

Abstract
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterized by progressive and selective loss of motor neurons. Causative genes for familial ALS (fALS), e.g. TARDBP or FUS/TLS, have been found, among which mutations within the profilin 1 (PFN1) gene have recently been identified in ALS18. To elucidate the mechanism whereby PFN1 mutations lead to neuronal death, we generated transgenic Drosophila melanogaster overexpressing human PFN1 in the retinal photoreceptor neurons. Overexpression of wild-type or fALS mutant PFN1 caused no degenerative phenotypes in the retina. Double overexpression of fALS mutant PFN1 and human TDP-43 markedly exacerbated the TDP-43-induced retinal degeneration, i.e. vacuolation and thinning of the retina, whereas co-expression of wild-type PFN1 did not aggravate the degenerative phenotype. Notably, co-expression of TDP-43 with fALS mutant PFN1 increased the cytoplasmic localization of TDP-43, the latter remaining in nuclei upon co-expression with wild-type PFN1, whereas co-expression of TDP-43 lacking the nuclear localization signal with the fALS mutant PFN1 did not aggravate the retinal degeneration. Knockdown of endogenous Drosophila PFN1 did not alter the degenerative phenotypes of the retina in flies overexpressing wild-type TDP-43. These data suggest that ALS-linked PFN1 mutations exacerbate TDP-43-induced neurodegeneration in a gain-of-function manner, possibly by shifting the localization of TDP-43 from nuclei to cytoplasm.
Keywords: amyotrophic lateral sclerosis (ALS) (Lou Gehrig disease), Drosophila, fused in sarcoma (FUS), neurodegeneration, neurodegenerative disease, profilin, TAR DNA-binding protein 43 (TDP-43) (TARDBP)
Introduction
Amyotrophic lateral sclerosis (ALS)2 is a progressive neurodegenerative disorder characterized by selective loss of upper and lower motor neurons. Approximately 10% of ALS cases are inherited as an autosomal dominant trait (familial ALS; fALS), and mutations in a number of causative genes, e.g. Cu/Zn superoxide dismutase (1) and TARDBP (2–5), have been identified in fALS cases. Recently mutations within the PFN1 gene were identified in pedigrees of ALS 18 (OMIM614808) (6, 7). Moreover, the E117G variant in PFN1 is also considered a genetic risk factor for ALS (8, 9). However, the mechanisms whereby PFN1 mutations induce degeneration and death of motor neurons remain elusive. PFN1 is a protein implicated in the regulation of actin assembly by binding to monomeric actin (10). Homozygous PFN1 knock-out mice die shortly after fertilization (11), and brain-specific conditional knock-out of the PFN1 gene during development led to cerebellar hypoplasia and abnormal neuronal migration (12), indicating the roles of PFN1 in development. However, there have been few reports on the pathological functions of fALS mutant PFN1; the level of PFN1-bound actin was shown to be reduced upon overexpression of the fALS mutant PFN1 compared with wild-type (wt) PFN1 in HEK293 cells (6). A recent report (13) showed that the fALS mutant PFN1 failed to restore growth of PFN1 mutant yeast. These data suggested a partial loss-of-function mechanism in neurodegeneration caused by fALS mutations in PFN1.
TAR DNA-binding protein of 43 kDa (TDP-43) is a member of heterogeneous nuclear ribonucleoproteins, which functions in a series of steps in DNA/RNA processing (14). TDP-43 was identified as a proteinaceous component of ubiquitin-positive inclusions found in the degenerating neurons of patients with frontotemporal lobar degeneration (FTLD) and ALS (15, 16). More than 30 missense mutations in the TARDBP gene encoding TDP-43 have been identified in fALS. Wu and colleagues (6) have reported that overexpression of fALS mutant PFN1 in primary cultured motor neurons co-aggregated with TDP-43. Importantly, recent autopsy studies of patients with ALS carrying PFN1 mutations revealed the presence of TDP-43-positive neuronal or glial cytoplasmic inclusions (8, 9), strongly suggesting the pathogenic mechanism of PFN1 mutations through TDP-43-induced neurodegeneration. However, the mechanisms whereby PFN1 and TDP-43 lead to neurodegeneration remain unclear. Targeted depletion of TDP-43 in the spinal motor neurons of mice has been shown to lead to a progressive motor dysfunction associated with motor neuron loss (17). We have previously reported that overexpression of wt or fALS mutant TDP-43 caused neurodegeneration through its RNA-binding motif in the retina of transgenic (TG) Drosophila melanogaster (18). These findings implicated malfunction of RNA processing in the mechanisms of neurodegeneration induced by TDP-43.
In the present study, we established a series of TG D. melanogaster lines overexpressing wt or fALS mutant PFN1 using the GAL4-UAS system, which we crossed with TDP-43 TG flies. We found that overexpression of the fALS mutant PFN1 in the retinal photoreceptor neurons significantly exacerbated the retinal degeneration of TDP-43 TG fly compared with that in TDP-43 single TG flies, whereas co-expression of wt PFN1 did not aggravate the phenotype caused by overexpression of TDP-43. Co-expression of TDP-43 with fALS mutant PFN1 increased the cytoplasmic localization of TDP-43 in retinal neurons, whereas it predominantly localized to nuclei upon co-expression with wt PFN1. Co-expression of TDP-43 lacking the nuclear localization signal with the fALS mutant PFN1 did not exacerbate retinal degeneration. Based on these findings, we propose a mechanism of neurodegeneration by fALS mutations of PFN1 through a shift in the localization of TDP-43 from nuclei to cytoplasm.
Results
Overexpression of wt or fALS Mutant PFN1 Did Not Cause Retinal Degeneration in the D. melanogaster
To elucidate the mechanism whereby fALS mutations in PFN1 cause neurodegeneration, we established D. melanogaster overexpressing human wt or fALS mutant (C71G or M114T) PFN1 using the GAL4-UAS system (19). We generated TG flies overexpressing UAS-wt PFN1, C71G, or M114T mutant PFN1 and crossed them with the gmr-Gal4 line to overexpress proteins in the retinal cells. In the eyes of adult Drosophila, each ommatidium contains a cluster of eight rhabdomeres (R1–R8) (20). External surface of eyes of 5-day-old TG flies revealed no degenerative phenotypes, e.g. necrotic patches or fusion of ommatidia, in the eye of wt, C71G, or M114T mutant PFN1 TG flies, as well as in TG flies expressing lacZ as a control protein (Fig. 1A). Histological observation of eyes of 5-day-old TG flies also revealed no apparent degenerative phenotype, e.g. vacuolation or disturbance in the ommatidial alignment, in the retina of lacZ TG flies, or two independent lines (transgenes inserted on chromosome 2 or 3) of wt or fALS mutant PFN1 TG flies (Fig. 1B). To evaluate the retinal degeneration, we measured the retinal thickness of TG fly lines and found that there was no significant difference between wt, C71G, or M114T mutant TG flies and lacZ TG flies (Fig. 1C, 66.7 ± 1.29 μm in LacZ, 67.0 ± 2.07 μm in wt PFN1 (p = 1.00), 65.9 ± 2.11 μm in C71G mutant PFN1 (p = 0.99), 65.3 ± 1.44 μm in M114T mutant PFN1 (p = 0.95)). Immunoblot analyses of the lysates of heads of TG flies showed that C71G or M114T mutant PFN1 proteins were expressed as ∼14-kDa polypeptides co-migrating with wt PFN1 in the retinal photoreceptor neurons, at similar expression levels to those in TG flies expressing wt PFN1 (Fig. 1D). These data showed that overexpression of wt or fALS mutant PFN1 did not cause any degeneration in the photoreceptor neurons of D. melanogaster.
FIGURE 1.

Overexpression of human PFN1 in the retina of Drosophila. A, external surface pictures of eyes of 5-day-old TG flies overexpressing gmr-driven LacZ, wt, C71G or M114T mutant PFN1. The enlarged view of each eye is shown in lower right corner. B, H&E-stained sections of eyes of 5-day-old TG flies overexpressing gmr-driven LacZ, wt, C71G or M114T mutant PFN1. Scale bar, 100 μm. C, retinal thickness measured in flies expressing LacZ, wt, C71G or M114T mutant PFN1. n = 10, mean ± S.E. D, immunoblot analyses of the heads of 5-day-old gmr-driven flies expressing LacZ, wt, C71G or M114T mutant PFN1 (upper panel). α-Tubulin levels are shown as a loading control (lower panel). Relative expression levels of PFN1 are indicated under the panels. n = 3, mean ± S.E.
