

Abstract
The first examples of 3,3‐diaryloxetanes are prepared in a lithium‐catalyzed and substrate dependent divergent Friedel–Crafts reaction. para‐Selective Friedel–Crafts reactions of phenols using oxetan‐3‐ols afford 3,3‐diaryloxetanes by displacement of the hydroxy group. These constitute new isosteres for benzophenones and diarylmethanes. Conversely, ortho‐selective Friedel–Crafts reactions of phenols afford 3‐aryl‐3‐hydroxymethyl‐dihydrobenzofurans by tandem alkylation–ring‐opening reactions; the outcome of the reaction diverging to structurally distinct products dependent on the substrate regioselectivity. Further reactivity of the oxetane products is demonstrated, suitable for incorporation into drug discovery efforts.
Keywords: carbocations, homogeneous catalysis, lithium, oxetanes, oxygen heterocycles
Introduction
The use of bioisosteres to replace problematic functionality whilst maintaining advantageous characteristics is an important strategy in drug discovery.1 The implementation of new isosteres provides access to novel chemical as well as intellectual property (IP) space, and presents interesting property modulation of biologically significant groups.2 Diarylketones display important biological activity and are prevalent in natural products (Figure 1).3, 4 However, their use in medicinal chemistry efforts suffers from issues of metabolic and chemical stability due to the electrophilic carbonyl, and lack of appropriate isosteres. Indeed, benzophenones are commonly exploited as photoactivatable cross‐linkers in chemical biology.5 Similarly, diarylmethane derivatives provide varied biological activity,6 for example tesmilifene has shown activity as a chemopotentiator in clinical studies including for metastatic breast cancer.7 However, the methylene group can often present a metabolic liability.8 The incorporation of blocking substituents, such as gem‐dimethyl groups, can improve metabolic stability, but deliver a negative impact on a compounds physicochemical properties, such as lipophilicity (LogD) and solubility. A similar motif appears in bisphenol A (BPA), used in bulk as a monomer in polycarbonates to ensure optically clarity, and in other diverse consumer products.9 This compound is controversial due to potential interference with the endocrine system and is banned in baby bottles, for example, in many countries. Due to these varied functions diaryl motifs have been the subject of wide ranging synthetic efforts.10
Figure 1.
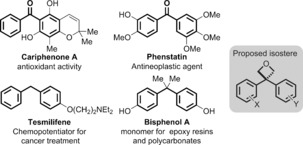
Some important benzophenones and diarylmethanes and proposed oxetane replacement.
Recently, Carreira has presented oxetanes as replacement motifs for gem‐dimethyl and carbonyl derivatives due to similar spatial properties and lone pair arrangements.11, 12 In drug discovery settings, the incorporation of the polar, low molecular weight oxetane moiety has often afforded compounds with enhanced properties such as improved metabolic stability, solubility and lipophilicity.13 As part of our interest in the synthesis of novel oxetane derivatives,14 we were intrigued by possible approaches to 3,3‐diaryloxetanes, designed to provide isosteres for diarylketone and diarylmethane structures with improved properties for drug discovery. At the outset of this work, there were no reported examples of disubstituted 3,3‐diaryloxetanes in the literature.15, 16 Indeed, internal efforts within Pfizer corroborated this observation, with this motif being recognized as difficult to access.
Herein we report the synthesis of 3,3‐diaryloxetanes, prepared by a lithium‐catalyzed Friedel–Crafts reaction of phenols with readily accessible oxetan‐3‐ols. We also disclose an efficient synthesis of 3‐aryl‐3‐hydroxymethyl‐dihydrobenzofuran derivatives using the same conditions, the reaction outcome being dependent on the regioselectivity of the Friedel–Crafts reaction. This presents a rare example of a divergent reaction whereby a regiochemical difference leads to structurally distinct products, so avoiding closely related isomers.17
Results and Discussion
We envisaged that the synthesis of 3,3‐diaryloxetanes might be achieved through a catalytic Friedel–Crafts reaction, directly displacing a hydroxy group from 3‐aryloxetan‐3‐ols. These substrates would be readily generated by the addition of aryl‐metal reagents to commercially available oxetan‐3‐one.11a However, this strategy posed interesting questions on the nature and feasibility of the required carbocationic intermediate due to the increased p‐character of the strained ring bonds, and the electron‐withdrawing nature of the oxygen atom. To date there were only two transformations to displace the hydroxy group of tert‐oxetanol derivatives, both on aryl derivatives; i) direct OH displacement with diethylaminosulfur trifluoride (DAST) to generate tertiary fluorides and ii) conversion to the tosylate and reduction with LiAlH4 to form the 3‐aryl oxetane.11a Furthermore, achieving the desired transformation under catalytic conditions (see Scheme 1), would require the activation of the hydroxyl group selectively, over coordination to the Lewis basic oxetane lone pairs18 and other oxygen containing species, and must prevent ring opening polymerization of the oxetanes under acidic conditions.19
Scheme 1.
