitive than WT (p < 0.01). Mean diameters of inhibition for katAH56A Δmsr and katAY339A Δmsr were significantly greater than those of their respective parental backgrounds, e.g. katAH56A and katAY339A (p < 0.01), confirming the importance of Msr in repairing HOCl-oxidized Met-O residues. The ΔkatA Δmsr double mutant strain was as sensitive to HOCl as the ΔkatA mutant strain, suggesting that absence of catalase protein is the single most important factor contributing to HOCl sensitivity.
FIGURE 4.

Sensitivity of wild-type and mutant strains to HOCl. Approximately 5 × 107 H. pylori cells were homogeneously spread onto an equivalent volume BA plates. NaOCl (10 μl of 1.4 m) was spotted onto a sterile paper disk (7.5 mm diameter) placed in the center of each plate. Cells were allowed to grow for 48 h, and the diameter of the inhibition zone was measured. A, results shown are mean and standard deviation of diameters from three to seven independent experiments, each with three to five replicates (n indicates the total number of measurements). Mutant strains that displayed higher sensitivity to HOCl (greater zone of inhibition) compared with WT (p < 0.01, ANOVA) are indicated with *. B, representative photographs of the experiment showing inhibition zones for the wild-type strain and the ΔkatA mutant. Photographs have been cropped and placed side by side to allow for direct comparison of the inhibition zones.
The ability of katAH56A and katAY339A mutant strains to colonize the gastric mucosa of mice was investigated. Each mutant were orally given to mice, and their colonization levels after 3 weeks were compared with those obtained with the wild-type strain (X47) and the ΔkatA catalase negative mutant (Fig. 5). The WT strain was able to colonize 11 mice (n = 14 total), with an average of 1.34 × 106 CFU recovered per g of colonized stomach. In contrast, the ΔkatA deletion mutant was detected in only one mouse (n = 10 total), and the colonization load was low (5.5 × 103 CFU per g). Interestingly, the site-directed katAH56A catalase mutant was able to colonize 10 mice (n = 12 total), with an average CFU number of 5.7 × 105 CFU per g of colonized stomach, although the site-directed katAY339A catalase (heme-deficient) mutant successfully colonized 5 out of 10 mice, with an average of 1.1 × 106 CFU per g of colonized stomach. Colonization counts for both katAH56A and katAY339A mutants were not significantly different from those obtained for the WT, while ΔkatA deletion mutants had significantly lower counts compared with the other three strains. To rule out an effect of the presence of the KSF cassette on the colonization-deficient phenotype of ΔkatA::KSF mutants, we constructed a markerless X47 ΔkatA deletion mutant strain (see under “Experimental Procedures”). As expected, those mutants had no measurable catalase activity, and catalase could not be detected by immunoblotting (data not shown). In addition, mouse colonization assays revealed those markerless ΔkatA mutants were unable to colonize mice (no CFU detected, n = 4), although the X47 parental strain (WT control) colonized all inoculated mice (n = 4, average colonization of 3.9 × 105 CFU per g of stomach), therefore confirming that the lack of colonization by ΔkatA::KSF was likely due to the absence of the katA gene, rather than the presence of the KSF cassette. Taken together, these results suggest that catalase is critical for combating oxidants in vivo, and this does not depend on H2O2 dismutation catalytic activity.
FIGURE 5.