Overexpression of fALS Mutant PFN1, but Not wt PFN1, Exacerbated TDP-43-induced Retinal Degeneration
We have previously reported that overexpression of TDP-43 caused a progressive neuronal degeneration in the retina of D. melanogaster (18). To investigate whether fALS mutation in PFN1 affects the TDP-43-induced neuronal degeneration, we generated TG fly lines doubly overexpressing human wt TDP-43 and human wt, C71G, or M114T mutant PFN1 in the retina, under the control of the gmr-Gal4 driver. The external surface of eyes of 5-day-old double TG flies revealed that C71G or M114T mutant PFN1/TDP-43 double TG flies exhibited severe degenerative phenotype, e.g. fusion of ommatidia compared with TDP-43 single TG flies or wt PFN1/TDP-43 double TG flies (Fig. 2A). Histological analysis also showed that overexpression of C71G or M114T mutant PFN1 exacerbated retinal degeneration and caused a significant reduction in the thickness of the retina in mutant PFN1/TDP-43 double TG flies compared with those in TDP-43 single TG flies (Fig. 2, B and C, 45.7 ± 2.40 μm in TDP-43 single, 22.8 ± 1.09 μm in C71G mutant PFN1/TDP-43 (p < 0.001), 23.9 ± 2.76 μm in M114T mutant PFN1/TDP-43 (p < 0.001)), whereas overexpression of wt PFN1 did not alter the retinal degeneration in wt PFN1/TDP-43 double TG flies (Fig. 2, B and C, 49.9 ± 2.52 μm in wt PFN1/TDP-43 (p = 0.65)). Immunoblot analysis of the lysates of heads of single or double TG flies showed that overexpression of wt, C71G, or M114T mutant PFN1 in TDP-43 TG flies did not affect the level of TDP-43 protein compared with that in TDP-43 single TG flies (Fig. 2D). To examine whether the level of severity of retinal degeneration is different among lines, we crossed 3 lines of PFN1 TG flies with different levels of PFN1 expression and with TDP-43 TG flies. We found that the 3 lines of wt PFN1/TDP-43 double TG flies exhibited similar levels of retinal degeneration to TDP-43 single TG flies (supplemental Fig. S1). We also found that 3 lines each of C71G or M114T mutant PFN1/TDP-43 double TG flies exhibited more severe retinal degeneration compared with TDP-43 single TG flies (supplemental Fig. S1). We did not observe significant differences in the severity of retinal degeneration among lines of C71G or M114T mutant PFN1/TDP-43 double TG flies.
FIGURE 2.

Overexpression of fALS mutant PFN1-exacerbated TDP-43-induced retinal degeneration. A, external surface pictures of eyes of 5-day-old gmr-driven TG flies overexpressing singly human TDP-43, or doubly TDP-43 and wt, C71G or M114T mutant PFN1. The enlarged view of each eye is shown in lower right corner. B, H&E-stained sections of eyes of 5-day-old gmr-driven TG flies overexpressing singly human TDP-43, or doubly TDP-43 and wt, C71G or M114T mutant PFN1. Scale bar, 100 μm. C, retinal thickness measured in TG flies overexpressing singly LacZ or TDP-43, or doubly TDP-43 and wt, C71G or M114T mutant PFN1. n = 10, mean ± S.E., ***, p < 0.001. D, comparison of the expression levels of TDP-43 (upper panel) and PFN1 (middle panel). Immunoblot analyses of the heads of 5-day-old gmr-driven TG flies expressing singly LacZ or TDP-43, or doubly TDP-43 and wt, C71G or M114T mutant PFN1. α-Tubulin levels are shown as a loading control (bottom panel). Relative expression levels of TDP-43 are indicated under the panels. n = 4, mean ± S.E.
To investigate whether fALS mutations in PFN1 specifically exacerbate neurodegeneration induced by TDP-43, we generated TG fly lines that doubly overexpress PFN1 and FUS under control of the gmr-Gal4 driver. Similar to TDP-43, FUS has been identified as a causative gene for familial ALS (21, 22), and the FUS-immunoreactive neuronal pathology is observed in ALS and FTLD (23, 24). We found that overexpression of human FUS in the retina of fly caused a progressive neuronal degeneration in a similar manner to TDP-43 TG flies (supplemental Fig. S2, B and C, 45.7 ± 1.80 μm in FUS TG (p < 0.001, compared with LacZ TG)). We found that the degree of retinal degeneration in double TG flies overexpressing wt, C71G, or M114T mutant PFN1 and FUS was at similar levels to those in FUS single TG flies (supplemental Fig. S2, A–C, 41.6 ± 2.36 μm in wt PFN1/FUS (p = 0.61), 45.7 ± 2.18 μm in C71G mutant PFN1/FUS (p = 1.00), 43.8 ± 2.17 μm in M114T mutant PFN1/FUS (p = 0.97)). Immunoblot analysis of the lysates of heads of TG flies showed that overexpression of wt, C71G, or M114T mutant PFN1 in FUS TG flies did not affect the levels of FUS proteins compared with those in FUS single TG flies (supplemental Fig. S2 D). These data suggested that the fALS mutant PFN1, but not wt PFN1, specifically exacerbated the retinal degeneration induced by TDP-43, but not by FUS.
FALS Mutant PFN1 Altered Subcellular Localization of Endogenous TDP-43 from Nucleus to Cytoplasm in HEK293 Cells and SK-N-SH Cells
To elucidate the mechanism whereby the fALS mutant PFN1 exacerbated retinal degeneration induced by TDP-43, we examined the effects of wt and fALS mutant PFN1 in culture cells. It has been reported that wt PFN1 was present predominantly in the Nonidet P-40-soluble fraction of Neuro-2A cells; in sharp contrast, the fALS mutant PFN1, including C71G and M114T, was detected in both soluble and insoluble fractions (6). We transfected EGFP, wt, C71G, or M114T mutant PFN1 into human embryonic kidney (HEK) 293 cells and found that wt PFN1 was predominantly present in the Nonidet P-40-soluble fraction, whereas C71G or M114T PFN1 were fractionated into both Nonidet P-40-soluble and -insoluble fractions (Fig. 3A). These data suggest that fALS mutations reduce the solubility of PFN1.
FIGURE 3.

Expression of fALS mutant PFN1 increased the cytoplasmic localization of endogenous TDP-43 in HEK293 cells and SK-N-SH cells. A, immunoblot analysis of Nonidet P-40 soluble (sol) and insoluble (ins) fractions of HEK293 cells transfected with EGFP or myc-tagged wt, C71G, or M114T mutant PFN1 using anti-myc antibody (upper panel) or anti-α-tubulin antibody (lower panel). B and F, immunofluorescence labeling of HEK293 cells transfected with EGFP or myc-tagged wt, C71G, or M114T mutant PFN1 using anti-TDP-43 antibody (green, upper panels), anti-myc antibody (red, middle panels), or DRAQ5 as a marker for cell nucleus (blue, surrounded by dashed lines). Scale bar, 50 μm. The enlarged view of a portion of cytoplasm surrounded by the white square is shown in the upper right corner. C, immunoblot analysis of nuclear (Nuc) and cytoplasmic (Cyto) protein fractions of HEK293 cells transfected with EGFP or myc-tagged wt, C71G, or M114T mutant PFN1 using anti-myc, anti-TDP-43, anti-FUS, and anti-histone H3 as a marker for nuclear protein, or anti-α-tubulin. D and E, quantitative cytoplasmic/nuclear ratio of endogenous TDP-43 protein (D) or endogenous FUS protein (E). n = 8 for TDP-43 (D) and n = 5 for FUS (E), mean ± S.E. *, p < 0.05. F, immunofluorescence labeling of SK-N-SH cells transfected with EGFP or myc-tagged wt, C71G, or M114T mutant PFN1 using anti-TDP-43 antibody (green, upper panels), anti-myc antibody (red, middle panels), or DRAQ5 as a marker for cell nucleus (blue, surrounded by dashed lines). Scale bar, 50 μm.
We further examined the subcellular localization pattern of PFN1 and TDP-43 in HEK293 cells and human neuroblastoma SK-N-SH cells. Wt PFN1 has been reported to exhibit a diffuse cytoplasmic localization pattern in HEK293 cells (25) or Neuro-2A cells (6). TDP-43 has been reported to shuttle between the cytoplasm and nuclei and reside predominantly in the nucleus (26). We transfected EGFP, wt, C71G, or M114T mutant PFN1 into HEK293 cells and observed that C71G or M114T mutant PFN1 localized in cytoplasmic aggregates, whereas wt PFN1 was diffusely distributed at the cytoplasm (Fig. 3B). Endogenous TDP-43 was predominantly localized at the nucleus in EGFP- or wt-PFN1-transfected HEK293 cells (Fig. 3B). Interestingly, however, endogenous TDP-43 was partially co-localized in the cytoplasmic aggregates with transfected C71G or M114T fALS mutant PFN1 in HEK293 cells (Fig. 3B). Subcellular fractionation analysis showed that the ratio of cytoplasmic TDP-43 to nuclear TDP-43 in C71G- or M114T-PFN1-transfected HEK293 cells was significantly increased compared with EGFP-transfected HEK293 cells, whereas the ratio of cytoplasmic TDP-43 to nuclear TDP-43 in wt-PFN1-transfected HEK293 cells was similar to that in EGFP-transfected HEK293 cells (Fig. 3, C and D). In contrast, the ratios of cytoplasmic FUS to nuclear FUS were comparable among EGFP-, wt-, C71G-, and M114T-PFN1-transfected HEK293 cells (Fig. 3, C and E). These data suggest that fALS mutant PFN1 specifically affected the subcellular localization of endogenous TDP-43. We also transfected EGFP, wt, C71G, or M114T mutant PFN1 into SK-N-SH cells and observed that endogenous TDP-43 was diffusely localized in both nucleus and cytoplasm in C71G- or M114T-PFN1-transfected SK-N-SH cells, whereas endogenous TDP-43 was predominantly localized in nucleus in EGFP- or wt-PFN1-transfected SK-N-SH cells (Fig. 3F). Unlike in HEK293 cells transfected with fALS mutant PFN1 (Fig. 3B), PFN1- or TDP-43-immunopositive cytoplasmic aggregates were not detected in the cytoplasm of SK-N-SH cells transfected with fALS mutant PFN1 (Fig. 3F). Taken together, we concluded that fALS mutations in PFN1 alter the subcellular localization of TDP-43 from nucleus to cytoplasm.