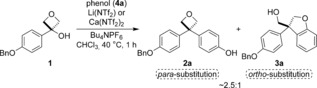
Friedel–Crafts alkylation of oxetane 1 with phenol.
We selected aryl‐oxetanol 1 for investigation being likely to stabilize a carbocationic intermediate, and with the potential to reveal phenol functionality for further derivatization on benzyl deprotection (Scheme 1). Inspired by recent developments of Friedel–Crafts reactions displacing hydroxy groups at tertiary centers,20, 21 we examined a variety of Lewis acids as potential catalysts with electron rich aromatic nucleophiles.22 We were delighted to observe that both calcium triflimide with Bu4NPF6 additive, as reported by Niggemann,21 and lithium triflimide/Bu4NPF6 successfully formed the 3,3‐diaryloxetane 2 a using excess phenol on heating at 40 °C in chloroform. This oxetane product was formed exclusively as the para‐substituted phenol derivative, in a ratio of around 2.5:1 with a minor product. Intriguingly, the minor product was identified as 2,3‐dihydrobenzofuran 3 a resulting from reaction at the ortho‐position and oxetane ring opening. The two heterocyclic products 2 a and 3 a were readily separated by flash column chromatography to give structurally distinct compounds. This suggested the potential for an unusual substrate controlled structurally divergent reaction based on the phenol regioselectivity.
To investigate and optimize the parameters of the reaction, a design‐of‐experiments (DOE) approach was adopted, and was conducted for both Ca and Li catalysts.22 Temperature, concentration, catalyst and additive loading, equivalents of nucleophile and reaction time were investigated (see Table 1 for representative results). Comparable results were obtained with Ca(NTf2)2 at 5 mol % and Li(NTf2) at 10 mol % loadings (entries 1 and 2). On optimization the best results were achieved using the Li(NTf2) catalyst (11 mol %) and five equivalents phenol affording a combined yield of 83 % for the two products after 1 h (entry 3). The Bu4NPF6 additive was needed, presumably to solubilize the catalyst, and a 2:1 ratio of Li(NTf2) to Bu4NPF6 gave the best yield (entries 3–5). Increasing the amount of phenol to 10 equivalents did not result in an improvement. A decrease in the yield of oxetane product was observed on further increasing the catalyst loading to 20 mol % (entry 7). Dichloroethane, chlorobenzene and hexane were also suitable solvents but delivered slightly reduced yields of oxetane (52–58 %). Both Li(NTf2) and Bu4NPF6 are inexpensive and easily handled powders. Increasing the scale of the reaction to 7.5 mmol gave comparable results, providing >1 g of oxetane 2 a (57 %) along with dihydrobenzofuran 3 a (21 %, entry 8). Oxetane 2 a was stored at room temperature in air for >6 months without any noticeable degradation.
Table 1.
Selected optimization for the reaction of 1 with phenol.
| Entry[a] | Catalyst | Cat./Bu4NPF6 [mol %] | Equiv. phenol | Yield 2 a [%][b] | Yield 3 a [%][b] |
|---|---|---|---|---|---|
| 1 | Ca(NTf2)2 | 5/5 | 3 | 44 | 19 |
| 2 | Li(NTf2) | 10/5 | 3 | 47 | 19 |
| 3 | Li(NTf2) | 11/5.5 | 5 | 63 (61)[c] | 20 (19)[c] |
| 4 | Li(NTf2) | 11/‐ | 5 | 0 | 0 |
| 5 | Li(NTf2) | 11/11 | 5 | 22 | 6[d] |
| 6 | Li(NTf2) | 11/5.5 | 10 | 65 | 21 |
| 7 | Li(NTf2) | 20/10 | 5 | 51 | 20 |
| 8[e] | Li(NTf2) | 11/5.5 | 5 | (57)[c] | (21)[c] |
[a] Conditions: 1 (0.25 mmol), CHCl3, 0.5 m, 40 °C, 1 h. [b] Yield determined by 1H NMR using 1,3,5‐trimethoxybenzene as internal standard. [c] Yield of isolated product in parenthesis. [d] 1 recovered in 46 %. [e] 7.5 mmol 1 used.