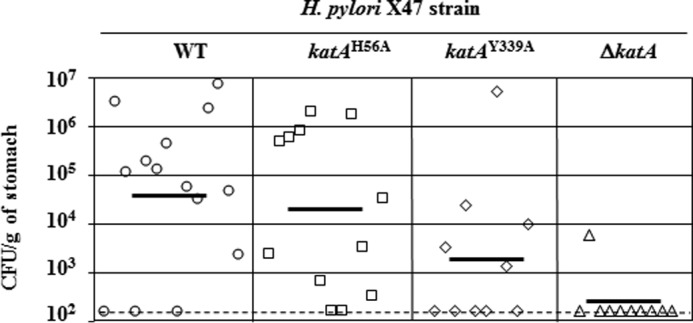
Mouse colonization by H. pylori X47 (parent) and X47 katA strains. Mice were inoculated with a dose of 1.5 × 108 viable cells. Colonization of the mouse stomachs was determined 3 weeks post-inoculation. Mouse stomachs were homogenized, and serial dilutions were plated. Data are presented as a scatter plot of numbers of CFU/g of stomach (log10 scale) as determined by plate counts. Each symbol represents the mean CFU count for one stomach (n = 14 for WT, n = 12 for katAH56A, n = 10 for katAY339A, and n = 10 for ΔkatA, respectively). Each horizontal bar represents the geometric mean of the colonization load for each group. The ΔkatA mutant (catalase null strain) geometric mean is significantly lower than the wild-type strain (p < 0.01, Student's t test), or the katAH56A mutant (p < 0.01), or the katAY339A mutant (p < 0.08), while there is no significant difference between average colonization loads (geometric mean) for WT, katAH56A, and katAY339A strains (p < 0.01). A dashed horizontal line shows the detection limit, which represents a count below 1.5 × 102 CFU/g of stomach.
Results from a previous study suggested that katA was only needed for long-term colonization (24 weeks) in mice, as there was no significant difference (between the wild-type strain and a ΔkatA null mutant) in colonization after 8 days or 12 weeks (27). However, a major difference between that study and ours is the H. pylori parent strain (mouse colonizer) used. SS1 was used as parental strain in the previous study, and this strain is known to preferentially colonize the mouse stomach antrum (27). By contrast, in this study, we used X47, a strain that is unrelated to SS1, and whose primary tropism is the mouse stomach corpus (28). Therefore, differences in oxidative stress levels between the corpus and the antrum, combined with genetic differences between SS1 and X47, are likely to account for the different outcomes in both colonization assays.
At least two independent studies have suggested that catalase is a good vaccine candidate for prevention of gastric ulcers and cancers (29, 30). In one such study, 90% of the mice were protected from H. pylori (SS1 strain) colonization by raising anti-KatA antisera (30). This strong protective immunization effect is another indication of the importance of this Met-containing enzyme. It would be of interest to assess whether the Helicobacter catalase could be detected in the mucus (i.e. extracellular to the bacterium) of infected animals, and if so, what downstream affects this catalase may have on the microflora.
More than 100 years after the initial report describing catalase activity (1), we now know its physiological importance in at least one organism is due to an additional mechanism. The finding that inactive catalase was protective to the bacterium was not expected, but it was nevertheless hypothesized based on the known susceptibility of its Met residues to oxidation and to (Msr-mediated) turnover. It is possible this new role can only be documented in Helicobacter, as the bacterium makes a large amount of catalase, and the mouse model is a facile way to assess viability in a natural stress environment. We do not know whether the cyclic turnover (oxidation and repair) of catalase occurs extracellularly or in the host mucous lining, but the oxidant quenching must occur to some extent, and both enzymes (Msr and catalase) are readily detected outside the (Helicobacter) cell (at least in vitro). Assessing the extent of extracellular repair (i.e. Met-O-Met turnover) must await repair assays and mass spectral studies to assess the Met(-O) state of the target using extracellular fractions.
Catalase in many organisms is up-expressed upon oxidative stress exposure, so it is reasonable that the protein would serve a secondary protective role. This secondary role, independent of activity, would be expected to operate even in cells that are iron- or heme-starved, conditions known to occur in host-infected tissue for many pathogens. Additionally, oxidative stress and protein oxidations are correlated with a number of human neurodegenerative diseases, including Parkinson's and Alzheimer's disease (31). Interestingly, the neurological tissues of concern here contain both catalase and methionine sulfoxide reductase.
In addition to catalase, other abundant and/or secreted proteins, especially ones that are repaired by Msr (such as GroEL and AhpC (26)) could also play an unexpected stress-combating role, e.g. quenching of oxidants to confer stress protection. Indeed, any Met-rich protein, and particularly if localized outside the cytoplasm, should be studied via mutagenesis or other approaches for roles in combating or quenching oxidants. Even if these proteins have an already known function, a secondary role as a protector from oxidants is a possibility. Of course, a Met-rich protein without an enzyme function could serve this role; in theory, only stability and MetO-forming ability would be required to be an antioxidant; Met recycling via recognition by Msr would be expected to aid this role. It seems possible this methionine-mediated antioxidant role within proteins may impact the physiology and survival of multiple organisms.