Overexpression of fALS Mutant PFN1 Increased the Cytoplasmic Localization of TDP-43 in the Retinal Cells of D. melanogaster
To examine whether fALS mutant PFN1 affects the subcellular localization of TDP-43 in the Drosophila retinal cells, we immunolabeled sections of heads of double TG flies overexpressing wt or fALS mutant PFN1 and TDP-43 in the retinal cells using two different anti-TDP-43 antibodies and found that TDP-43 was predominantly localized at nuclei of retinal cells in wt PFN1/TDP-43 double TG flies, in a similar manner to those in TDP-43 single TG flies (Fig. 4, A and C). Notably, TDP-43 was localized both to the nuclei and cytoplasm in retinal cells of double TG flies overexpressing C71G or M114T mutant PFN1 and TDP-43 (Fig. 4, A and C). Fluorescence intensity profiles of immunolabeling with the anti-TDP-43 antibodies also revealed the nuclear localization pattern of TDP-43 in the retinal cells of wt PFN1/TDP-43 double TG flies, whereas both nuclear and cytoplasmic localization patterns of TDP-43 in the retinal cells was observed in the fALS mutant PFN1/TDP-43 double TG flies (Fig. 4, B and D), suggesting that fALS mutations in PFN1 lead to the redistribution of TDP-43 from the nucleus to the cytoplasm in the Drosophila retinal cells. We found that nuclear localization of FUS in the retinal cells was not altered by overexpression of wt or fALS mutant PFN1 in double TG flies (data not shown). Wt or fALS mutant PFN1 has been reported to predominantly localize at the cytoplasm (6, 25). We also found that wt or C71G, M114T mutant PFN1 localized at cytoplasm in the HEK293 cells (Fig. 3, B and C) or in the retinal cells of PFN1/TDP-43 double TG flies (data not shown). These data suggest that the increase in cytoplasmic localization of TDP-43 in fALS mutant PFN1/TDP-43 double TG flies may contribute to the exacerbation of retinal degeneration induced by TDP-43.
FIGURE 4.

Overexpression of fALS mutant PFN1 increased the cytoplasmic localization of TDP-43. A and C, immunofluorescence histochemistry of the retina of 5-day-old gmr-driven TG flies overexpressing singly human TDP-43, or doubly TDP-43 and wt, C71G or M114T mutant PFN1 labeled by anti-TDP-43 (rabbit polyclonal (A) or mouse monoclonal (C)) and DRAQ-5 as a marker for nucleus (blue, surrounded by dashed lines). Note that co-expression of fALS mutant PFN1 (C71G, M114T) with TDP-43 increased cytoplasmic TDP-43 staining (white arrowheads). Scale bar, 10 μm. B and D, immunofluorescence intensity profiles of TDP-43 (green, rabbit polyclonal anti-TDP-43 antibody (B), mouse monoclonal anti-TDP-43 (D)) and nuclear (blue) measured at the dashed arrows in the bottom panels of A and C using ImageJ software. Black arrows indicate the cytoplasmic localization of TDP-43.
To test this idea, we crossed TG flies overexpressing wt or fALS mutant PFN1 with TG flies expressing nuclear localization signal (NLS) mutant TDP-43 (18). We have previously shown that TG flies overexpressing NLS mutant TDP-43, in which TDP-43 is localized to the cytoplasm due to the replacement of 6 amino acids within the NLS of TDP-43 with alanine (27), exhibited more severe retinal degeneration compared with TG flies expressing wt TDP-43 (18). TG flies overexpressing the NLS mutant TDP-43 exhibited more severe retinal degeneration compared with wt TDP-43 TG flies (Fig. 5, A–C, 31.8 ± 1.47 μm in NLS mutant TDP-43 (p < 0.001)), as described (18). Co-expression of NLS mutant TDP-43 with wt PFN1 also exhibited more severe degenerative phenotypes in the retina than those expressing wt TDP-43 and wt PFN1 (Fig. 5, A–C, 29.7 ± 2.28 μm in wt PFN1/NLS mutant TDP-43 (p < 0.001)). However, co-expression of the NLS mutant TDP-43 with C71G or the M114T mutant PFN1 exhibited retinal degeneration at similar levels to those co-expressing wt TDP-43 with C71G or M114T mutant PFN1, respectively (Fig. 5, A–C, 23.4 ± 1.50 μm in C71G mutant PFN1/NLS mutant TDP-43 (p = 0.82), 22.4 ± 0.76 μm in M114T mutant PFN1/NLS mutant TDP-43 (p = 0.60)). Immunoblot analysis of lysates of heads revealed that overexpression of wt, C71G, or M114T mutant PFN1 did not affect the expression level of NLS mutant TDP-43 (Fig. 5D). These data led us to speculate that fALS mutations in PFN1 exacerbated the TDP-43-induced neurodegeneration in the retina through an increase in cytoplasmic localization of TDP-43.
FIGURE 5.

Overexpression of fALS mutant PFN1 did not alter NLS mutant TDP-43-induced retinal degeneration. A, external surface pictures of eyes of 5-day-old gmr-driven TG flies overexpressing singly human NLS mutant TDP-43, or doubly NLS mutant TDP-43 and wt, C71G or M114T mutant PFN1. The enlarged view of each eye is shown in the lower right corner. B, H&E-stained sections of eyes of 5-day-old gmr-driven TG flies overexpressing singly human NLS mutant TDP-43, or doubly NLS mutant TDP-43 and wt, C71G or M114T mutant PFN1. Scale bar, 100 μm. C, thickness of retina measured in TG flies doubly expressing NLS mutant TDP-43 and wt, C71G or M114T mutant PFN1. n = 10, mean ± S.E., *, p < 0.05; **, p < 0.01; ***, p < 0.001. D, comparison of the protein levels of TDP-43 (upper panel) and PFN1 (middle panel). Immunoblot analyses of the heads of 5-day-old gmr-driven TG flies overexpressing singly LacZ, human TDP-43, or NLS mutant TDP-43, or doubly NLS mutant TDP-43 and wt, C71G or M114T mutant PFN1. α-Tubulin levels are shown as a loading control (bottom panel).
To rule out the possibility that the degree of degenerative phenotypes of retina caused by overexpression of NLS mutant TDP-43 showed ceiling effect and was too severe to exhibit additional worsening in C71G or M114T mutant PFN1/NLS mutant TDP-43 double TG flies, we investigated the dose dependence of the effect of NLS mutant TDP-43 in heterozygous or homozygous NLS mutant TDP-43 TG flies. 5-Day-old homozygoous NLS mutant TDP-43 TG flies expressed ∼2-fold higher levels of NLS mutant TDP-43 and exhibited more severe retinal degeneration compared with 5-day-old heterozygous NLS mutant TDP-43 single TG flies (data not shown). This suggests that the retinal degeneration in heterozygous NLS mutant TDP-43 has not reached the maximal level, and that the similar degree of retinal degeneration observed in C71G or M114T mutant PFN1/NLS mutant TDP-43 double TG flies and in NLS mutant TDP-43 flies implies the lack of exacerbating effects by additional expression of mutant PFN1.
Because endogenous TDP-43 was predominantly localized to nuclei in HEK293 cells, mislocalization of TDP-43 to the cytoplasm may be a cause of dysfunction of TDP-43. TDP-43 functions in a series of steps in DNA/RNA processing (14). To test this hypothesis, we examined the exon 31 skipping in MADD gene. MADD gene is one of the previously identified, direct targets of TDP-43, and TDP-43 knockdown in HEK293 cells has been reported to increase the level of exon-skipped MADD gene (28). We transfected EGFP, wt, C71G or M114T mutant PFN1 into HEK293 cells and observed that C71G or M114T mutant PFN1 significantly increased the levels of the exon 31-skipped MADD gene, whereas wt PFN1 did not change the splicing profiles of the MADD gene (Fig. 6, A and B, 1.55 ± 0.22% in EGFP, 0.96 ± 0.23% in wt PFN1 (p = 0.83), 6.39 ± 0.81% in C71G PFN1 (p = 0.0001), 3.92 ± 0.47% in M114T PFN1 (p = 0.0086)). We also examined exon 7 skipping in the FNIP1 gene, exon 20 skipping in the BRD8 gene, or exon 3 skipping in the SKAR gene (28, 29), although no changes in the splicing profiles of these genes were observed among HEK293 cells transfected with EGFP, wt, C71G, or M114T mutant PFN1 (data not shown). Human HDAC6 mRNA has been reported to be down-regulated in TDP-43-silenced HEK293 cells (30). We transfected EGFP, wt, C71G or M114T mutant PFN1 into HEK293 cells and observed that C71G mutant PFN1 significantly decreased, whereas wt or M114T mutant PFN1 did not change the levels of HDAC6 mRNA (Fig. 6C, 1.00 ± 0.02% in EGFP, 1.13 ± 0.05% in wt PFN1 (p = 0.32), 0.74 ± 0.07% in C71G PFN1 (p = 0.0036), 0.98 ± 0.05% in M114T PFN1 (p = 0.98)). To investigate whether cytoplasmic mislocalization of TDP-43 induced by the fALS mutant PFN1 caused impairment of the function of TDP-43 in Drosophila, we examined the expression level of TBPH mRNA, a Drosophila homologue of TDP-43. We have previously shown a significant reduction of TBPH mRNA in the eyes of TDP-43 TG flies through its RNA recognition motif (18). Here, we found that TDP-43 single, or wt PFN1/TDP-43 double TG flies exhibited a significant reduction in the expression levels of TBPH mRNA compared with LacZ TG flies, whereas C71G or M114T PFN1/TDP-43 double TG flies did not (Fig. 6D). These results support the notion that overexpression of mutant PFN1, but not wt PFN1, partially suppressed the physiological function of TDP-43 through an increase in the cytoplasmic mislocalization of TDP-43.