With optimized conditions in hand, the scope of the reaction to selectively form oxetane products was investigated. Pleasingly, the use of ortho‐substituted phenols gave excellent selectivity for the diaryloxetane products, which were formed exclusively (Scheme 2, <3 % dihydrobenzofuran for all examples by 1H NMR of the crude reaction). Electron‐rich phenols gave high yields of diaryloxetanes 2 a–c. Electron‐withdrawing and electron‐neutral ortho‐substituents were tolerated on phenol (2 d–f, h), though reduced yields were obtained for the halide examples. This is likely to be due to decomposition of the unstable carbocation with lower rates of addition for these nucleophiles. In all cases the starting material was consumed within the 1 h reaction time. Catechol and 1‐naphthol also proved to be suitable nucleophiles with the diaryloxetanes 2 g and 2 i formed good yield (63 and 44 %, respectively). Furthermore, dimethoxybenzene provided an example without phenol functionality (2 j), albeit in low yield.
Scheme 2.
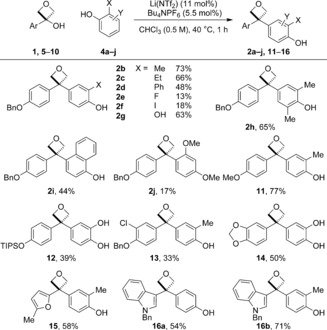
Scope of 3,3‐diaryloxetanes using ortho‐substituted phenols.
Alternative 3‐aryl‐oxetan‐3‐ol substrates were prepared (5–10), each with a para‐donating aromatic groups (see the Supporting Information). Using either catechol or 2‐methylphenol gave the desired oxetane products in each case (11–14) in 33–77 % yields. Importantly, heterocycles were also suitable as the preinstalled aromatic group. 2‐Methylfuran was capable of stabilizing the carbocationic intermediate affording oxetane 15 in 58 % yield, providing another functionalizable handle. Furthermore, indole‐containing oxetanes 16 a and 16 b were formed in high yields under the same conditions. As such, by using a combination of different nucleophiles and preinstalled aryl groups a variety of 3,3‐diaryl‐oxetanes can be rapidly prepared, with attractive functionality for further elaboration.
The dihydrobenzofuran product 3 a would exhibit the diaryl motif in an alternative defined orientation and represented an interesting compound in its own right. This scaffold is present in a multitude of natural products and synthetic compounds including marketed drugs with wide ranging biological activity.23, 24 The intramolecular ring opening of oxetane products to generate other heterocycles has received interest in recent years,25 including the formation of certain dihydrobenzofuran derivatives.26 Herein little studied 3‐aryl‐3‐hydroxymethyl‐dihydrobenzofurans are prepared in a short sequence,27 whereby oxetan‐3‐one functions as a ‘triple electrophile’ over 2 steps.
We anticipated that blocking the phenol para‐position would afford these products exclusively, which was proven correct (Scheme 3). Dihydrobenzofurans were prepared using a wide selection of para‐ and meta‐substituted phenols and related compounds. para‐Substituted phenols (4 k–o) were successful using an extended reaction time of 20 h. As with the oxetane products, more electron donating substituents gave good yields, and lower yields for halide substituted examples. A 98 % yield of sesamol derivative 3 q was obtained, formed as a single regioisomer. 3‐Methoxyphenol also gave exclusively the dihydrobenzofuran product 3 r, the steric difference between hydroxy and methoxy proving sufficient to completely prevent addition at the 4‐position of the phenol. Resorcinols proved to be good substrates forming the dihydrobenzofuran products exclusively in yields of 67–74 % (3 s–v). With 5‐methylresorcinol, a mixture of dihydrobenzofuran regioisomers were obtained in 73 % combined yield (3 w and 3 w’). 2‐Napthol derivatives gave excellent yields of single regioisomers of derivatives 3 x and 3 y. Substrates in which the preinstalled aromatic group was varied were successful upon reaction with p‐methylphenol to give dihydrobenzofuran derivatives 17–21.
Scheme 3.