Experimental Procedures
Bacterial Strains and Plasmids
Strains and plasmids are described in supplemental Table S1. E. coli TOP10 (Invitrogen) was used for all cloning experiments, and E. coli BL21(DE3)RIL (Novagen) was used to express recombinant H. pylori KatA proteins. H. pylori wild-type strain 26695 was used as a source of DNA for PCR. All plasmids and polymerase chain reaction (PCR) products were sequenced at the Georgia Genomics Facility, University of Georgia, Athens, GA, and compared with DNA sequences from strain 26695 (32) to ensure that no error had been introduced following PCR amplification, as well as to verify the presence of engineered site-directed mutations within katA.
Growth Conditions
E. coli was grown aerobically in Luria-Bertani (LB) broth or on LB plates at 37 °C. Ampicillin (100 μg/ml), chloramphenicol (30 μg/ml), and isopropyl β-d-thiogalactopyranoside (0.5 mm) were added as needed. H. pylori was routinely grown on Brucella agar (BA) plates supplemented with 10% defibrinated sheep blood at 37 °C under microaerophilic conditions with 4% O2, 5% CO2, and 91% N2. Kanamycin (30 μg/ml), chloramphenicol (30 μg/ml), bacitracin (50 μg/ml), amphotericin B (10 μg/ml), nalidixic acid (10 μg/ml), vancomycin (10 μg/ml), and sucrose (5%) were added as needed. For liquid cultures, brain heart infusion supplemented with 0.4% β-cyclodextrin was used.
Construction of H. pylori katA Deletion and Site-directed Mutants
Markerless chromosomal katAH56A and katAY339A mutants were constructed following a two-step kanamycin-sucrose (selection-counter selection) method, as described previously (33). Briefly, a 1720-bp DNA sequence containing the katA gene (hp0875 in strain 26695) and flanking sequences was amplified by PCR using genomic DNA from H. pylori strain 26695 as template and primers KatA-KpnI and KatA-BamHI (supplemental Table S2). The PCR product was digested with KpnI and BamHI and ligated into similarly digested pBS-KS plasmid to generate pKS-katA. Next, a unique HindIII site located within katA was used to insert a 3-kb KanR-sacB-PflaA (KSF) cassette previously excised from pKSF-II plasmid (33). Insertion of the KSF cassette is not expected to have any polar effect, because the gene downstream of katA (kapA, hp0874 in strain 26695) has its own promoter. The resulting plasmid, pKS-katA::KSF, was introduced into H. pylori strain 43504, and mutants were selected on BA plates supplemented with kanamycin. Colonies appeared after 3–5 days and were shown to be sucrose-sensitive as well as catalase-negative. Those mutants, ΔkatA::KSF, were used as negative controls (absence of katA) for all experiments. In addition, they were used as parental strains to generate site-directed point mutants following transformation with plasmid pKS-katAH56A or pKS-katAY339A. Each point mutation was generated using a Splicing by Overlap Extension-PCR method. Briefly, external primers KatA-KpnI and KatA-BamHI were used concomitantly with internal primers designed to introduce a mutation in the open reading frame of katA, resulting in either a H56A or a Y339A substitution (supplemental Table S2). After a final round of amplification with purified PCR products and external primers, each 1720-bp-long PCR product (containing the desired mutation) was digested with KpnI and BamHI and ligated into pKS to yield plasmid pKS-katAH56A or pKS-katAY339A. Each plasmid was introduced separately into the ΔkatA::KSF mutant, and transformants (site-directed mutants) were isolated after 3–5 days on BA supplemented with 5% sucrose. Those sucrose-resistant katAH56A and katAY339A chromosomal mutants were shown to be kanamycin-sensitive as well as catalase-negative. The same procedure was used to generate ΔkatA::KSF, katAH56A, and katAY339A mutations in the mouse colonizing strain X47 (33). Finally, to rule out an effect of the presence of the KSF cassette on the colonization-deficient phenotype of ΔkatA::KSF mutants, we constructed a markerless ΔkatA deletion mutant. Briefly, plasmid pKS-katA was digested with HindIII, treated with T4 polymerase, and ligated onto itself, to generate a new plasmid, pKS-katA*. This treatment introduces a frameshift in the katA gene after the Val-55 codon, leading to early translation termination. Transformation of ΔkatA::KSF mutants with plasmid pKS-katA* generated ΔkatA markerless mutants that were sucrose-resistant, kanamycin-sensitive, and catalase-negative. The chromosomal disruption of katA by the KSF cassette, the frameshift of katA in ΔkatA mutants, and the presence of chromosomal mutation katAH56A or katAY339A in each H. pylori strain were confirmed by DNA sequencing.