FIGURE 6.

Overexpression of fALS mutant PFN1, but not wt PFN1, suppressed function of TDP-43. A, semi-quantitative RT-PCR analyses of the splicing pattern of MADD gene in control siRNA-treated, TDP-43 siRNA-treated, EGFP, or myc-tagged wt, C71G, or M114T mutant PFN1 stably expressed HEK293 cells. 200-Base pair DNA size marker was indicated at the left of the panel. B, quantitative ratio of exon 31 skipping of MADD gene. n = 6, mean ± S.E., *, p < 0.05; ***, p < 0.001. C, quantitative RT-PCR analyses of expression level of HDAC6 mRNA in EGFP, or myc-tagged wt, C71G, or M114T mutant PFN1 stably expressed HEK293 cells. n = 6, mean ± S.E., *, p < 0.05. D, quantitative RT-PCR analyses of the expression level of TBPH mRNA in the compound eyes of 1–3-day-old gmr-driven TG flies expressing singly human TDP-43, or doubly TDP-43 and wt, C71G or M114T mutant PFN1. n = 4, mean ± S.E., **, p < 0.01.
Knockdown of Endogenous PFN1 Did Not Alter the TDP-43-induced Retinal Degeneration
A previous report showed that fALS mutations (C71G, M114T, G118V) in PFN1 reduced the levels of PFN1-bound actin relative to wt PFN1 in HEK293 cells (6). We found that overexpression of mutant PFN1, but not wt PFN1, partially suppressed the physiological function of TDP-43 (Fig. 6). These observations led one to speculate that partial loss in the regulation of actin dynamics may be the pathomechanism caused by fALS mutant PFN1. To elucidate whether the loss of PFN1 function affects the TDP-43-induced neurodegeneration in the retina, we crossed TDP-43 TG flies with RNAi lines of Drosophila homologue of PFN1 (chickadee, chic), in which the expression of chic mRNA in the eyes was reduced (Fig. 7A). Histological examination of the retina of chic RNAi flies revealed no degenerative phenotypes, and the retinal thickness of chic RNAi flies was similar to that of LacZ TG flies (Fig. 7, B–D). Notably, knockdown of chic in TDP-43 TG flies exhibited retinal degeneration at a similar degree to those in TDP-43 TG flies (Fig. 7, B–D, 67.6 ± 1.06 μm in chic RNAi (p = 0.98), 49.6 ± 1.71 μm in chic RNAi/TDP-43 (p = 0.39)). Immunoblot analyses of the heads of chic RNAi/TDP-43 TG flies revealed similar levels of expression of TDP-43 or TBPH compared with that in TDP-43 TG flies or chic RNAi flies, respectively (Fig. 7, E and F). We immunolabeled sections of heads of chic RNAi/TDP-43 TG flies and found that TDP-43 was dominantly localized at nuclei in the retinal cells, in a similar manner to TDP-43 TG flies (Fig. 7G). Fluorescence intensity profiles of immunolabeling with anti-TDP-43 antibodies also revealed a nuclear localization pattern of TDP-43 in the retinal cells of chic RNAi/TDP-43 TG flies (Fig. 7H). These data indicated that the loss of PFN1 function did not affect the neurodegeneration induced by TDP-43 in Drosophila retina.
FIGURE 7.

RNAi knockdown of endogenous PFN1 did not alter TDP-43-induced retinal degeneration. A, semi-quantitative RT-PCR analyses of chic expression of the 1–3-day-old compound eyes of gmr-driven TG flies expressing LacZ or chic RNAi (upper panel). Levels of Rp49 mRNA are shown as an internal control (lower panel). B, external surface pictures of eyes of 5-day-old gmr-driven TG flies expressing chic RNAi, or doubly chic RNAi and human TDP-43. The enlarged view of each eye is shown in lower right corner. C, H&E-stained sections of eyes of 5-day-old gmr-driven TG flies expressing chic RNAi, or doubly chic RNAi and human TDP-43. Scale bar, 100 μm. D, thickness of retina measured in flies expressing chic RNAi or chic RNAi and TDP-43. Note that knockdown of chic caused neither retinal degeneration nor exacerbation of TDP-43-induced degeneration. n = 10, mean ± S.E. E, expression levels of TDP-43. Immunoblot analyses of the heads of 5-day-old gmr-driven TG flies expressing TDP-43 or chic RNAi and TDP-43 (upper panel). α-Tubulin levels are shown as a loading control (lower panel). Relative expression levels of TDP-43 are indicated under the panels. n = 3, mean ± S.E. F, expression levels of TBPH. Immunoblot analyses of the compound eyes of 5-day-old gmr-driven TG flies expressing LacZ or chic RNAi (upper panel). α-Tubulin levels are shown as a loading control (lower panel). Relative expression levels of TBPH are indicated under the panels. n = 4, mean ± S.E. G, immunofluorescence histochemistry of the retina of 5-day-old gmr-driven TG flies expressing TDP-43 or chic RNAi and TDP-43 immunolabeled by an anti-TDP-43 antibody (green) and DRAQ-5 (blue). Note that knockdown of chic did not alter the subcellular localization of TDP-43. Scale bar, 10 μm. H, immunofluorescence intensity profiles of TDP-43 (green) and nuclear (blue) measured at the dashed arrows in the bottom panels (G).
To investigate whether the loss of PFN1 function affects the pattern of subcellular distribution of TDP-43, we knocked down endogenous PFN1 in HEK293 cells by siRNAs. Immunoblot analyses of lysates of HEK293 cells treated with PFN1 siRNA revealed a similar expression level of endogenous TDP-43 compared with control siRNA-treated HEK293 cells (Fig. 8, A and B). Immunofluorescence labeling of PFN1 siRNA-treated HEK293 cells showed a predominant nuclear localization of TDP-43 as in HEK293 cells treated with a control siRNA (Fig. 8C). Subcellular fractionation analysis also showed that the ratio of cytoplasmic to nuclear TDP-43 in PFN1 siRNA-treated HEK293 cells was similar to that in control siRNA-treated HEK293 cells (Fig. 8, D and E). These data indicated that the loss of PFN1 function did not affect the pattern of subcellular distribution of TDP-43 in HEK293 cells.
FIGURE 8.

RNAi knockdown of endogenous PFN1 did not change the subcellular localization pattern of endogenous TDP-43 in HEK293 cells. Immunoblot analyses of the lysate of control or PFN1 siRNA (#1 or #2)-treated HEK293 cells using anti-PFN1 (upper panel), anti-TDP-43 (middle panel), or anti-α-tubulin (lower panel, loading control) antibodies. B, quantification of the relative expression level of TDP-43 in A. n = 6, mean ± S.E. C, immunofluorescence labeling of TDP-43 in control or PFN1 siRNA (#1 or #2)-treated HEK293 cells by anti-TDP-43 antibody (green, top panels), anti-PFN1 antibody (red, middle panels), or DRAQ5 (blue, bottom panels). Scale bar, 25 μm. D, immunoblot analysis of nuclear (Nuc) and cytoplasmic (Cyto) protein fractions of control or PFN1 siRNA (#2)-treated HEK293 cells using anti-TDP-43, anti-PFN1, anti-histone H3 as a marker of nuclear protein, or anti-α-tubulin. E, quantitative cytoplasmic/nuclear ratio of endogenous TDP-43 protein in D. n = 5, mean ± S.E.