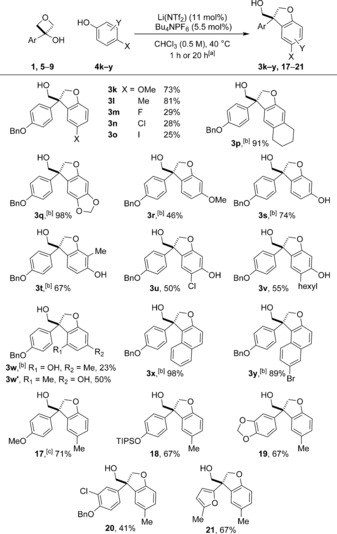
Scope of dihydrobenzofurans using meta and para‐substituted phenols. [a] Reaction time of 20 h unless stated. [b] Reaction time of 1 h. [c] Reaction time of 6 h.
Only a small selection of meta‐substituted phenols gave rise to mixtures of product types, with the product ratio affected by the bulk of the meta‐substituent and the presence of an additional ortho‐substituent (Scheme 4). Importantly the structural isomers were readily separable in each case. With 3‐methylphenol as the nucleophile, dihydrobenzofuran 3 z was formed as the major product yield (2 z:3 z;1:2) in 86 % combined yield. 2,3‐Dimethylphenol led to 3,3‐diaryloxetane 2 aa as the major product. The reaction with 3‐fluorophenol showed a slight preference for oxetane 2 ab, and dihydrobenzofuran 3 ab was formed as a mixture of 6‐F and 4‐F isomers.
Scheme 4.
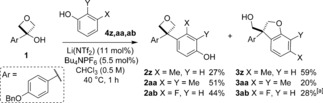
Some meta‐substituted phenols gave separable mixtures of oxetane and dihydrobenzofuran products. [a] Formed as a mixture of 6‐F and 4‐F dihydrobenzofurans (85:15).
With a selection of 3,3‐diaryloxetanes available, we demonstrated the removal of the benzyl group to reveal the phenol functionality. Simple hydrogenolysis of 2 a using Pd/C under H2 gave oxetane 22 in quantitative yield (Scheme 5). Oxetane 22 is a novel derivative of BPA, in which the gem‐dimethyl is linked with an oxygen atom, which may present interesting properties as a BPA alternative.
Scheme 5.
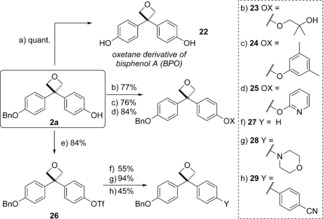
Derivatization of 3,3‐diaryloxetane 2 a. a) Pd/C (10 % w/w), H2 (1 atm), EtOH, 0.1 m, 25 °C, 14 h. b) 1,1‐Dimethyloxirane, K2CO3, DMF, 0.5 m, 100 °C, 16 h. c) 5‐Iodo‐m‐xylene, CuI (5 mol %), N,N‐dimethylglycine.HCl (18.5 mol %), Cs2CO3, dioxane, 0.5 m, 90 °C, 20 h. d) 2‐iodopyridine, CuI (5 mol %), picolinic acid (10 mol %), K3PO4, DMSO, 0.5 m, 90 °C, 23 h. e) Tf2O, pyridine, CH2Cl2, 0.5 m, 25 °C, 2 h. f) Pd(OAc)2 (2 mol %), 1,1′‐ferrocenediyl‐bis(diphenylphosphine) (=dppf) (2 mol %), Et3N, HCO2H, DMF, 0.5 m, 60 °C, 1 h. g) Morpholine, Pd(OAc)2 (1 mol %), JohnPhos (2 mol %), K3PO4, THF, 0.5 m, 65 °C, 24 h. h) 4‐Cyanoboronic acid, Pd(OAc)2 (5 mol %), SPhos (10 mol %), K3CO3, THF, 0.5 m, 65 °C, 18 h.
To exploit the phenol functionality, the derivatization of oxetane 2 a was explored through a range of reactions, intended to demonstrate the stability of the oxetane unit. Alkylation by ring opening of 1,1‐dimethyloxirane under basic conditions gave oxetane 23 in 77 % yield. Copper‐catalyzed Ullmann couplings proceeded in good yields with both 1‐iodo‐3,5‐dimethylbenzene and 2‐iodopyridine (24 and 25),28 indicating good stability of the oxetane over extended reaction times at elevated temperatures. Conversion of oxetane 2 a to triflate 26 was successful under standard conditions (Tf2O, pyridine). From triflate 26, palladium‐catalyzed deoxygenation afforded phenyl substituted oxetane 27 (X=H).29 Buchwald–Hartwig amination gave morpholine substituted oxetane 28 in an excellent 94 % yield.30 Finally, Suzuki–Miyaura cross‐coupling using Pd(OAc)2 and SPhos gave biaryl 29.