Construction of H. pylori Δmsr Mutants
Δmsr deletion mutants were generated using Splicing by Overlap Extension-PCR. Briefly, primers Δmsr-cat1 and Δmsr-cat2 were used to amplify a 412-bp-long DNA sequence located upstream of msr (hp0224 in strain 26695). Primers Δmsr-cat3 and Δmsr-cat4 were used to amplify a 411-bp-long sequence located downstream of hp0224. The final amplification step included each purified PCR product, a cat (CmR) cassette (0.8 kb) and primers Δmsr-cat1 and Δmsr-cat4. The resulting 1620-bp-long PCR product was introduced into either H. pylori wild-type strain 43504 or ΔkatA, katAH56A, or katAY339A mutant strain, to generate Δmsr single mutant, or ΔkatA Δmsr, katAH56A Δmsr, or katAY339A Δmsr double mutants, respectively (supplemental Table S1). H. pylori cells were transferred after 16 h onto BA plates supplemented with 25 μg/ml chloramphenicol. Colonies appeared after 3–5 days of incubation. The concomitant chromosomal deletion of msr and the insertion of cat were confirmed by PCR using genomic DNA from each strain and primers Δmsr-cat1 and Δmsr-cat4 and by DNA sequencing.
Cloning, Expression, and Purification of Recombinant Native and Variant KatA Proteins
Recombinant native and variant KatA proteins were expressed in E. coli BL21 (DE3)-RIL (Novagen). Briefly, in three separate PCRs, primers KatA-NdeI and Kat-BamHI were used with three different plasmid templates, e.g. pKS-katA, pKS-katAH56A, or pKS-katAY339A to amplify a 1540-bp-long DNA sequence containing either the native katA gene, the katAH56A mutated gene, or the katAY339A mutated gene, respectively. Each PCR product was digested with NdeI and XhoI, gel-purified, and cloned into similarly digested pET21b plasmid, generating plasmids pET-KatAWT, pET-KatAH56A, and pET-KatAY339A. E. coli BL21-RIL cells transformed with the appropriate pET-KatA plasmid were grown at 37 °C in 500 ml of LB supplemented with ampicillin and chloramphenicol until an absorbance of 0.5 at 600 nm was reached. Gene expression was induced by the addition of 0.5 mm isopropyl β-d-thiogalactopyranoside followed by incubation (at 37 °C) for 3–5 h. Recombinant native and variant catalase proteins were purified as reported previously for the H. pylori native catalase, by using a stepwise dual approach combining anion-exchange and size exclusion chromatography (34). Briefly, cells were centrifuged and washed with 200 ml of 25 mm Na2HPO4 (pH 7.5), 50 mm NaCl buffer (buffer A), and pellets were suspended in 5 ml of buffer A. Protease inhibitor (cOmplete Mini, Roche Applied Science) was added, and cells were disrupted by passage through a French pressure cell at 18,000 pounds/inch2, and the lysate was centrifuged at 17,000 × g for 15 min to remove cell debris. The supernatant was then collected and subjected to ultracentrifugation at 100,000 × g for 1 h. The membrane-free supernatant was applied to an SP-Sepharose cation-exchange column (GE Healthcare) that had been equilibrated with buffer A, and the protein was eluted with a linear gradient of 0.05–1 m NaCl in buffer A. Catalase-containing fractions were selected, pooled, concentrated using Amicon Ultra-4 devices with a 10-kDa molecular mass cutoff (Merck Millipore, Billerica, MA) and further purified by size exclusion chromatography using a HiLoad 16/60 Superdex 75 column (GE Healthcare) in buffer A with 0.3 m NaCl. Catalase-containing fractions were selected, pooled, concentrated, and stored at 4 °C protected from light. Upon purification, recombinant native catalase (KatAWT) was light brown in solution, purified KatAH56A variant was dark brown, and purified KatAY339A variant was colorless. The protein purity was assessed by SDS-PAGE, and the final protein concentration was determined using the BCA kit (ThermoFisher Pierce).