Discussion
Using TG flies that overexpress PFN1 and/or TDP-43 in the retinal photoreceptor neurons, we have shown the following: 1) overexpression of wt or fALS mutant PFN1 did not cause retinal degeneration (Fig. 1); 2) co-expression of fALS mutant PFN1, but not wt PFN1, together with TDP-43 exacerbated retinal degeneration, associated with a shift in localization of TDP-43 to cytoplasm (Figs. 2 and 4); 3) the levels of retinal degeneration upon co-expression of fALS mutant PFN1 with NLS mutant TDP-43 or wt TDP-43 were similar (Fig. 5); and 4) silencing of endogenous PFN1 affected neither the retinal degeneration induced by TDP-43 (Fig. 7) nor nuclear localization of TDP-43 (Fig. 8). We have also shown in PFN1/FUS double TG flies that: 5) overexpression of wt or fALS mutant PFN1 did not affect the retinal degeneration induced by FUS (supplemental Fig. S2); last, 6) fALS mutant, but not wt PFN1, altered subcellular localization of endogenous TDP-43 from the nucleus to cytoplasm in mammalian HEK293 and SK-N-SH cells (Fig. 3) and suppressed the physiological function of TDP-43 in HEK293 cells (Fig. 6). These data collectively support the notion that fALS mutations in PFN1 exacerbate the TDP-43-induced neurodegeneration by shifting the localization of TDP-43 to the cytoplasm, where disease-related interactions of TDP-43 with RNAs and proteins might take place (Fig. 9).
FIGURE 9.
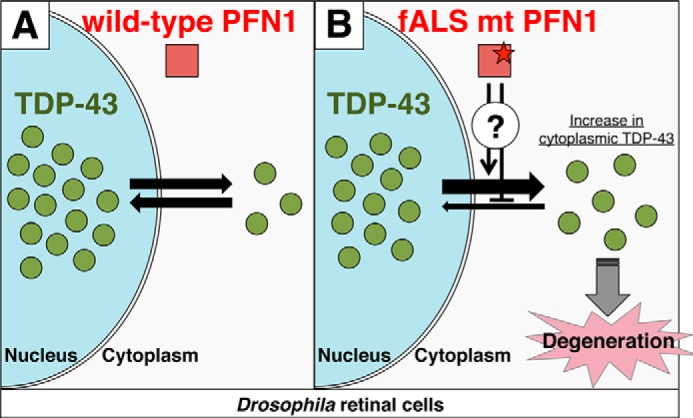
Schematic model of the hypothetical mechanism for fALS mutation of PFN1. A, in the presence of wt PFN1, TDP-43 shuttles between the nucleus and cytoplasm and predominantly localizes at the nucleus in retinal cells. B, in the presence of fALS mutant PFN1, mutant PFN1 may impede the transport of TDP-43 from the cytoplasm to nucleus, or promote that of TDP-43 from the nucleus to cytoplasm by as yet unknown mechanism, resulting in an increase in the cytoplasmic TDP-43 and leading to degeneration.
Although TDP-43 predominantly localizes at the nucleus in normal conditions, TDP-43-positive pathological inclusions are typically found in the cytoplasm of neurons in the brains and spinal cord of patients with ALS or FTLD (15, 16). This strongly supports the idea that abnormal cytoplasmic localization of TDP-43 or loss of TDP-43 in the nucleus may be causative to neuronal degeneration and death. TDP-43 harbors both NLS and nuclear exporting signal, and is shown to shuttle between the cytoplasm and nucleus (26). Overexpression of NLS mutant TDP-43 in the murine primary culture neurons led to aggregate formation in the neuronal perikarya and neurites (27). It has been shown in multiple lines of TG flies that overexpression of NLS mutant TDP-43 caused more severe retinal degeneration compared with those caused by overexpression of wt TDP-43 (18, 31, 32). These data support the view that abnormal cytoplasmic localization of TDP-43 exacerbates neuronal degeneration. In the present study, we found that fALS mutant PFN1, but not wt PFN1, shifted the intracellular localization of TDP-43 to the cytoplasm in HEK293 and SK-N-SH cells or Drosophila retinal cells (Figs. 3 and 4), and simultaneously exacerbated the degeneration induced by TDP-43 in the retina (Fig. 2). Our finding that the degree of degeneration upon co-expression of fALS mutant PFN1 with NLS mutant TDP-43 was similar to that upon co-expression with wt TDP-43 may be interpreted that the nuclear exclusion effect of fALS mutant PFN1 might have been saturated, without allowing additional cytoplasmic relocation of TDP-43. Taken together with the observation that TDP-43 immunopositive neuronal cytoplasmic inclusions were found in motor neurons of spinal cord of ALS patients with the E117G mutation in PFN1 gene (9, 33), we speculate that fALS mutant PFN1 aggravates neurodegeneration by enhancing the abnormal cytoplasmic localization of TDP-43 (Fig. 9).
The mechanism whereby fALS mutations in PFN1 alter the subcellular distribution of TDP-43 is unknown. It has previously been reported that TDP-43 is involved in the regulation of stress granules, and that its association with stress granules is increased after arsenite treatment (34–36). Stress granules are cytoplasmic dense granules composed of mRNAs and RNA-binding proteins, whose formation is triggered by environmental stress, e.g. heat shock or exposure to oxidants (37, 38). Recent reports suggest that protein components of stress granules, e.g. poly(A)-binding protein, modulates TDP-43-induced toxicity in the Drosophila model (39), implicating stress granule formation in the TDP-43-induced toxicity. Furthermore, it has been reported that PFN1 is also associated with stress granules in the cytoplasm under arsenite treatment or heat shock stimulation, and is involved in the regulation of the dynamics of stress granules (13); also, fALS mutant PFN1 (C71G, M114T, or T109M) has impaired ability in the regulation of the stress granule dynamics. These results lead one to speculate that the intracellular transport of TDP-43 in relationship to stress granule assembly is impeded within the cytoplasm by fALS mutant PFN1, resulting in an increase in the cytoplasmic localization of TDP-43. Our finding that silencing of endogenous PFN1 did not alter the nuclear localization of TDP-43 may suggest that the mechanism whereby mutant PFN1 altered the subcellular localization pattern of TDP-43 is distinct from those occurring upon inhibition of the physiological function of endogenous PFN1 (Figs. 7G and 8). We found that Nonidet P-40 solubility of fALS mutant PFN1 expressed in HEK293 cells was reduced, suggesting that the fALS mutations in PFN1 may lead to self-aggregation, resulting in sequestration of endogenous TDP-43 in the cytoplasm. It has recently been reported that C71G, M114T, E117G, or G118V mutant PFN1 transiently expressed in SH-SY5Y cells increased the insolubility and phosphorylation of TDP-43, possibly by relocation of TDP-43 from the nucleus to cytoplasm and providing a scaffold for the conformational change of TDP-43 (40). These data also lead one to speculate that fALS mutations in PFN1 may cause conformational changes, forming cytoplasmic aggregates and leading to the sequestration of TDP-43 into the aggregates. The interaction between PFN1 and TDP-43 is still elusive. A recent report showed an interaction of wt PFN1 with TDP-43 by a co-immunoprecipitation experiment (40), although it is still unclear whether mutant PFN1 interacts with TDP-43.
Our observations are relevant to the effects of fALS-linked mutations in the valosin-containing protein (VCP) or C9orf72 gene. VCP mutations cause an autosomal-dominant inclusion body myopathy and Paget disease of bone and frontotemporal dementia (IBMPFD) (41), and familial ALS (42), in which TDP-43 positive ubiquitinated inclusions are also detected (43). Overexpression of disease-related mutant VCP, but not of wt VCP, has been shown to cause redistribution of TDP-43 to the cytoplasm in mouse primary neurons and retina of Drosophila (31). Overexpression of FTLD-linked mutant VCP in HEK293 cells caused relocalization of endogenous TDP-43 to speckles or granules within the cytoplasm through the interaction between mutant VCP and TDP-43 (44). These data suggest that mutant VCP also enhances neurotoxicity of TDP-43 by increasing the cytoplasmic localization of TDP-43. Expanded GGGGCC hexanucleotide repeat in the noncoding region of C9orf72, known as the most common cause associated with familial ALS and frontotemporal dementia (45, 46) has recently been shown to disrupt the nuclear localization of TDP-43 through an interaction with RanGAP, a key regulator of nucleocytoplasmic transport, in Drosophila, or in C9-ALS patient-derived induced pluripotent stem cells (47). Taken together with our results on the effects of fALS mutations in PFN1, it is strongly suggested that the increase in the cytoplasmic localization of TDP-43 underlies the mechanism of neurodegeneration in ALS and FTLD.
In this study, we found that silencing of endogenous PFN1 did not affect the retinal degeneration induced by TDP-43 (Fig. 7). The facts that endogenous PFN1, chickadee, was originally identified as a female sterile mutation in Drosophila (48) and that stranded, a P-element inserted mutant in Drosophila PFN1 gene, as an abnormal embryonic motoneuron projection mutant (49) suggest that Drosophila PFN1 is crucial for the development of retinal photoreceptor neurons. However, our finding that knockdown of endogenous PFN1 did not exacerbate retinal degeneration induced by TDP-43 supports the notion that fALS mutations in PFN1 exacerbate TDP-43-induced degeneration in a gain-of-function fashion.