Mechanistically, we propose a catalytic cycle involving initial activation of the Li(NTf2) pre‐catalyst through disruption of the tetrahedrally coordinated solid‐state lattice,31 aided by the ammonium salt additive (Scheme 6). Coordination and activation of the tertiary‐hydroxy group is likely to generate the key carbocationic intermediate. Addition of phenol through the para‐ or ortho‐position would generate intermediates 30 or 31, respectively (boxed). From intermediate 30, rearomatization would yield the 3,3‐diaryloxetane product 2 a, with a loss of water regenerating the catalyst. Conversely, if ortho‐addition into the carbocation occurs, rearomatization and coordination of the Lewis acid catalyst to the oxetane oxygen initiates intramolecular cyclization to dihydrobenzofuran 3 a. This is rapid and the ortho‐substituted diaryloxetane was not observed in any dihydrobenzofuran forming example. On this path, the Li catalyst displays selective sequential activation of the hydroxy group for Friedel–Crafts alkylation and then activation of the oxetane to ring opening.
Scheme 6.
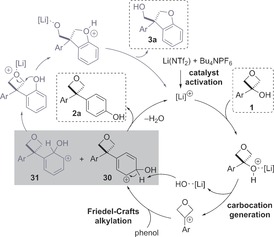
Proposed catalytic cycle.
Conclusion
We have developed a rapid approach to 3,3‐diaryloxetane and 2,3‐dihydrobenzofuran scaffolds through a Friedel–Crafts reaction on oxetanols with phenols, using an inexpensive and widely available lithium catalyst. The reaction is divergent to give the two heterocyclic classes which can be accessed with complete selectivity dependent on the ortho‐ or para‐regioselectivity of the phenol substrates. We present successful conditions for the efficient synthesis of 3,3‐diaryloxetanes, the first examples of this motif, designed as an isostere for diarylmethanes and diaryl ketones. High yields were achieved using ortho‐substituted phenols and related derivatives, with complete selectivity for the 3,3‐diaryloxetane product observed. Remarkably, the lithium catalyst for the Friedel–Crafts reaction selectively activates the tertiary‐hydroxy group of oxetan‐3‐ols without being impeded by coordination to other oxygen containing species present, and without causing ring opening or polymerization of the oxetanes. Furthermore, good reactivity and stability of the products to a number of synthetically useful transformations such as alkylation, deoxygenation and metal‐catalyzed cross‐coupling reactions, was demonstrated, suitable for their use in medicinal chemistry programs. In all cases the oxetane derivatives showed very high stability to the reaction conditions. Under the same conditions, ortho‐selective alkylation afforded the synthesis of dihydrobenzofurans, also a biologically important motif, by one‐pot Friedel–Crafts alkylation and oxetane ring opening. The use of meta‐ and para‐substituted phenols, resourcinols and 2‐napthol derivatives enabled formation of these novel scaffolds in high yields. Both product classes present novel substitution patterns on the O‐heterocycles and a wide scope of derivatives is prepared. The reaction likely proceeds via an unusual carbocationic intermediate on the oxetane ring formed through selective activation of the hydroxyl group of oxetan‐3‐ols. Future studies on the properties of the 3,3‐diaryloxetanes, and application to other ring systems will be reported in due course.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
For financial support we gratefully acknowledge The Royal Society for a University Research Fellowship (to J.A.B.), the EPSRC [CAF to J.A.B. (EP/J001538/1), and Impact Acceleration Account (EP/K503733/1)], and Imperial College London for a PhD Scholarship (to R.A.C). We acknowledge the EPSRC National Mass Spectrometry Facility, Swansea. The authors would also like to thank Y. Fobian, M. Bunnage, and D. Owen for their support and insightful discussions.
R. A. Croft, J. J. Mousseau, C. Choi, J. A. Bull, Chem. Eur. J. 2016, 22, 16271.
Contributor Information
Rosemary A. Croft, http://www3.imperial.ac.uk/people/j.bull
Dr. James A. Bull, Email: j.bull@imperial.ac.uk.