Catalase Assays
Catalase assays were done spectrophotometrically using whole cells or purified catalase proteins, in phosphate-buffered saline (PBS) containing 15 mm H2O2, as described previously (7).Briefly, for whole cells assays, cells were washed and resuspended in PBS to a final A600 of 1.0. Five μl of whole cells were mixed with 495 μl of PBS containing 15 mm H2O2, and the initial H2O2 disappearance (decrease in A240) was followed for up to 1 min. Catalase assays with purified proteins were carried out using 0.05 to 0.1 μg of purified KatAWT and 1 to 10 μg of purified KatAH56A or KatAY339A.
UV-visible Spectrometry
Measurements were recorded at 22 °C on a SpectraMax plus spectrophotometer (Molecular Devices, Sunnyvale, CA) using a final volume of 500 μl in a quartz cuvette with a 1-cm path length. Each purified catalase protein (native KatAWT, KatAH56A, or KatAY339A variant) was diluted to a final concentration of 10 μm in size exclusion chromatography buffer (see above). UV-visible spectroscopy scan was run from 250 to 600 nm. Scans were run three times, with three independent batches of purified proteins.
Immunoblotting Experiments
For detection of KatA in crude extracts, cells were grown on BA, harvested, and resuspended in loading buffer. All samples were subjected to SDS-12.5% PAGE using a Mini-Protean II apparatus (Bio-Rad), according to the method of Laemmli (35) and transferred to a nitrocellulose membrane (0.2-μm pore size; Bio-Rad). The membrane was blocked by incubation in 20 mm Tris-HCl (pH 7.6), 137 mm NaCl buffer (Tris-buffered saline, TBS) supplemented with 3% gelatin (Mallinckrodt Baker). This was followed by a 1-h incubation along with a 1:1,000 dilution of anti-KatA (rabbit polyclonal) antiserum, in TBS buffer with 0.1% Tween 20 (TTBS), 1% gelatin. When needed as control, anti-UreA (rabbit polyclonal, Santa Cruz Biotechnology, Dallas, TX) antiserum was used with a 1:2,000 dilution. The membrane was washed with TTBS and then incubated for 1 h with the secondary antibody (goat anti-rabbit immunoglobulin G coupled with alkaline phosphatase, Bio-Rad) diluted 1:2,000 in TTBS, 1% gelatin. The membrane was again washed with TTBS buffer. Bound antibodies were detected following addition of the chromogenic reagents nitro blue tetrazolium (0.25 mg/ml) and 5-bromo-4-chloro-3-indolyl phosphate (0.125 mg/ml) (Sigma) in 10 mm Tris-HCl (pH 9.5), 150 mm NaCl.
HOCl Challenge of H. pylori in Presence of Purified Proteins
H. pylori wild-type strain 43504 cells were grown for 24–36 h on BA plates, harvested, spun down, and suspended in sterile PBS to a final A600 of 1.1 in PBS buffer. Then 0.9-ml aliquots of this suspension were incubated for 60 min at 37 °C (no shaking) with either 0.1 ml of PBS, 0.1 ml of PBS with NaOCl (10–15% available chlorine; Sigma), or 0.1 ml of PBS/NaOCl that had been previously incubated for 15 min at 37 °C in the presence of purified protein KatWT, KatH56A, KatY339A, HypC, or UreE. Both HypC and UreE were expressed and purified as described previously (26, 36). Final protein and NaOCl concentrations were 0.25 and 200 μm, respectively, and final A600 was equal to 1 in each tube (∼5 × 108 cells per ml). After 60 min at 37 °C (no shaking), cells were serially diluted in sterile PBS, and 10 μl of each dilution was spotted in triplicate on BA plates. CFU were counted after 3 days of incubation at 37 °C under microaerophilic conditions (4% O2). Results (remaining CFU after 1 h) represent means and standard deviations from three independent challenge experiments and serial dilutions spotted in triplicate.