Overexpression of wt or fALS mutant PFN1 did not affect the retinal degeneration induced by FUS (supplemental Fig. S2). FUS is also a DNA/RNA-binding protein that is predicted to function through its RNA recognition motif (14) and identified as the causative gene for autosomal-dominant familial ALS type 6 (21, 22). Although the mechanism whereby mutation or abnormality in FUS leads to neurodegeneration remains elusive, our findings suggest that fALS mutant PFN1 is not involved in the neurodegeneration induced by FUS. Co-aggregation of mutant PFN1 (C71G or G118V) with TDP-43, but not with FUS, has been reported in primary motor neurons upon overexpression (6), which may be consistent with our results.
In summary, we conclude that PFN1 exacerbates TDP-43-induced neurodegeneration by increasing the cytoplasmic localization of TDP-43. Further studies of our TG Drosophila models expressing wt or mutant PFN1 and TDP-43, combined with mammalian studies, will unravel the mechanism whereby abnormal cytoplasmic localization of TDP-43 is induced by PFN1, leading to neurodegeneration in ALS and FTLD.
Experimental Procedures
Fly Stocks and Generation of TG Flies
The GAL4-UAS expression system was used for the generation of TG Drosophila. Constructs encoding human wt or fALS mutant C71G, M114T PFN1, or human FUS, were injected into w1118 embryos to produce TG flies. TG flies expressing UAS-wt or -NLS mutant TDP-43 were previously generated (18). At least four independent transformant lines were obtained per each construct. gmr-GAL4 and UAS-lacZ lines were obtained from the Bloomington Drosophila stock center. UAS-chickadee (chic) RNAi line was obtained from NIG-Fly. Fly stocks were raised on standard Drosophila medium at 20 °C. Crosses between the Drosophila strains were carried out at 25 °C using standard procedures.
Cell Culture and Transfection
HEK293 cells and SK-N-SH cells were obtained from American Type Culture Collection. HEK293 cells and SK-N-SH cells were cultured in Dulbecco's modified Eagle's medium (Sigma) supplemented with 10% fetal bovine serum and 100 units/ml of penicillin/streptomycin. Plasmid DNA was introduced into HEK293 cells by transfection with Polyethylenimine “Max” (PolySciences, Inc.) and into SK-N-SH cells by transfection with Lipofectamine 3000 (Life Technologies) according to the manufacturer's procedures. siRNA was introduced into HEK293 cells by transfection with Lipofectamine RNAiMAX (Life Technologies) according to the manufacturer's procedures.
Plasmid Construction
The human PFN1 cDNA clone in pENTRTM221 (pENTRTM221-PFN1) expression vector was purchased from Life Technologies. Site-directed mutagenesis of pENTRTM221-PFN1 was carried out to substitute Cys-71 to Gly (C71G), Met-114 to Thr (M114T). Primer sequences were as follows: C71G, 5′-CTTGGGGGCCAGAAAGGTTCGGTGATCCG-3′, M114T, 5′-ACGCTAGTCCTGCTGACGGGCAAAGAAGGTG-3′. To generate PFN1 TG flies, EcoRI restriction site at the 5′ end and XbaI restriction site at the 3′ end were added by PCR. Primer sequences were as follows: forward 5′-AAAAAGAATTCATGGCCGGGTGGAACGCCTAC-3′, reverse 5′-AAAAATCTAGATCAGTACTGGGAACGCCGAAGG-3′. The EcoRI/XbaI fragment containing full-length wt or mutant PFN1 was cloned into the EcoRI/XbaI site of the pUAST vector and verified by sequencing. The human FUS cDNA clone was purchased from Life Technologies. XhoI restriction site at the 5′ end and HindIII restriction site at the 3′ end were added by PCR. Primer sequences were as follows: forward 5′-CGGTGCTCGAGGGTGTTGGAACTTC-3′, reverse 5′-AAGCTTTTCCAGAACCTGGGGAGCC-3′. The XhoI/HindIII fragment containing full-length FUS was inserted into the XhoI/HindIII site of the cold shock expression pColdI vector (Takara). This product was used to verify protein expression as a His-tagged FUS in Escherichia coli and digested with XhoI/XbaI and subcloned into the pUAST vector and verified by sequencing. To generate expression plasmids for transfection into mammalian cells, EcoRV restriction site at the 5′ end and XbaI restriction site at the 3′ end were inserted by PCR. Primer sequences were as follows: forward 5′-AAAAAGATATCATGGCCGGGTGGAACGCC-3′, reverse 5′-AAAAATCTAGATCAGTACTGGGAACGCC-3′. The EcoRV/XbaI fragment containing full-length wt or mutant PFN1 was cloned into the EcoRV/XbaI site of the pcDNA3.1 vector, inserted Myc-tag encoding DNA at HindIII/EcoRV site, and verified by sequencing. Oligonucleotide sequences were as follows: top strand 5′-AGCTATGGAACAAAAACTCATCTCAGAAGAGGATCTG-3′, bottom strand 5′-CAGATCCTCTTCTGAGATGAGTTTTTGTTCCAT-3′.
Histology and Immunohistochemistry of TG Flies
Heads of 5-day-old adult TG Drosophila were dissected, briefly washed in phosphate-buffered saline (PBS), and fixed with 4% paraformaldehyde containing 0.1% Triton X-100 at room temperature for 2 h. Tissues were dehydrated by graded ethanol, cleared in buthanol, and embedded in paraffin. Tissue blocks were cut in coronal sections at 4-μm thickness. Sections were stained with hematoxylin and eosin (H&E). For immunostaining, antigen retrieval pretreatment was performed on deparaffinized sections by microwave treatment (550 W, 10 min) in citrate buffer (pH 6.0). After blocking of nonspecific reactions in 10% calf serum in PBS, sections were incubated with an anti-TDP-43 rabbit polyclonal antibody (1:2,500, Proteintech 10782-2-AP), an anti-TDP-43 monoclonal antibody (1:500, ABNOVA, clone 2E2-D3, H00023435-M01), an anti-PFN rabbit monoclonal antibody (1:100, Abcam, clone EPR6304, ab124904), or an anti-PFN1 mouse monoclonal antibody (1:100, Abcam, clone 1D5, ab118983) overnight at room temperature. For immunofluorescence labeling, sections were incubated by a mixture of Alexa fluorophore-conjugated secondary antibodies against mouse or rabbit IgG (1:1,000), and DRAQ5 as a nuclear marker and observed with a Leica confocal microscope. The retinal thickness in H&E-stained sections was measured using ImageJ software as an average of two measurements at the central area per a fly as described (18). Ten eye sections cut at the center of retina perpendicularly to the eye surface were measured per line.
Immunofluorescence Analysis of Cell Cultures
HEK293 cells or SK-N-SH cells were plated on glass coverslips for 48 h after transfection. Cells were fixed for 20 min in 4% paraformaldehyde and then permeabilized with 0.1% Triton X-100 for 30 min. After blocking of nonspecific reactions in 10% calf serum in PBS, cells were incubated with an anti-TDP-43 rabbit polyclonal antibody (1:2500, Proteintech) or anti-Myc antibody (1:2000, Cell Signaling Technology, clone 9B11, number 2276) overnight at room temperature. For immunofluorescence labeling, cells were incubated by a mixture of Alexa fluorophore-conjugated secondary antibodies against mouse or rabbit IgG (1:1000), and DRAQ5 (Biostatus) as a nuclear marker and observed with a Leica TCS SP5 confocal microscope.
Separation of Cellular Proteins
Separation of soluble and insoluble proteins in HEK293 cells were performed as described previously (6). Briefly, cells were collected, lysed in Nonidet P-40 buffer (1% Nonidet P-40, 20 mm Tris-HCl, 150 mm NaCl, 5 mm EDTA, 10% glycerol, 1 mm DTT, 10 mm sodium fluoride, 1 mm sodium orthovanadate, 5 mm sodium pyrophosphate, pH 7.4) and rotated for 30 min at 4 °C. After centrifugation at 20,000 × g for 15 min, the supernatant was collected as Nonidet P-40 soluble fraction. The pellet was washed with Nonidet P-40 buffer twice, and sonicated in urea-SDS buffer (8 m urea and 3% SDS in Nonidet P-40 buffer). After centrifugation at 20,000 × g for 15 min, the supernatant was collected as Nonidet P-40-insoluble fraction. Complete protease inhibitor (Roche Applied Science) was added in every sample to avoid protein degradation. Subcellular fractionation assays of HEK293 cells were performed using an NE-PER Nuclear and Cytoplasmic Extraction Reagents kit (Thermo Fisher Scientific) according to the manufacturer's procedures.
Immunoblotting
To compare the expression levels of endogenous proteins, 5-day-old flies were freeze-dried in acetone and 10 compound eyes were dissected. To compare the expression levels of TG fly lines, 10 heads of 5-day-old flies were dissected. Samples were lysed in Laemmli sample buffer for SDS-PAGE containing 2% SDS. Separated samples of HEK293 cells were also mixed with Laemmli sample buffer. Samples were separated by 10 or 15% SDS-PAGE, transferred to polyvinylidene difluoride membranes, and probed with anti-TDP-43 (1:1000, Proteintech), anti-PFN1 (1:2000, Abcam clone 1D5), anti-FUS (1:5000, Bethyl A300–302A), anti-TBPH (1:2500, (18)), anti-α-tubulin (1:2500, Sigma, clone DM1A, T9026), anti-Myc (1:2000, Cell Signaling Technology, clone 9B11), or anti-Histone H3 (1:1000, Cell Signaling Technology, clone D1H2, number 4499) antibody. The immunoblots were developed using a chemiluminescence kit (Wako) or SuperSignal West Femto Chemiluminescent Substrate (Thermo Scientific) and visualized by LAS-4000 (Fujifilm).