References
- 1. Meanwell N. A., J. Med. Chem. 2011, 54, 2529–2591. [DOI] [PubMed] [Google Scholar]
- 2. Goldberg F. W., Kettle J. G., Kogej T., Perry M. W. D., Tomkinson N. P., Drug Discovery Today 2015, 20, 11–17. [DOI] [PubMed] [Google Scholar]
- 3. Wu S.-B., Long C., Kennelly E. J., Nat. Prod. Rep. 2014, 31, 1158–1174. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a.For cariphenone: Bernardi A. P. M., Ferraz A. B. F., Albring D. V., Bordignon S. A. L., Schripsema J., Bridi R., Dutra-Filho C. S., Henriques A. T., von Poser G. L., J. Nat. Prod. 2005, 68, 784–786; [DOI] [PubMed] [Google Scholar]
- 4b.For phenstatin: Pettit G. R., Toki B., Herald D. L., Verdier-Pinard P., Boyd M. R., Hamel E., Pettit R. K., J. Med. Chem. 1998, 41, 1688–1695. [DOI] [PubMed] [Google Scholar]
- 5. Preston G. W., Wilson A. J., Chem. Soc. Rev. 2013, 42, 3289–3301. [DOI] [PubMed] [Google Scholar]
- 6. Mondal S., Panda G., RSC Adv. 2014, 4, 28317–28358. [Google Scholar]
- 7.
- 7a. Brandes L. J., Hermonat M. W., Biochem. Biophys. Res. Commun. 1984, 123, 724–728; [DOI] [PubMed] [Google Scholar]
- 7b. Reyno L. L., Seymour L., Tu D., Dent S., Gelmon K., Walley B., Pluzanska A., Gorbunova V., Garin A., Jassem J., Pienkowski T., Dancey J., Pearce L., MacNeil M., Marlin S., Lebwohl D., Voi M., Pritchard K., J. Clin. Oncol. 2003, 22, 269–276; [DOI] [PubMed] [Google Scholar]
- 7c. Walter F. R., Veszelka S., Pásztói M., Péterfi Z. A., Tóth A., Rákhely G., Cervenak L., Ábrahám C. S., Deli M. A., J. Neurochem. 2015, 134, 1040–1054. [DOI] [PubMed] [Google Scholar]
- 8.For studies on oxidation, see: Sterckx H., De Houwer J., Mensch C., Caretti I., Tehrani K. A., Herrebout W. A., Van Doorslaer S., Maes B. U. W., Chem. Sci. 2016, 7, 346–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Geens T., Goeyens L., Covaci A., Int. J. Hyg. Environ. Health 2011, 214, 339–347. [DOI] [PubMed] [Google Scholar]
- 10.For selected recent synthetic strategies towards diarylketones:
- 10a. Weires N. A., Baker E. L., Garg N. K., Nat. Chem. 2015, 8, 75–79; [DOI] [PubMed] [Google Scholar]
- 10b. Bechara W. S., Pelletier G., Charette A. B., Nat. Chem. 2012, 4, 228–234. Towards diarylmethanes: [DOI] [PubMed] [Google Scholar]
- 10c. Barluenga J., Tomás-Gamasa M., Aznar F., Valdés C., Nat. Chem. 2009, 1, 494–499; [DOI] [PubMed] [Google Scholar]
- 10d. Endo K., Ishioka T., Ohkubo T., Shibata T., J. Org. Chem. 2012, 77, 7223–7231; [DOI] [PubMed] [Google Scholar]
- 10e. Zhang F.-L., Hong K., Li T.-J., Park H., Yu J.-Q., Science 2016, 351, 252–256; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10f. Mo X., Yakiwchuk J., Dansereau J., McCubbin J. A., Hall D. G., J. Am. Chem. Soc. 2015, 137, 9694–9703. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Wuitschik G., Rogers-Evans M., Müller K., Fischer H., Wagner B., Schuler F., Polonchuk L., Carreira E. M., Angew. Chem. Int. Ed. 2006, 45, 7736–7739; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 7900–7903; [Google Scholar]
- 11b. Wuitschik G., Rogers-Evans M., Buckl A., Bernasconi M., Märki M., Godel T., Fischer H., Wagner B., Parrilla I., Schuler F., Schneider J., Alker A., Schweizer W. B., Müller K., Carreira E. M., Angew. Chem. Int. Ed. 2008, 47, 4512–4515; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 4588–4591; [Google Scholar]
- 11c. Wuitschik G., Carreira E. M., Wagner B., Fischer H., Parrilla I., Schuler F., Rogers-Evans M., Müller K., J. Med. Chem. 2010, 53, 3227–3246; [DOI] [PubMed] [Google Scholar]
- 11d. Burkhard J. A., Wuitschik G., Rogers-Evans M., Müller K., Carreira E. M., Angew. Chem. Int. Ed. 2010, 49, 9052–9067; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 9236–9251. [Google Scholar]