Disk Inhibition Assays
H. pylori wild-type, ΔkatA or Δmsr single mutant, or ΔkatA Δmsr double mutant cells were grown on BA for 24 h before being resuspended to a final A600 of 1 in sterile PBS buffer. Then 0.1 ml of cells were homogenously spread on top of 25-ml standardized BA plates (3–5 replicates per strain). A sterile paper disk (7.5 mm diameter) was placed in the center of each plate, and 10 μl of undiluted NaOCl (Sigma, 1.4 m, with NaClO ϵ292 = 350 m−1 cm−1) was added onto the disk. Cells were allowed to grow for 48 h, and the diameter of the inhibition zone was measured. Results shown are means and standard deviations from three to seven independent growth experiments, each with three to five replicates (n = total number of measurements). ANOVA was used to compare diameter means between strains.
Mouse Colonization Experiments
All procedures were approved by the Institutional Animal Care and Use Committee of the University of Georgia. H. pylori X47 (mouse-adapted, parental strain) and X47 ΔkatA, X47 katAH56A, or X47 katAY339A mutant strains were grown for 24 h or less on BA plates, harvested, and resuspended in sterile PBS buffer (pH 7.4) to a final A600 of 2. Five- to 6-week-old female C57BL/6NCr mice (NCI, Frederick, MD) were infected via oral gavage with 0.15 ml of bacterial suspension (∼1.5 × 108 H. pylori cells per mouse). Mice were sacrificed by CO2 asphyxiation and cervical dislocation 3 weeks post-inoculation. Stomachs were quickly removed, weighed, and gently homogenized in 5 ml of sterile PBS using a Dounce hand homogenizer. Dilutions were made in sterile PBS and plated (0.1 ml) in duplicate on plates supplemented with amphotericin B, bacitracin, nalidixic acid, and vancomycin. Plates were incubated for 5–7 days at 37 °C in a 4% O2 partial pressure atmosphere for colony counting. Data are expressed as CFU recovered per g of mouse stomach. The detection limit of the assay is 150 CFU per g of stomach. Student's t test was used to compare geometrical means of colonization between strains.
Author Contributions
R. J. M. and S. L. B. both conceived the study, designed the experiments, and wrote the paper. S. L. B. performed most of the experiments, and R. J. M. did the animal work. Both authors reviewed and organized the results and approved of the final version.
Supplementary Material
Acknowledgments
We thank Susan Maier for technical assistance with the mouse colonization assays and Marie-Anaïs Benoit for help with the construction of site-directed mutants.
This work was supported by National Institutes of Health Grant 1R21AI121181 and by the University of Georgia Foundation. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article was selected as a Paper of the Week.

This article contains supplemental Tables S1 and S2.
- Met-O
- methionine sulfoxide
- Msr
- methionine sulfoxide reductase
- ANOVA
- analysis of variance
- ROS
- reactive oxygen species
- BA
- Brucella agar.