Quantitative or Semi-quantitative RT-PCR Analysis
1- to 3-day-old flies were freeze-dried in acetone and compound eyes of gmr-driven RNAi flies were dissected. 30 compound eyes were used to average individual difference. Total RNA from compound eyes or HEK293 cells was extracted using IsoGen (Nippon Gene) and converted to cDNA using ReverTra Ace Quantitative PCR RT Master Mix with gDNA remover (TOYOBO). The primer sequences used for PCR of fly cDNA were as follows: 5′-GAGGAGCTCTCCAAACTGATCAG-3′ and 5′-GATCTATTCTCCTAGTACCCGCAAG-3′ for chic, 5′-GAGCAACCAGTGAATGCTCA-3′ and 5′-CTTCCGTCCACCAAAGTTGT-3′ for TBPH, and 5′-ATACAGGCCCAAGATCGTGAAGAAG-3′ and 5′-GCTTGTTCGATCCGTAACCGATG-3′ for rp49. The primer sequences used for PCR for HEK293 cDNA were as follows: 5′-GACCTGAATTGGGTGGCGAGTTCCCT-3′ and 5′-CATTGGTGTCTTGTACTTGTGGCTC-3′ for MADD, and 5′-CCACGATAGACCAGCTGTAG-3′ and 5′-TCTTGGGGAGTTGCAAAGG-3′ for HDAC6, and 5′-GTGGTCTCCTCTGACTTCAACAG-3′ and 5′-GTCTTACTCCTTGGAGGCCATG-3′ for GAPDH. For quantitative analysis, real-time PCR was performed using LightCycler480 II (Roche Applied Science) and SYBR Green Real-time PCR Master Mix (TOYOBO). For semi-quantitative analysis of exon exclusion rate, PCR products were separated by 2% agarose gel. Band intensity was quantified using ImageJ software.
Statistical Analysis
All results were shown as mean ± S.E. Statistical analysis was performed with one-way or two-way analysis of variance followed by post hoc test comparison using Tukey-Kramer test. Significance was established at p < 0.05.
Author Contributions
K. M., T. H., T. W., and T. I. designed the project. K. M., T. H., and T. I. wrote manuscript. K. M., T. H., T. M., and R. I. performed experiments. K. M., T. H., and T. I. analyzed data. T. C. and M. M. gave technical support and advice.
Supplementary Material
Acknowledgment
We thank NIG-Fly Stock Center for chic knockdown line.
This work was supported by The Nakabayashi Trust for ALS Research (to T. H.) and Grants-in-Aid for Scientific Research (C) (to T. H.) and for JSPS Fellows (to K. M.) from the Ministry of Education, Culture, Sports, Science and Technology, Japan. The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Figs. S1 and S2.
- ALS
- amyotrophic lateral sclerosis
- fALS
- familial ALS
- PFN1
- profilin 1
- TDP-43
- TAR DNA-binding protein of 43 kDa
- FTLD
- frontotemporal lobar degeneration
- TG
- transgenic
- NLS
- nuclear localization signal
- chic
- chickadee
- VCP
- valosin-containing protein.
References
- 1. Rosen D. R., Siddique T., Patterson D., Figlewicz D. A., Sapp P., Hentati A., Donaldson D., Goto J., O'Regan J. P., Deng H. X., Rahmani Z., Krizus A., McKenna-Yasek D., Cayabyab A., Gaston S. M., et al. (1993) Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62 [DOI] [PubMed] [Google Scholar]
- 2. Sreedharan J., Blair I. P., Tripathi V. B., Hu X., Vance C., Rogelj B., Ackerley S., Durnall J. C., Williams K. L., Buratti E., Baralle F., de Belleroche J., Mitchell J. D., Leigh P. N., Al-Chalabi A., et al. (2008) TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319, 1668–1672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gitcho M. A., Baloh R. H., Chakraverty S., Mayo K., Norton J. B., Levitch D., Hatanpaa K. J., White C. L. 3rd., Bigio E. H., Caselli R., Baker M., Al-Lozi M. T., Morris J. C., Pestronk A., et al. (2008) TDP-43 A315T mutation in familial motor neuron disease. Ann. Neurol. 63, 535–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yokoseki A., Shiga A., Tan C. F., Tagawa A., Kaneko H., Koyama A., Eguchi H., Tsujino A., Ikeuchi T., Kakita A., Okamoto K., Nishizawa M., Takahashi H., and Onodera O. (2008) TDP-43 mutation in familial amyotrophic lateral sclerosis. Ann. Neurol. 63, 538–542 [DOI] [PubMed] [Google Scholar]
- 5. Kabashi E., Valdmanis P. N., Dion P., Spiegelman D., McConkey B. J., Vande Velde C., Bouchard J. P., Lacomblez L., Pochigaeva K., Salachas F., Pradat P. F., Camu W., Meininger V., Dupre N., and Rouleau G. A. (2008) TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 40, 572–574 [DOI] [PubMed] [Google Scholar]
- 6. Wu C. H., Fallini C., Ticozzi N., Keagle P. J., Sapp P. C., Piotrowska K., Lowe P., Koppers M., McKenna-Yasek D., Baron D. M., Kost J. E., Gonzalez-Perez P., Fox A. D., Adams J., Taroni F., et al. (2012) Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature 488, 499–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ingre C., Landers J. E., Rizik N., Volk A. E., Akimoto C., Birve A., Hübers A., Keagle P. J., Piotrowska K., Press R., Andersen P. M., Ludolph A. C., and Weishaupt J. H. (2013) A novel phosphorylation site mutation in profilin 1 revealed in a large screen of US, Nordic, and German amyotrophic lateral sclerosis/frontotemporal dementia cohorts. Neurobiol. Aging 34, 1708.e1–e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fratta P., Charnock J., Collins T., Devoy A., Howard R., Malaspina A., Orrell R., Sidle K., Clarke J., Shoai M. Lu C. H., Hardy J., Plagnol V., and Fisher E. M. (2014) Profilin1 E117G is a moderate risk factor for amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Phychiatry 85, 506–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Smith B. N., Vance C., Scotter E. L., Troakes C., Wong C. H., Topp S., Maekawa S., King A., Mitchell J. C., Lund K., Al-Chalabi A., Ticozzi N., Silani V., Sapp P., Brown R. H. Jr., et al. (2015) Novel mutations support a role for Profilin 1 in the pathogenesis of ALS. Neurobiol. Aging 36, 1602.e17–1602.e27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schafer D. A., and Cooper J. A. (1995) Control of actin assembly at filament ends. Annu. Rev. Cell Dev. Biol. 11, 497–518 [DOI] [PubMed] [Google Scholar]
- 11. Witke W., Sutherland J. D., Sharpe A., Arai M., and Kwiatkowski D. J. (2001) Profilin I is essential for cell survival and cell division in early mouse development. Proc. Natl. Acad. Sci. U.S.A. 98, 3832–3836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kullmann J. A., Neumeyer A., Gurniak C. B., Friauf E., Witke W., and Rust M. B. (2012) Profilin 1 is required for glial cell adhesion and radial migration of cerebellar granule neurons. EMBO Rep. 13, 75–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Figley M. D., Bieri G., Kolaitis R. M., Taylor J. P., and Gitler A. D. (2014) Profilin 1 associates with stress granules and ALS-linked mutations alter stress granule dynamics. J. Neurosci. 34, 8083–8097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ling S. C., Polymenidou M., and Cleveland D. W. (2013) Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79, 416–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Neumann M., Sampathu D. M., Kwong L. K., Truax A. C., Micsenyi M. C., Chou T. T., Bruce J., Schuck T., Grossman M., Clark C. M., McCluskey L. F., Miller B. L., Masliah E., Mackenzie I. R., Feldman W., et al. (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133 [DOI] [PubMed] [Google Scholar]
- 16. Arai T., Hasegawa M., Akiyama H., Ikeda K., Nonaka T., Mori H., Mann D., Tsuchiya K., Yoshida M., Hashizume Y., and Oda T. (2006) TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 351, 602–611 [DOI] [PubMed] [Google Scholar]
- 17. Wu L. S., Cheng W. C., and Shen C. K. (2012) Targeted depletion of TDP-43 expression in the spinal cord motor neurons leads to the development of amyotrophic lateral sclerosis. J. Biol. Chem. 287, 27335–27344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ihara R., Matsukawa K., Nagata Y., Kunugi H., Tsuji S., Chihara T., Kuranaga E., Miura M., Wakabayashi T., Hashimoto T., and Iwatsubo T. (2013) RNA binding mediates neurotoxicity in the transgenic Drosophila model of TDP-43 proteinopathy. Hum. Mol. Genet. 22, 4474–4484 [DOI] [PubMed] [Google Scholar]
- 19. Brand A. H., and Perrimon N. (1993) Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118, 401–415 [DOI] [PubMed] [Google Scholar]
- 20. Kumar J. P. (2001) Signaling pathways in drosophila and vertebrate retinal development. Nat. Rev. Genet. 2, 846–857 [DOI] [PubMed] [Google Scholar]
- 21. Kwiatkowski T. J. Jr., Bosco D. A., Leclerc A. L., Tamrazian E., Vanderburg C. R., Russ C., Davis A., Gilchrist J., Kasarskis E. J., Munsat T. Valdmanis P., Rouleau G. A., Hosler B. A., Cortelli P., et al. (2009) Mutations in FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323, 1205–1208 [DOI] [PubMed] [Google Scholar]
- 22. Vance C., Rogelj B., Hortobágyi T., De Vos K. J., Nishimura A. L., Sreedharan J., Hu X., Smith B., Ruddy D., Wright P., Ganesalingam J., Williams K. L., Tripathi V., Al-Saraj S., Al-Chalabi A., et al. (2009) Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis 6. Science 323, 1208–1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Neumann M., Rademakers R., Roeber S., Baker M., Kretzschmar H. A., and Mackenzie I. R. (2009) A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain 132, 2922–2931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kobayashi Z., Tsuchiya K., Arai T., Aoki M., Hasegawa M., Ishizu H., Akiyama H., and Mizusawa H. (2010) Occurrence of basophilic inclusions and FUS-immunoreactive neuronal and glial inclusions in a case of familial amyotrophic lateral sclerosis. J. Neurol. Sci. 293, 6–11 [DOI] [PubMed] [Google Scholar]
- 25. Shao J., and Diamond M. I. (2012) Protein phosphatase 1 dephosphorylates profilin-1 at Ser-137. PLoS ONE 7, e32802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ayala Y. M., Zago P., D'Ambrogio A., Xu Y. F., Petrucelli L., Buratti E., and Baralle F. E. (2008) Structural determinants of the cellular localization and shuttling of TDP-43. J. Cell Sci. 121, 3778–3785 [DOI] [PubMed] [Google Scholar]
- 27. Winton M. J., Igaz L. M., Wong M. M., Kwong L. K., Trojanowski J. Q., and Lee V. M. (2008) Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. J. Biol. Chem. 283, 13302–13309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. De Conti L., Akinyi M. V., Mendoza-Maldonado R., Romano M., Baralle M., and Buratti E. (2015) TDP-43 affects splicing profiles and isoform production of genes involved in the apoptotic and mitotic cellular pathways. Nucleic Acids Res. 43, 8990–9005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fiesel F. C., Weber S. S., Supper J., Zell A., and Kahle P. J. (2012) TDP-43 regulates global translational yield by splicing of exon junction complex component SKAR. Nucleic Acids Res. 40, 2668–2682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fiesel F. C., Voigt A., Weber S. S., Van den Haute C., Waldenmaier A., Görner K., Walter M., Anderson M. L., Kern J. V., Rasse T. M., Schmidt T., Springer W., Kirchner R., Bonin M., Neumann M., et al. (2010) Knockdown of transactive response DNA-binding protein (TDP-43) downregulates histone deacetylase 6. EMBO J. 29, 209–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ritson G. P., Custer S. K., Freibaum B. D., Guinto J. B., Geffel D., Moore J., Tang W., Winton M. J., Neumann M., Trojanowski J. Q., Lee V. M., Forman M. S., and Taylor J. P. (2010) TDP-43 mediates degeneration in a novel Drosophila model of disease caused by mutations in VCP/p97. J. Neurosci. 30, 7729–7739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Miguel L., Frébourg T., Campion D., and Lecourtois M. (2011) Both cytoplasmic and nuclear accumulations of the protein are neurotoxic in Drosophila models of TDP-43 proteinopathies. Neurobiol. Dis. 41, 398–406 [DOI] [PubMed] [Google Scholar]
- 33. van Blitterswijk M., Baker M. C., Bieniek K. F., Knopman D. S., Josephs K. A., Boeve B., Caselli R., Wszolek Z. K., Petersen R., Graff-Radford N. R., Boylan K. B., Dickson D. W., and Rademakers R. (2013) Profilin-1 mutations are rare in patients with amyotrophic lateral sclerosis and frontotemporal dementia. Amyotroph. Lateral Scler. Frontotemporal Degener. 14, 463–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu-Yesucevitz L., Bilgutay A., Zhang Y. J., Vanderwyde T., Citro A., Mehta T., Zaarur N., McKee A., Bowser R., Sherman M., Petrucelli L., and Wolozin B. (2010) Tar DNA binding protein-43 (TDP-43) associates with stress granules: analysis of cultured cells and pathological brain tissue. PLoS ONE 5, e13250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. McDonald K. K., Aulas A., Destroismaisons L., Pickles S., Beleac E., Camu W., Rouleau G. A., and Vande Velde C. (2011) TAR DNA-binding protein 43 (TDP-43) regulates stress granule dynamics via differential regulation of G3BP and TIA-1. Hum. Mol. Genet. 20, 1400–1410 [DOI] [PubMed] [Google Scholar]
- 36. Bentmann E., Neumann M., Tahirovic S., Rodde R., Dormann D., and Haass C. (2012) Requirements for stress granule recruitment of fused in sarcoma (FUS) and TAR DNA-binding protein of 43 kDa (TDP-43). J. Biol. Chem. 287, 23079–23094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Anderson P., and Kedersha N. (2008) Stress granules: the Tao of RNA triage. Trends Biochem. Sci. 33, 141–150 [DOI] [PubMed] [Google Scholar]
- 38. Kedersha N., Ivanov P., and Anderson P. (2013) Stress granules and cell signaling: more than just a passing phase. Trends Biochem. Sci. 38, 494–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kim H. J., Raphael A. R., LaDow E. S., McGurk L., Weber R. A., Trojanowski J. Q., Lee V. M., Finkbeiner S., Gitler A. D., and Bonini N. M. (2014) Therapeutic modulation of eIF2α phosphorylation rescues TDP-43 toxicity in amyotrophic lateral sclerosis disease models. Nat. Genet. 46, 152–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tanaka Y., Nonaka T., Suzuki G., Kametani F., and Hasegawa M. (2016) Gain-of-function profilin 1 mutations linked to familial amyotrophic lateral sclerosis cause seed-dependent intracellular TDP-43 aggregation. Hum. Mol. Genet. 25, 1420–1433 [DOI] [PubMed] [Google Scholar]
- 41. Watts G. D., Wymer J., Kovach M. J., Mehta S. G., Mumm S., Darvish D., Pestronk A., Whyte M. P., and Kimonis V. E. (2004) Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused mutant valosin-containing protein. Nat. Genet. 36, 377–381 [DOI] [PubMed] [Google Scholar]
- 42. Johnson J. O., Mandrioli J., Benatar M., Abramzon Y., Van Deerlin V. M., Trojanowski J. Q., Gibbs J. R., Brunetti M., Gronka S., Wuu J., Ding J., McCluskey L., Martinez-Lage M., Falcone D., Hernandez D. G., et al. (2010) Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 68, 857–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Neumann M., Mackenzie I. R., Cairns N. J., Boyer P. J., Markesbery W. R., Smith C. D., Taylor J. P., Kretzschmar H. A., Kimonis V. E., and Forman M. S. (2007) TDP-43 in the ubiquitin pathology of frontotemporal dementia with VCP gene mutations. J. Neuropathol. Exp. Neurol. 66, 152–157 [DOI] [PubMed] [Google Scholar]
- 44. Gitcho M. A., Strider J., Carter D., Taylor-Reinwald L., Forman M. S., Goate A. M., and Cairns N. J. (2009) VCP mutations causing frontotemporal lobar degeneration disrupt localization of TDP-43 and induce cell death. J. Biol. Chem. 284, 12384–12398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. DeJesus-Hernandez M., Mackenzie I. R., Boeve B. F., Boxer A. L., Baker M., Rutherford N. J., Nicholson A. M., Finch N. A., Flynn H., Adamson J., Kouri N., Wojtas A., Sengdy P., Hsiung G. Y., Karydas A., et al. (2011) Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Renton A. E., Majounie E., Waite A., Simón-Sánchez J., Rollinson S., Gibbs J. R., Schymick J. C., Laaksovirta H., van Swieten J. C., Myllykangas L., Kalimo H., Paetau A., Abramzon Y., Remes A. M., Kaganovich A., et al. (2011) A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang K., Donnelly C. J., Haeusler A. R., Grima J. C., Machamer J. B., Steinwald P., Daley E. L., Miller S. J., Cunningham K. M., Vidensky S., Gupta S., Thomas M. A., Hong I., Chiu S. L., Huganir R. L., et al. (2015) The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 525, 56–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cooley L., Verheyen E., and Ayers K. (1992) Chickadee encodes a profilin required for intercellular cytoplasm transport during Drosophila oogenesis. Cell 69, 173–184, [DOI] [PubMed] [Google Scholar]
- 49. Wills Z., Marr L., Zinn K., Goodman C. S., and Van Vactor D. (1999) Profilin and the Abl tyrosine kinase are required for motor axon outgrowth in the Drosophila embryo. Neuron 22, 291–299 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.