- 12.For oxetane peptide isosteres, see:
- 12a. McLaughlin M., Yazaki R., Fessard T. C., Carreira E. M., Org. Lett. 2014, 16, 4070–4073; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12b. Powell N. H., Clarkson G. J., Notman R., Raubo P., Martin N. G., Shipman M., Chem. Commun. 2014, 50, 8797–8800; [DOI] [PubMed] [Google Scholar]
- 12c. Lopez-Ortega B., Jenkinson S. F., Claridge T. D. W., Fleet G. W. J., Tetrahedron: Asymmetry 2008, 19, 976–983. [Google Scholar]
- 13.For selected applications, see:
- 13a. Stepan A. F., Karki K., McDonald W. S., Dorff P. H., Dutra J. K., DiRico K. J., Won A., Subramanyam C., Efremov I. V., O'Donnell C. J., Nolan C. E., Becker S. L., Pustilnik L. R., Sneed B., Sun H., Lu Y., Robshaw A. E., Riddell D., O'Sullivan T. J., Sibley E., Capetta S., Atchison K., Hallgren A. J., Miller E., Wood A., Obach R. S., J. Med. Chem. 2011, 54, 7772–7783; [DOI] [PubMed] [Google Scholar]
- 13b. Dowling J. E., Alimzhanov M., Bao L., Block M. H., Chuaqui C., Cooke E. L., Denz C. R., Hird A., Huang S., Larsen N. A., Peng B., Pontz T. W., Rivard-Costa C., Carlos Saeh J., Thakur K., Ye Q., Zhang T., Lyne P. D., ACS Med. Chem. Lett. 2013, 4, 800–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.
- 14a. Davis O. A., Bull J. A., Angew. Chem. Int. Ed. 2014, 53, 14230–14234; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 14454–14458; [Google Scholar]
- 14b. Morgan K. F., Hollingsworth I. A., Bull J. A., Chem. Commun. 2014, 50, 5203–5205; [DOI] [PubMed] [Google Scholar]
- 14c. Davis O. A., Croft R. A., Bull J. A., Chem. Commun. 2015, 51, 15446–15449; [DOI] [PubMed] [Google Scholar]
- 14d. Morgan K. F., Hollingsworth I. A., Bull J. A., Org. Biomol. Chem. 2015, 13, 5265–5272; [DOI] [PubMed] [Google Scholar]
- 14e. Morgan K. F., Doran R., Croft R. A., Hollingsworth I. A., Bull J. A., Synlett 2016, 27, 106–110. [Google Scholar]
- 15.One example occurs in a patent, formed in a 6 % yield: B. L. Hodous, J. L. Kim, K. J. Wilson, D. Wilson, Y. Zhang, Compositions useful for treating disorders related to KIT. US20150111887A1, 2015.
- 16.There are limited examples of tetrasubstituted 3,3-diaryloxetanes through photochemical Paternò-Büchi reactions. See:
- 16a. Kubo Y., Tojo S., Suto M., Toda R., Araki T., Chem. Lett. 1984, 13, 2075–2078; [Google Scholar]
- 16b. Kubo Y., Suto M., Araki T., Mazzocchi P. H., Klingler L., Shook D., Somich C., J. Org. Chem. 1986, 51, 4404–4411; [Google Scholar]
- 16c. Aoyama H., Hatori H., Omote Y., J. Org. Chem. 1989, 54,, 2359–2364; [Google Scholar]
- 16d. Xue J., Zhang Y., Wu T., Fun H.-K., Xu J.-H., J. Chem. Soc. Perkin Trans. 1 2001, 183–191; [Google Scholar]
- 16e. Matsumura K., Mori T., Inoue Y., J. Am. Chem. Soc. 2009, 131, 17076–17077; [DOI] [PubMed] [Google Scholar]
- 16f. Matsumura K., Mori T., Inoue Y., J. Org. Chem. 2010, 75, 5461–5469. [DOI] [PubMed] [Google Scholar]
- 17.For examples of structurally distinct products from divergent reactions
- 17a. Cahard E., Male H. P. J., Tissot M., Gaunt M. J., J. Am. Chem. Soc. 2015, 137, 7986–7989; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17b. Guo C., Fleige M., Janssen-Müller D., Daniliuc C. G., Glorius F., Nat. Chem. 2015, 7, 842–847; [DOI] [PubMed] [Google Scholar]
- 17c. Webster R., Böing C., Lautens M., J. Am. Chem. Soc. 2009, 131, 444–445; [DOI] [PubMed] [Google Scholar]
- 17d. Liddon J. T. R., James M. J., Clarke A. K., O'Brien P., Taylor R. J. K., Unsworth W. P., Chem. Eur. J. 2016, 22, 8777–8780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. West R., Powell D. L., Lee M. K. T., Whatley L. S., J. Am. Chem. Soc. 1964, 86, 3227–3229. [Google Scholar]
- 19. Kudo H., Nishikubo T., J. Polym. Sci. Part A 2007, 45, 709–726. [Google Scholar]
- 20.