References
- 1. Loew O. (1900) A new enzyme of general occurrence in organismis. Science 11, 701–702 [DOI] [PubMed] [Google Scholar]
- 2. Masters C., and Crane D. (1995) The Peroxisome: A Vital Organelle, Griffith University, Queensland, Australia [Google Scholar]
- 3. Mahawar M., Tran V., Sharp J. S., and Maier R. J. (2011) Synergistic roles of Helicobacter pylori methionine sulfoxide reductase and GroEL in repairing oxidant-damaged catalase. J. Biol. Chem. 286, 19159–19169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Boonjakuakul J. K., Syvanen M., Suryaprasad A., Bowlus C. L., and Solnick J. V. (2004) Transcription profile of Helicobacter pylori in the human stomach reflects its physiology in vivo. J. Infect. Dis. 190, 946–956 [DOI] [PubMed] [Google Scholar]
- 5. Kim G., Weiss S. J., and Levine R. L. (2014) Methionine oxidation and reduction in proteins. Biochim. Biophys. Acta 1840, 901–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Alamuri P., and Maier R. J. (2006) Methionine sulfoxide reductase in Helicobacter pylori: interaction with methionine-rich proteins and stress-induced expression. J. Bacteriol. 188, 5839–5850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Benoit S. L., and Maier R. J. (2014) Twin-arginine translocation system in Helicobacter pylori: TatC, but not TatB, is essential for viability. mBio 5, e01016–01013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Smith T. G., Lim J. M., Weinberg M. V., Wells L., and Hoover T. R. (2007) Direct analysis of the extracellular proteome from two strains of Helicobacter pylori. Proteomics 7, 2240–2245 [DOI] [PubMed] [Google Scholar]
- 9. Harris A. G., and Hazell S. L. (2003) Localisation of Helicobacter pylori catalase in both the periplasm and cytoplasm, and its dependence on the twin-arginine target protein, KapA, for activity. FEMS Microbiol. Lett. 229, 283–289 [DOI] [PubMed] [Google Scholar]
- 10. Levine R. L., Berlett B. S., Moskovitz J., Mosoni L., and Stadtman E. R. (1999) Methionine residues may protect proteins from critical oxidative damage. Mech. Ageing Dev. 107, 323–332 [DOI] [PubMed] [Google Scholar]
- 11. Levine R. L., Mosoni L., Berlett B. S., and Stadtman E. R. (1996) Methionine residues as endogenous antioxidants in proteins. Proc. Natl. Acad. Sci. U.S.A. 93, 15036–15040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Blaser M. J. (1995) The role of Helicobacter pylori in gastritis and its progression to peptic ulcer disease. Aliment. Pharmacol. Ther. 9, 27–30 [DOI] [PubMed] [Google Scholar]
- 13. Sipponen P., Hyvärinen H., Seppälä K., and Blaser M. J. (1998) Review article: Pathogenesis of the transformation from gastritis to malignancy. Aliment. Pharmacol. Ther. 12, 61–71 [DOI] [PubMed] [Google Scholar]
- 14. Bagchi D., Bhattacharya G., and Stohs S. J. (1996) Production of reactive oxygen species by gastric cells in association with Helicobacter pylori. Free Radic. Res. 24, 439–450 [DOI] [PubMed] [Google Scholar]
- 15. Ramarao N., Gray-Owen S. D., and Meyer T. F. (2000) Helicobacter pylori induces but survives the extracellular release of oxygen radicals from professional phagocytes using its catalase activity. Mol. Microbiol. 38, 103–113 [DOI] [PubMed] [Google Scholar]
- 16. Davies G. R., Simmonds N. J., Stevens T. R., Sheaff M. T., Banatvala N., Laurenson I. F., Blake D. R., and Rampton D. S. (1994) Helicobacter pylori stimulates antral mucosal reactive oxygen metabolite production in vivo. Gut 35, 179–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Weiss S. J. (1989) Tissue destruction by neutrophils. N. Engl. J. Med. 320, 365–376 [DOI] [PubMed] [Google Scholar]
- 18. Handa O., Naito Y., and Yoshikawa T. (2010) Helicobacter pylori: a ROS-inducing bacterial species in the stomach. Inflamm. Res. 59, 997–1003 [DOI] [PubMed] [Google Scholar]
- 19. Hawkins C. L., Pattison D. I., and Davies M. J. (2003) Hypochlorite-induced oxidation of amino acids, peptides and proteins. Amino Acids 25, 259–274 [DOI] [PubMed] [Google Scholar]
- 20. Achilli C., Ciana A., and Minetti G. (2015) The discovery of methionine sulfoxide reductase enzymes: an historical account and future perspectives. BioFactors 41, 135–152 [DOI] [PubMed] [Google Scholar]
- 21. Loewen P. C., Carpena X., Rovira C., Ivancich A., Perez-Luque R., Haas R., Odenbreit S., Nicholls P., and Fita I. (2004) Structure of Helicobacter pylori catalase, with and without formic acid bound, at 1.6 A resolution. Biochemistry 43, 3089–3103 [DOI] [PubMed] [Google Scholar]
- 22. Alfonso-Prieto M., Vidossich P., and Rovira C. (2012) The reaction mechanisms of heme catalases: an atomistic view by ab initio molecular dynamics. Arch. Biochem. Biophys. 525, 121–130 [DOI] [PubMed] [Google Scholar]
- 23. Kleanthous H., Tibbitts T. J., Gray H. L., Myers G. A., Lee C. K., Ermak T. H., and Monath T. P. (2001) Sterilizing immunity against experimental Helicobacter pylori infection is challenge-strain dependent. Vaccine 19, 4883–4895 [DOI] [PubMed] [Google Scholar]
- 24. Hazell S. L., Evans D. J. Jr., and Graham D. Y. (1991) Helicobacter pylori catalase. J Gen. Microbiol. 137, 57–61 [DOI] [PubMed] [Google Scholar]
- 25. Aruoma O. I., and Halliwell B. (1987) Action of hypochlorous acid on the antioxidant protective enzymes superoxide dismutase, catalase and glutathione peroxidase. Biochem. J. 248, 973–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Benoit S. L., Bayyareddy K., Mahawar M., Sharp J. S., and Maier R. J. (2013) Alkyl hydroperoxide reductase repair by Helicobacter pylori methionine sulfoxide reductase. J. Bacteriol. 195, 5396–5401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Harris A. G., Wilson J. E., Danon S. J., Dixon M. F., Donegan K., and Hazell S. L. (2003) Catalase (KatA) and KatA-associated protein (KapA) are essential to persistent colonization in the Helicobacter pylori SS1 mouse model. Microbiology 149, 665–672 [DOI] [PubMed] [Google Scholar]
- 28. Akada J. K., Ogura K., Dailidiene D., Dailide G., Cheverud J. M., and Berg D. E. (2003) Helicobacter pylori tissue tropism: mouse-colonizing strains can target different gastric niches. Microbiology 149, 1901–1909 [DOI] [PubMed] [Google Scholar]
- 29. Miyashita M., Joh T., Watanabe K., Todoroki I., Seno K., Ohara H., Nomura T., Miyata M., Kasugai K., Tochikubo K., Itoh M., and Nitta M. (2002) Immune responses in mice to intranasal and intracutaneous administration of a DNA vaccine encoding Helicobacter pylori-catalase. Vaccine 20, 2336–2342 [DOI] [PubMed] [Google Scholar]
- 30. Radcliff F. J., Hazell S. L., Kolesnikow T., Doidge C., and Lee A. (1997) Catalase, a novel antigen for Helicobacter pylori vaccination. Infect. Immun. 65, 4668–4674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Johnson W. M., Wilson-Delfosse A. L., Chen S. G., and Mieyal J. J. (2015) The roles of redox enzymes in Parkinson's disease: focus on glutaredoxin. Ther. Targets Neurol. Dis. 2, e790. [PMC free article] [PubMed] [Google Scholar]
- 32. Tomb J. F., White O., Kerlavage A. R., Clayton R. A., Sutton G. G., Fleischmann R. D., Ketchum K. A., Klenk H. P., Gill S., Dougherty B. A., Nelson K., Quackenbush J., Zhou L., Kirkness E. F., Peterson S., et al. (1997) The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 388, 539–547 [DOI] [PubMed] [Google Scholar]
- 33. Copass M., Grandi G., and Rappuoli R. (1997) Introduction of unmarked mutations in the Helicobacter pylori vacA gene with a sucrose sensitivity marker. Infect. Immun. 65, 1949–1952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang G., Conover R. C., Benoit S., Olczak A. A., Olson J. W., Johnson M. K., and Maier R. J. (2004) Role of a bacterial organic hydroperoxide detoxification system in preventing catalase inactivation. J. Biol. Chem. 279, 51908–51914 [DOI] [PubMed] [Google Scholar]
- 35. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 36. Benoit S. L., Mehta N., Weinberg M. V., Maier C., and Maier R. J. (2007) Interaction between the Helicobacter pylori accessory proteins HypA and UreE is needed for urease maturation. Microbiology 153, 1474–1482 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.