- 20a. Chen L., Yin X.-P., Wang C.-H., Zhou J., Org. Biomol. Chem. 2014, 12, 6033–6048; [DOI] [PubMed] [Google Scholar]
- 20b. Dryzhakov M., Richmond E., Moran J., Synthesis 2016, 48, 935–959. [Google Scholar]
- 21. Niggemann M., Meel M. J., Angew. Chem. Int. Ed. 2010, 49, 3684–3687; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 3767–3771. [Google Scholar]
- 22.See Supporting Information for further detail and discussion on reaction development and optimization.
- 23.For reviews:
- 23a. Sheppard T. D., J. Chem. Res. 2011, 35, 377–385; [Google Scholar]
- 23b. Bertolini F., Pineschi M., Org. Prep. Proced. Int. 2009, 41, 385–418. [Google Scholar]
- 24.
- 24a. Duan J. A., Wang L., Qian S., Su S., Tang Y., Arch. Pharmacal Res. 2008, 31, 965–969; [DOI] [PubMed] [Google Scholar]
- 24b. Huang H. Y., Ishikawa T., Peng C. F., Tsai I. L., Chen I. S., J. Nat. Prod. 2008, 71, 1146–1151; [DOI] [PubMed] [Google Scholar]
- 24c. Tripathi R. P., Yadav A. K., Ajay A., Bisht S. S., Chaturvedi V., Sinha S. K., Eur. J. Med. Chem. 2010, 45, 142–148; [DOI] [PubMed] [Google Scholar]
- 24d. Zhang H., Qiu S., Tamez P., Tan G. T., Aydogmus Z., Van Hung N., Cuong N. M., Angerhofer C., Doel Soejarto D., Pezzuto J. M., Fong H. H. S., Pharm. Biol. 2002, 40, 221–224; [Google Scholar]
- 24e. Huang Z., Cui Q., Xiong L., Wang Z., Wang K., Zhao Q., Fuchun B., Wang Q., J. Agric. Food Chem. 2009, 57, 2447–2456. [DOI] [PubMed] [Google Scholar]
- 25.
- 25a. Malapit C. A., Howell A. R., J. Org. Chem. 2015, 80, 8489–8495; [DOI] [PubMed] [Google Scholar]
- 25b. Wang Z., Chen Z., Sun J., Org. Biomol. Chem. 2014, 12, 6028–6032. [DOI] [PubMed] [Google Scholar]
- 26.
- 26a. Loy R. N., Jacobsen E. N., J. Am. Chem. Soc. 2009, 131, 2786–2787; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26b. Boxall R., Grainger R., Aricò C., Ferris L., Synlett 2008, 25–28. [Google Scholar]
- 27.For the only related example in the synthesis of diazonamide A see: Nicolaou K. C., Huang X., Giuseppone N., Rao P. B., Bella M., Reddy M. V., Snyder S. A., Angew. Chem. Int. Ed. 2001, 40, 4705–4709; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2001, 113, 4841–4845. [Google Scholar]
- 28.
- 28a. Ma D., Cai Q., Org. Lett. 2003, 5, 3799–3802; [DOI] [PubMed] [Google Scholar]
- 28b. Maiti D., Buchwald S. L., J. Org. Chem. 2010, 75, 1791–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cacchi S., Ciattini P. G., Morera E., Ortar G., Tetrahedron Lett. 1986, 27, 5541–5544. [Google Scholar]
- 30. Wolfe J. P., Tomori H., Sadigho J. P., Yin J., Buchwald S. L., J. Org. Chem. 2000, 65, 1158–1174. [DOI] [PubMed] [Google Scholar]
- 31. Nowinski J. L., Lightfoot P., Bruce P. G., J. Mater. Chem. 1994, 4, 1579–1580. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary